

Abstract
Insulin promotes glucose uptake by triggering the translocation of glucose transporter type 4 (GLUT4) from intracellular vesicles to the plasma membrane through exocytosis. GLUT4 exocytosis is a vesicle fusion event involving fusion of GLUT4-containing vesicles with the plasma membrane. For GLUT4 vesicle fusion to occur, GLUT4 vesicles must first be tethered to the plasma membrane. A key tethering factor in exocytosis is a heterooctameric protein complex called the exocyst. The role of the exocyst in GLUT4 exocytosis, however, remains incompletely understood. Here we first systematically analyzed data from a genome-scale CRISPR screen in HeLa cells that targeted virtually all known genes in the human genome, including 12 exocyst genes. The screen recovered only a subset of the exocyst genes, including exocyst complex component 7 (Exoc7/Exo70). Other exocyst genes, however, were not isolated in the screen, likely because of functional redundancy. Our findings suggest that selection of an appropriate exocyst gene is critical for genetic studies of exocyst functions. Next we developed an inducible adipocyte genome editing system that enabled Exoc7 gene deletion in adipocytes without interfering with adipocyte differentiation. We observed that insulin-stimulated GLUT4 exocytosis was markedly inhibited in Exoc7 KO adipocytes. Insulin signaling, however, remained intact in these KO cells. These results indicate that the exocyst plays a critical role in insulin-stimulated GLUT4 exocytosis in adipocytes. We propose that the strategy outlined in this work could be instrumental in genetically dissecting other membrane-trafficking pathways in adipocytes.
Keywords: exocytosis, membrane fusion, membrane protein, membrane trafficking, vesicles, glucose transporter type 4 (GLUT4), CRISPR genome editing, exocyst, exocyst complex component 7 (Exoc7), insulin signaling
Introduction
A major function of insulin is to promote glucose uptake into adipocytes and skeletal muscles, a process mediated by the glucose transporter GLUT4 (1–3). Under basal conditions, GLUT4 is sequestered in intracellular storage vesicles (1, 4–6). Insulin stimulation activates a cascade of signaling events that ultimately triggers fusion of GLUT4 vesicles with the plasma membrane (GLUT4 exocytosis), delivering GLUT4 to the cell surface to facilitate glucose uptake (1, 7, 8). Like other intracellular vesicle fusion events, GLUT4 vesicle fusion is driven by a conserved engine composed of soluble NSF attachment proteins (SNAREs)3 and Sec1/Munc18 (SM) proteins (9–17).
To form the SNARE-SM vesicle fusion machinery, exocytic vesicles must first be tethered to the plasma membrane (18–20). An evolutionarily conserved tether factor is the exocyst, a heterooctameric complex comprised of Exoc1/Sec3, Exoc2/Sec5, Exoc3/Sec6, Exoc4/Sec8, Exoc5/Sec10, Exoc6/Sec15, Exoc7/Exo70, and Exoc8/Exo84 (Fig. 1A) (21–26). As a member of the multisubunit tethering complex family, the exocyst interacts with lipids, small GTPases (e.g. Rabs), and SNAREs to promote vesicle tethering in a broad range of eukaryotic exocytic pathways (27–34).
Figure 1.

Exocyst genes exhibit disparate phenotypes in a genome-wide CRISPR screen. A, illustration of Rab10 and exocyst anchored to membrane bilayers. The model is based on the structures of exocyst (PDB codes 5YFP and 3HIE) (22), and guanosine 5′-[β,γ-imido]triphosphate (GMP-PNP)-bound Rab10 (PDB code 5LPN) (67). The model was prepared using PyMOL (DeLano Scientific LLC, San Carlos, CA). Rab10, a known mediator of GLUT4 exocytosis (42, 51), interacts with the Exoc6/Sec15 subunit in yeast two-hybrid screens (36). The Exoc1 and Exoc7 subunits bind to phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), whereas Rab10 is anchored to the membrane through prenyl groups. Because the structures of the Rab10–exocyst complex and exocyst–membrane association are still unavailable, the positions of exocyst and Rab10 in the model are arbitrary. The proteins are shown to scale whereas the lipids and membranes are not. Besides Rab GTPases, the exocyst also interacts with other molecules, such as RalA and SNAREs (not shown) (29–31). B, ranking of exocyst genes in a genome-wide CRISPR screen of insulin-stimulated GLUT4 exocytosis in HeLa cells (42). The significance of a gene was calculated based on enrichment of its corresponding gRNAs in the screen using the MAGeCK algorithm (46). Genes above the horizontal cutoff line (p = 0.01) are significant hits. Only exocyst genes are shown. Of the 12 exocyst genes included in the CRISPR library, EXOC1, EXOC7, and EXOC8 were recovered as significant hits. C, a CRISPR score of a gene is calculated based on fold changes in the abundance of its corresponding gRNAs by comparing a passage control population of HeLa mutant cells (without stimulation or selection) with the initial CRISPR library. Genes with CRISPR scores below the horizontal cutoff line (CRISPR score, −0.25) are considered essential genes. Only exocyst genes are shown. CRISPR score ranking of all genes in the CRISPR library is shown in Table S2. D, diagram of the HA-GLUT4-GFP reporter used to monitor insulin-stimulated GLUT4 exocytosis. The GLUT4 reporter faithfully recapitulates trafficking of endogenous GLUT4 proteins (42, 68, 69). E, normalized surface levels of the GLUT4 reporter in WT and mutant HeLa cells. The cells were either left untreated or treated with 100 nm insulin for 30 min before surface GLUT4 reporters were labeled using anti-HA antibodies and APC-conjugated secondary antibodies. GFP and APC fluorescence was measured using flow cytometry. To inhibit insulin signaling, 100 nm wortmannin was added. Datasets were normalized to untreated WT cells. Data are presented as mean ± S.D. n = 3. ***, p < 0.001; n.s., not significant, p > 0.05.
In previous genetic studies, the exocyst was suggested to regulate tethering of GLUT4 vesicles (29, 35–38). In another study, however, manipulations of the exocyst had little effect on GLUT4 exocytosis (39). These genetic studies, however, mainly relied on RNAi and overexpression of dominant-negative mutants, which often suffer insufficient gene silencing as well as off-target effects. Moreover, mutations of the exocyst impair differentiation of adipocytes (40), complicating functional analysis. To definitively establish the role of the exocyst in GLUT4 exocytosis, these limitations needed to be overcome.
In this work, we first analyzed data from a genome-scale CRISPR genetic screen performed in HeLa cells. We found that only a subset of mammalian exocyst genes, including Exoc7, were isolated as significant hits in the screen. By contrast, other exocyst genes did not exhibit a phenotype. Next we developed an inducible adipocyte genome editing system to ablate Exoc7 expression without interfering with adipocyte differentiation. We observed that Exoc7 KO markedly inhibited insulin-stimulated GLUT4 exocytosis in adipocytes. Insulin signaling, however, remained intact in the KO cells. This study established a critical role of the exocyst in insulin-stimulated GLUT4 exocytosis in adipocytes.
Results
Genome-scale genetic analysis reveals disparate KO phenotypes of exocyst genes
Genome-wide CRISPR genetic screens are a powerful, unbiased approach to interrogate gene functions in mammalian cells (41). The GeCKO V2 CRISPR library contains guide RNAs (gRNAs) targeting virtually all known genes in the human genome, including 12 exocyst genes (42, 43). These 12 exocyst genes included the three paralogues of EXOC3 (EXOC3L1, EXOC3L2, and EXOC3L4) and EXOC6B, a paralogue of EXOC6 (Fig. 1B). Each protein-encoding gene in the library is targeted by six independent gRNAs (42, 43). In a cell population mutagenized by the CRISPR library, each cell usually receives a single gRNA so that only one gene is mutated (43).
Although GLUT4 is not expressed in HeLa cells, exogenously expressed GLUT4 is sorted into insulin-responsive vesicles in HeLa cells and undergoes insulin-stimulated exocytosis (42, 44, 45). HeLa cells expressing a GLUT4 reporter (HA-GLUT4-GFP) were mutagenized by the genome-wide CRISPR library. After insulin stimulation, the mutant cell population was labeled using anti-HA antibodies and allophycocyanin (APC)–conjugated secondary antibodies. The cells were sorted by flow cytometry based on APC staining (surface reporters) and GFP fluorescence (total reporters) to enrich mutants defective in insulin-stimulated GLUT4 exocytosis (42). The abundance of gRNAs in the final sorted population was analyzed by deep sequencing and compared with that in a control CRISPR mutant population (without insulin stimulation or cell sorting). Systematic analysis of gRNA enrichment showed that only three exocyst genes (EXOC1, EXOC7, and EXOC8) were recovered as significant hits in the HeLa cell screen (Fig. 1B). By contrast, other exocyst genes did not show up as significant hits (Fig. 1B).
We next examined whether exocyst genes are essential to cell viability or growth. KO of an essential gene leads to cell death or growth arrest, which precludes accurate characterization of its function. We cultured the HeLa mutant cell population continuously for 4 weeks without sorting or insulin treatment. The abundance of gRNAs in the mutant cell population was then compared with that in the original CRISPR library. Essential genes were identified based on depletion (dropout) of its corresponding gRNAs in the mutant cell population (46). A total of 1,391 essential genes were identified, encompassing a range of pathways critical to cell physiology, such as protein translation, RNA transcription, and cell cycle regulation (Table S2). However, none of the 12 exocyst genes was among the essential genes (Fig. 1C and Tables S1 and S2), indicating that mutations of exocyst genes do not impair cell viability or growth.
To validate the results of the genome-wide genetic analysis, we used CRISPR genome editing to delete the EXOC7 gene, a significant hit in the HeLa cell screen (Fig. 1B) (42). We then used a flow cytometry–based assay to quantitatively measure insulin-regulated exocytosis of the HA-GLUT4-GFP reporter in individual cells (Fig. 1D) (42). Insulin stimulation strongly accelerated GLUT4 exocytosis, leading to translocation of the GLUT4 reporter to the cell surface (Fig. 1E and Fig. S1). We observed that insulin-stimulated GLUT4 translocation was abrogated in EXOC7 KO HeLa cells (Fig. 1E and Fig. S1). KO of EXOC3, which was not a significant hit in the screen (Fig. 1B), had no effect on insulin-stimulated GLUT4 translocation (Fig. 1E). These genome editing data confirmed the results of the genetic screen.
These results demonstrated that the majority of mammalian exocyst genes exhibit no phenotype in CRISPR KO studies, likely because of functional redundancy (see “Discussion”). Thus, to genetically determine exocyst functions, it is critical to select an appropriate exocyst gene. In this work, we focused on EXOC7, an exocyst gene with the strongest phenotype in the HeLa cell genetic screen (Fig. 1B).
Doxycycline (dox)–induced KO of Exoc7 in adipocytes
Although HeLa cells are a facile platform for initial analysis of exocyst genes, the role of the exocyst in insulin-stimulated GLUT4 exocytosis needs to be directly examined in adipocytes, a physiologically relevant insulin-responsive cell type. Because the exocyst is required for adipocyte differentiation (40), we developed an inducible adipocyte genome editing system in which a dox-controlled Cas9 expression cassette was stably integrated into mouse preadipocytes (Fig. 2A). Dox was added to transgenic preadipocytes shortly before onset of differentiation so that the gradually diminishing exocyst proteins could support adipocyte differentiation (Fig. 2B). As shown below (Fig. 3), preadipocytes differentiated normally into mature adipocytes when Exoc7 was deleted using this inducible CRISPR system. Because mutant preadipocytes cannot be cloned out, two distinct gRNAs were used to target the Exoc7 gene (Fig. 2A). Simultaneous introduction of two distinct gRNAs efficiently abrogates the expression of a candidate gene in a pooled cell population (42, 44). Indeed, we observed that Exoc7 expression was strongly reduced in adipocytes using this inducible CRISPR system (Fig. 2C).
Figure 2.
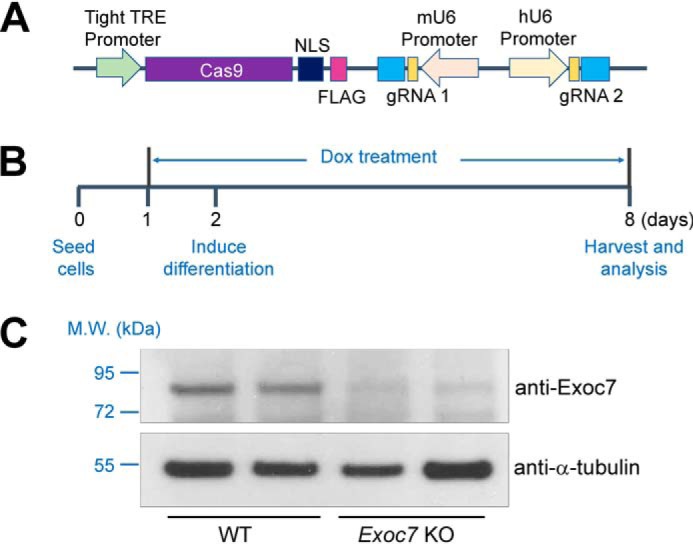
Development of an inducible CRISPR system for adipocyte genome editing. A, diagram of an inducible CRISPR KO system in which Cas9 expression is under the control of a tight tetracycline response element (TRE). Expression of the first single guide RNA is driven by a mouse U6 (mU6) promoter, whereas expression of the second sgRNA is driven by a human U6 (hU6) promoter. NLS, nuclear localization signal. B, timeline of adipocyte differentiation and dox-induced genome editing. Dox was added to preadipocytes at a final concentration of 2 μg/ml to induce Cas9 expression 24 h before addition of an adipocyte differentiation mixture. Unless indicated otherwise, all functional assays were performed using differentiated adipocytes. C, representative immunoblots showing the expression of the indicated proteins in WT or Exoc7 KO adipocytes. Two independent samples are shown for each cell line. M.W., molecular weight.
Figure 3.

Inducible KO of Exoc7 permits normal adipocyte differentiation. A, representative images of preadipocytes and adipocytes. Dox was added to adipocytes but not preadipocytes. Lipid droplets in the cells were stained with Nile red, and the images were captured using a ×20 objective on an Olympus IX81 microscope. Scale bars = 50 μm. B, quantification of lipid droplet–positive cells (WT or Exoc7 KO). Cells were stained with Nile red and the fluorescence of Nile red was measured using flow cytometry. Data are presented as mean ± S.D. n = 3. n.s., not significant, p > 0.05. C, representative immunoblots showing expression of the indicated proteins in WT and Exoc7 KO adipocytes. Two independent samples were prepared for each cell line. M.W., molecular weight.
Inducible Exoc7 KO permits normal adipocyte differentiation
A hallmark of adipocyte differentiation is accumulation of lipid droplets, which could be stained with the Nile red dye (Fig. 3A) (2, 47). After extensive testing, we identified a dox induction condition (Fig. 2B) that permitted Exoc7-targeted preadipocytes to differentiate normally into mature adipocytes, as indicated by analysis of Nile red–stained lipid droplets using microscopy and flow cytometry (Fig. 3, A and B). Moreover, the overall morphology of Exoc7 KO adipocytes was indistinguishable from that of WT cells (Fig. 3A). The expression levels of PPARγ, a master regulator of adipocyte differentiation (48), were comparable between WT and Exoc7 KO cells (Fig. 3C). These data demonstrated that adipocyte differentiation occurred normally when Exoc7 was deleted using the inducible genome editing system.
Insulin-stimulated GLUT4 exocytosis is inhibited in Exoc7 KO adipocytes
We then examined the effect of Exoc7 KO on the GLUT4 exocytic pathway. We observed that insulin-stimulated GLUT4 translocation was markedly inhibited in Exoc7 KO adipocytes (Fig. 4A and Fig. S2). Further kinetic analysis showed that the exocytosis rate of GLUT4 was markedly reduced in Exoc7 KO adipocytes (Fig. 4B), correlating with GLUT4 translocation defects in these cells (Fig. 4A and Fig. S2). Next, we examined the responses of Exoc7 KO adipocytes to various concentrations of insulin. We found that GLUT4 translocation was diminished in KO cells at all the insulin concentrations we tested (Fig. 4C). These results demonstrated that Exoc7 is essential for insulin-stimulated GLUT4 exocytosis in adipocytes.
Figure 4.

Insulin-stimulated GLUT4 exocytosis is inhibited in Exoc7 KO adipocytes. A, normalized surface levels of the GLUT4 reporter in WT and Exoc7 KO adipocytes. After treatment with 100 nm insulin for the indicated periods, surface levels of the GLUT4 reporter were measured using flow cytometry as described in Fig. 1E. Data are presented as mean ± S.D.; n = 3. B, initial exocytosis rates of the GLUT4 reporter in WT and Exoc7 KO adipocytes treated with insulin. The initial exocytosis rates were calculated based on increases in surface levels of the GLUT4 reporter during the first 10 min of insulin treatment shown in A. Datasets are normalized to untreated WT cells. Data are presented as mean ± S.D. n = 3. ***, p < 0.001. C, dose dependence of insulin-stimulated translocation of the GLUT4 reporter in WT and Exoc7 KO adipocytes. Cells were treated with the indicated concentrations of insulin for 30 min before the surface levels of the reporter were measured by flow cytometry. NT, not treated. Datasets are normalized to untreated WT cells. Data are presented as mean ± S.D.; n = 3.
Intact insulin signaling in Exoc7 KO adipocytes
Next we examined whether Exoc7 KO influences insulin signaling. Insulin treatment induced the phosphorylation and activation of AKT/PKB, a downstream serine/threonine kinase in the insulin signaling cascade controlling GLUT4 trafficking (Fig. 5) (49–51). We observed that insulin-induced AKT phosphorylation remained unchanged in Exoc7 KO adipocytes (Fig. 5). Likewise, insulin-induced phosphorylation of AS160, a Rab-binding protein connecting insulin signaling to GLUT4 exocytosis (52), occurred normally in KO cells (Fig. 5). The expression level of sortilin 1 (Sort1), a molecule involved in post-Golgi GLUT4 sorting (53, 54), was not altered in Exoc7 KO adipocytes either (Fig. 5). Thus, the exocyst is dispensable for insulin signaling and Sort1 expression, consistent with its role in the downstream vesicle tethering step of GLUT4 trafficking.
Figure 5.
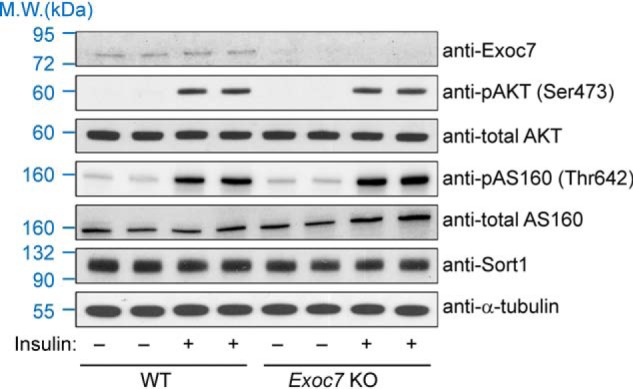
Insulin signaling remains intact in Exoc7 KO adipocytes. Representative immunoblots showing expression of the indicated proteins in WT and Exoc7 KO adipocytes. For insulin stimulation, cells were treated with 10 nm insulin for 10 min before the cells were harvested for analysis. Two independent samples were prepared for each condition. M.W., molecular weight.
Tight regulation of adipocyte genome editing using the inducible CRISPR system
Next we further characterized the inducible adipocyte genome editing system. We observed that Cas9 was expressed only in dox-treated adipocytes, correlating with loss of Exoc7 expression in these cells (Fig. 6A). In contrast, Exoc7 was expressed at WT levels in adipocytes that contained the CRISPR cassette but were not induced with dox (Fig. 6A). Thus, Cas9 expression and genome editing were strictly dependent on dox induction.
Figure 6.

Tight regulation of adipocyte genome editing using the inducible CRISPR system. A, representative immunoblots showing expression of the indicated proteins in WT and Exoc7-targeted adipocytes with or without dox induction. B, representative immunoblots showing expression of the indicated proteins in Exoc7 KO adipocytes with or without expression of a human EXOC7 rescue gene. For insulin stimulation, cells were treated with 10 nm insulin for 10 min. C, normalized surface levels of the GLUT4 reporter in WT and Exoc7 KO adipocytes with or without expression of the human EXOC7 rescue gene. The cells were left untreated or treated with 100 nm insulin for 30 min. Data are presented as mean ± S.D.; n = 3. ***, p < 0.001; n.s., not significant, p > 0.05.
Finally, we sought to rule out an off-target effect of the Exoc7 KO phenotype observed in adipocytes. To this end, we expressed the human EXOC7 gene in Exoc7 KO preadipocytes (Fig. 6B). We observed that the human EXOC7 gene, which was not targeted by the mouse Exoc7 gRNAs, fully restored insulin-stimulated GLUT4 exocytosis in Exoc7 KO adipocytes (Fig. 6C and Fig. S2). Thus, the defect of GLUT4 exocytosis in KO cells was caused by loss of Exoc7 expression. These results also suggest that the inducible CRISPR system permits specific gene ablation in adipocytes.
Discussion
In this work, we provide direct evidence of a critical role of the exocyst in insulin-stimulated GLUT4 exocytosis in adipocytes. This genetic study was made possible by an inducible adipocyte genome editing system that overcame the limitations of previous genetic assays. A major advantage of this system is that the timing of genome editing can be coordinated with adipocyte differentiation. Upon dox induction, the Exoc7 gene was deleted, terminating new protein production. Existing Exoc7 proteins were gradually degraded, but the remaining proteins were able to support differentiation of adipocytes. Exoc7 proteins were eventually eliminated upon completion of adipocyte differentiation. In this system, Cas9 expression and adipocyte genome editing are tightly regulated by dox induction. No leaky genome editing was observed, even after prolonged culture of uninduced cells.
The exocyst promotes vesicle tethering by interacting with molecules on exocytic vesicles (27, 28). The small GTPase Rab10 is anchored to GLUT4 vesicles and is known to bind to the Exoc6 subunit of the exocyst complex (36). It is conceivable that the Rab10–exocyst interaction facilitates tethering of GLUT4 vesicles to the plasma membrane. RalA, another vesicle-associated GTPase, also binds to the exocyst and is expected to contribute to GLUT4 vesicle tethering (29, 34, 55, 56). Although vesicle tethering is clearly a key function of the exocyst, in yeasts, the exocyst also directly interacts with SNAREs and SM proteins (30, 31, 57, 58), suggesting a more active role of the exocyst in the vesicle fusion reaction. Further studies will be needed to determine whether the exocyst directly regulates the vesicle fusion machinery and whether it is involved in other steps of GLUT4 trafficking, such as movement of GLUT4 vesicles on cytoskeletons.
Insulin-stimulated GLUT4 exocytosis was markedly inhibited but not abolished in Exoc7 KO adipocytes. KO of Exoc7 using two independent gRNAs was highly efficient so that the partial inhibition of GLUT4 exocytosis was not due to incomplete gene ablation. Instead, the partial inhibition phenotype was likely due to the existence of a parallel tethering mechanism or the unique features of adipocytes. The narrow cytoplasm of adipocytes may enable GLUT4 vesicles to partially bypass the requirement of the exocyst in vesicle tethering. Another notable observation from this study is that GLUT4 exocytosis increased with higher concentrations of insulin in both WT and Exoc7 KO cells. This finding suggests that the exocyst is not a direct target of insulin stimulation in GLUT4 trafficking.
An unexpected observation of this study is that the majority of mammalian exocyst genes exhibit no phenotype in CRISPR KO studies. A lack of phenotype is likely due to compensation by (a) redundant gene(s). Indeed, both Exoc3 and Exoc6 have paralogous genes that are expected to be functionally redundant (Fig. 1B). Exoc2, Exoc4, and Exoc5 may also have functionally redundant paralogues that remain to be uncovered. Alternatively, these three exocyst genes might not be fully targeted in the CRISPR screen. In the CRISPR library, six independent gRNAs were designed to target constitutive early exons of a protein-encoding gene (43). With lentiviral expression and prolonged cell culture (∼4 weeks), all alleles of a gene are efficiently edited. However, it remains possible that an alternatively spliced RNA transcript(s) of an exocyst gene skips the exons targeted by all six gRNAs. In this regard, the functional redundancy occurs at the RNA level. These redundancy issues need to be taken into consideration when designing genetic experiments to examine exocyst functions. The disparate KO phenotypes of exocyst genes could also be explained by a function conferred by an exocyst subcomplex rather than the full exocyst octamer. Interestingly, exocyst subunits can be found in subcomplexes, although there is no evidence that these subcomplexes themselves are biologically active (27, 28, 59, 60). Future research is required to further characterize exocyst gene redundancy and to examine the functional roles of exocyst subcomplexes.
We anticipate that the strategy described in this work could guide studies of additional GLUT4 exocytic regulators or other membrane trafficking pathways in adipocytes. Notably, adipocytes release a variety of molecules, including leptin, adiponectin, and exosomes, to modulate the metabolic activities of other organs in the body (2, 61, 62). The molecular basis of these exocytic pathways remains largely unknown. It is challenging to genetically identify the components of an exocytic pathway because there is a substantial level of redundancy in membrane trafficking genes (Fig. 1) (42, 44). A lack of KO phenotype cannot rule out a role of a candidate gene in a trafficking pathway. It is often necessary to test multiple genes in a gene family to identify an appropriate candidate gene exhibiting a significant loss-of-function phenotype. When a candidate gene is selected, its expression can be eliminated using the inducible adipocyte genome editing system developed in this study. Because different proteins are degraded at markedly distinct rates in the cell, the timing of dox induction needs to be experimentally determined for each candidate gene to abrogate its expression while maintaining normal adipocyte differentiation.
Experimental procedures
Analysis of genome-wide CRISPR screens
HeLa cells were grown in DMEM supplemented with 10% FB essence (FBE; Seradigm, 3100-500) and penicillin/streptomycin (Sigma, P4333). HeLa cells expressing the HA-GLUT4-GFP reporter were mutagenized using the GeCKO V2 CRISPR Library (Addgene, 1000000048) (42). Mutant cells with reduced insulin-stimulated GLUT4 exocytosis were sorted consecutively for three rounds using flow cytometry (42). The abundance of gRNAs in the sorted mutant population was analyzed by Illumina deep sequencing. The enrichment of gRNAs was analyzed by comparing the sorted population with a passage control mutant population (without sorting) using the Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) algorithm (https://sourceforge.net/projects/ mageck/).4
To identify essential genes, the unsorted passage control mutant population of HeLa cells was cultured continuously for 4 weeks. The abundance of gRNAs in the passage control population was analyzed by deep sequencing as described above. The depletion of gRNAs was calculated by comparing the passage control population with the initial GeCKO V2 CRISPR DNA library. A CRISPR score was calculated for each gene based on fold changes in the abundance of its corresponding gRNAs using an established algorithm (46). A CRISPR score of −0.25 was set as the cutoff, corresponding to a total of 1,391 essential genes in these cells. This number is consistent with the essential genes identified in previous genome-wide genetic studies (46, 63, 64).
Genome editing in HeLa cells
Two independent gRNA sequences were selected to target early constitutive exons of a candidate gene. One of the gRNAs was subcloned into the pLenti-CRISPR-V2 vector (Addgene, 52961). The other gRNA was subcloned into a modified version of the pLentiGuide-Puro vector (Addgene, 52963), in which the puromycin selection marker was replaced with a hygromycin selection marker. CRISPR plasmids were transfected into 293T cells along with pAdVAntage (Promega, E1711), pCMV-VSVG (Addgene, 8454), and psPax2 (Addgene, 12260) as described previously (42). The 293T culture media containing lentiviral particles were harvested daily for 4 days and centrifuged in a Beckman SW28 rotor at 25,000 rpm for 1.5 h. Viral pellets were resuspended in PBS and used to infect HeLa cells. Infected HeLa cells were consecutively selected using 1 μg/ml puromycin (Sigma, 3101118) and 500 μg/ml hygromycin B (Thermo, 10687010).
The sequence of gRNAs targeting the human EXOC3 gene was CGCCCGGACCAGCTGGACA (guide 1), and CAAAGACATCCAGCAGTCGC (guide 2). The sequence of gRNA targeting the human EXOC7 gene was TTCTCCGTCTGCTTGTGCAC (guide 1), and AATCTTGGCCATGCTTCCC (guide 1).
Measurement of insulin-stimulated GLUT4 exocytosis
Insulin-stimulated GLUT4 exocytosis was measured using flow cytometry as described previously (42, 44, 65). Briefly, cells were washed three times with KRH buffer (121 mm NaCl, 4.9 mm KCl, 1.2 mm MgSO4, 0.33 mm CaCl2, and 12 mm HEPES (pH 7.0)). After incubation in KRH buffer for 2 h, the cells were treated with 100 nm insulin for 30 min. When applicable, 100 nm wortmannin (Sigma, W1628) was added to the adipocytes 10 min before insulin treatment. After insulin stimulation, the cells were rapidly chilled, and surface GLUT4 reporters were stained using anti-HA antibodies (BioLegend, 901501) and APC-conjugated secondary antibodies (eBioscience, 17-4014). Subsequently, the cells were dislodged using Accutase (Innovative Cell Technologies, AT 104), and GFP and APC fluorescence was measured on a CyAN ADP analyzer (Beckman Coulter). To calculate normalized surface levels of GLUT4 reporters, the mean APC fluorescence (surface staining) was divided by mean GFP fluorescence (total reporters), and the obtained values were normalized to those of untreated WT samples. Data from populations of ∼5,000 cells were analyzed using the FlowJo software (FlowJo, LLC, v10) based on experiments run in biological triplicates.
To visualize GLUT4 exocytosis using confocal microscopy, Hela cells and preadipocytes were cultured as described above. After incubation in KRH buffer for 2 h, the cells were stimulated with 100 nm insulin for 30 min. The cells were then fixed using 4% paraformaldehyde (Sigma, P6148) and blocked using 2% BSA (Thermo, 064985). Surface GLUT4 reporters were stained using anti-HA antibodies and Alexa Fluor 568–conjugated secondary antibodies (Thermo, A-11004). Nuclei were stained with Hoechst 33342 (Sigma, D9642). Images were captured using a ×100 oil immersion objective on a Nikon A1 laser-scanning confocal microscope.
Inducible adipocyte genome editing
To engineer an inducible genome editing system, we first created a TLCV2-DelGFP construct (deposited to Addgene, 133302) by deleting the GFP sequence from TLCV2, a lentiviral vector (Addgene, 87360) (66), using site-directed mutagenesis. Next, one gRNA targeting the mouse Exoc7 gene was subcloned into the BsmbI site of the TLCV2-DelGFP vector. The second gRNA targeting mouse Exoc7 was subcloned into the BbsI site of the pmU6-gRNA vector (Addgene, 53187). Next, the expression cassette of the second gRNA, including the mU6 promoter, the gRNA, and the gRNA scaffold, was amplified by PCR with KpnI sites introduced at both ends. The PCR product was subcloned into the KpnI site of the TLCV2-DelGFP vector containing the first gRNA. The resulting construct, TLCV2-Exoc7-Ex5&10 (deposited into Addgene, 133303), was transfected into 293T cells to produce lentiviruses as described above for HeLa cells. Lentiviral particles were used to infect preadipocytes, which were derived from inguinal white adipose tissues. These preadipocytes were a spontaneously arising immortal cell line (a gift from Dr. Shingo Kajimura) that could be passaged for at least 13 generations without losing differentiation capacity. The transduced preadipocytes were selected using 3.5 μg/ml puromycin.
The primers for site-directed mutagenesis of the TLCV2 vector were CGAGGAGAATCCTGGCCCAGCTAGCTAAATCCGGGCAGAGCGCACATC (forward) and GATGTGCGCTCTGCCCGGATTTAGCTAGCTGGGCCAGGATTCTCCTCG (reverse). The sequence of gRNA targeting the mouse Exoc7 gene was AGAAGCTGCTGTTTGAGCGA (guide 1), and TTCTAGAGCTTTGGCCCCAA (guide 2). The primers for amplification of the expression cassette of the second gRNA were CGCGGTACCACCGGTATAGATCCGACGCCGCCA (forward) and GCGACCGGTGGTACCGCGGCGTCTCAGTACAAA (reverse).
Preadipocytes were cultured in DMEM supplemented with 10% FBE and penicillin/streptomycin to ∼95% confluence before a differentiation mixture was added at the following concentrations: 5 μg/ml insulin (Sigma, I0516), 1 nm tri-iodo-l-thyronine (T3, Sigma, T2877), 125 μm indomethacin (Sigma, I-7378), 5 μm dexamethasone (Sigma, D1756), and 0.5 mm 3-isobutyl-1-methylxanthine (Sigma, I5879). After 2 days, the cells were switched to DMEM supplemented with 10% FBE, 5 μg/ml insulin, and 1 nm T3. After another 2 days, fresh DMEM containing 10% FBE and 1 nm T3 was added. Adipocytes were usually analyzed 6 days after addition of the differentiation mixture. To induce Cas9 expression, dox (Sigma, D3447) was added 24 h before differentiation at a final concentration of 2 μg/ml. Dox was included in the cell culture media throughout the differentiation process.
Expression of the EXOC7 rescue gene
To express the EXOC7 rescue gene, we first created a SHC003BSD construct (deposited into Addgene, 133300) by replacing the puromycin selection marker of the SHC003 vector (Sigma, SHC003) with a blasticidin selection marker. We then created a SHC003BSD-DelGFP construct (deposited into Addgene, 133301) by deleting the GFP sequence from SHC003BSD using site-directed mutagenesis, introducing a SalI site at the same time. The human EXOC7 gene was amplified using PCR with a SalI site and three tandem copies of FLAG tags introduced at the 5′ end. A NheI site was introduced to the 3′ end of the PCR product. The EXOC7 PCR product was then subcloned into the SalI and NheI sites of the SHC003BSD-DelGFP vector. The resulting SHC003BSD-EXOC7 construct (deposited into Addgene, 133298) was transfected into 293T cells to produce lentiviral particles, which were used to infect preadipocytes. The transduced preadipocytes were selected using 10 μg/ml of blasticidin (Thermo Fisher Scientific, BP2647).
The primers for site-directed mutagenesis of the SHC003BSD vector were CAGATCCGCTAGCGCTACCGTCGACCCCCAGCATCCTGCAGAACG (forward) and CGTTCTGCAGGATGCTGGGGGTCGACGGTAGCGCTAGCGGATCTG (reverse). The Primers for amplification of the human EXOC7 gene were CGCGCTAGCCTAGCCACCATGGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATCGATTACAAGGATGACGATGACAAGATGATTCCCCCACAGGAG (forward) and GCGGTCGACTTAGGCAGAGGTGTCGAAAA (reverse).
Immunoblotting
Cells grown in 24-well plates were lysed in a SDS protein sample buffer. Cell lysates were resolved on 8% BisTris SDS-PAGE and probed using primary antibodies and horseradish peroxidase–conjugated secondary antibodies. Primary antibodies used in immunoblotting included mouse monoclonal anti-Exoc7 antibodies (Santa Cruz Biotechnology, sc-365825), mouse monoclonal anti-Sort1 antibodies (Santa Cruz Biotechnology, sc-376576), mouse monoclonal anti-PPARγ antibodies (Santa Cruz Biotechnology, sc-7273), mouse monoclonal anti-α-tubulin (DSHB, clone 12G10), rabbit monoclonal anti-Rab10 antibodies (Cell Signaling Technology, 8127), rabbit polyclonal anti-phospho-AKT (Ser-473) antibodies (Cell Signaling Technology, 9271), rabbit polyclonal anti-AKT antibodies (Cell Signaling Technology, 9272), rabbit monoclonal anti-AS160 antibodies (Cell Signaling Technology, 2670), rabbit monoclonal anti-phospho-AS160 (Thr-642) antibodies (Cell Signaling Technology, 8881), and mouse monoclonal anti-FLAG antibodies (Sigma, F1804).
Lipid droplet staining and analysis
To analyze lipid droplets using microscopy, preadipocytes and adipocytes grown in clear-bottom 96-well plates were fixed using 4% paraformaldehyde. To stain lipid droplets, Nile red (Sigma, N3013) was added to the cells at a final concentration of 5 μg/ml in PBS. After 15 min, the cells were washed three times with PBS and visualized on an Olympus IX81 microscope using a ×20 objective. Nile red fluorescence was detected using the FITC channel. For flow cytometry analysis of lipid droplets, cells were stained with Nile red in the presence of 10 μg/ml Hoechst 33342. Subsequently, the cells were detached using Accutase, and the fluorescence of Nile red and Hoechst 33342 was measured on a CyAN ADP analyzer. Data from populations of ∼5,000 cells were analyzed using FlowJo software based on experiments run in biological triplicates. Fluorescence of unstained WT preadipocytes was used as a blank control.
Statistical analysis
Statistical significance was calculated using datasets from at least three independent experiments. Student's t test was used to compare two experimental groups, whereas one-way analysis of variance was used to compare more than two groups.
Author contributions
S. W., L. C., and J. S. data curation; S. W., L. C., J. M., and J. S. formal analysis; S. W. validation; S. W., L. C., J. M., H. Y., and J. S. methodology; S. W. and J. S. writing-original draft; S. W., L. C., I. D., D. R. G., H. Y., and J. S. writing-review and editing; L. C., J. M., H. Y., and J. S. investigation; I. D., D. R. G., Y. T., Q. Y., and J. S. resources; I. D., D. R. G., Y. T., and Q. Y. software; D. R. G. and J. S. supervision; Y. T. and Q. Y. visualization; J. S. conceptualization; J. S. funding acquisition; J. S. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Gus Lienhard and Xiaowei Chen for insightful discussions, Dr. Shingo Kajimura for providing preadipocytes, Drs. Wei Guo and Fei Sun for providing exocyst genes, and Yan Ouyang for technical assistance. We thank the Light Microscopy Facility at University of Colorado Boulder for assistance with confocal microscopy.
This work was supported by National Institutes of Health Grants DK095367 (to J. S.), GM126960 (to J. S.), and AI108793 (Q. Y.); an American Diabetes Association basic science award (to J. S.); a fellowship from the Postdoctoral Overseas Training Program at Beijing University of Chinese Medicine (to S. W.); National Institutes of Health Institutional Predoctoral Training Grant GM088759 (to L. C.), an American Heart Association predoctoral fellowship (to D. R. G.), and a fellowship from the Undergraduate Research Opportunities Program at University of Colorado Boulder (to J. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2 and Tables S1 and S2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- SNARE
- soluble NSF attachment protein
- SM
- Sec1/Munc18
- gRNA
- guide RNA
- APC
- allophycocyanin
- dox
- doxycycline
- GMP-PNP
- guanosine 5′-[β,γ-imido]triphosphate.
References
- 1. Bryant N. J., Govers R., and James D. E. (2002) Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell Biol. 3, 267–277 10.1038/nrm782 [DOI] [PubMed] [Google Scholar]
- 2. Leto D., and Saltiel A. R. (2012) Regulation of glucose transport by insulin: traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 13, 383–396 10.1038/nrm3351 [DOI] [PubMed] [Google Scholar]
- 3. Antonescu C. N., McGraw T. E., and Klip A. (2014) Reciprocal regulation of endocytosis and metabolism. Cold Spring Harb. Perspect. Biol. 6, a016964 10.1101/cshperspect.a016964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bogan J. S., and Kandror K. V. (2010) Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Curr. Opin. Cell Biol. 22, 506–512 10.1016/j.ceb.2010.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang S., and Czech M. P. (2007) The GLUT4 glucose transporter. Cell Metab. 5, 237–252 10.1016/j.cmet.2007.03.006 [DOI] [PubMed] [Google Scholar]
- 6. Patki V., Buxton J., Chawla A., Lifshitz L., Fogarty K., Carrington W., Tuft R., and Corvera S. (2001) Insulin action on GLUT4 traffic visualized in single 3T3-l1 adipocytes by using ultra-fast microscopy. Mol. Biol. Cell 12, 129–141 10.1091/mbc.12.1.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vassilopoulos S., Esk C., Hoshino S., Funke B. H., Chen C. Y., Plocik A. M., Wright W. E., Kucherlapati R., and Brodsky F. M. (2009) A role for the CHC22 clathrin heavy-chain isoform in human glucose metabolism. Science 324, 1192–1196 10.1126/science.1171529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olson A. L., Knight J. B., and Pessin J. E. (1997) Syntaxin 4, VAMP2, and/or VAMP3/cellubrevin are functional target membrane and vesicle SNAP receptors for insulin-stimulated GLUT4 translocation in adipocytes. Mol. Cell. Biol. 17, 2425–2435 10.1128/MCB.17.5.2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Südhof T. C., and Rothman J. E. (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477 10.1126/science.1161748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen J., Tareste D. C., Paumet F., Rothman J. E., and Melia T. J. (2007) Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 128, 183–195 10.1016/j.cell.2006.12.016 [DOI] [PubMed] [Google Scholar]
- 11. Jewell J. L., Oh E., and Thurmond D. C. (2010) Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. Am. J. Physiol. Regu.l Integr. Comp. Physiol. 298, R517–R531 10.1152/ajpregu.00597.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brunger A. T., Weninger K., Bowen M., and Chu S. (2009) Single-molecule studies of the neuronal SNARE fusion machinery. Annu. Rev. Biochem. 78, 903–928 10.1146/annurev.biochem.77.070306.103621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwartz M. L., and Merz A. J. (2009) Capture and release of partially zipped trans-SNARE complexes on intact organelles. J. Cell Biol. 185, 535–549 10.1083/jcb.200811082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. James D. J., and Martin T. F. (2013) CAPS and Munc13: CATCHRs that SNARE vesicles. Front. Endocrinol. 4, 187 10.3389/fendo.2013.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baker R. W., and Hughson F. M. (2016) Chaperoning SNARE assembly and disassembly. Nat. Rev. Mol. Cell Biol. 17, 465–479 10.1038/nrm.2016.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rizo J., and Südhof T. C. (2012) The Membrane Fusion Enigma: SNAREs, Sec1/Munc18 Proteins, and Their Accomplices-Guilty as Charged? Annu. Rev. Cell Dev. Biol. 28, 279–308 10.1146/annurev-cellbio-101011-155818 [DOI] [PubMed] [Google Scholar]
- 17. Wickner W. (2010) Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu. Rev. Cell Dev. Biol. 26, 115–136 10.1146/annurev-cellbio-100109-104131 [DOI] [PubMed] [Google Scholar]
- 18. Pfeffer S., and Aivazian D. (2004) Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 5, 886–896 10.1038/nrm1500 [DOI] [PubMed] [Google Scholar]
- 19. Whyte J. R., and Munro S. (2002) Vesicle tethering complexes in membrane traffic. J. Cell Sci. 115, 2627–2637 [DOI] [PubMed] [Google Scholar]
- 20. Hughson F. M., and Reinisch K. M. (2010) Structure and mechanism in membrane trafficking. Curr. Opin. Cell Biol. 22, 454–460 10.1016/j.ceb.2010.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lepore D. M., Martínez-Núñez L., and Munson M. (2018) Exposing the elusive exocyst structure. Trends Biochem. Sci. 43, 714–725 10.1016/j.tibs.2018.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mei K., Li Y., Wang S., Shao G., Wang J., Ding Y., Luo G., Yue P., Liu J. J., Wang X., Dong M. Q., Wang H. W., and Guo W. (2018) Cryo-EM structure of the exocyst complex. Nat. Struct. Mol. Biol. 25, 139–146 10.1038/s41594-017-0016-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo W., Roth D., Walch-Solimena C., and Novick P. (1999) The exocyst is an effector for Sec4p, targeting secretory vesicles to sites of exocytosis. EMBO J. 18, 1071–1080 10.1093/emboj/18.4.1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin-Urdiroz M., Deeks M. J., Horton C. G., Dawe H. R., and Jourdain I. (2016) The exocyst complex in health and disease. Front. Cell Dev. Biol. 4, 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Picco A., Irastorza-Azcarate I., Specht T., Böke D., Pazos I., Rivier-Cordey A. S., Devos D. P., Kaksonen M., and Gallego O. (2017) The in vivo architecture of the exocyst provides structural basis for exocytosis. Cell 168, 400–412.e18 10.1016/j.cell.2017.01.004 [DOI] [PubMed] [Google Scholar]
- 26. Hsu S. C., Hazuka C. D., Roth R., Foletti D. L., Heuser J., and Scheller R. H. (1998) Subunit composition, protein interactions, and structures of the mammalian brain sec6/8 complex and septin filaments. Neuron 20, 1111–1122 10.1016/S0896-6273(00)80493-6 [DOI] [PubMed] [Google Scholar]
- 27. Wu B., and Guo W. (2015) The exocyst at a glance. J. Cell Sci. 128, 2957–2964 10.1242/jcs.156398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heider M. R., and Munson M. (2012) Exorcising the exocyst complex. Traffic 13, 898–907 10.1111/j.1600-0854.2012.01353.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen X. W., Leto D., Xiao J., Goss J., Wang Q., Shavit J. A., Xiong T., Yu G., Ginsburg D., Toomre D., Xu Z., and Saltiel A. R. (2011) Exocyst function regulated by effector phosphorylation. Nat. Cell Biol. 13, 580–588 10.1038/ncb2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen D., Yuan H., Hutagalung A., Verma A., Kümmel D., Wu X., Reinisch K., McNew J. A., and Novick P. (2013) The synaptobrevin homologue Snc2p recruits the exocyst to secretory vesicles by binding to Sec6p. J. Cell Biol. 202, 509–526 10.1083/jcb.201211148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dubuke M. L., Maniatis S., Shaffer S. A., and Munson M. (2015) The exocyst subunit Sec6 interacts with assembled exocytic SNARE complexes. J. Biol. Chem. 290, 28245–28256 10.1074/jbc.M115.673806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rivera-Molina F., and Toomre D. (2013) Live-cell imaging of exocyst links its spatiotemporal dynamics to various stages of vesicle fusion. J. Cell Biol. 201, 673–680 10.1083/jcb.201212103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Polgar N., and Fogelgren B. (2018) Regulation of cell polarity by exocyst-mediated trafficking. Cold Spring Harb. Perspect. Biol. 10, a031401 10.1101/cshperspect.a031401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen X. W., Leto D., Xiong T., Yu G., Cheng A., Decker S., and Saltiel A. R. (2011) A Ral GAP complex links PI 3-kinase/Akt signaling to RalA activation in insulin action. Mol. Biol. Cell 22, 141–152 10.1091/mbc.e10-08-0665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Inoue M., Chang L., Hwang J., Chiang S. H., and Saltiel A. R. (2003) The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature 422, 629–633 10.1038/nature01533 [DOI] [PubMed] [Google Scholar]
- 36. Sano H., Peck G. R., Blachon S., and Lienhard G. E. (2015) A potential link between insulin signaling and GLUT4 translocation: association of Rab10-GTP with the exocyst subunit Exoc6/6b. Biochem. Biophys. Res. Commun. 465, 601–605 10.1016/j.bbrc.2015.08.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ewart M. A., Clarke M., Kane S., Chamberlain L. H., and Gould G. W. (2005) Evidence for a role of the exocyst in insulin-stimulated Glut4 trafficking in 3T3-L1 adipocytes. J. Biol. Chem. 280, 3812–3816 10.1074/jbc.M409928200 [DOI] [PubMed] [Google Scholar]
- 38. Inoue M., Chiang S. H., Chang L., Chen X. W., and Saltiel A. R. (2006) Compartmentalization of the exocyst complex in lipid rafts controls Glut4 vesicle tethering. Mol. Biol. Cell 17, 2303–2311 10.1091/mbc.e06-01-0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lizunov V. A., Lisinski I., Stenkula K., Zimmerberg J., and Cushman S. W. (2009) Insulin regulates fusion of GLUT4 vesicles independent of Exo70-mediated tethering. J. Biol. Chem. 284, 7914–7919 10.1074/jbc.M806460200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Inoue M., Akama T., Jiang Y., and Chun T. H. (2015) The exocyst complex regulates free fatty acid uptake by adipocytes. PLoS ONE 10, e0120289 10.1371/journal.pone.0120289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shalem O., Sanjana N. E., and Zhang F. (2015) High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 16, 299–311 10.1038/nrg3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gulbranson D. R., Davis E. M., Demmitt B. A., Ouyang Y., Ye Y., Yu H., and Shen J. (2017) RABIF/MSS4 is a Rab-stabilizing holdase chaperone required for GLUT4 exocytosis. Proc. Natl. Acad. Sci. U.S.A. 114, E8224–E8233 10.1073/pnas.1712176114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gulbranson D. R., Crisman L., Lee M., Ouyang Y., Menasche B. L., Demmitt B. A., Wan C., Nomura T., Ye Y., Yu H., and Shen J. (2019) AAGAB controls AP2 adaptor assembly in clathrin-mediated endocytosis. Dev. Cell 50, 436–446.e5 10.1016/j.devcel.2019.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kioumourtzoglou D., Pryor P. R., Gould G. W., and Bryant N. J. (2015) Alternative routes to the cell surface underpin insulin-regulated membrane trafficking of GLUT4. J. Cell Sci. 128, 2423–2429 10.1242/jcs.166561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li W., Xu H., Xiao T., Cong L., Love M. I., Zhang F., Irizarry R. A., Liu J. S., Brown M., and Liu X. S. (2014) MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 10.1186/s13059-014-0554-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Greenspan P., Mayer E. P., and Fowler S. D. (1985) Nile red: a selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 100, 965–973 10.1083/jcb.100.3.965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Spiegelman B. M. (1998) PPAR-γ: adipogenic regulator and thiazolidinedione receptor. Diabetes 47, 507–514 10.2337/diabetes.47.4.507 [DOI] [PubMed] [Google Scholar]
- 49. Bai L., Wang Y., Fan J., Chen Y., Ji W., Qu A., Xu P., James D. E., and Xu T. (2007) Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metab. 5, 47–57 10.1016/j.cmet.2006.11.013 [DOI] [PubMed] [Google Scholar]
- 50. Humphrey S. J., Yang G., Yang P., Fazakerley D. J., Stöckli J., Yang J. Y., and James D. E. (2013) Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 17, 1009–1020 10.1016/j.cmet.2013.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sano H., Eguez L., Teruel M. N., Fukuda M., Chuang T. D., Chavez J. A., Lienhard G. E., and McGraw T. E. (2007) Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 5, 293–303 10.1016/j.cmet.2007.03.001 [DOI] [PubMed] [Google Scholar]
- 52. Eguez L., Lee A., Chavez J. A., Miinea C. P., Kane S., Lienhard G. E., and McGraw T. E. (2005) Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2, 263–272 10.1016/j.cmet.2005.09.005 [DOI] [PubMed] [Google Scholar]
- 53. Pan X., Zaarur N., Singh M., Morin P., and Kandror K. V. (2017) Sortilin and retromer mediate retrograde transport of Glut4 in 3T3-L1 adipocytes. Mol. Biol. Cell 28, 1667–1675 10.1091/mbc.e16-11-0777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shi J., and Kandror K. V. (2005) Sortilin is essential and sufficient for the formation of Glut4 storage vesicles in 3T3-L1 adipocytes. Dev. Cell 9, 99–108 10.1016/j.devcel.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 55. Skorobogatko Y., Dragan M., Cordon C., Reilly S. M., Hung C. W., Xia W., Zhao P., Wallace M., Lackey D. E., Chen X. W., Osborn O., Bogner-Strauss J. G., Theodorescu D., Metallo C. M., Olefsky J. M., and Saltiel A. R. (2018) RalA controls glucose homeostasis by regulating glucose uptake in brown fat. Proc. Natl. Acad. Sci. U.S.A. 115, 7819–7824 10.1073/pnas.1801050115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fukai S., Matern H. T., Jagath J. R., Scheller R. H., and Brunger A. T. (2003) Structural basis of the interaction between RalA and Sec5, a subunit of the sec6/8 complex. EMBO J. 22, 3267–3278 10.1093/emboj/cdg329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yue P., Zhang Y., Mei K., Wang S., Lesigang J., Zhu Y., Dong G., and Guo W. (2017) Sec3 promotes the initial binary t-SNARE complex assembly and membrane fusion. Nat. Commun. 8, 14236 10.1038/ncomms14236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morgera F., Sallah M. R., Dubuke M. L., Gandhi P., Brewer D. N., Carr C. M., and Munson M. (2012) Regulation of exocytosis by the exocyst subunit Sec6 and the SM protein Sec1. Mol. Biol. Cell 23, 337–346 10.1091/mbc.e11-08-0670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ahmed S. M., Nishida-Fukuda H., Li Y., McDonald W. H., Gradinaru C. C., and Macara I. G. (2018) Exocyst dynamics during vesicle tethering and fusion. Nat. Commun. 9, 5140 10.1038/s41467-018-07467-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Heider M. R., Gu M., Duffy C. M., Mirza A. M., Marcotte L. L., Walls A. C., Farrall N., Hakhverdyan Z., Field M. C., Rout M. P., Frost A., and Munson M. (2016) Subunit connectivity, assembly determinants and architecture of the yeast exocyst complex. Nat. Struct. Mol. Biol. 23, 59–66 10.1038/nsmb.3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Czech M. P. (2017) Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 23, 804–814 10.1038/nm.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thomou T., Mori M. A., Dreyfuss J. M., Konishi M., Sakaguchi M., Wolfrum C., Rao T. N., Winnay J. N., Garcia-Martin R., Grinspoon S. K., Gorden P., and Kahn C. R. (2017) Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542, 450–455 10.1038/nature21365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Patel S. J., Sanjana N. E., Kishton R. J., Eidizadeh A., Vodnala S. K., Cam M., Gartner J. J., Jia L., Steinberg S. M., Yamamoto T. N., Merchant A. S., Mehta G. U., Chichura A., Shalem O., Tran E., et al. (2017) Identification of essential genes for cancer immunotherapy. Nature 548, 537–542 10.1038/nature23477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang T., Birsoy K., Hughes N. W., Krupczak K. M., Post Y., Wei J. J., Lander E. S., and Sabatini D. M. (2015) Identification and characterization of essential genes in the human genome. Science (New York, N.Y.) 350, 1096–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Menasche B. L., Crisman L., Gulbranson D. R., Davis E. M., Yu H., and Shen J. (2019) Fluorescence activated cell sorting (FACS) in genome-wide genetic screening of membrane trafficking. Curr. Protoc. Cell Biol. 82, e68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Barger C. J., Branick C., Chee L., and Karpf A. R. (2019) Pan-cancer analyses reveal genomic features of FOXM1 overexpression in cancer. Cancers 11, E251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rai A., Oprisko A., Campos J., Fu Y., Friese T., Itzen A., Goody R. S., Gazdag E. M., and Müller M. P. (2016) bMERB domains are bivalent Rab8 family effectors evolved by gene duplication. Elife 5, e18675 10.7554/eLife.18675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Blot V., and McGraw T. E. (2008) Use of quantitative immunofluorescence microscopy to study intracellular trafficking: studies of the GLUT4 glucose transporter. Methods Mol. Biol. 457, 347–366 10.1007/978-1-59745-261-8_26 [DOI] [PubMed] [Google Scholar]
- 69. Jewell J. L., Oh E., Ramalingam L., Kalwat M. A., Tagliabracci V. S., Tackett L., Elmendorf J. S., and Thurmond D. C. (2011) Munc18c phosphorylation by the insulin receptor links cell signaling directly to SNARE exocytosis. J. Cell Biol. 193, 185–199 10.1083/jcb.201007176 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.