

SUMMARY
Dysregulated axonal trafficking of mitochondria is linked to neurodegenerative disorders. We report a high-content screen for small-molecule regulators of the axonal transport of mitochondria. Six compounds enhanced mitochondrial transport in the sub-micromolar range, acting via three cellular targets: F-actin, Tripeptidyl peptidase 1 (TPP1), or Aurora Kinase B (AurKB). Pharmacological inhibition or small hairpin RNA (shRNA) knockdown of each target promotes mitochondrial axonal transport in rat hippocampal neurons and induced pluripotent stem cell (iPSC)-derived human cortical neurons and enhances mitochondrial transport in iPSC-derived motor neurons from an amyotrophic lateral sclerosis (ALS) patient bearing one copy of SOD1A4V mutation. Our work identifies druggable regulators of axonal transport of mitochondria, provides broadly applicable methods for similar image-based screens, and suggests that restoration of proper axonal trafficking of mitochondria can be achieved in human ALS neurons.
Graphical Abstract
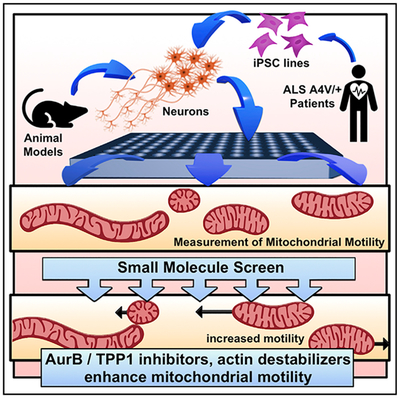
In Brief
Shlevkov et al. establish a high-content screen for enhancers of axonal mitochondrial trafficking. Identified compounds act through three cellular targets: F-Actin, Tripeptidyl peptidase 1, and Aurora Kinase B. Motor neurons derived from a SOD1+/A4VALS patient have decreased mitochondrial motility, which can be reversed by inhibitors of these targets.
INTRODUCTION
The extraordinary architecture of neurons, with vast arbors of axons and dendrites, represents a challenge for the adequate supply and proper distribution of mitochondria. Axonal trafficking of mitochondria ensures that the neuron matches its needs for local energy production and calcium buffering capacity to appropriate spatial locations throughout this extended architecture. Axonal trafficking also plays a role in mitochondrial quality control by allowing newly synthesized mitochondria to replace or fuse with stationary, older mitochondria within the arbor, including those that are resident at synapses (Misgeld and Schwarz, 2017; Saxton and Hollenbeck, 2012).
Defective axonal transport of mitochondria has been linked to several neurodegenerative disorders in animal and cellular models, including, but not limited to, multiple sclerosis (Joshi et al., 2015; Sorbara et al., 2014), amyotrophic lateral sclerosis (ALS) (Kiskinis et al., 2014), Charcot-Marie-Tooth type 2 disease (Misko et al., 2010), and Parkinson’s disease (Hsieh et al., 2016; Wang et al., 2011; Weihofen et al., 2009). Defective axonal trafficking of mitochondria can impair mitochondrial function, produce energetic deficits, and lead to loss of appropriate synaptic signaling and neuronal degeneration (Guo et al., 2005; López-Doménech et al., 2018; Russo et al., 2009; Stowers et al., 2002). Therefore, enhancing mitochondrial transport may be a rational therapeutic strategy with potentially broad therapeutic impact. However, testing this hypothesis is hindered at present by our limited understanding of the signaling pathways that regulate the axonal transport of mitochondria as well as the lack of pharmacological tools for their manipulation.
Mitochondria are trafficked in animal cells by the Miro-milton motor-adaptor complex (Glater et al., 2006; Guo et al., 2005; Russo et al., 2009; Stowers et al., 2002). Miro (RhotT1/2) is a mitochondrial GTPase on the outer membrane that interacts with the adaptor Milton (Trak1/2) (Glater et al., 2006). Milton recruits kinesin heavy chain (Kif5b) and the dynein-dynactin complexes to mitochondria (Glater et al., 2006). The Miro-milton complex receives inputs from signaling pathways that adjust mitochondrial transport to cellular needs. Such signals include Ca2+ (Saotome et al., 2008; Wang and Schwarz, 2009b; Yi et al., 2004), hypoxia (Li et al., 2009), mitochondrial damage (Liu et al., 2011; Shlevkov et al., 2016; Wang et al., 2011), the glucose/O-GlcNAc Transferase pathway (Cheung et al., 2008; Iyer and Hart, 2003; Pekkurnaz et al., 2014), and Armcx (López-Doménech et al., 2012). Mitochondrial motility is further refined by the abundance or localization of anchoring proteins, which can be either actin-myosin based (Chada and Hollenbeck, 2003, 2004; Pathak et al., 2010) or microtubule-syntaphilin based (Courchet et al., 2013; Kang et al., 2008). The interplay of motors and anchors creates two pools of neuronal mitochondria: a predominantly motile pool that moves either anterograde or retrograde and a long-term stationary pool (Misgeld and Schwarz, 2017).
To explore the pathways that regulate axonal transport of mitochondria and to identify potentially druggable targets, we developed a high-content screening assay based on time-lapse imaging of mitochondria. We used this platform to perform an unbiased compound screen using a “mechanism of action” library. We found enhancers and suppressors of axonal trafficking of mitochondria and proceeded to identify relevant cellular targets of our enhancer molecules using both rodent and human neurons. Finally, we also show that manipulating these targets can reverse defective mitochondrial trafficking in motor neurons generated from a patient-derived induced pluripotent stem cell (iPSC) line carrying an SOD1+/A4V ALS mutation.
RESULTS
PATHS: Particle Analysis and Tracking for High-Throughput Screening
Analysis of axonal transport typically involves constructing kymographs, two-dimensional representations of movement, where the location of a mitochondrion (or other particle) is coded along the x axis and the time is represented along the y axis (Wang and Schwarz, 2009a). While kymographs provide rich information about the motile behavior of individual particles, their analysis is laborious, time consuming, restricted to individual axons, potentially subject to bias in the selection of axons, and only captures the behavior of tens of mitochondria per axon. Kymography, therefore, cannot be used as a readout in large-scale, unbiased screening; consequently, we set out to develop a particle-tracking method for automated quantitation of axonal transport that would be compatible with high-throughput screening.
Particle tracking is a hard task for automation; challenges include high particle density, particle merging and splitting, and temporal disappearance (Jaqaman and Danuser, 2009). Many of these problems can be addressed by careful optimization of the fluorescent label density and time-lapse frequency, as well as by the choice of the tracking algorithm. The linear assignment problem (LAP) tracking method (Jaqaman et al., 2008) is an algorithm well suited for automated screening. The open-source CellProfiler biological analysis package is one of the few options for combining cellular identification and the ability to process large numbers of time-lapse movies for high-throughput screening (Carpenter et al., 2006) that also incorporates LAP-based tracking. Therefore, we used CellProfiler to develop a pipeline for automated tracking of mitochondria.
We dissociated E18 rat hippocampal neurons and cultured them on 96-well plates for 6–7 days (DIV6-7) before transfecting a subset of the cells (approximately 20%–30%) with MitoDsRed, a red fluorescent protein localized to the mitochondrial matrix (see STAR Methods; Figure S1B). Two days later (DIV8-9), labeled mitochondrial movement was observable (Video S1) and a set of time-lapse series was collected on an automated imaging platform. We used these images to create an analysis pipeline within CellProfiler combined with a custom MATLAB script that we called PATHS (Particle Analysis and Tracking for High-throughput Screening). PATHS starts by identifying mitochondria in each frame of a time-lapse sequence (Figure 1A). In order to focus our analysis exclusively on mitochondria traveling along neurites, we used a size-based mask to identify the small (1–10 μm in size) individual mitochondria that are typically observed in axons and secondary dendrites and to eliminate from the analysis larger mitochondria and mitochondrial networks, which correspond to the highly fused mitochondria of somato-dendritic regions (Chang et al., 2006). The mask also eliminated non-neuronal cells and fluorescent debris (Figure S1A).
Figure 1. High-Content Assay to Screen for Regulators of Axonal Mitochondrial Trafficking.

(A) Rat hippocampal neurons transfected with Mito-DsRed to label mitochondria showing the original channel (left) and the masked channel (right) in which large objects such as somata and cell debris have been eliminated.
(B) Representative image of a portion of the time-lapse series where a mask based on minimum intensity projection is used to identify stationary (outlined in green) mitochondria. These mitochondria are subtracted from the image to facilitate tracking of the motile objects (orange).
(C) Time-lapse sequence of a small portion of an imaged field, with stationary (red) and motile (green) mitochondria identified based on the minimum intensity projection. Objects that exhibit significant movement are marked with an arrow.
(D) Results of tracking of mitochondria in the field shown in (C). A threshold for a minimum amount of movement has been applied to classify “wiggling” mitochondria as stationary, and the ”bona fide” motile mitochondria have been tracked using LAP algorithms (blue lines). Arrows indicated the tracks that represent the motile mitochondria identified in (C). See also Video S2.
(E). Graphic representation, as in (D), of mitochondrial tracks in neurons either treated with DMSO(mock) or 1 μM calcimycin. Calcimycin reduced movement (blue lines) and increased the number of stationary objects (red dots).
(F) Percent motile mitochondria as quantified by the CellProfiler pipeline, represented as Tukey’s box plot, and a heatmap of Z scores that was created using PATHS (see STAR Methods).
(G) Integrated distance of the tracked objects expressed as cumulative distribution plots to compare either two sets of DMSO-treated cultures (top) or DMSO versus calcimycin cultures (bottom). Heat maps are shown for KS values that compare the distributions of integrated distances. The displacement parameter was similarly altered by calcimycin (KS value ~0.3; Z score ~19).
(H, I, and J) Same analyses and representation as in (E), (F), and (G), respectively, but for a comparison between murine SNPH+/+ and SNPH−/− neurons. Two-tailed t test was used to obtain p values in (F) and (I) upon determination of normality on D’Agostino-Pearson test.
The ability to track faithfully the paths of moving mitochondria from frame to frame depends critically on the spacing between objects; the more cluttered the field, the greater the difficulty in tracking (Jaqaman and Danuser, 2009). Because the majority of mitochondria (typically 70%–80%) in rat hippocampal cultures remain stationary for long periods of time (Misgeld and Schwarz, 2017), we found that masking the stationary mitochondria from the images substantially improved the accuracy of tracking the moving mitochondria. For (x,y) locations located within a stationary mitochondrion, the fluorescent intensity values will be consistently high across all frames, whereas for motile mitochondria, the intensity will be transiently high at an (x,y) location as the mitochondria pass through. PATHS takes advantage of this property to sort objects into motile and stationary categories by taking a minimum-intensity projection of the time-lapse series (i.e., the minimum intensity value across the frames at each (x,y) location), such that only the stationary mitochondria remain visible (Figure S1C). Subsequently, PATHS identifies and masks the stationary mitochondria from each frame of the time-lapse series (Figure 1B) and then tracks the motile mitochondria through the decluttered fields of view using the LAP algorithm (Figures 1C and 1D; Video S2).
Our algorithms extract three descriptors of motility: percent motile (the proportion of motile organelles per well), the integrated distance traveled for each motile organelle (the length of travel, or the sum of all movements, including changes in direction), as well as the displacement (absolute distance between the point of initiation of travel and the final destination) for each motile organelle. We evaluated all three parameters using the Z-prime statistic to identify which parameter is most sensitive to changes in mitochondrial motility by known factors that enhance or decrease their axonal transport. To decrease motility as a positive control, we used calcimycin, a calcium ionophore that arrests mitochondria (Wang and Schwarz, 2009a). We seeded 96-well plates with rat hippocampal neurons, transfected them with mito-DSred at DIV07 and incubated 48 of the wells for a brief period (15 min) with 10 mM calcimycin at DIV09. In each well, approximately 40,000 mitochondria were imaged. As expected, PATHS detected fewer mitochondrial tracks in the calcimycin-treated wells (Figure 1E, blue lines) and a reduced percent of motile mitochondria and Z score of percent motile (Figure 1F). Calcimycin treatment, by decreasing the integrated distances and displacement, enhanced Kolmogorov-Smirnov (KS) values and Z scores for both parameters (Figure 1G). We then optimized cell culture conditions, plate type, transfection protocol, control molecules, and assay kinetics with a target of Z-prime factor Z′ > 0.5 (Figure S1B). When we calculated the Z prime for percent motile, the KS of integrated distance, and the KS of displacement, we found that the KSs of integrated distance and displacement were the most sensitive descriptors, superior to percent motile and yielding Z-prime values higher than 0.5, indicating that the ability of these parameters to distinguish treated and untreated wells was sufficiently robust for screening. The ability to monitor and quantify the movement of approximately 40,000 mitochondria was crucial to the sensitivity of the assay.
To test our ability to detect enhanced motility, we used neurons derived from syntaphillin knockout mice (SNPH−/−) (Kang et al., 2008), which show large increases in axonal trafficking of mitochondria relative to control C57Bl6 mice. We seeded 48 wells of a 96-well plate with hippocampal neurons dissected from each of these genotypes, transfected with Mito-DsRed at DIV06, and imaged them at DIV08. We checked plates under a fluorescent microscope before running the assay to ensure transfection and also monitored cell viability by taking a short time-lapse sequence of a ”negative control” well, making sure mitochondria were motile. Upon analysis by our automated pipeline, SNPH−/− neurons showed a significantly higher percentage of motile mitochondria as well as integrated distance and displacement as compared to control SNPH+/+ neurons (Figures 1H–J). Again, the KS integrated distance and displacement were equally sensitive, and both were more sensitive than percent motile. Thus, the platform can identify both enhanced and diminished mitochondrial trafficking along neurites, can detect changes in response to genetic or chemical perturbation, and is robust enough to be used for high-content screening.
Compound Screening Identifies Six Enhancers of Mitochondrial Transport
We screened two bioactive chemical libraries: the Biomol collection (480 compounds) and the Selleck Bioactive collection (2,800 compounds). Both libraries contain well-characterized, potent compounds covering a large collection of druggable targets (see STAR Methods). Each chemical library was screened in two replicates. We dissociated rat hippocampal neurons from E18 embryos, cultured the neurons on 96-well plates, transfected with Mito-DsRed at DIV6-7, and imaged at DIV8-9. At this stage the neurons are still growing, but already have functional synapses (Basarsky et al., 1994; Harrill et al., 2015). Compounds were added at a single concentration (3 μM) 1 h prior to imaging. For imaging, time-lapse series of ~50 frames were acquired with a 20× objective from four different fields in each well of a 96-well plate. With this protocol, each plate was imaged for a total of 2 h (see STAR Methods for details). An average Z score of 3 or higher in both rounds of screening was taken as the threshold for each descriptor to qualify a compound as a hit. We used the KS of integrated distance as the descriptor for Z-score calculations. During the screen, we continued to use calcimycin as a positive control, which consistently yielded an overall Z-prime value of 0.5 or higher. At the end of the assay, the cells were incubated with the mitochondrial potentiometric dye tetramethylrhodamine methyl ester (TMRM). Levels of TMRM fluorescence were used as an orthogonal screen to filter out hits that were decreasing motility via mitochondrial depolarization rather than via regulatory pathways.
The distribution of Z scores, as presented in Figures 2A and 2B, identified 38 suppressors and 8 enhancers of motility. Suppressors covered a variety of chemical classes (Figure S2A), but all but two suppressing compounds either were known to depolymerize microtubules (vincristine and vinblastine) or significantly decreased mitochondrial membrane potential as determined from TMRM intensity (Figure S2B). The latter are likely to suppress motility by depleting axonal ATP or activating the PINK1/Parkin pathway (Wang et al., 2011). The compounds that did not affect TMRM or microtubules were NMS-873, a valosin-containing protein (VCP)/p97 inhibitor (Magnaghi et al., 2013), and NH125, a regulator of eEF-2 kinase (Chen et al., 2011; Devkota et al., 2012) (Figures S2C and S2D). Their mechanism is unclear, as these had not previously been identified as suppressors of mitochondrial transport.
Figure 2. Compound Screen and Identification of Hits.

(A and B) 2D plots where each axis corresponds to one replicate of the library of Z scores (KS of integrated distance) obtained in the screen. The cutoff levels of Z score = 3 are marked as dotted lines. Red box in (A) represents enlarged data in (B). Compounds that enhanced (green) or suppressed (red) transport were defined as hits if their KS of integrated distance was equal to or above Z = 3. Non-hits are gray. Enlargement in (B) shows the location of the hits that increased mitochondrial trafficking. The correlation coefficient for the two replicates was 0.75 (including positive and negative controls and test compounds) or 0.38 (test compounds only).
(C) Z scores for all three parameters for each confirmed enhancer of motility. Red dotted line corresponds to Z score = 3
(D-I) For each confirmed hit, the effect on the percent of motile mitochondria is shown together with a dose-response curve, for which the enhancement of motility was expressed as a percent of maximal enhancement. Panels correspond to Latrunculin B (D), Cytochalasin B (E), Ala-Ala-Phe-CMK (F), BMS-754807 (G), Hesperadin (H), and TAK-901 (I).
Error bars in (C) represent SDs. Data represented in (D)–(I) as Tukey’s box plots as well as normalized dose-response curves, where error bars represent SD. Two-tailed t test was used to obtain p values. n = 3 wells/plate from a minimum of two plates for (D)–(I).
We focused on the compounds that increased motility. We found, and subsequently confirmed by retesting, two actin depolymerizers (Latrunculin B, effective concentration [EC]50 = 800 nM, and Cytochalasin B, EC50 = 300 nM), three kinase inhibitors (BMS-754807, EC50 = 350 nM; Hesperadin, EC50 = 230 nM; and TAK-901, EC50 = 540 nM), and one peptidase inhibitor (Ala-Ala-Phe-chloromethylketone [AAF-CMK], EC50 =1.9 μM) (Figure 2C). Two other initial positives, paclitaxel and the bioactive lipid 24(S)-hydroxycholesterol, failed upon retesting. Dose response curves with the six active enhancers showed that all compounds acted at sub-micromolar concentrations, with the exception of AAF-CMK, which acted at low micromolar concentrations (Figures 2D–2I). F-actin has been observed previously to inhibit axonal transport of mitochondria (Chada and Hollenbeck, 2004) and thus identification of Latrunculin B and Cytochalasin B validated the screen.
TPP1 Is a Negative Regulator of Mitochondrial Motility in Hippocampal Axons
AAF-CMK is a serine protease inhibitor that reversibly inhibits the lysosomal enzyme Tripeptidyl Peptidase 1 (TPP1) (Ezaki et al., 2000) and irreversibly inhibits the post-proteasome exopeptidase tripeptidyl peptidase II (TPPII) (Geier et al., 1999). AAF-CMK also marginally inhibits the chymotrypsin-like activity of the proteasome (Figure 3A) (Johnson et al., 1988). To distinguish among these potential targets, we used available small molecule inhibitors for each and small hairpin RNA (shRNA) against TPP1 and TPP2. We excluded TPP2 as the relevant target because an effective TPP2 shRNA (Figure S3A) neither enhanced mitochondrial transport nor occluded the effect of AAF-CMK (Figures 3B and 3C). Furthermore, butabindide, a potent inhibitor of TPP2 with a Ki = 7 nM (Rose et al., 1996), failed to mimic the efficacy of AAF-CMK (Figure 3D). We similarly ruled out the contribution of the proteasome because inhibitors of the chymotrypsin-like activity of the proteasome such as carfilzomib (Figure 3E) did not enhance mitochondrial motility. In contrast, two shRNA against TPP1 (Figure S3B) potently enhanced mitochondrial transport and occluded any further effect of AAF-CMK (Figures 3F and 3G). Conversely, overexpression of TPP1 suppressed mitochondrial movement (Figure 3H). Thus, the lysosomal enzyme TPP1 is a negative regulator of mitochondrial trafficking, although the mechanism by which it influences mitochondria remains unclear.
Figure 3. TPP1 Is the Relevant Substrate of AAF-CMK.

(A) Known cellular targets of AAF-CMK.
(B and C) Z score of percent motile (B) and KS integrated distance (C) in neurons transfected for at least 3 days with either scrambled or TPP2 shRNA (TPP2-i) and treated with 10 μM AAF-CMK 1 h prior to and during time-lapse imaging. TPP2-i neither mimicked nor occluded the effect of AAF-CMK.
(D and E) Lack of effect of 2-h incubation with TPP2 inhibitor butabindide at 10 μM (D) and proteasome inhibitor carfilzomib at 5 μM (E) on percent motile and lack of dose-dependent response.
(F and G) Z scores for percent motile (F) and KS integrated distance (Id) (G) in wells transduced either with scrambled shRNA or with TPP1 shRNAs and treated with either solvent or 10 μM AAF-CMK for 2 h before time-lapse imaging. Two non-overlapping sequences targeting TPP1 were used (TPP1-i 1 and 2) (Z scores of KS displacement were similar).
(H) Effect of TPP1 over expression (OE) on the three motility descriptors.
At least 4 wells per condition (each well representing the traces collected from more than 10,000 mitochondria) were used for the calculation of Z scores. Statistical significance between groups of Z scores was determined using an unpaired two-tailed t test. ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns = p > 0.05. Data in all panels represented as Tukey’s box plots.
Aurora B Regulates Mitochondrial Motility in Hippocampal Axons
The three kinase inhibitors (BMS-754807, Hesperadin, and TAK-901) share targets within the Aurora kinase family (Aurora A, B, and C). BMS-754807 was originally developed as an insulin-like growth factor (IGFR) inhibitor with an inhibitory concentration (IC)50 of 1.8 nM/1.7 nM in cell free assays, although it can inhibit other kinases, including Aurora Kinase A (AurKA) with IC50 = 9 nM and Aurora Kinase B (AurKB) with an IC50 = 25 nM (Carboni et al., 2009). Two selective inhibitors of IGFR (Picopodophyllin and OSI-906) were included in the screen but did not enhance mitochondrial motility. Hesperadin inhibits Aurora Kinase B with IC50 = 250 nM in a cell-free assay (Hauf et al., 2003). TAK-901 inhibits Aurora Kinase A with IC50 = 21 nM and Aurora Kinase B with IC50 = 15 nM. We therefore focused on the Aurora family as the likely relevant cellular substrate for these hits. To test this hypothesis, we first re-examined our initial screen to determine the effects of all the Aurora family inhibitors that had been present in our libraries. Three additional compounds, Danusertib, CYC116, and AMG-900 (Figures S4A and S4B), had enhanced mitochondrial transport in both replicates although they had not met our original selection criteria because in at least one of the replicates the enhancement of motility did not reach the threshold of Z score = 3. All 6 positives were either pan-Aurora inhibitors or Aurora Kinase B inhibitors. Aurora Kinase C is expressed primarily in the germline and so we used shRNAs against Aurora Kinases A and B (Figure S4C) to determine the relevant cellular target. Knockdown of Aurora Kinase B, but not of Aurora Kinase A, both mimicked and occluded the effect of the Aurora Kinase B inhibitor Hesperadin on percent motile (Figures 4A–4D) and integrated distance (Figures 4E–4G). Furthermore, Barasertib, a selective Aurora Kinase B inhibitor with IC50 = 0.37 nM in a cell-free assay and ~3,700-fold more selective for Aurora Kinase B over Aurora Kinase A (Yang et al., 2007) also enhanced mitochondrial transport in a dose-dependent manner (Figure 4H). In contrast, the more selective Aurora Kinase A inhibitors in the compound screen did not enhance mitochondrial motility (Figures S4A and S4B). The effect of Aurora Kinase B knockdown on mitochondrial transport was reversed by overexpression of an shRNA-resistant Aurora Kinase B construct, but not by the kinase-dead form of Aurora Kinase B (Figure 4I). Conversely, Aurora Kinase B overexpression suppressed mitochondrial transport (Figures 4J–4L). Thus, Aurora Kinase B is a negative regulator of mitochondrial transport in hippocampal neurons.
Figure 4. Aurora B is the Target of the Kinase Inhibitor Class of Hits.

(A) Z score of percent motile in neurons transfected with either scrambled or Aurora Kinase B shRNA and treated with 3 μM Hesperadin 1 h prior to and during time-lapse imaging.
(B) Z score of percent motile in neurons transfected with either scrambled or Aurora Kinase A shRNA and treated with 3 μM Hesperadin 1 h prior to and during time-lapse imaging.
(C) Representative image of mitochondrial traces in a field of neurons either transfected with scrambled shRNA or with Aurora Kinase B shRNA.
(D) Percent of motile mitochondria per well in neurons expressing either scrambled shRNA or shRNA against Aurora Kinase B.
(E and F) Z scores of KS-integrated distance of neurons treated as in (A) and (B), respectively.
(G) Integrated distance of mitochondrial traces in a representative well either transfected with scrambled shRNA or shRNA against Aurora Kinase B represented as a histogram and as a cumulative distribution. Each dot in the histogram corresponds to a single mitochondrial trajectory.
(H) Dose-response curve of the effect of Barastertib on percent motility; values were normalized to the maximum percentage motility at 2 μM Barastertib.
(I) Z score of percent motile of rat AurKB-i in combination with either wild-type (WT) or a kinase dead form of the RNAi-resistant human Aurora Kinase B.
(J) Representative images of mitochondrial traces in a field of neurons either transfected with empty plasmid or with a plasmid expressing rat Aurora Kinase B.
(K and L) Effect of Aurora Kinase B overexpression on percent motility (K) and integrated distance traveled (L).
At least 4 wells per condition (each well containing traces collected from over 10,000 mitochondria on average) were used for the calculation of Z scores. Statistical significance between groups of Z scores was determined using an unpaired two-tailed t test. ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns = p > 0.05. Data in (A), (B), (D)–(F), (I), and (K) represented as Tukey’s box plots. Data in (G) and (L) represented as dot-plots and cumulative distributions of integrated distance, and p values are given for Mann-Whitney test. Dispersion bars in (H) represent SD.
Because PATHS cannot unambiguously differentiate between axonal and dendritic mitochondria, nor between anterograde and retrograde movement, we tested the effects of Aurora Kinase B inhibition using traditional kymography in axons (Figure 5A). Hesperadin, a selective Aurora Kinase B inhibitor, was added 1 h before acquiring 5-min videomicroscopy series from which kymographs were constructed. Hesperadin enhanced the percent of axonal mitochondria that were motile as well as the average time spent in retrograde motion (Figures 5B–5D). No changes in anterograde motility or mitochondrial density were observed (Figure 5E). Neurons transfected with shRNA against Aurora Kinase B also showed an increased percent motile and time mitochondria spent in motion (Figures 5F and 5G); however, this long-term reduction in Aurora Kinase B enhanced both anterograde and retrograde movements (Figures 5H and 5I) and reduced the density of stationary mitochondria (Figure 5J). Neither Hesperadin nor knockdown of Aurora Kinase B altered the motility of late endosomes as determined by the tracking of late endosomes (Figures S5A–S5D).
Figure 5. Kymography of Aurora Kinase B Inhibition of Axonal Mitochondrial Transport.

(A) Representative kymographs of segments of an axon expressing Mito-DsRed treated with 3 μM Hesperadin or DMSO for 1 h prior to and during the imaging. Mitochondrial trajectories during the time-lapse movie (~3 min) correspond to white lines with location represented along the x axis and time proceeding downward along the y axis. Diagonal lines indicate moving mitochondria, with retrograde movement to the left and anterograde movement to the right.
(B) Hesperadin effect on percent of motile mitochondria per segment of axon as well as the average time a mitochondrion spends in motion represented as histograms as well as cumulative distribution curves.
(C and D) Effects of Hesperadin on the direction of mitochondrial movement expressed as percent time in motion (C) and overall percent of events (D).
(E) Density of all mitochondria (motile+stationary) per μM of axon as well as density of only stationary mitochondria per μM of axon.
(F–J) Neurons transfected with either scrambled shRNA or shRNA against Aurora Kinase B analyzed as in (A)–(E).
(F) Representative kymographs of segments of an axon expressing Mito-DsRed and Aurora Kinase B shRNA or scrambled shRNA.
(G) Effect of shRNA against Aurora Kinase B on percent motile mitochondria per segment of axon, as well as the average time a mitochondrion spends in motion, represented as histograms as well as cumulative distribution curves.
(H and I) Effect of shRNA on the direction of mitochondrial movement expressed as percent time in motion (H) and overall percent of events (I).
(J) Density of all mitochondria (motile+stationary) per μM of axon as well as density of only stationary mitochondria per μM of axon.
At least 100 mitochondria from at least 9 axons coming from three independent transfections were used for all quantifications. D, DMSO; H, Hesperadin; s, scrambled; and i, shRNA against Aurora Kinase B. Scale bars, 10 μM. Data in (B) (percent motile) and (E) and (G) (percent motile) represented as Tukey’s box plots. Dispersion bars in (B) (time spent in motion) and (G) (time spent in motion) represent SD. Mann-Whitney test was used to obtain p values upon determination of normality on D’Agostino-Pearson test.
Hits Enhance Mitochondrial Transport in Human Cortical Neurons and Alleviate Mitochondrial Transport Defects in ALS Motor Neurons
We next asked if the screen was able to identify mechanisms relevant to axonal transport in human neurons. We first tested whether the pathways identified in our screen were conserved in neurons differentiated from iPSCs with a protocol that generates ~90% pure cortical neurons (Zhang et al., 2013; Figure 6A). Neurons were transduced with lentivirus encoding Mito-DsRed, and treated with a representative compound of each class of hit (Latrunculin, Hesperadin, and AAF-CMK) before automated imaging and analysis with PATHS. All three compounds significantly enhanced mitochondrial transport (Figures 6B and 6C). Subsequent kymography of axons revealed that inhibition of Aurora Kinase B increased both anterograde and retrograde mitochondrial motility (Figures 6D and 6E). Thus, the three classes of hits from our screen (inhibitors of F-actin, TPP1, and Aurora Kinase B) appear to act on mitochondrial motility through mechanisms that are conserved between rat hippocampal and human cortical neurons.
Figure 6. Activity of Hits in Human iPSC-Derived Neurons.

(A) Representative images of control iPSCs differentiated by neurogenin2-induction into cortical neurons. Nuclei were labeled with Hoechst 33258. GFP expression is driven by viral overexpression alongside the neurogenin2 vector. Neurons were labeled with β3-tubulin (red) to label all somata and neurites. Scale bar, 400 μm.
(B and C) Z scores of percent motile (B) and KS values of integrated distance (C) for mitochondrial transport in human cortical neurons transduced with CMV-Mito-DsRed and treated with 3 μM Hesperadin, 1 μM Latrunculin A, and 10 μM AAF-CMK for 1 h before and during time-lapse imaging.
(D and E) Kymographs were used to establish the average time a mitochondrion spends in motion (D) as well as its direction (E) in human cortical neurons transduced with CMV-Mito-DsRed and treated with either DMSO or Hesperadin.
(F) Generation and differentiation of human motor neurons from induced pluripotent stem cells (iPSCs) derived from an ALS SOD1+/A4V patient and from isogenic TALEN-corrected cells. HB9-GFP positive motor neurons were isolated by flow cytometry and co-cultured with mouse primary glial cells for 14 days. These cells maintained their motor neuron identity as indicated by Isl1 expression. Scale bar, 100 μm.
(G and H) Z scores of percent of motile mitochondria (G) and KS value of integrated distance (H) in +/A4V neurons and +/+ isogenic control motor neurons.
(I) Effect of Hesperadin, Latrunculin, and AAF-CMK on percent of motile mitochondria in +/A4V motor neurons.
(J and K) Average time a mitochondrion spends in motion in +/A4V neurons treated with DMSO or with 3 μM Hesperadin represented as histograms (J) or as cumulative distribution plots (K).
Dispersion bars in (B)–(E), (I), and (J) represent SD. Data in (G) and (H) are represented as Tukey’s box plots. Mann-Whitney test was used to obtain p values upon determination of normality on D’Agostino-Pearson test.
Previous studies have demonstrated that the dominant ALS mutation SOD1A4V reduces mitochondrial transport in motor-neuron axons (Kiskinis et al., 2014). To determine whether the hit compounds have the potential to restore defective mitochondrial trafficking in diseased neurons, we examined transport in human motor neurons derived from an iPSC line from a SOD1+/A4V patient (Li et al., 2009). We differentiated motor neurons from the SOD1+/A4V line as well as from the transcription activator-like effector nuclease (TALEN)-corrected isogenic control, transduced neurons with a lentivirus coding for mito-DSred, and performed imaging on our platform followed by PATHS analysis (Figure 6F). Our automated analysis confirmed the defect in mitochondrial motility that had previously been described in these cells by kymographic analysis (Kiskinis et al., 2014) (Figures 6G and 6H). Treatment with latrunculin, Hesperadin, and AAF-CMK each increased mitochondrial motility in the +/A4V neurons (Figure 6I). A more detailed analysis by kymography found that Aurora Kinase B inhibition with Hesperadin restored mitochondrial transport back to the levels of the TALEN-corrected control (Figures 6J and 6K).
DISCUSSION
We performed a screen to identify druggable regulators of axonal transport and thereby uncover cellular targets that influence mitochondrial movement. We show that (1) axonal transport can be used as an endpoint in an image-based screening campaign; (2) three classes of compounds and their targets can enhance mitochondrial transport in primary neurons; and (3) the targets of active compounds are functionally conserved in human neurons and can be targeted to reverse mitochondrial transport defects in ALS motor neurons.
The development of PATHS, which combines a tracking algorithm and data analysis package, enabled exploration of the druggable space for regulators of mitochondrial transport. This method allowed precise quantitative description of motility parameters for tens of thousands of individual mitochondria per condition, a scale that is orders of magnitude higher in sampling power than kymography can achieve. Kymography serves as a complementary secondary assay because, although limited in throughput, it offers information on the direction of transport, and can unambiguously identify specifically axonal regulators because mitochondria traveling along secondary dendrites are excluded in the analysis.
The results of our screen validate the approach. As expected, mitochondrial motility was arrested by compounds that depolarized mitochondria and by vincristine and vinblastine, which disrupt microtubules (Jordan and Wilson, 2004). F-actin is known to inhibit mitochondrial trafficking (Chada and Hollenbeck, 2003; Kruppa et al., 2018; Pathak et al., 2010) and we identified as enhancers of motility two compounds that destabilize the F-actin cytoskeleton: latrunculin, which sequesters globular monomeric actin, and cytochalasin, which blocks the addition of G-actin to the growing end of the microfilament. An attractive hypothesis is that mitochondria use F-actin as a structural scaffold onto which they can anchor in response to particular stop signals. As appropriate for such a function, F-actin is found in periodic rings along the axonal shaft (Xu et al., 2013) and is highly enriched at nascent presynaptic sites (Spence and Soderling, 2015). It is important to note, however, that in our study, as in previous studies, the effect is subtle: it does not mobilize the majority of the stationary mitochondria and much remains to be learned about the relative importance and interplay of different mechanisms that arrest or anchor mitochondria. Indeed, none of the compounds found in the screen alter mitochondrial motility as dramatically as the knockout of syntaphilin (Kang et al., 2008), a major mitochondrial anchor. This likely reflects that there are many mechanisms working in parallel to govern the balance between stationary and motile mitochondria. These pathways will include F-actin, myosins, Ca2+, glucose availability, syntaphilin, PINK1, and more. A drug that impacts a single pathway among the many involved is likely to cause only a modest change in the percent motile. The pathways we found are thus likely to modulate mitochondrial motility without fundamentally disrupting the key mechanisms of motility and anchoring.
The screening methods described here can have applications beyond the particulars of our screen. Although we chose to meet the need for large numbers of neurons of consistent quality by screening on rodent hippocampal neurons, we confirmed that our screen identified hits and druggable targets that are also effective on human neurons (Figure 6). The screening method is applicable to genetic screens as well as the acute drug effects we assayed. PATHS would, for example, be very suitable for using axonal transport as an endpoint in a CRISPR-based genetic screen in which iPSC-progenitor lines are mutated and then differentiated to neurons prior to screening. In addition, the algorithms are applicable to cargos other than mitochondria, as long as the experiment is designed with attention to object density and speed so as to allow robust automated tracking (Jaqaman and Danuser, 2009). The screen is also applicable to libraries larger than those we tested. However, because the screen involves taking thousands of short time-lapse images from each well, escalation to a larger plate size would need to take into account the delay between the moment a compound is added and the moment where mitochondria are imaged. Larger compound screens would thus require careful design of drug dosage and duration of treatment. Also, because the overall level of motility declines as the population of synaptically anchored mitochondria increases (Lewis et al., 2016), additional optimization of PATHS would be required to image mitochondrial motility in more mature cultures.
Although we focused on enhancers of motility, two compounds arrested mitochondria without affecting TMRM intensity and these may point to regulatory pathways of mitochondrial motility: NMS-873 is a VCP/p97 inhibitor (Magnaghi et al., 2013) and NH-125 is a putative regulator of eEF2 kinase (Chen et al., 2011; Devkota et al., 2012). VCP/p97 is implicated in mitochondrial clearance by the PINK1/Parkin pathway and in removal of mitofusin (Tanaka et al., 2010), but no mechanistic connection of either NMS-873 or NH125 to mitochondrial transport has not been reported at this time.
TPP1 inhibition by AAF-CMK or knockdown by RNAi enhanced mitochondrial motility. TPP1 is a lysosomal enzyme, and loss of function mutations in the gene are causally linked to a familial lysosomal storage disorder (Ezaki et al., 2000). How TPP1 regulates mitochondrial transport remains unclear but lysosomal dysfunction and mitochondrial transport share linkages to Parkinson’s disease (Gegg and Schapira, 2018; Hsieh et al., 2016; Shlevkov et al., 2016). The pathological consequences of loss of TPP1 activity suggest that it is not itself an attractive therapeutic target in ALS, but further elucidation of the pathway by which it regulates mitochondrial motility might identify targets downstream that did not cause lysosomal storage defects.
By both pharmacologic and knockdown methods, we found that Aurora Kinase B is a regulator of mitochondrial transport. Aurora Kinase B has a well-characterized role in orchestrating cell division and cytokinesis, but the role of Aurora Kinase B in differentiated cells is largely unexplored. Recently, Aurora Kinase B has been found to be expressed in post-mitotic neurons of the zebrafish brain and spinal cord, where it regulates axonal outgrowth and regeneration (Gwee et al., 2018). Aurora Kinase B is significantly upregulated upon axotomy (Ng et al., 2012) and may mediate regenerative sprouting, and yet, paradoxically, axon regeneration involves enhanced mitochondrial trafficking (Cartoni et al., 2016, 2017; Sekine et al., 2018) and yet Aurora Kinase B decreased mitochondrial traffic in our study. An intriguing possibility is that Aurora Kinase B participates in a pathway that positions stationary mitochondria at critical places such as branch points, nascent synapses, and growth cones (Courchet et al., 2013). As such, it could act in regenerating axons to capture the increased axonal flux of mitochondria at necessary junctures. Acute pharmacological inhibition of Aurora Kinase B preferentially stimulated retrograde transport (Figures 5C and 5D), while sustained inhibition with shRNA promoted both directions (Figures 5J and 5K). Therefore, it is possible that AuKB has a preferred substrate in the retrograde motor and a less direct, or possibly compensatory, effect on anterograde movement. Thus, Aurora Kinase B may promote axon regeneration by biasing transport down the axon via suppression of retrograde movement. Aurora Kinase A has been shown to disrupt the association of kinesin with mitochondria in dividing cells (Chung et al., 2016), but the mechanism by which Aurora Kinase B regulates mitochondrial trafficking is presently unclear.
Extensive evidence links neurodegeneration to defects in mitochondrial transport, including the evidence presented here for the SOD1A4V/+ neurons. An appealing hypothesis, therefore, is that improving mitochondrial transport and thereby improving the energy supply and Ca2+-buffering capacity of axons may prolong the life and function of affected neurons and ameliorate progression of the disease. The particular targets identified here, however, may not be ideal therapeutics: TPP1 inhibition is likely to cause deleterious side effects (Ezaki et al., 2000) and Aurora Kinase B inhibitors, though investigated for chemotherapeutic purposes, have counterindicating toxicities due to their effects on dividing cells. The success of the screen in identifying pathways and in demonstrating that hit compounds can be effective in improving transport in human neurons and in a pathogenic genetic background indicates that a broader application of the screening protocol may be a productive line of investigation. The arrest of mitochondria at important sites, such as synapses, is likely to be as important for neuronal function as sufficient levels of trafficking. A drastic increase in mitochondrial motility, as in the case of syntaphilin knockout mice, might therefore diminish mitochondrial supply at crucial sites and this may explain the failure of syntaphilin knockout to be effective in a mouse model of ALS (Zhu and Sheng, 2011). The ability of the hits identified here to restore normal levels of transport rather than to mobilize most axonal mitochondria (Zhu and Sheng, 2011) may be a valuable feature of a pharmacological approach. Nevertheless, there is not, as yet, a critical test of the hypothesis that improving mitochondrial transport can extend neuronal survival in ALS and therefore the therapeutic potential of this approach remains uncertain.
In summary, we developed a unique assay that overcomes the inherent difficulty of tracking motile objects in high throughput. The assay is sensitive and successfully identified regulators of axonal transport of mitochondria. The finding that TPP1 and Aurora Kinase B kinase play a previously unsuspected role in neurons by regulating mitochondrial trafficking has created an opportunity to explore the cell biological mechanisms of these pathways. Finally, because defective axonal transport in ALS motor neurons is restorable upon treatment with the hit compounds here identified, the necessary tools are now available to test the potential of modulating axonal transport as a neuro-protective intervention.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Materials Availability Statement
Plasmids generated in this study are available upon request. Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Thomas L. Schwarz (Thomas.schwarz@childrens.harvard.edu). An MTA agreement prevents us from sharing the Syntaphilin KO mouse (Kang et al., 2008) and the GON0515-03 #5 iPSC line.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Models
Time-pregnant SAS Sprague Dawley rats were obtained from Charles River Laboratories. Syntaphilin KO homozygous mice received from Zu-Hang Sheng (C57BL/BJ background) were crossed to C57BL/BJ mice (#000664, The Jackson Laboratory). Heterozygotes were then crossed to obtain homozygotes littermates. SNPH KO or WT/WT homozygous littermates were intercrossed again and pregnant females were dissected obtain mouse primary hippocampal cultures from embryos. All animal procedures were approved by the Institutional Animal Care Committee at the Boston Children’s Hospital.
Primary Cells, Cell Lines and iPSCs
Primary hippocampal neurons were isolated from E18 embryos according to standard procedures and cultured in Neurobasal Medium (#21103049) supplemented with GIBCO B27 in 5% CO2 at 37C. HEK293T cells were obtained from ATCC and cultured in DMEM+5%FBS. Cells were used between P1-P13. The iPSC line used to generate cortical neurons, GON0515-03 #5, was derived from peripheral blood mononuclear cells of a normal individual using CytoTune-iPS Sendai Reprogramming Kit (ThermoFisher, #A1378001) at the BCH Human Neuron Core. This iPSC line has a normal karyotype, and expresses pluripotency markers, and was negative for mycoplasm (Lonza MycoAlert readings A = 158, B = 77, B/A ratio = 0.487). The iPSC lines 39b SOD+/A4V and 39b SOD+/+, previously described in Wainger et al. (2014) were tested negative for mycoplasma. The lines were maintained at 37°C, 5% CO2 on Matrigel (BD Biosciences) with mTeSR1 media (Stem Cell Technologies). iPSCs were karyotyped before genome engineering. Culture Medium was changed every 24 h and cells were passaged by accutase (Innovative Cell Technologies) as required.
METHOD DETAILS
Plasmids
Mito-DsRed (pDsRed2-Mito) was a gift of G. Hajnoczky. Rab7-GFP was a gift from Richard Pagano (Addgene plasmid # 12605). FLAG-TPP1, human AurKB and rat AurKB were purchased from OriGene (9620 Medical Center Drive, Suite 200, Rockville, MD 20850). CMV-Mito-DsRed and Synapsin-Mito-DsRed expressing lentivirus were produced at the BCH viral Core. Human Aurora B K106R mutant (kinase dead) was synthetized using pCMV-Myc-DDK-AurkB backbone using Q5® Site-Directed Mutagenesis Kit (NEB). shRNA expressing lentivirus and coding plasmids were obtained from Origene. Rat TPP1, TPP2 and AurKB shRNAs were selected for their efficacy based on western blots (Figures S3A, S3B, and S4C). Transfection efficiency of shRNAs was verified by fluorescent microscopy, as the plasmids also express GFP. Co-transfection efficiency with Mito-DsRed was almost 90% in primary neuronal cultures. All plasmids were sequence-verified prior to use. The lentiviral FS_MitoDsRed plasmid was generated from the pLV-MitoDsRed (Addgene #44386) and a homemade FSGW backbone where a human synapsin promoter (GGCCCTGCGTATGAGTGCAAGTGGGTTTTAGGACCAGGATGAGGCGGGGTGGGGGTGCCTACCTGACGACCGACCCCGACCCACTGGACAAGCACCCAACCCCCATTCCCCAAATTGCGCATCCCCTATCAGAGAGGGGGAGGGGAAACAGGATGCGGCGAGGCGCGTGCGCACTGCCAGCTTCAGCACCGCGGACAGTGCCTTCGCCCCCGCCTGGCGGCGCGCGCCACCGCCGCCTCAGCACTGAAGGCGCGCTGACGTCACTCGCCGGTCCCCCGCAAACTCCCCTTCCCGGCCACCTTGGTCGCGTCCGCGCCGCCGCCGGCCCAGCCGGACCGCACCACGCGAGGCGCGAGATAGGGGGGCACGGGCGCGACCATCTGCGCTGCGGCGCCGGCGACTCAGCGCTGCCTCAGTCTGCGGTGGGCAGCGGAGGAGTCGTGTCGTGCCTGAGAGCGCAG) was cloned to the FUGW backbone to replace the original ubiquitin promoter. The MitoDsRed fragment was PCR amplified from pLV-MitoDsRed, and assembled with the FSGW backbone using Gibson cloning (NEB: E2611S). Lentiviral particles expressing the human synapsin promoter driven MitoDsRed reporter were packed and concentrated through Boston Children’s Hospital Viral Core.
Western Blotting
Cells were harvested in lysis buffer (0.05M Tris pH7.5,150mM NaCl, 1% Triton, 0.1mM PMSF, 1:500 Calbiochem protease inhibitor cocktail set III). For quantification, membranes were incubated with fluorescent secondary antibodies and images were acquired on Typhoon laser scanner (GE Healthcare, Wilmington, MA) within the linear range. If not indicated, western blots are representative of at least three independent transfections.
Compound Screen
E18 rat hippocampal neurons were routinely seeded at 30,000 cells/well on Greiner Cellstar 96 well plates, cat# 655090 (4238 Capital Drive, Monroe, NC 28110) coated with 20 μg/mL poly-L-Lysine (Sigma Aldrich) and 3.5 ug/mL laminin (ThermoFisher Scientific) overnight at room temperature, and maintained in Neurobasal medium (ThermoFisher Scientific) supplemented with B27 (GIBCO), L-glutamine, and penicillin/streptomycin. Cells in each well were transfected with 0.3 μg Mito-DSred at DIV6-7 using 0.8 μL Lipofectamine 2000 (ThermoFisher Sientific, Waltham, MA 02451) and imaged at DIV8-9. Small molecule libraries were obtained from ICCB-Longwood (https://iccb.med.harvard.edu/). Compounds were pin-transferred into Greiner 384 well plates (cat#784201) at the ICCB-Longwood facility and immediately added to the cells using Agilent Bravo Liquid Handling Platform (Agilent, 5301 Stevens Creek Blvd, Santa Clara, CA 95051). Cells were incubated with compounds at 3μM for 1 h before and during time-lapse imaging. The plates were dosed and imaged one at a time. Z-scores were calculated for each parameter using the formula Z = (x-μ)/σ. A cutoff of Z-score > 3 in KS integrated distance was used to identify hits. Dose-response curves were performed using either six to eight quarter-log concentration increments with maximum values reaching the concentration used in the screen (3 μM). Concentrations (in nM) were transformed to log nM and values were internally normalized from low to high as percentage activity. Curve fitting was performed in GraphPad Prism using log(agonist) versus normalized response – variable slope –curve fitting. Values presented in Figure 2 were collected from an average of three to six well replicates from one 96-well plate.
Kymography
Rat hippocampal neurons were isolated according to standard procedures and cultured as in Pekkurnaz et al. (2014). Briefly, neurons were obtained from E18 embryos, plated at 150 × 103 on 12-mm glass coverslips (Bellco Glass) or on Greiner Cellstar 96 well plates coated and maintained as above. Neurons were transfected at DIV07-8 and imaged at DIV9-10 at 5% CO2 and 37C using NikonTiE Eclipse 20X. Time-lapse imaging was performed every 2 s for ~300 s. Movies were analyzed using the Kymolyzer macro for ImageJ developed by the laboratory. In all kymographs, anterograde motility is to the right, retrograde motility is to the left.
Immunofluorescence
Neurogenin2-induced cortical neurons were fixed with 4% paraformaldehyde (PFA) by adding 50ul 8% PFA to 50ul media and incubating for 20 min at room temperature. After three washes with 1X PBS, the cells were blocked with blocking reagent (2% Bovine Serum Albumin, 5% normal goat serum, 0.1% Triton X-100 in 1XPBS) for 1 h at room temperature and immunostained with primary antibody overnight at 4C. The next day, cells were washed three times with 1X PBS before staining with secondary antibodies. After a final three washes with 1X PBS, cells were stored in 1X PBS + 0.02% sodium azide.
Time-Lapse High-Content Imaging of Mitochondrial Transport
Neurons were imaged on an ArrayScan XTI imaging platform (ThermoFisher Sientific, Waltham, MA 02451). 30 frames per field were acquired at the maximum speed of 2 Hz. Four fields were imaged per well of a 96-well plate containing 30,000 rat hippocampal neurons/well at DIV08-09 using a 20X objective with 2x2 binning. To minimize well to well differences in exposure to compounds, the fields were imaged according to this sequence: field 1 was imaged in every well before returning to the start of the plate and imaging field 2 in every well, and similarly for fields 3 and 4. Imaging one field per well for 96 wells took ~30 min when acquiring 30 frames at 2Hz in each well. As a result, collecting a series from each of the 4 fields took a total of 2 h per 96-well plate. This protocol was used for the primary screen, compound retesting, and dose response curves in Figure 2, as well as for all experiments in Figures 3, 4, and 6A–6C. During imaging, neurons were maintained in conditioned media at 37C and 5%CO2. In the primary screen the plate was incubated with Tetramethylrhodamine, methyl ester (TMRM, ThermoFisher Scientific, cat#T668), and with Hoechst dye upon completion of the time-lapse sequence and then live-imaged again to determine mitochondrial membrane potential.
Image Analysis
In brief, data are processed by two CellProfiler pipelines: the first pipeline creates the minimum intensity projections of the time-lapse sequences, and the second pipeline employs the projections to create a mask of the stationary objects and performs the tracking of the motile objects using the Linear Assignment Problem method (Jaqaman et al., 2008). The outcome of the Cell Profiler analysis was processed in MATLAB® (MathWorks, 1 Apple Hill Drive, Natick, MA 01760) using a custom script with a graphic user interface that represents data at the field and plate levels, creates heat-maps, and performs statistical computations such as Z-scores and KS statistics. TMRM levels were analyzed using ImageJ. Briefly, TMRM-labeled mitochondria were analyzed from areas of the wells lacking Mito-DsRed due to the low transfection efficiency. Cell bodies were outlined based on the Hoechst signal, and the intensity of TMRM per cell was quantified. 100-150 cells were analyzed per compound.
Cortical Neuron Differentiation
iPSCs were maintained in mTeSR-1 media (STEMCELL Technologies, #85850) on Geltrex (ThermoFisher, #A1413301), and passaged about once a week with Gentle Cell Dissociation Reagent (STEMCELL Technologies, #07174), but were not used for differentiation past passage number 30. Cortical neurons were differentiated according to a protocol published by Zhang et al. (2013) with minor modifications described below. iPSCs (under passage 10 after karyotyping) were treated with Accutase (Innovative Cell Technologies, #AT104) and plated at 90,000 cells/cm2 in mTeSR-1 media supplemented with 10uM ROCK inhibitor, Y-27632 (Cayman Chemical, #10005583) on Geltrex-coated 12-well plates. On day 2, no mouse glia cells were added and we used Ara-C (Sigma-Aldrich, #C1768) at a final concentration of 2uM. To label mitochondria in neurons, a lentivirus carrying CMV-Mito-DsRed was added to the media on day 5, supplemented with 8ug/mL polybrene (Sigma-Aldrich, #TR-1003-G). Mitochondrial motility was assayed after day 14.
Motor Neuron Differentiation
The iPSC lines 39b SOD+/A4V and 39b SOD+/+The cells lines were differentiated between passage 8 to passage 14. Transfection with the HB9::GFP reporter was performed as described in Wainger et al. (2014). A 1kb HB9 promoter fragment (gift from Hynek Wichterle) controlling the expression of myristoylated GFP was inserted into a donor plasmid specific for the AAVS1 locus (Sigma). Subsequently, 2.5 million iPS cells were single-cell dissociated using accutase and electroporated using the Neon transfection system (100ul tip; 1600V Voltage, 20ms Width, 1 Pulse; Life Technologies) with 2 g of AAVS1 ZFN plasmid and 6 g of modified AAVS1 donor plasmid. After nucleofection cells were plated on matrigel with mTeSR1 in the presence of ROCK inhibitor. After 48hrs, puromycin selection was applied and surviving clonal colonies were individually passaged and gDNA was extracted. PCR was used to confirm proper targeting of the cassette. Primer sequences are available upon request. Faithful expression of the reporter was verified using expression of the motor neuron marker Isl1 (Abcam (Ab109517), 1:1000) and the pan-neuronal marker Map2 (Milipore (MAB378), 1:1000). Stem cell cultures were differentiated into motor neurons as previously described in Kiskinis et al. (2014). Briefly, iPSCs were dissociated to single cells and plated in suspension in low-adherence flasks (400k/mL), in mTeSR media with 10uM ROCK inhibitor. Media was gradually diluted (50% on day 3 and 100% on day 4) to KOSR (DMEM/F12, 10% KOSR) between days 1-4 and to a neural induction medium (NIM: DMEM/F12 with L-glutamine, NEAA, Heparin (2 ug/mL), N2 supplement (Invitrogen) for days 5-24. From days 1-6 cells were cultured in the presence of SB431542 (10 uM, Sigma Aldrich) and Dorsmorphin (1 uM, Stemgent), and from days 5-24 with BDNF (10 ng/mL, R&D), ascorbic acid (AA, 0.4 ug/mL, Sigma), Retinoic Acid (RA, 1 uM, Sigma) and Smoothened Agonist 1.3 (SAG 1.3, 1 uM, Calbiochem). On day 24 floating cell aggregates were dissociated into single cells with Papain/DNase (Worthington Bio) and HB9::GFP positive cells isolated using fluorescence activated cell sorting (FACS). Subsequently, 30k motor neurons were plated onto poly-D-lysine/laminin-coated 96 well plates together with 30k mouse primary glial cells (P1). Cell cultures were fed every 2-3 days with complete Neurobasal media containing L-glutamine, NEAA, glutamax, N2 and B27, with BDNF/CNTF/GDNF (10 ng/mL, R&D) and ascorbic acid (0.2 ug/mL, Sigma). After 14 days, the cultures were transduced with synapsin-MitoDSred lentivitrus and used for subsequent mitochondrial movement kinetics experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Statistical analysis was conducted in GraphPad Prism v6.0 and MATLAB®. Data were represented either using R (https://www.R-project.org) or GraphPad Prism v6.0 (GraphPad Software, 7825 Fay Avenue, Suite 230 La Jolla, CA 02037). If not indicated in the figures, **** = p < 0.0001; *** = p < 0.001; ** = p < 0.01; * = p < 0.05; ns = p > 0.05. Z-scores were calculated for each parameter using the formula Z = (x-μ)/σ. Because the values of integrated distance did not follow a normal distribution, we turned to the Kolmogorov-Smirnov (KS) statistic and used it as a descriptor (Perlman et al., 2004). The KS statistic quantifies a distance between the distribution functions of samples, and is one of the most useful nonparametric methods for comparing two samples, as it is sensitive to differences in both location and shape of cumulative distribution functions, in this case distances traveled by each mitochondrion in the fields. No difference between distributions leads to KS = 0, while significant changes in the curves generate KS > 0. We then transformed the KS values per well into Z-scores.
DATA AND CODE AVAILABILITY
The datasets (primary screen data) as well as codes generated during this study are available at GitHub:
https://github.com/ThomasSchwarzLab/ScreeningData_Shlevkov_CellReports_2019
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-beta3-tubulin | Millipore Sigma | T8660; RRID:AB_477590 |
| Rabbit anti-TPP1 | Abcam | Ab96498, RRID; AB_10680725 |
| Mouse αnti-Myc, clone 4A6 | EMD Millipore | 05724, RRID; AB_309938 |
| Rabbit monoclonal anti-Isl1 | Abcam | Ab109517, RRID; AB_10866454 |
| Mouse anti-MAP2A,2B | EMD Millipore | MAB378, RRID; AB_94967 |
| Bacterial and Virus Strains | ||
| CMV-mitoDSred LV | Orian Shririhai, UCLA | N/A |
| Synapsin-mitoDSred LV | This study | N/A |
| pTet-O-Ngn2-puro | Zhang et al., 2013 | https://www.addgene.org/52047/ |
| Tet-O-FUW-EGFP | Vierbuchen et al., 2010 | https://www.addgene.org/30130/ |
| FUW-M2rtTA | Hockemeyer et al., 2008 | https://www.addgene.org/20342/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Biomol ICCBL – 2012 library (480) | ICCB-Longwood | https://iccb.med.harvard.edu/biomol-iccb-l-known-bioactives-2012 |
| Selleck Bioactive compound library (1,902) | ICCB-Longwood | https://iccb.med.harvard.edu/selleck-bioactive-compound-library |
| Calcimycin | Millipore Sigma | A23187 |
| Latrunculin B | Tocris | Cat # 3974 |
| Latrunculin A | Tocris | Cat # 3973 |
| Cytochalasin B | Tocris | Cat # 5474 |
| Ala-Ala-Phe-CMK | Enzo | BML-PI123-0025 |
| BMS-754807 | Selleck Chemicals | Cat # S1124 |
| Hesperadin | Selleck Chemicals | Cat # S1529 |
| TAK-901 | Selleck Chemicals | Cat # S2718 |
| Barasertib | Selleck Chemicals | Cat # S1147 |
| Paclitaxel | Tocris | Cat# 1097 |
| 24(S)-Hydroxycholesterol | Millipore Sigma | Cat # SML1648 |
| Butabindide oxalate | Tocris | Cat# 1323 |
| Carfilzomib (PR-171) | Selleck Chemicals | Cat # S2853 |
| Y-27632(ROCK inhibitor) | Cayman Chemicals | Cat# 10005583 |
| Tetramethylrhodamine, methyl ester | ThermoFisher | Cat #T668 |
| Deposited Data | ||
| Primary screen data were deposited at GitHub | This study | https://github.com/ThomasSchwarzLab/ScreeningData_Shlevkov_CellReports_2019 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | ATCC CRL-3216, RRID:CVCL_0063 |
| GON0515-03 #5 iPSC line | This study | N/A |
| 39b SOD+/A4V iPSC line | Wainger et al., 2014 | N/A |
| 39b SOD+/+ iPSC line | Wainger et al., 2014 | N/A |
| Experimental Models: Organisms/Strains | ||
| Syntaphilin KO mouse | Kang et al., 2008 | N/A |
| Oligonucleotides | ||
| Primer AurkB K106R (forward): GTGGCGTCAGGGTCCTCTTG | This study | N/A |
| Primer AurkB K106R (reverse): GATGAAATGGCTTTTCTTCTCCC | This study | N/A |
| Primer synapsin-mitoDSred Lenti (forward): GCCTGAGAGCGCAGTAGCGCTACCGGACTCAGA | This study | N/A |
| Primers for synapsin-mitoDSred Lenti (reverse): CCGCTAGCTTCGAAGATTATGATCTAGAGTCGCGGC | This study | N/A |
| Recombinant DNA | ||
| Mito-DsRed (pDsRed2-Mito) | G. Hajnoczky, Thomas Jefferson University | N/A |
| GFP-rab7 WT | Choudhury et al., 2002 | Addgene #12605 |
| pLV-CMV-mitoDSred | This study | N/A |
| pLV-Synapsin-mitoDSred | This study | N/A |
| pGFP-C-shLenti-Tpp1 (rat) | OriGene | TL711322 |
| pCMV6-Myc-DDK-Tpp1 (human) | OriGene | RC204471 |
| pGFP-C-shLenti-Tpp2 (rat) | OriGene | TL711260 |
| pGFP-C-shLenti-Aurkb (rat) | OriGene | TL712117 |
| pCMV6-Myc-DDK-Aurkb (human) | OriGene | RC210288 |
| pCMV6-Myc-DDH-AurkBK106R (kinase dead) | This study | N/A |
| pGFP-C-shLenti-Aurka (rat) | OriGene | TL320538 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| MATLAB® | MathWorks | https://www.mathworks.com |
| CellProfiler | Carpenter et al., 2006 | https://cellprofiler.org |
| R | R Core Team, 2014 | https://www.R-project.org |
| GraphPad Prism v6.0 | GraphPad Software | https://www.graphpad.com |
| Other | ||
| Cell Profiler pipeline for motility analysis and PATHS: MATLAB-based code for processing of Cell Profiler data for motility analysis, deposited at GitHub | This study | https://github.com/ThomasSchwarzLab/ArrayScanCodes |
Highlights.
High-content imaging and object tracking enables a screen for mitochondrial motility
Aurora Kinase B or TPP1 inhibition or knockdown enhanced axonal transport of mitochondria
Actin depolymerization enhanced axonal transport of mitochondria
Hit compounds improve mitochondrial transport in SOD1+/A4V ALS patient-derived neurons
ACKNOWLEDGMENTS
We thank Drs. Joseph Gonzalez-Heydrich and Catherine Brownstein (Boston Children’s Hospital) for providing the iPSC line used to generate human cortical neurons, Dr. Thomas Sudhof (Stanford University) for providing the lentiviral plasmids to the BCH’s Translational Neuroscience Center, and Dr. Zu-Hang Shen (NINDS) for sharing the Syntaphilin KO mouse strain. We thank members of the ICCB-L Screening Facility and the Boston Children’s Hospital Translational Neuroscience Center for their support in performing the HCS and related data analysis. We also thank Lala Mkhitaryan for technical support. This project was supported by the Parkinson’s Disease Foundation, United States (E.S.); grants from the Massachusetts Life Sciences Consortium, United States; the Mathers Foundation, United States; NIH, United States R01GM069808 (T.L.S.); NIH, United States MIRA R35GM122547 (A.E.C.); Boston Children’s Hospital-Broad Institute Collaboration Grant, United States (T.L.S. and A.E.C.); ALS Association, United States (O.W.); and NIH, United States P30 HD018655 to the IDDRC Translational Neuroscience, Screening, Molecular Genetics, and Cellular Imaging Cores.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.035.
DECLARATION OF INTERESTS
M.-A.B. is an employee and shareholder of Novartis. K.K. is an employee and shareholder of Wayfair. J.L.M.S. is an employee and shareholder of Regeneron. O.W. is an employee and shareholder of Amgen. K.R. is an employee and shareholder of Quralis. K.E. is founder of Q-State and Quralis and a member of their scientific advisory boards.
REFERENCES
- Basarsky TA, Parpura V, and Haydon PG (1994). Hippocampal synaptogenesis in cell culture: developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J. Neurosci 14, 6402–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J, et al. (2009). BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther 8, 3341–3349. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, Zhang Y, Gabel CV, Schwarz TL, and He Z (2016). The Mammalian-Specific Protein Armcx1 Regulates Mitochondrial Transport during Axon Regeneration. Neuron 92, 1294–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartoni R, Pekkurnaz G, Wang C, Schwarz TL, and He Z (2017). A high mitochondrial transport rate characterizes CNS neurons with high axonal regeneration capacity. PLoS ONE 12, e0184672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chada SR, and Hollenbeck PJ (2003). Mitochondrial movement and positioning in axons: the role of growth factor signaling. J. Exp. Biol 206, 1985–1992. [DOI] [PubMed] [Google Scholar]
- Chada SR, and Hollenbeck PJ (2004). Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr. Biol 14, 1272–1276. [DOI] [PubMed] [Google Scholar]
- Chang DT, Honick AS, and Reynolds IJ (2006). Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci 26, 7035–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Gopalakrishnan SM, Bui MH, Soni NB, Warrior U, Johnson EF, Donnelly JB, and Glaser KB (2011). 1-Benzyl-3-cetyl-2-methylimida-zolium iodide (NH125) induces phosphorylation of eukaryotic elongation factor-2 (eEF2): a cautionary note on the anticancer mechanism of an eEF2 kinase inhibitor. J. Biol. Chem 286, 43951–43958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung WD, Sakabe K, Housley MP, Dias WB, and Hart GW (2008). O-linked beta-N-acetylglucosaminyltransferase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins.J. Biol. Chem 283, 33935–33941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury A, Dominguez M, Puri V, Sharma DK, Narita K, Wheatley CL, Marks DL, and Pargano RE (2002). Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Invest 109, 1541–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JY, Steen JA, and Schwarz TL (2016). Phosphorylation-Induced Motor Shedding Is Required at Mitosis for Proper Distribution and Passive In-heritance of Mitochondria. Cell Rep. 16, 2142–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchet J, Lewis TL Jr., Lee S, Courchet V, Liou DY, Aizawa S, and Polleux F (2013). Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell 153, 1510–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota AK, Tavares CD, Warthaka M, Abramczyk O, Marshall KD, Kaoud TS, Gorgulu K, Ozpolat B, and Dalby KN (2012). Investigating the kinetic mechanism of inhibition of elongation factor 2 kinase by NH125: evidence of a common in vitro artifact. Biochemistry 51, 2100–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezaki J, Takeda-Ezaki M, and Kominami E (2000). Tripeptidyl peptidase I, the late infantile neuronal ceroid lipofuscinosisgene product, initiatesthe lysosomal degradation of subunit c of ATP synthase. J. Biochem 128, 509–516. [DOI] [PubMed] [Google Scholar]
- Gegg ME, and Schapira AHV (2018). The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 285, 3591–3603. [DOI] [PubMed] [Google Scholar]
- Geier E, Pfeifer G, Wilm M, Lucchiari-Hartz M, Baumeister W, Eichmann K, and Niedermann G (1999). A giant protease with potential to substitute for some functions of the proteasome. Science 283, 978–981. [DOI] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, and Schwarz TL (2006). Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol 173, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, and Zinsmaier KE (2005). The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. [DOI] [PubMed] [Google Scholar]
- Gwee SSL, Radford RAW, Chow S, Syal MD, Morsch M, Formella I, Lee A, Don EK, Badrock AP, Cole NJ, et al. (2018). Aurora kinase B regulates axonal outgrowth and regeneration in thespinal motor neurons of developing zebrafish. Cell. Mol. Life Sci 75, 4269–4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrill JA, Chen H, Streifel KM, Yang D, Mundy WR, and Lein PJ (2015). Ontogeny of biochemical, morphological and functional parameters of synaptogenesis in primarycultures of rat hippocampal and cortical neurons. Mol. Brain 8, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, and Peters JM (2003). The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol 161, 281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Cook EG, Gao Q, Mitalipova M, and Jaenisch R (2008). A drug-inducible system for direct reproramming of human somatic cells to pluripotency. Cell Stem Cell 3, 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, Schule B, Krainc D, Palmer TD, and Wang X (2016). Functional Impairment in Miro Degradation and Mitophagy IsaShared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 19, 709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SP, and Hart GW (2003). Roles ofthe tetratricopeptide repeat domain in O-GlcNAc transferase targeting and protein substrate specificity. J. Biol. Chem 278,24608–24616. [DOI] [PubMed] [Google Scholar]
- Jaqaman K, and Danuser G (2009). Computational image analysisofcellular dynamics: a case study based on particle tracking. Cold Spring Harb. Protoc 2009, pdb.top65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, and Danuser G (2008). Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 5, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Moon KE, and Eisenberg M (1988). Inactivation of chymotrypsin and human skin chymase: kinetics of time-dependent inhibition in the presence of substrate. Biochim. Biophys. Acta 953, 269–279. [DOI] [PubMed] [Google Scholar]
- Jordan MA, and Wilson L (2004). Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Joshi DC, Zhang CL, Lin TM, Gusain A, Harris MG, Tree E, Yin Y, Wu C, Sheng ZH, Dempsey RJ, et al. (2015). Deletion of mitochondrial anchoring protects dysmyelinating shiverer: implications for progressive MS. J. Neurosci 35, 5293–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J-S, Tian J-H, Pan P-Y, Zald P, Li C, Deng C, and Sheng Z-H (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruppa AJ, Kishi-Itakura C, Masters TA, Rorbach JE, Grice GL, Kendrick-Jones J, Nathan JA, Minczuk M, and Buss F (2018). Myosin VI-Dependent Actin Cages Encapsulate Parkin-Positive Damaged Mitochondria. Dev. Cell 44, 484–499.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL Jr., Turi GF, Kwon SK, Losonczy A, and Polleux F (2016). Progressive Decrease of Mitochondrial Motility during Maturation of Cortical Axons In Vitro and In Vivo. Curr. Biol 26, 2602–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lim S, Hoffman D, Aspenstrom P, Federoff HJ, and Rempe DA (2009). HUMMR, a hypoxia- and HIF-1alpha-inducible protein, alters mitochondrial distribution and transport. J. Cell Biol 185, 1065–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Acín-Peréz R, Geghman KD, Manfredi G, Lu B, and Li C (2011). Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. USA 108, 12920–12924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Doménech G, Serrat R, Mirra S, D’Aniello S, Somorjai I, Abad A, Vitureira N, García-Arumí E, Alonso MT, Rodriguez-Prados M, et al. (2012). The Eutherian Armcx genes regulate mitochondrial trafficking in neurons and interact with Miro and Trak2. Nat. Commun 3, 814. [DOI] [PubMed] [Google Scholar]
- López-Doménech G, Covill-Cooke C, Ivankovic D, Halff EF, Sheehan DF, Norkett R, Birsa N, and Kittler JT (2018). Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 37, 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnaghi P, D’Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, et al. (2013). Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol 9, 548–556. [DOI] [PubMed] [Google Scholar]
- Misgeld T, and Schwarz TL (2017). Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 96, 651–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misko A, Jiang S,Wegorzewska I, Milbrandt J, and Baloh RH (2010). Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci 30, 4232–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng JM, Chen MJ, Leung JY, Peng ZF, Manikandan J, Qi RZ, Chuah MI, West AK, Vickers JC, Lu J, et al. (2012). Transcriptional insights on the regenerative mechanics of axotomized neurons in vitro. J. Cell. Mol. Med 16, 789–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak D, Sepp KJ, and Hollenbeck PJ (2010). Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J. Neurosci 30, 8984–8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekkurnaz G, Trinidad JC, Wang X, Kong D, and Schwarz TL (2014). Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 158, 54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman ZE, Slack MD, Feng Y, Mitchison TJ, Wu LF, and Altschuler SJ (2004). Multidimensional drug profiling by automated microscopy. Science 306, 1194–1198. [DOI] [PubMed] [Google Scholar]
- R Core Team. (2014). R: A language en environment for statistical computing. R Foundation for Statistical Computing; (Vienna, Austria: ). http://www.R-project.org. [Google Scholar]
- Rose C,Vargas F, Facchinetti P, Bourgeat P, Bambal RB, Bishop PB, Chan SM, Moore AN, Ganellin CR, and Schwartz JC (1996). Characterization and inhibition of a cholecystokinin-inactivating serine peptidase. Nature 380,403–409. [DOI] [PubMed] [Google Scholar]
- Russo GJ, Louie K, Wellington A, Macleod GT, Hu F, Panchumarthi S, and Zinsmaier KE (2009). Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J. Neurosci 20, 5443–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, and Hajnoczky G (2008). Bidirectional Ca2+-dependent control ofmitochondrial dynamics bythe Miro GTPase. Proc. Natl. Acad. Sci. USA 105,20728–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WM, and Hollenbeck PJ (2012). The axonal transport of mitochondria. J. Cell Sci 125, 2095–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine Y, Lin-Moore A, Chenette DM, Wang X, Jiang Z, Cafferty WB, Hammarlund M, and Strittmatter SM (2018). Functional Genome-wide Screen Identifies Pathways Restricting Central Nervous System Axonal Regeneration. Cell Rep. 24, 269. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlevkov E, Kramer T, Schapansky J, LaVoie MJ, and Schwarz TL (2016). Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. USA 113, E6097–E6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorbara CD, Wagner NE, Ladwig A, Nikic I, Merkler D, Kleele T, Marinkovic P, Naumann R, Godinho L, Bareyre FM, et al. (2014). Pervasive axonal transport deficits in multiple sclerosis models. Neuron 84, 1183–1190. [DOI] [PubMed] [Google Scholar]
- Spence EF, and Soderling SH (2015). Actin Out: Regulation ofthe Synaptic Cytoskeleton. J. Biol. Chem 290, 28613–28622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, and Schwarz TL (2002). Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36, 1063–1077. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, and Youle RJ (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol 191, 1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, SOhof TC, and Wernig M (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7,1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, and Schwarz TL (2009a). Imaging axonal transport of mitochondria. Methods Enzymol. 457, 319–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, and Schwarz TL (2009b). The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, and Schwarz TL (2011). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, and Selkoe DJ (2009). Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 48, 2045–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zhong G, and Zhuang X (2013). Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ikezoe T, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, Komatsu N, Bandobashi K, Togitani K, Koeffler HP, et al. (2007). AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood 110, 2034–2040. [DOI] [PubMed] [Google Scholar]
- Yi M, Weaver D, and Hajnoczky G (2004). Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol 167, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YB, and Sheng ZH (2011). Increased axonal mitochondrial mobility does not slow amyotrophic lateral sclerosis (ALS)-like disease in mutant SOD1 mice. J. Biol. Chem 286, 23432–23440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets (primary screen data) as well as codes generated during this study are available at GitHub:
https://github.com/ThomasSchwarzLab/ScreeningData_Shlevkov_CellReports_2019