

In this study, Greenstein et al. set out to define the spatial encoding signals within euchromatin that act to limit heterochromatin spreading. Using molecular and cell-based assays in fission yeast, the authors report that heterochromatin repulsion is locally encoded by Set1/COMPASS on certain actively transcribed genes and that this protective role is most prominent at heterochromatin islands, small domains interspersed in euchromatin that regulate cell fate specifiers.
Keywords: H3K4 methylation, Set1/COMPASS, facultative heterochromatin, gene orientation, heterochromatin spreading
Abstract
Protection of euchromatin from invasion by gene-repressive heterochromatin is critical for cellular health and viability. In addition to constitutive loci such as pericentromeres and subtelomeres, heterochromatin can be found interspersed in gene-rich euchromatin, where it regulates gene expression pertinent to cell fate. While heterochromatin and euchromatin are globally poised for mutual antagonism, the mechanisms underlying precise spatial encoding of heterochromatin containment within euchromatic sites remain opaque. We investigated ectopic heterochromatin invasion by manipulating the fission yeast mating type locus boundary using a single-cell spreading reporter system. We found that heterochromatin repulsion is locally encoded by Set1/COMPASS on certain actively transcribed genes and that this protective role is most prominent at heterochromatin islands, small domains interspersed in euchromatin that regulate cell fate specifiers. Sensitivity to invasion by heterochromatin, surprisingly, is not dependent on Set1 altering overall gene expression levels. Rather, the gene-protective effect is strictly dependent on Set1's catalytic activity. H3K4 methylation, the Set1 product, antagonizes spreading in two ways: directly inhibiting catalysis by Suv39/Clr4 and locally disrupting nucleosome stability. Taken together, these results describe a mechanism for spatial encoding of euchromatic signals that repel heterochromatin invasion.
Heterochromatin is a conserved nuclear ultrastructure (Rea et al. 2000) that enacts genome partitioning by repressing transcription and recombination at repetitive sequences and structural elements, as well as genetic information not pertaining to the specified cell fate. Once seeded at specific sequences (Hall et al. 2002; Jia et al. 2004; Reyes-Turcu et al. 2011), heterochromatin is subsequently propagated in cis over qualitatively distinct regions of the chromosome in a process termed spreading. Positional regulation of heterochromatin is key to determining and remembering cell fate decisions. Boundary regions often separate adjacent heterochromatin and euchromatin domains, reinforcing the distinct signals and functional environments on each side and countering the intrinsic propensity for heterochromatin to invade and silence genes. Major mechanisms of boundary formation fall into three broad classes: (1) recruitment of factors that directly antagonize the opposite state (for example, by removal of state-specific signals on chromatin) (Ayoub et al. 2003; Schlichter and Cairns 2005; Lan et al. 2007; Trewick et al. 2007; Braun et al. 2011), (2) promotion of the original state by either depositing or protecting such signals (Wang et al. 2013, 2015; Sadeghi et al. 2015; Verrier et al. 2015), or (3) structural constraint via recruitment of DNA-binding proteins that tether heterochromatin regions to the nuclear periphery (Bell and Felsenfeld 1999; Kurukuti et al. 2006; Noma et al. 2006). Despite the varied modalities used in boundary formation, containment is not absolute. This is evidenced by the observation that boundaries can be overcome by modest dosage changes in heterochromatin factors (Noma et al. 2006; Ceol et al. 2011), which leads to the silencing of genes critical to normal cellular function.
In addition to constitutive heterochromatin found at centromeres, telomeres, and other repetitive sequences, repressed domains also form at additional genomic locations in response to developmental and environmental signals (Wen et al. 2009; Zofall et al. 2012; Zhu et al. 2013). These facultative heterochromatin domains are often embedded in euchromatic regions and silence developmental genes in a lineage-specific manner (Wen et al. 2009). Resulting from response to changing stimuli, the final extent of facultative domains can change over time, expanding to different degrees (Wen et al. 2009) and even contracting (McDonald et al. 2011) in genomic space, though how this is achieved is not well understood. Facultative domain size may be tuned at the level of the heterochromatin spreading reaction (Hathaway et al. 2012) and/or the activities promoting its containment or disassembly. While little is known about the former, several models, beyond those known to operate at constitutive boundaries (Guelen et al. 2008; Zofall et al. 2012), could be invoked to explain the latter.
How might euchromatin regulate heterochromatin spreading at facultative sites or respond to its expansion beyond constitutive domains? One of the defining features of euchromatin is the presence of active genes. It is thought that transcription from active genes is incompatible with heterochromatin formation (Scott et al. 2006). Multiple direct effects of transcription have been proposed to interfere with heterochromatin assembly. These include nucleosome turnover (eviction) by transcribing polymerase, formation of nucleosome-depleted regions at transcriptional units, or steric interference by transcription-associated complexes (Noma et al. 2006; Garcia et al. 2010; Aygün et al. 2013). Furthermore, we understand that unique molecular signatures characterize euchromatin and heterochromatin states and are critical to their formation. Heterochromatin is marked by methylation of histone 3 at lysine 9 or lysine 27 (H3K9me and H3K27me, respectively) and hypoacetylation of various histone lysine residues. In contrast, euchromatin features H3K4me, H3K36me, and histone hyperacetylation (Nielsen et al. 2001; Guelen et al. 2008). Multiple studies have documented the apparent mutual exclusion of H3K9me- and H3K4me-marked regions (Litt et al. 2001; Noma et al. 2001; Cam et al. 2005; Guelen et al. 2008) and the requirement for removal of signals associated with the opposite state (Lan et al. 2007; Li et al. 2008). While we are beginning to understand how this dichotomy is formed, it still remains unclear whether this is a cause or consequence of separating heterochromatin and euchromatin.
We aimed to investigate the role of euchromatic signals in regulating the extent of spreading in fission yeast, a well-characterized model system for the study of heterochromatin formation, which shares critical features with the processes found in metazoans. Fission yeast form constitutive heterochromatin marked by H3K9me at centromeres, telomeres, and the mating type (MAT) locus. Boundary formation occurs at pericentromeric regions and the MAT locus via at least two mechanisms: tethering to the nuclear periphery through binding of TFIIIC proteins to B-box element sequences in boundary regions (Noma et al. 2006) as well as specific enrichment of a JmjC domain-containing protein, Epe1 (Ayoub et al. 2003; Zofall and Grewal 2006; Trewick et al. 2007; Braun et al. 2011), which recruits additional downstream boundary effectors. In addition to these constitutive sites, facultative heterochromatin forms at developmentally regulated meiotic genes in regions surrounded by canonical euchromatin, which are partially dependent on Epe1 for containment (Zofall et al. 2012; Wang et al. 2015). Using the well-characterized MAT locus boundary as a model for euchromatic invasion, we found that active gene units could repel spreading and that this function depends on the H3K4 methylase complex Set1/COMPASS. Set1 is the catalytic subunit of COMPASS and is responsible for monomethylation, dimethylation, and trimethylation of H3K4 in vivo. It is recruited by RNA polymerase and forms a characteristic pattern of H3K4 methylation states over genes, with H3K4me3 near the transcription start site (TSS) and H3K4me2 in the gene body (for review, see Shilatifard 2012). We show that rather than acting as a global antagonist of spreading, like Epe1 or the histone acetyltransferase Mst2 (Wang et al. 2015), Set1 regulates spreading at gene-rich environments such as heterochromatin islands. Set1 does not exert its euchromatin protective function by modulating steady-state transcript levels. Rather, it acts via two separate mechanisms, both dependent on its catalytic activity: (1) the disruption of nucleosome stability and (2) catalytic inhibition of the sole fission yeast H3K9 methylase Suv39/Clr4, by the Set1 product H3K4me. This study provides a mechanism for the encoding of spatial cues within euchromatin that contain heterochromatin expansion.
Results
Genes can function as a barrier to heterochromatin spreading
To investigate heterochromatin invasion into euchromatin, we used our previously described heterochromatin spreading sensor (HSS) (Al-Sady et al. 2016; Greenstein et al. 2018) in the euchromatic region proximal to the MAT inverted repeat right (IR-R) boundary (Ayoub et al. 2000). This HSS system contains two central components: (1) the spreading sensor, a monomeric Kusabira-Orange 2 fluorescent protein driven by the validated ade6 promoter, hereafter referred to as “orange,” integrated 0.7 kb outside IR-R, and (2) the control, a E2Crimson fluorescent protein driven by the same promoter, hereafter referred to as “red,” integrated at a constitutive euchromatic locus (Fig. 1A; Supplemental Table 1; Greenstein et al. 2018). The IR-R is a well-described boundary system that can be easily manipulated (Garcia et al. 2015). Precisely controlling its disruption leads to an excellent model system for identifying determinants within euchromatin that regulate heterochromatin spreading. With the HSS system, we used flow cytometry to capture information from tens of thousands of single cells. We divided “orange” by “red” for each cell to normalize for cell-to-cell transcription and translation noise, allowing us to quantify heterochromatin-specific gene silencing at the “orange” reporter over the population.
Figure 1.

Genes repel heterochromatin across boundaries in a manner dependent on Set1/COMPASS. (A) An overview of the heterochromatin spreading sensor (HSS) outside the MAT locus IR-R boundary with transcriptional reporters encoding fluorescent proteins as sensor (“orange”) and control (“red”). IR-R (depicted as purple arrow) uses at least two independent pathways dependent on Epe1 and TFIIIC, respectively, to contain spreading of H3K9 methylation via Suv39/Clr4. IR-R function can be abrogated by deletion of epe1 and removing the B-box-binding sequences for TFIIIC. (B) Histogram of “orange” signal in a WT boundary background normalized to Δclr4. (C) Histogram of “orange” signal in boundaryC (Δepe1) background normalized to the corresponding WT (epe1+) strain. (D) Histogram of normalized “orange” signal in Δboundary (Δepe1 ΔB-box) background as in C. (E) Illustration depicting genetic screen for modulators of gene-mediated heterochromatin repulsion. An HSS variant at the ura4 locus was crossed to ∼400 gene deletions. The resulting strains were analyzed by flow cytometry. (F) Histograms plotted as in C of normalized “orange” signal in nucleation-gated cells in WT and Δset1. (G) Data table. (Left) Fraction of cells that experienced silencing at “orange.” Two thresholds were applied: a cutoff for nucleation at “green” and a cutoff for silencing at “orange.” Cells that met both criteria were counted as repressed. (Right) Odds ratio, calculated by Fisher's exact test, comparing the odds of being the silenced “off” state for a cell in the Set1C mutant relative to wild-type populations. (H) Histogram of normalized “orange” signal Δset1 in the WT boundary background as in C. (I) Histogram of normalized “orange” signal in boundaryCΔset1 background as in C. (J) Histogram of normalized “orange” signal in WT ΔboundaryΔset1 background as in C. All 1D histograms are plotted as the mean ± 3SD of 300 bootstrap iterations for combined data from the indicated number of biological isolates (n). Signal is normalized to the median signal from a Δclr4 or corresponding WT (epe1+) strain control to represent the maximum fluorescence in the absence of heterochromatin (x = 1). A threshold for silencing (dashed red line) represents the mean signal of the WT strain less 2SD with the exception of F, where the threshold for silencing in nucleation+ cells was determined as mean less 1SD of the “orange” signal from the Δclr4 strain. The faction of cells below this cutoff was calculated (%off).
We first examined the normalized orange fluorescence of a strain with a WT boundary (epe1+, B-box+) and detected no silencing in the population distribution (Fig. 1B), as expected (see legend for Fig. 1; Greenstein et al. 2018). We define a threshold for silencing as the mean of the appropriate WT (epe1+) strain less two standard deviations (see dashed red line). We next compromised one or both of the pathways required for containment of spreading at IR-R (Ayoub et al. 2003; Trewick et al. 2007; Garcia et al. 2015) and assessed the effect on “orange” silencing. Consistent with previous results (Garcia et al. 2015), little silencing is detected in Δepe1 isolates harboring a partially compromised boundary (referred to hereafter as boundaryC) (Fig. 1C, solid line histograms). In a fully compromised boundary, absent both epe1 and the five B-box sequence elements contained within IR-R (referred to hereafter as Δboundary) (Noma et al. 2006), we detected increased silencing (Fig. 1D, dashed line histograms). However, even in the Δboundary background, >80% of cells in the population fully express “orange.” Given this result, and the observation that H3K9me2 spreading declines sharply over endogenous IR-R-bordering genes (Garcia et al. 2015), we wondered whether other activities beyond boundaries, possibly centered on active genes, repel spreading.
Set1/COMPASS regulates genic protection from heterochromatin spreading
In order to identify potential factors that regulate gene-mediated repulsion of heterochromatin spreading, we designed a genetic screen to query the effect of gene deletions on silencing measured via our reporters. We conducted the screen in the context of the HSS embedded at the euchromatic ura4 locus (Greenstein et al. 2018), downstream from an ectopically placed RNAi-based heterochromatin nucleator (Fig. 1E; Marina et al. 2013). This construct can generate spreading up to 8 kb downstream over two endogenous and two reporter genes (Supplemental Fig. S1A; Greenstein et al. 2018), representing about one-third the size of the MAT locus. Importantly, this 8-kb region is not protected by a natural cis-encoded boundary, eliminating the need to remove any boundary factors, which avoids confounding global effects on growth in the screen. Since nucleation is less robust at this locus than at endogenous heterochromatin domains (Greenstein et al. 2018), we exploited the presence of a nucleator-proximal third reporter cassette encoding “green” at this locus. Based on this reporter, we can apply a computational gate to isolate successfully nucleated cells (greenOFF) (described in Greenstein et al. 2018) and assess their spreading state at the “orange” reporter, 3 kb downstream from “green.” In the WT background, the nucleation gated “orange” signal in this strain resembles the behavior seen in the Δboundary IR-R HSS strain (Fig. 1, cf. F [black line] and D), exhibiting both gene silencing and fully expressed states.
We crossed this ura4-HSS background strain to a curated ∼400-gene subset of the S. pombe deletion library enriched for nuclear factors (Fig. 1E) and measured reporter fluorescence from the resultant strains via flow cytometry. For each strain, we plotted a 2D histogram of red-normalized orange versus green fluorescence (Supplemental Fig. S1B) and calculated the fraction of cells that experienced silencing at “orange.” Silencing in this context is defined as the fraction of all cells that met both the greenOFF criteria for nucleation (blue line) and had orange signal below the mean less one standard deviation of the matched Δclr4 strain (red line).
Upon analysis of this data set, we noticed five genes whose absence had the same characteristic effect of increased silencing at the spreading reporter: ash2, swd1, swd3, spf1, and set1 (Fig. 1F,G; Supplemental Fig. S1B). To probe the significance of increased silencing in these mutants, we performed a Fisher's exact test and found the odds of being in the “off” state for the mutants to be three to four times higher than for wild type (Fig. 1G). In contrast, this odds ratio comparing the other mutants with Δset1 was close to 1 (Supplemental Fig. S1B), indicating a similar likelihood of silencing. These genes are five members of the Set1/COMPASS complex, which catalyzes H3K4me and deposits H3K4me3 at active gene promoters (Miller et al. 2001; Noma and Grewal 2002; Santos-Rosa et al. 2002; Roguev et al. 2003). Of the remaining complex members, Δswd2 did not grow and Δsdc1 was not in the screen, while Δshg1 showed no phenotype, consistent with other studies, which denote it as marginally associated with the complex (Roguev et al. 2003). All five gene deletions were validated by independent knockout in the parental reporter background (Supplemental Fig. S1B).
Given this result, we sought to test whether the removal of Set1C might have a similar effect at the boundary-proximal locus. While there was not a major effect of Δset1 on reporter strains with a WT boundary (Fig. 1H), both boundaryC-proximal (Fig. 1I) and Δboundary-proximal (Fig. 1J) reporters experienced a significant increase in silencing in Δset1, supporting the hypothesis that Set1/COMPASS enacts a heterochromatin-protective function.
Endogenous IR-R-proximal genes regulate H3K9me2 spreading and silencing
In order to probe the effect of Δset1 on euchromatic invasion at heterochromatic sites genome-wide, we performed chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq) with antibodies against H3K4me3 and H3K9me2 in WT, Δepe1, and Δepe1Δset1 strains that contained no reporters (Fig. 2A). We did not perform H3K4me3 ChIP-seq for Δset1 isolates due to the absence of H3K4me, which we validated by ChIP-qPCR (Supplemental Fig. S2A). Signal tracks for each genotype are plotted as mean and 95% confidence interval of two to four replicates.
Figure 2.

Set1 regulates H3K9me spreading at the IR-R-proximal region. (A) Overview of ChIP-seq experiment. (B, top and middle tracks) Input-normalized (see the Materials and Methods) ChIP-seq signal tracks and gene annotations for the IR-R-proximal region. (Bottom track) H3K9me2 ChIP-seq data sets were independently normalized to signal from a sample containing merged data from both WT isolates. Tracks are represented as mean (line) and 95% confidence interval (shaded region) per genotype (WT n = 2, Δepe1 n = 3, Δepe1Δset1 n = 4; each n represents a single colony deriving from a parental knockout for each genotype). P-value track represents regions above a threshold for H3K9me2 enrichment over background (gray boxes, 300-bp bins; absence of box indicates values below threshold). P-values for differences between genotypes are indicated in colors according to the scale. (C) RT-qPCR analysis for genes in the IR-R-proximal region. Error bars represent 1SD of three replicate cultures from single colonies deriving from one parent isolate. (n.s.) P > 0.05; (**) P < 0.01 (t-test). (D, top) Overview of the rpl401p:HSS. (Middle) Histogram plots as in Figure 1 of normalized “orange” signal from set1+ (purple) and Δset1 (blue) rpl401p:HSS boundaryC isolates. (Bottom) Histogram plots of normalized “orange” signal from rpl401p:HSS Δboundary isolates. (E, top) Overview of the mtd1p:HSS. (Bottom) Histogram plots of normalized “orange” signal from mtd1p:HSS boundaryC isolates.
Given that our above results show set1-dependent heterochromatin containment at our reporter gene, we asked whether the removal of set1 would affect H3K9me2 spreading beyond IR-R (Fig. 2B). Unlike WT (black line), both Δepe1 (purple line) and Δepe1Δset1 (blue line) display similar and significant enrichment for H3K9me2 immediately next to IR-R, as seen by their closely superimposed means and confidence intervals (for clarity, we did not plot MAT internal traces). As distance increases from IR-R, the traces begin to separate, with H3K9me2 signal from Δepe1Δset1 strains exceeding that from Δepe1 and WT. This separation is most evident over the open reading frame of rpl401 (Fig. 2B, inset) and is statistically significant as indicated by the separation of the 95% confidence bounds and the P-value analysis below the traces. Interestingly, this gene is also highly enriched for H3K4me3. This increase in H3K9me2 spreading significantly affects the transcript levels of genes proximal to the separation of the H3K9me2 traces in Δepe1Δset1 versus Δepe1 strains (Fig. 2C), but not genes either immediately by the compromised boundary or beyond rpl401. We wanted to test the role of endogenous gene promoters in effecting the Set1-dependent decline in H3K9me2 spreading and chose two genes, mtd1 and rpl401, around which spreading is most strongly impaired. To do so, we first modified the original ade6p:HSS to express “orange” from the rpl401 promoter at the same locus (Fig. 2D). The rpl401 gene promoter effectively repels spreading in the context of a compromised (boundaryC) (Fig. 2D, middle) or fully abrogated (Δboundary) (Fig. 2D, bottom) IR-R boundary. However, the removal of set1 (Δset1) resulted in complete rpl401p:HSS repression in a Δboundary context (Fig. 2D). In the case of mtd1p, instead of inserting it at the original reporter locus, we replaced the endogenous mtd1 open reading frame with “orange” to generate an mtd1p:HSS (Fig. 2E), which is located 2.5 kb from the edge of IR-R. Just like ade6p:HSS and rpl401p:HSS at the IR-R-proximal locus, the mtd1p:HSS also displays genic barrier function that is set1-dependent (Fig. 2E). Thus, for all the promoters tested, formation of a spreading barrier is highly sensitive to the presence of Set1. Given these results, we conclude that Set1 contributes to the containment of spreading into the euchromatic region outside of IR-R in the case of boundary failure.
Set1 contributes to spreading containment at facultative but not constitutive heterochromatin
We next examined other constitutive heterochromatin loci, centromeres and telomeres, for set1-mediated spreading effects. Broadly, Δset1 did not significantly increase the extent of spreading already evident in Δepe1 at such loci. Marginally increased spreading was detected in Δepe1Δset1 beyond the boundaries of pericentromeric heterochromatin on chromosomes II and III (Fig. 3A; Supplemental Fig. S2B), while at the right subtelomere I and at the pericentromere of chromosome I spreading was in fact reduced in Δepe1Δset1 relative to Δepe1 (Fig. 3A; Supplemental Fig. S2B,C).
Figure 3.

Set1 regulates spreading at euchromatic heterochromatin islands. (A) A global analysis comparing H3K9me2 accumulation measured by ChIP-seq in Δepe1Δset1 and Δepe1 genotypes. The mean value of input-normalized H3K9me2 ChIP signal per 300-bp bin was calculated for each genotype. For bins containing H3K9me2 signal above 1.5 times the global background, the log2 ratio of Δepe1Δset1 over Δepe1 is plotted by chromosome (black line). Bins where this ratio exceeds a cutoff of three times enriched [red lines at y = ±log2(3)] in Δepe1Δset1 (blue) or Δepe1 (purple) are plotted as individual points. Pericentromeres, centromeres, and telomeres are demarcated by red-shaded boxes. H3K9me2 merged peak calls from the WT strains are annotated in red along the chromosome. An asterisk denotes regions where mean signal is increased due to disproportionate enrichment in a single isolate. (B) Signal tracks analysis at euchromatic heterochromatin islands and ectopic domains for H3K4me3 and H3K9me2 ChIP-Seq as in Figure 2B. P-values calculated as in Figure 2B except with 1200-bp bins. (C) H3K9me2 ChIP-qPCR measured at heterochromatin islands and ectopic domains in wild type (black) and Δset1 (light blue). Error bars represent 1SD from two technical replicate ChIPs. Replicate values are plotted as individual points.
Given the major role of Set1/COMPASS at genes and the enrichment of H3K4me in canonical euchromatin (Noma et al. 2001), we wondered whether Set1 might regulate spreading at facultative heterochromatin sites, islands of H3K9me embedded in gene-rich euchromatin (Zofall et al. 2012; Gallagher et al. 2018). In our relatively stringent ChIP-seq analysis (see the Materials and Methods), we detected ∼10 heterochromatin islands (8 and 13 in both WT replicates). These islands display TSS-proximal H3K4me and low to intermediate levels of H3K9me2 (Fig. 3B; Zofall et al. 2012). Our analysis found an increase in the number of known heterochromatin islands and novel ectopic H3K9me2 peaks (sites where WT shows no significant H3K9me2 enrichment) in both Δepe1 and Δepe1Δset1 mutants (Fig. 3A; Supplemental Fig. S3A). However, we found that heterochromatin spreading is exacerbated significantly in Δepe1Δset1 compared with Δepe1 at several sites, which include several known heterochromatin islands (Fig. 3B). Importantly, the ability of Set1 to antagonize H3K9me heterochromatin does not strictly depend on Epe1, as we could observe enhanced H3K9me2 enrichment at heterochromatin islands and ectopic sites in Δset1 alone (Fig. 3C). Since transcription at islands and island-proximal genes is already extremely low in wild type (cf. Fig. 2C and Supplemental Fig. S3B), partly due to locally acting RNA processing pathways (Lee et al. 2013; Egan et al. 2014; Sugiyama et al. 2016), it is not surprising that we observed only a mild further effect on transcript levels in Δepe1Δset1 (Supplemental Fig. S3B). These results describe a critical role for set1 in spreading containment at gene-rich euchromatin with prominent H3K4me3 peaks, but not at gene-poor constitutive heterochromatin regions.
Set1 functions in spreading containment independent of regulating steady-state transcription
What might be the mechanisms by which Set1 confers barrier activity to genes? We considered three pathways that could account for this activity: (1) regulation of steady state transcription by Set1, where altered frequency of RNA polymerase II passage would disrupt spreading; (2) interference with heterochromatin spreading by the Set1/COMPASS enzymatic product, H3K4me; or (3) a nonenzymatic effect of chromatin-bound Set1, consistent with prior reports (Lorenz et al. 2014; Mikheyeva et al. 2014). We summarize the possible mechanisms in Figure 4A and in what follows we test whether and how mechanisms 1–3 contribute to the observed Set1-dependent barrier activity.
Figure 4.
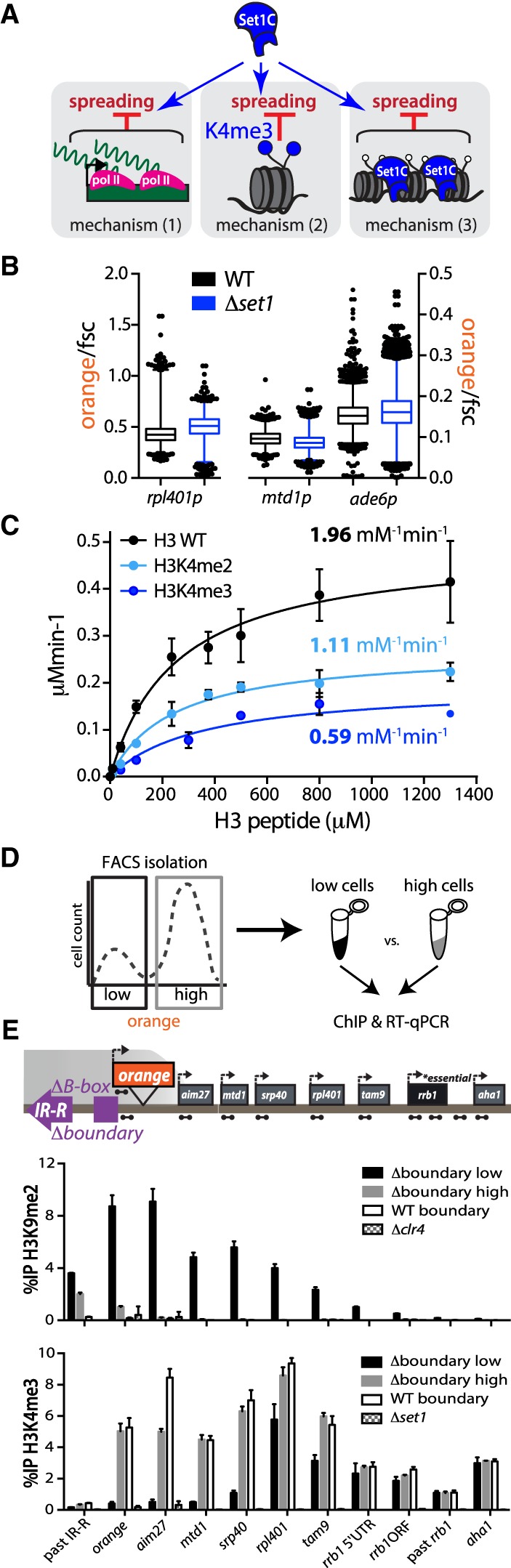
The gene-protective activity of Set1 is independent of mean transcription levels and is rooted in catalytic inhibition of Suv39/Clr4 by H3K4me2/3. (A) Possible mechanisms by which Set1 repels heterochromatin spreading: (1) maintaining a level of transcription that is refractory to heterochromatin invasion due to local RNA polymerase activity and associated cycles of nucleosome eviction; (2) interference of H3K4me3, the Set1 product, with heterochromatin spreading; and (3) noncatalytic effect of Set/COMPASS, including its occupancy on chromatin. (B) Box and whisker plots of “orange” signal normalized to forward scatter (fsc) for rpl401p:HSS, mtd1p:HSS, and ade6p:HSS in set1+ (black) and Δset1 (blue) backgrounds. One percent to 99% of the data are included within the whiskers. Outliers are plotted as individual points. (C) Histone methyltransferase assay with Clr4-SET and H3(1–20) peptides with modifications as indicated. Error bars represent 1SD from three replicate experiments. kcat/KM (specificity constant) values are derived from measurement of the kcat and KM (see Supplemental Fig. S4F). (D) Cartoon overview depicting FACS isolation of “low” and “high” Δboundary 5′ ade6p-“orange” cells followed by ChIP and RT-qPCR. (E) ChIP-qPCR data for FACS-sorted cells: H3K9me2 (top) and H3K4me3 (bottom). Amplicons for each qPCR are depicted as dumbbells on cartoon locus. Error bars represent 1SD from three technical replicate ChIPs.
Previous reports have described both transcription-activating and -repressive roles for Set1/COMPASS (Buratowski and Kim 2010; Mikheyeva et al. 2014; D'Urso et al. 2016). To directly test the involvement of mechanism 1, we examined the “orange” signal expressed from rpl401p, mtd1p, and ade6p in a set1+ or Δset1 backgrounds in a WT boundary context (Fig. 4B). “Orange” signal was normalized to forward scatter (fsc). This parameter tracks with the size of a cell and has been used extensively to estimate the cell volume, which is a central parameter to normalize RNA and protein between single cells. The use of fsc bypasses any confounding effect Δset1 might have on our ade6p-driven “red” control. We did not detect any major decrease in “orange” in Δset1 isolates (Fig. 4B). We confirmed this result by RT-qPCR analysis, where we normalized ade6p “orange,” mtd1, and rpl401 transcripts to an act1 control (Supplemental Fig. S4A, left). In the normalization, we adjusted for the Δset1 effect on this act1 control (Materials and Methods; Supplemental Fig. S4A, right). Together, these results argue against Set1 regulating the mean level of RNA polymerase II-mediated transcription at these genes. Thus, mechanism 1 is an unlikely avenue for the Δset1-dependent phenotype in reporter silencing.
H3K4me directly interferes with Suv39/Clr4 catalysis
Set1 is the only H3K4 methylase in fission yeast (Noma and Grewal 2002) and H3K4me and H3K9me appear mutually exclusive (Noma et al. 2001). Hence, we hypothesized one implementation of mechanism 2 could be direct interference with Suv39/Clr4 activity. This could potentially occur via two mechanisms—either by directly impacting catalysis of H3K9 methylation by Suv39/Clr4 or by disrupting the “read–write” positive feedback characteristic of histone methyl transferases. This spreading feedback mechanism is mediated by the binding of Suv39/Clr4 enzyme to its own product via the chromodomain (CD), which stimulates the catalysis of H3K9 methylation on proximal nucleosomes via the SET domain (Zhang et al. 2008a; Margueron et al. 2009; Al-Sady et al. 2013; Müller et al. 2016). Clr4-CD recognition of H3K9me has been shown to be sensitive to acetylation (ac) of the H3K4 residue (Xhemalce and Kouzarides 2010).
We tested whether the Clr4-CD's ability to recognize H3K9me is impacted by H3K4me3. We purified the Clr4-CD (Supplemental Fig. S4B) and performed fluorescence polarization with modified histone tail peptides. We found that the Clr4-CD has a similar binding affinity for H3K9me3 and H3K4me3K9me3 tail peptides (Supplemental Fig. S4C), which is recapitulated by the full-length Clr4 enzyme (Supplemental Fig. S4D). Thus, the presence of H3K4me3, unlike H3K4ac, does not disrupt the “read–write” feedback mechanism.
Our understanding of the effects of H3K4me on H3K9me catalysis by various enzymes are based mostly on endpoint analysis and have yielded conflicting results. Two previous studies using endpoint analysis indicated no obvious effect of H3K4me2 or a K4A mutation on Suv39/Clr4 activity (Nakayama et al. 2001; Kusevic et al. 2017), yet a number of other studies document a range of effects of H3K4me2 or H3K4me3 on H3K9 methyl transferases, and these results do not always agree (Wang and Zhang 2001; Nishioka et al. 2002; Chin et al. 2005; Binda et al. 2010). To definitively determine any effect of H3K4me2 or H3K4me3 may have on Suv39/Clr4 catalysis, we performed multiple turnover Michaelis-Menten kinetic analysis using N-terminal truncation of Clr4 comprising residues 192–490 (Collazo et al. 2005; Dirk et al. 2007), which includes the catalytic SET domain (Fig 4C; Supplemental Fig. S4E). The masses of the H3K4me0, H3K4me2, and H3K4me3 peptides used were verified by MADLI-TOF analysis (Supplemental Fig. S9). We determined kcat, KM, and specificity constant (kcat/KM) values (Fig. 4C; Supplemental Fig. S4F) and, interestingly, found that H3K4me3 and H3K4me2 reduce Clr4's kcat/KM by 3.3 times and 1.8 times, respectively, relative to an H3K4me0 (WT) peptide. This derives mostly from an adverse effect on Suv39/Clr4's kcat rather than on the KM (see Supplemental Fig. S4F). We confirmed that this effect is reflected in the full-length enzyme under kcat/KM conditions (Supplemental Fig. S4G), where we found H3K4me3 to reduce kcat/KM by 4.6 times, in good agreement with the Michaelis-Menten parameters extracted with the SET domain. These results were confirmed with an independently produced set of H3K4me0 and H3K4me3 peptides (data not shown). In conclusion, these results demonstrate that Suv39/Clr4 catalysis (Fig. 4C; Supplemental Fig. S4F,G), but not its product recognition (Supplemental Fig. S4C,D), is inhibited by the presence of H3K4me3 and, to a milder extent, by H3K4me2. These results support a role for mechanism 2 and make the following two predictions: First, if H3K4me3 is directly involved in repelling spreading, genes downstream from an H3K4me3 peak are protected from heterochromatin invasion if the peak correlates with effective disruption of silencing. Second, chromatin recruitment of Set1 is insufficient for barrier activity, which requires Set1's catalytic activity.
Protection of downstream genes by H3K4me
To test the first prediction, we used fluorescence-assisted cell sorting (FACS) to isolate both repressed (“low”) and expressed (“high”) populations of 5′ ade6p:HSS Δboundary cells (Fig. 4D) and then assessed their chromatin and transcriptional state via ChIP and RT-qPCR, respectively (Fig. 4E; Supplemental Fig. S4H). While both populations evidenced H3K9me2 accumulation upstream of the reporter, H3K9me2 signal could not be detected at any point beyond “orange” in the “high” cells (gray bars). This immediate drop coincides with the ade6p H3K4me3 peak in the “high” cells, and H3K4me3 is enriched at the downstream gene promoters comparable with WT levels. Consistent with this H3K4me3 distribution, transcription levels are similar to the no heterochromatin (Δclr4) state. This result, in conjunction with our above findings (Figs. 1I,J, 4C), suggest that H3K4me3 accumulation at ade6p protects downstream transcriptional units. On the other hand, the “low” population (black bars) displays high levels of H3K9me2 at and beyond “orange,” while H3K4me3 is severely reduced (Fig. 4E). H3K9me2 levels eventually decline towards the essential rrb1 gene, concomitant with a rise in H3K4me3 enrichment. The discrepancy between the H3K4me3 signal in the “low” and “high” populations thus eventually decreases with distance. In cells where ade6p-localized H3K4me3 is overcome, downstream transcriptional units therefore appear to succumb to repressive H3K9me2. Thus, these data are consistent with a model where encounter of a substantial and/or persistent H3K4me3 peak disrupts spreading, protecting downstream gene units.
Catalytic activity of Set1, and not chromatin recruitment alone, underpins heterochromatin containment
To test the second prediction concerning catalytic activity of Set1, we constructed an allelic series of H3K4 methylation Set1 hypomorphs, based on sequence alignments with the Saccharomyces cerevisiae Set1 ortholog, and published catalytic mutants within this gene (Schlichter and Cairns 2005). We introduced tagless C862A (Set1C862A) and G852S (Set1G852S) (Fig. 5A) into S. pombe Set1 within its native gene context, marked with a nourseothricin resistance (NATR) gene, and produced a corresponding wild-type (wt-Set1) control. We produced a separate set of strains where Set1C862A and Set1G852S and wt-Set1 version were N-terminally 2xFlag tagged and inserted at the native set1 locus to test for expression by Western blot. N-terminally Flag tagged Set1 has been shown to retain function (Mikheyeva et al. 2014). We found mutants and wt-Set1 to accumulate to similar levels by two independent extraction methods (Supplemental Fig. S5A). Next, we probed for H3K4me3 accumulation by Western blot and found that both mutants show a defect in H3K4me3 accumulation, with the C862A mutant showing almost no H3K4me3 by Western blot and G852S accumulating significantly reduced amounts (Fig. 5B). We next moved to directly test above prediction, and found that both Set1C862A and Set1G852S mutants are significantly impaired in their ability to protect rpl401p:HSS from invasion by heterochromatin compared with wt-Set1 (Fig. 5C). Set1/COMPASS, similar to Suv39/Clr4, contains a positive feedback loop with the enzyme recognizing its product (Roguev et al. 2003; Kirmizis et al. 2007), and it is possible that abrogation of catalytic function leads to reduction of Set1 recruitment to chromatin. We used our 2xFlag Set1 constructs to test whether Set1C862A and Set1G852S are still normally recruited to chromatin at the TSS (Fig. 5D) and indeed found no major difference in enrichment of Set1C862A or Set1G852S versus wt-Set1. Importantly, this data allows us to exclude that the recruitment of Set1, part of a megadalton complex (Miller et al. 2001), itself or associated H3K4me-independent functions, repel heterochromatin spreading (mechanism 3) (Fig. 4A). Instead, these data offer further support for mechanism 2, showing in vivo that Set1 catalytic activity is required for containment of heterochromatin spreading, likely in part via direct interference with Suv39/Clr4 catalysis.
Figure 5.
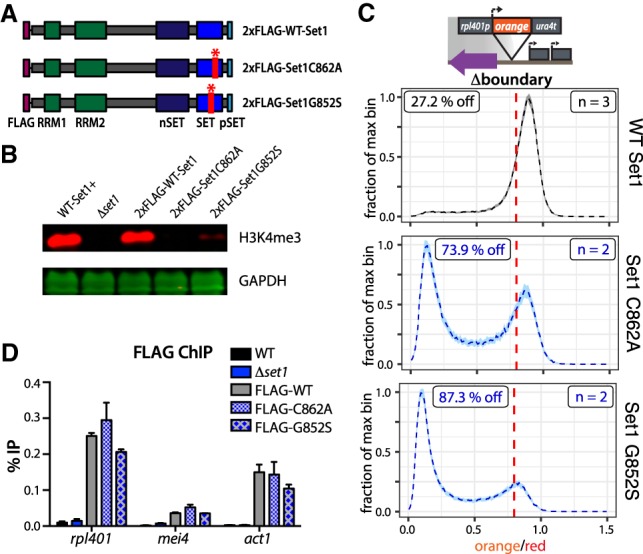
Set1 catalytic activity but not its recruitment to chromatin is required for its gene protective function. (A) Diagram of Set1 constructs including WT-Set1 and two point mutations in the catalytic SET domain: C862A and G852S. Constructs are expressed from the set1 promoter at the native set1 locus with an N-terminal 2xFlag tag. (B) Licor Western blot for H3K4me3 with GAPDH loading control of whole-cell extracts from wild-type untagged Set1, the Δset1 parent of the 2xFlag constructs, 2xFlag-wtSet1, 2xFlag-Set1C862A, and 2xFlag-Set1G852S. (C) Histogram plots as in Figure 1 of normalized “orange” signal from rpl401p:HSS Δboundary isolates that were transformed with either untagged wt-Set1, Set1C862A, or Set1G852S. (D) Anti-Flag ChIP-qPCR data in genetic backgrounds as in B. Error bars represent 1SD from two technical replicate ChIPs.
The distribution of H3K4me3 and nucleosome occupancy over genes correlate with orientation dependence of genic heterochromatin boundary function
H3K4me3 is enriched near the TSSs of genes (Santos-Rosa et al. 2002; Pokholok et al. 2005), and in fission yeast, heterochromatin silencing can proceed in a cotranscriptional manner (Bühler et al. 2006, 2008). This led us to hypothesize that encountering a gene first at the promoter (5′ end) versus the terminator (3′ end) will more effectively protect against gene silencing, since heterochromatin will be antagonized before cotranscriptional silencing mechanisms can proceed. To our surprise, while our ade6p:HSS is clearly more effective in the 5′-proximal orientation (Fig. 6A; Supplemental Fig. S6A), rpl401p:HSS shows much less bias (Fig. 6B; Supplemental Fig. S6B). In both 5′- and 3′-proximal orientations, rpl401p:HSS effectively disrupts spreading in a set1-dependent manner (Fig. 6B). We wondered whether this discrepancy can be explained by the profile of H3K4me3 over the native gene. Indeed, we found that rpl401 has significantly elevated H3K4me3 over middle and, critically, 3′ of the gene, while it was strongly diminished at the 3′ of ade6 (Fig. 6C). We found a similar distribution pattern for the respective HSS cassettes (Supplemental Fig. S6D). The difference in orientation bias between ade6p:HSS and rpl401p:HSS can thus be partially accounted for by the H3K4me3 profile. However, we wondered whether additional, nonetheless H3K4me-dependent, mechanisms beyond the direct catalytic interference we document above, underlie the striking difference in gene orientation effect.
Figure 6.

H3K4me3 disrupts local nucleosome occupancy. (A) Locus cartoon for ade6p-driven “orange” reporters in either the 3′ or 5′ (shaded) orientation with respect to IR-R (cartoon). Histograms for normalized “orange” signal in Δboundary context as in Figure 1. 3′ ade6p:HSS is plotted in full color, while 5′ ade6p:HSS is shaded (data depicted in Fig. 1, redrawn for comparison). epe1 and set1 genotypes are as indicated. (B) Locus cartoons and histogram plots as in A for 3′ and 5′ rpl401p:HSS. Shaded lines reproduced from Figure 2D. (C) H3K4me3 ChIP-qPCR over the gene body of ade6 and rpl401 open reading frames in wild-type untagged Set1, Δset1, 2xFlag-wtSet1, 2xFlag-Set1C862A, and 2xFlag-Set1G852S. (D) H3 ChIP-qPCR over the gene body of ade6 and rpl401 open reading frames in genotypes as in C. (E) H3 ChIP-qPCR in WT (black) and Δset1 (blue). Constitutive heterochromatin targets (boxed in gray) (F) H3 ChIP-qPCR at mei4, iec1, and act1 in genotypes as in C. In E H3 ChIP-qPCR Error bars represent 1SD from four replicates, each representing a single colony deriving from each genotype. For ChIPs in C, D, and F, error bars represent 1SD from two technical replicate ChIPs.
We focused on regulation of nucleosome occupancy, known to adversely affect spreading (Garcia et al. 2010; Aygün et al. 2013). We first assessed nucleosome occupancy in set1+ and Δset1 strains by H3 ChIP in log phase cultures (Fig. 6D–F; Supplemental Fig. S6E), but also in G2-stalled cells to exclude cell cycle passage effects (Supplemental Fig. S6F). Intriguingly, nucleosome occupancy is highly elevated at the 3′ of ade6 but remains low throughout rpl401 (Fig. 6D; Supplemental Fig. S6E). Low nucleosome occupancy is strongly antagonistic to spreading (Garcia et al. 2010; Aygün et al. 2013). Therefore, the data showing that ade6 retains high nucleosome occupancy at its 3′, provide an additional explanation why ade6 and not rpl401 is vulnerable to heterochromatin invasion from the 3′. More broadly, we observed increases in nucleosome occupancy at heterochromatin islands and the IR-R-proximal genes in Δset1 (Fig. 6E,F) and across active genes distributed on the three S. pombe chromosomes (Supplemental Fig. S6E), but not on heterochromatin targets (Fig. 6E, gray box). This effect therefore likely represents a general feature of Set1 activity. Importantly, we found that the catalytic activity of Set1 is required for this regulation of nucleosome occupancy, as the catalytic Set1C862A and Set1G852S mutants partially or fully mirror the Δset1 phenotype at ade6, rpl401, and heterochromatin islands (Fig. 6D,F). The extent to which the catalytic mutants recapitulate the Δset1 phenotype correlates both with the global H3K4me3 accumulation defect in each hypomorph (Fig. 5B), as well as the residual H3K4me3 at any given gene-internal location relative to the wt-Set1 (Fig. 6C,D).
However, these data do not explain how heterochromatin can overcome the TSS-localized H3K4me3 peak when invading a gene like ade6 from 3′ and then enact stable repression. We hypothesized that for this to occur, 3′-invading heterochromatin would need to be able to (1) partially invade the gene, (2) down-regulate transcription without fully reaching the promoter, consistent with cotranscriptional gene silencing, and, finally, (3) reduce H3K4me3, the key spreading antagonizing signal, likely via a reduction in transcription (Shilatifard 2012). To address these hypotheses, we built a variant of the 3′ade6p:HSS reporter construct that would permit spreading to proceed into the gene unit but hinder its ability to reach the promoter. To achieve this, we fused the “orange” and “green” coding sequences by an in-frame linker containing five B-box elements (Supplemental Fig. S7A), multimers of which have been shown to confer synthetic boundary activity (Noma et al. 2006). Signal from “green” and “orange” in WT, boundaryC, and Δboundary contexts, as well as their RNA levels (Supplemental Fig. S7A,B), were well correlated in each isolate. This indicates that the entire transcriptional unit is uniformly regulated, despite presence of the synthetic B-box boundary midway through the tandem gene unit. We next assessed the chromatin state at “green” and “orange” by ChIP. H3K9me2 is significantly reduced at “orange” compared with “green” across all isolates from both boundaryC and Δboundary contexts (Supplemental Fig. S7C) supporting that the 5x B-box sequence was functioning as a synthetic roadblock to spreading. The difficulty of separating nucleosomes by shearing within heterochromatin likely prevented us from documenting any potentially sharper drops across the synthetic barrier. Surprisingly, H3K4me3 ChIP revealed that boundaryC and Δboundary had significantly reduced methylation levels compared with WT at the “orange” TSS (Supplemental Fig. S7D). These results demonstrate that invasion of a gene from the 3′ end can reduce both inhibitory H3K4me levels and transcription, despite not fully reaching the gene promoter. This mechanism would presumably not operate in the 5′ orientation, since H3K4me3 would be encountered first.
Histone acetylation links H3K4me3 to Set1-dependent regulation of nucleosome occupancy
Our data show both that the catalytic activity of Set1, hence production of H3K4me, is required for containment of heterochromatin spreading and that regulation of nucleosome mobilization tightly correlates with this containment function. However, the question remains of how regulation of nucleosome turnover is tied to H3K4me. Previous studies have identified a role for Set1/COMPASS and H3K4me in promoting global histone acetylation at various residues (Noma and Grewal 2002; Taverna et al. 2006; Ginsburg et al. 2014). To validate this finding in our system, we performed ChIP against H3K9ac, as well as H3 and H4 acetylation broadly, and found indeed that in Δset1 acetylation was similarly reduced (Fig. 7A–C) at all of the genes tested, whether at heterochromatin islands or canonical euchromatin. The fact that we found a robust decrease in H3 and H4 acetylation as well as H3K9ac specifically, indicates the involvement of multiple histone acetyltransferase (HAT) complexes (Buratowski and Kim 2010; Woo et al. 2017). Since HAT mutants or knock-downs have broad effects on heterochromatin (Gomez et al. 2005; Tong et al. 2012; Wang et al. 2013, 2015), we chose not to pursue mutational analysis of the catalytic subunits. S. pombe contains two genes that are orthologs of H3K4me3-specific PHD reader modules within HAT complexes: png1, which associates with Mst1 in S. pombe (Chen et al. 2010), and png2, which does not impact H4 acetylation or have known HAT associations in S. pombe (Chen et al. 2010). To test whether recruitment of HATs to H3K4me3 specifically is involved in providing protection against heterochromatin invasion, we deleted either the H3K4me3-reading PHD fingers of png1 or png2 in the context of the 3′-oriented rpl401p:HSS. While png2ΔPHD did not have an effect on rpl401p:HSS in the Δboundary context, we found png1ΔPHD to have a subtle but highly reproducible effect, resulting in elevated silencing at the otherwise highly efficient barrier forming 3′- rpl401p:HSS (Fig. 7D). This phenotype was recovered after outcrossing to wild type and retesting nine bone fide Δepe1 png1ΔPHD resulting progeny (Supplemental Fig. S8A). Additionally, the phenotype was recovered upon reintroduction of Δepe1 into epe1+ png1ΔPHD isolates, indicating it is a stable phenotype (Fig. 7D). png1ΔPHD importantly does not affect basal expression of rpl401p:HSS in the presence of epe1+ (Fig. 7D, dashed gray line). The fact that the phenotype is significantly weaker than Δset1 is expected, since it appears that more than one HAT is involved in maintaining elevated acetylation in response to Set1 activity (Fig. 7A–C). The Δset1 png1ΔPHD double mutant has very similar degree of silencing as Δset1, 86% versus 82% of cells, respectively, (Supplemental Fig. S8B), indicating that the effect of png1ΔPHD on silencing is likely not additive with Set1. These data provide evidence for Png1's involvement in H3K4me-dependent heterochromatin containment, possibly recruiting Mst1, which likely acts redundantly with other HAT complexes (Buratowski and Kim 2010).
Figure 7.

Role of histone acetylation in heterochromatin containment. (A) H3 (pan) acetyl ChIP-qPCR in WT and Δset1. ChIP is normalized to H3 signal to account for differences in nucleosome occupancy. (B) H3K9ac ChIP-qPCR plotted as in A. (C) H4 (pan) acetyl ChIP-qPCR plotted as in A. For A–C, error bars represent 1SD from four replicates, each representing a single colony deriving from each genotype. (D) Cartoon depicting Png1 and Png2 containing H3K4me3 reading PHD finger domains. Png1 is associated with NuA4. Histograms as in Figure 1 of normalized “orange” signal from 3′ rpl401p:HSS Δboundary isolates from png1ΔPHD (brown), or WT boundary with png1ΔPHD (grey) and png1+ (black). (E) Model for the contribution of Set1/COMPASS to gene-mediated heterochromatin repulsion. (Top) In 5′ invasion, Set1-dependent TSS-proximal H3K4me3 repels heterochromatin spreading via direct Suv39/Clr4 inhibition and nucleosome destabilization. (Bottom) Broader distributions of Set1-dependent H3K4me3 in bodies of some genes and the ensuing increased nucleosome destabilization repels 3′ heterochromatin invasion. Histone acetyltransferase complexes (HATs) attracted to the H3K4me3 via reader proteins acetylate locally and contribute to nucleosome destabilization.
Discussion
Two paradigms have emerged for heterochromatin domain regulation, which when taken together present an intriguing paradox. On one hand is the ability for heterochromatin domains to expand beyond their borders when containment mechanisms are compromised (Noma et al. 2006; Zofall and Grewal 2006; Trewick et al. 2007; Zofall et al. 2012; Wang et al. 2013, 2014; Garcia et al. 2015). On the other hand is the widespread dispersion of factors, activities, and posttranslational modifications embedded in euchromatin, which are known to antagonize the establishment and maintenance of heterochromatic domains (Lan et al. 2007; Sugiyama et al. 2007; Garcia et al. 2010; Aygün et al. 2013; Wang et al. 2013, 2015). Why then is heterochromatin spreading able to overcome these negative regulators and expand into euchromatin? Part of the answer may lie in the activities inherently associated with the spreading machinery, including HDACs (Grewal et al. 1998; Shankaranarayana et al. 2003; Yamada et al. 2005; Sugiyama et al. 2007), nucleosome remodelers (Sugiyama et al. 2007; Taneja et al. 2017), and H3K4 – demethylase complexes (Li et al. 2008), which apparently can overpower euchromatin. However, how and why heterochromatin spreading is halted at specific euchromatic locations is not understood.
In this work we investigated the signals within local active euchromatin that define spatial limits to heterochromatin spreading in fission yeast. The key principles that derive from this work are as follows: (1) Euchromatic barrier signals depend on Set1/COMPASS activity at active genes. (2) High gene transcript levels are not intrinsically refractory to heterochromatin invasion. (3) Set1-dependent repulsion of heterochromatin acts via two pathways downstream from H3K4 methylation: direct catalytic inhibition of Clr4/Suv39 and nucleosome mobilization. (4) The ability to repel heterochromatin can be gene orientation-specific, directed by the distribution of H3K4me over the gene.
Mechanisms regulating facultative heterochromatin domain size
We found Set1/COMPASS enacts a heterochromatin containment signal at gene-rich regions, including facultative heterochromatin in fission yeast that responds to environmental conditions (Figs. 2,3; Zofall et al. 2012; Sugiyama et al. 2016; Gallagher et al. 2018). These findings are in contrast to previously identified spreading regulators that function globally, such as Epe1, Leo1, Paf1, and Mst2 (Trewick et al. 2007; Zofall et al. 2012; Kowalik et al. 2015; Sadeghi et al. 2015; Verrier et al. 2015; Wang et al. 2015; Flury et al. 2017). The containment function of Set1 is localized to specific euchromatic regions, heterochromatin islands, and euchromatin exposed to boundary failure at IR-R (Figs. 2,3), but is not prominent at constitutive heterochromatin (Supplemental Fig. S2C).
Critically, containment of heterochromatin spreading does not require a change in mean transcript level (Fig. 4B), but specifically its ability to methylate H3K4. This is remarkable as active transcription in fission yeast and other systems leads to formation of nucleosome-free regions (NFRs) at the TSS (Lantermann et al. 2010), and NFRs are thought to be refractory to heterochromatin spreading (Garcia et al. 2010; Lantermann et al. 2010). Our data point to NFRs still being intact in Δset1, as is evident from the coinciding dips in the H3K9me2 tracks in Δepe1 and Δepe1Δset1 (Fig. 2B). This is consistent with findings that formation of NFRs alone is insufficient to block spreading (Oki and Kamakaka 2005). Just as heterochromatin overcomes the TSS-proximal NFR it can overcome the presence of Set1 on chromatin, even though it is part of a megadalton complex. This conclusion is supported by the normal chromatin localization, but defective H3K4 methylation and heterochromatin repulsion, of the Set catalytic hypomorphs (Fig. 5D). The crucial heterochromatin repelling signal is, therefore, the H3K4me mark. This modification takes two parallel tracks to push back against encroaching heterochromatin: (1) Catalytic interference. In contrast to prior findings obtained by endpoint analysis (Nakayama et al. 2001; Kusevic et al. 2017), we found that H3K9me catalysis by Suv39/Clr4 H3K9 is directly inhibited by Set1 products, most strongly by H3K4me3. This finding represents a rare example of direct regulation of the Suv39/Clr4 SET domain active site, beyond autoinhibition (Iglesias et al. 2018), but is consistent with the effect H3K4me can have on other H3K9 methylases (Wang and Zhang 2001; Nishioka et al. 2002; Binda et al. 2010). (2) Locally decreased nucleosome occupancy. We found that the distribution of H3K4me3 tracks with a Set1-dependent decrease in nucleosome occupancy. It is known that specific nucleosome stabilizing factors are required for constitutive heterochromatin assembly (Yamane et al. 2011; Taneja et al. 2017), and we recently showed that repression of turnover is critical to stable spreading (Greenstein et al. 2018). Thus, the disruption of nucleosome occupancy by Set1 will antagonize heterochromatin formation. Increased mobilization is dependent on Set1's catalytic activity (Fig. 6D,F), raising the question of how increased mobilization is instructed. In principle, this could occur via direct recruitment of nucleosome remodelers, or via changes in the chromatin landscape that increase nucleosome turnover. Set1 has been shown to increase histone acetylation (Noma and Grewal 2002; Ginsburg et al. 2014), which has long been linked to decreased nucleosome occupancy (Reinke and Hörz 2003; Wirén et al. 2005) and stability (Ausio and Van Holde 1986; Brower-Toland et al. 2005). We observed strong, Set1-dependent increases in pan-H3ac and H4ac, as well as H3K9ac in most genes tested, including all those acting as spreading boundaries in our system (Fig. 7A–C). The H3K4me3 HAT targeting pathways in S. pombe are not well understood. However, the involvement of HAT targeting downstream from H3K4me3 is evidenced by the moderate loss of heterochromatin containment at rpl401p:HSS in the in-frame deletion of the Png1, but not Png2, PHD finger, a conserved H3K4me3 targeting module (Fig. 7D,E). This result is consistent with the observation that in S. pombe, only Png1 and not Png2 associates with a HAT (Chen et al. 2010). We believe that this phenotype indicates significant contribution of Png1 in containment given that (1) rpl401 features a very large H3K4me3 peak (Fig. 2B) and the 3′-oriented HSS we used is our strongest barrier construct, and (2) HATs likely act additively in implementing the H3K4me3 signal, as we observed increases in H3ac and H4ac, which are known to be mediated by a number of HATs including SAGA, Mst1, Mst2, and Hat1. Further, it remains possible that direct recruitment of chromatin remodelers by H3K4me works in concert with histone acetylation. Collectively, our data point to catalytic interference and reduced nucleosome occupancy working synergistically in the containment of heterochromatin spread downstream of Set1. Of note, unlike in fission yeast as documented here, in budding yeast, Set1 has a more global heterochromatin-antagonizing role, in concert with H2A.Z (Venkatasubrahmanyam et al. 2007). This suggests that Set1's role in constraining heterochromatin in euchromatin specifically may have coevolved with H3K9me-marked heterochromatin systems, with other factors regulating constitutive domains (see above).
Regulation of active and repressed chromatin states by Set1 and COMPASS
How do the mechanisms of heterochromatin regulation we describe for Set1/COMPASS relate to its known roles in transcriptional regulation? The recruitment of Set1/COMPASS to chromatin requires H2B monoubiquitination mediated by Rad6 and Bre1 as well as interaction with the Paf1 elongation complex (Paf1C), which engages RNA polymerase and is additionally responsible for activation of Rad6 and Bre1 function on chromatin. Set1/COMPASS also associates with elongating RNA polymerase, giving rise to a characteristic pattern of H3K4 methylation states (see above). Interestingly, previous studies in fission yeast have described a role for Paf1C components Paf1 and Leo1 in antagonizing heterochromatin spreading through promoting increased histone turnover and H4K16 acetylation (Sadeghi et al. 2015; Verrier et al. 2015). Both studies tested, but did not identify, a role for Set1 in their respective systems at loci (IRC1L of the centromere and IR-L of MAT), where we also do not detect an effect of Δset1 even in the sensitized Δepe1 genetic background (Supplemental Fig. S2).
Several additional data support a model where Set1 and Paf1/Leo1 act in separate pathways to regulate heterochromatin spreading: (1) set1 was not found to be epistatic to leo1 in a genome-wide genetic interaction study for heterochromatin spreading using an IRC1L reporter (Verrier et al. 2015). (2) Global H4K16 acetylation levels did not change in response to Δset1 (Noma and Grewal 2002), whereas acetyl marks such as H3K9 and H3K14 were reduced in this background (Fig. 7B; Noma and Grewal 2002). (3) In our repelling factor screen, Δleo1 did not result in the characteristic spreading phenotype seen for Set1/COMPASS complex deletions (data not shown). Taken together, these results describe separate mechanisms for spreading regulation by Paf1/Leo1 and Set1/COMPASS. Additionally, since the observations on heterochromatin containment are dependent on Set1's catalytic activity (Fig. 5), they are unlikely to be related to the gene-repressive functions of Set1 that are independent of its H3K4me catalytic activity (Lorenz et al. 2014).
The role of gene orientation in heterochromatin repulsion
The nucleosome mobilizing effect of Set1 we document is generally strongest close to the TSS, as is evident from ade6, as well as the very long sib1 gene, where we observed lowest occupancy and Set1 dependence at the 5′ end (Supplemental Fig. S6E). However, in the case of rpl401, both H3K4me3 and the concomitant decrease in occupancy is much more broadly distributed. This phenomenon is especially true for H3K4me2, which is evenly distributed throughout the rpl401p:HSS reporter (Supplemental Fig. S6D). While recruitment of HATs via PHD fingers is H3K4me3-specific (Li et al. 2006; Taverna et al. 2006), Suv39/Clr4 catalysis is still impacted by H3K4me2 (Fig. 4C), implying that both methylation states could work in concert through both catalytic and nucleosome mobilization pathways to repel spreading. The differential distributions of H3K4me and nucleosome occupancy changes we observed across genes give rise to an orientation bias in the ability of a gene to repel heterochromatin (Fig. 6A). If gene orientation can influence containment effectiveness, an orientation bias may emerge at genomic sites where containment of silencing is critical. Such a case has indeed been documented in mammals. Lamina-associated domains (LADs) are gene-repressive chromatin domains associated with the nuclear periphery that contain both H3K9 and H3K27 methylation (for review, see van Steensel and Belmont 2017) and regions immediately flanking LADs are enriched for 5′ oriented genes and concomitant H3K4 methylation (Guelen et al. 2008). It is not surprising that mammalian genomes may require use of 5′ orientation more than fission yeasts, which lack such a bias at boundaries of constitutive heterochromatin domains (Supplemental Fig. S8C): Yeast genes are very small, at a median length of ∼1.8 kb, with the H3K4me3 peak comprising, on average, 25% of the gene, while mouse and human genes have a median length of 16 and 20 kb, respectively (Supplemental Fig. S8D), yet preserve a similar TSS-localized H3K4me3 peak (Guenther et al. 2007). Thus, it is plausible that H3K4me3 signals distribute far enough across a gene to make effective boundaries in either orientation for fission yeast genes, but explains a 5′ bias for mammalian genes where the H3K4me3 peak is restricted to a narrow fraction of the gene.
Our above results lead to a model (Fig. 7E) for how facultative heterochromatin domains can be delimited in a manner that is specific in genomic space. It remains to be determined why only some, but not other, euchromatically embedded heterochromatin domains require Set1 for their containment, and we believe this may be encoded in the relative rates of local heterochromatin spreading and availability of limiting factors (Nakayama et al. 2000; Noma et al. 2006; Kagansky et al. 2009). Regardless, the gene-centered role of Set1/COMPASS we document here in constraining heterochromatin spreading gives insight into the mechanisms of locus encoded and potentially cell-type specific restriction of facultative gene-repressive domains, as opposed to the global means of delimiting heterochromatin that have been described to date.
Materials and methods
Strain and plasmid construction
Plasmids used to generate genomic integration constructs were assembled using in vivo recombination. S. pombe transformants were selected as described (Greenstein et al. 2018). XFP reporters were targeted to specific genomic locations as described (Greenstein et al. 2018). Direct gene knockout constructs were generated using long primer PCR to amplify resistance cassettes with homology to the regions surrounding the open reading frame of the target. Genomic integrations were confirmed by PCR.
Flow cytometry and FACS sorting
Cells were grown for flow cytometry experiments as described (Greenstein et al. 2018). Flow cytometry was performed using a Fortessa X20 dual machine (Becton Dickinson) and high-throughput sampler (HTS) module. Approximately 20,000–100,000 cells were collected, dependent on strain growth and volume collected. Fluorescence detection, compensation, and data analysis were as described (Al-Sady et al. 2016; Greenstein et al. 2018).
For the FACS experiment, cells were grown overnight from OD = 0.05 in YES and in the morning concentrated into a smaller volume (∼3–5×) and filtered with 35–40-µm mesh (Corning) to achieve 5000–7000 events/sec on the cytometer and reduce potential for clogs. Cells were first gated for size (forward and side scatter), removal of doublet cells, and the presence of the control “red” signal and then sorted into low and high populations for “orange.” Low “orange” population was defined by signal overlapping a control with no fluors. High “orange” population was defined by signal overlapping the matched background Δclr4 control. For each population, 16 × 106 to 18 × 106 cells were collected for chromatin immunoprecipitation and 3 × 106 cells were collected for RT-qPCR. Cells were processed for downstream analysis immediately following sorting.
Repelling factor screen
An h− reporter strain with “green” and “orange” at the ura4 locus (natMX marked) and “red” at the leu1 locus (hygMX marked) was crossed to a 408 strain subset of the Bioneer haploid deletion library (kanMX marked). Crosses were performed as described (Verrier et al. 2015; Barrales et al. 2016) with limited modifications. Briefly, crosses were arrayed onto SPAS plates using a RoToR HDA colony pinning robot (Singer) and mated for 4 d at room temperature. The plates were incubated for 4 d at 42°C following mating to remove haploid and diploid cells, retaining spores. Resultant spores were germinated on YES medium with added hygromycin B, G418, and nourseothricin for selection of both reporter loci and the appropriate gene deletion. The resultant colonies were passaged into liquid YES and grown overnight for flow cytometry as described above. In the morning, cells were diluted again into YES medium and grown 4–6 h at 32°C prior to analysis via flow cytometry.
RNA extraction and quantification
Cells from log phase cultures or FACS-sorted cells were pelleted, and supernatant was decanted and flash-frozen in liquid nitrogen. Pellets were stored at −80°C. RNA extraction was performed as described (Greenstein et al. 2018). cDNA synthesis was performed with either SuperScript RTIII or IV (Invitrogen) and an oligo dT primer (Fig. 2C; Supplemental Figs. S1A, S3B, S4H) or SuperScript RTIV (Invitrogen) and random hexamers (Supplemental Figs. S4A, S7C) via the manufacturer's protocol. cDNA samples were quantified by RT-qPCR as described (Greenstein et al. 2018). Values from cDNA targets were normalized to act1 or pyk1. Samples in Supplemental Figures S1A, S4H, and S7C were normalized to the target/actin value for the Δclr4 strain of a matched background. For Supplemental Figure S4A (left), given that signal from act1p-driven “red” increases by ∼50% in Δset1 backgrounds, the target/actin values in Δset1 samples were multiplied by the mean ratio Δset1/WT of act1p driven “red” signal from the four WT and mutant pairs in Supplemental Figure S4A (right). This adjusts the normalization for the up-regulation of actin observed in this background.
Chromatin immunoprecipitation
Chromatin Immunoprecipitation (ChIP) followed by qPCR was performed essentially as described (Greenstein et al. 2018) with the following modifications. For Figure 4E, 16 × 106 to –18 × 106 cells of both “low” and “high” FACS populations, as well as controls, were collected and processed for ChIP. Prior to lysis, 50 × 106 cells of independently fixed S. cerevisiae W303 strain were added to each population as carrier. ChIP experiments with bulk populations of log phase cells were performed as described (Greenstein et al. 2018) without the addition of W303 carrier. In Supplemental Figure S6F, Hht2-HA cells were grown at 25°C and 225 rpm in YES + hygromycin B from OD = 0.05. After cells reached OD = 0.2, G2 stall was induced by shifting the temperature to 37°C for 3 h prior to fixation. Following lysis, sonication was performed using a Diagenode BioRuptor Pico for 20–28 rounds of 30 sec on/30 sec rest or Diagenode BioRuptor standard on high for 30–40 for rounds of 30 sec on/30 sec rest. Cleared chromatin was split into equal volumes per IP after a small fraction (5%–10%) was set aside as input/WCE. One microliter of the following antibodies was added per ChIP sample: H3K9me2 (Abcam, ab1220), H3K4me3 (Active Motif 39159), H3K4me2 (Active Motif, 39141), H3K9ac (Active Motif, 39137), H3(pan)ac (Active Motif, 39064), H4(pan)ac (Active Motif, 39140), HA (Abcam, ab9110); 1.5 µL of anti-Flag M2 antibody (Sigma) was added per ChIP sample, and 1.4 μg of H3 antibody (Active Motif, 39064) was added per ChIP sample. Immune complexes were collected with Protein A Dynabeads (Thermo Fisher) for all ChIP samples except the anti-Flag ChIP samples, which were collected with Protein G Dynabeads (Thermo Fisher). DNA was quantified by RT-qPCR, and percentage IP (ChIP DNA/input DNA) was calculated as described (Greenstein et al. 2018).
ChIP-seq sample and library preparation
Sample preparation and ChIP prior to sequencing was performed essentially as described (Greenstein et al. 2018) with the following modifications: 50 mL of cells was grown to OD = 0.6–0.8 overnight from OD = 0.025. Biological duplicate samples were generated for WT, biological triplicate samples were generated for Δepe1, and four biological samples were generated for Δepe1Δset1 genotypes. Based on OD measurements, 300 × 106 cells per sample were fixed and processed for ChIP. Shearing was performed with 20 cycles of 30 sec on/30 sec rest. Samples were not precleared. Sonication efficiency was determined for each sample and only samples where DNAs averaged 200–300 bp were used. Chromatin was split into two samples after 8% was set aside as input. Three microliters of H3K9me2 (Abcam,1220) or H3K4me3 (Active Motif, 39159) antibodies was added per tube and incubated overnight at 4°C with rotation. (Only H3K9me2 ChIP was performed for Δset1 strains. The absence of H3K4me3 was validated by ChIP qPCR in Supplemental Fig. S2A.) Immune complexes were collected with 30 μL of twice-washed Protein A Dynabeads (Invitrogen) for 3 h at 4°C. Beads were washed as above with the exception that the wash buffer step was performed twice. Following incubation for 20 min at 70°C, DNA was eluted in 100 μL of TE + 1%SDS and the beads were washed and eluted a second time with 100 μL of TE + 1% SDS + 5 μL of 20 mg/mL Proteinase K (Roche). Following overnight incubation at 65°C, ChIP and input samples were purified using Machery Nagel PCR cleanup kit. Library preparation for sequencing was performed as described (Inada et al. 2016; Parsa et al. 2018). Samples were sequenced on a HiSeq 4000 platform (Illumina) with a single-end 50 run.
ChIP-seq data analysis
Sliding window quality filtering and adapter trimming were carried out using Trimmomatic 0.38 (Bolger et al. 2014) before the reads were aligned to the S. pombe genome (Wood et al. 2002) with Bowtie2 2.3.4.2 (Langmead and Salzberg 2012) using standard end-to-end sensitive alignment. Indexed bam files were generated using SAMtools 1.9 (Li et al. 2009) “view,” “sort,” and “index” functions. Combined input files and WT H3K9me2 ChIP files were generated with SAMtools “merge” function for use in normalization. Input or WT normalized signal tracks were generated using the MACS2 version 2.1.1.20160309 (Zhang et al. 2008b; Feng et al. 2012) callpeak function to generate reads per million normalized bedGraph files with the following flags: -g 1.26e7 --nomodel --extsize 200 --keep-dup auto -B --SPMR -q 0.01. The resulting pileup was normalized with the bdgcmp function via the fold enrichment method (m –FE). The resulting normalized signal track files were trimmed back to the length of the genome and converted to bigwig format using UCSCtools bedClip and bedGraphToBigWig functions. BigWig files were imported into R 3.5.1 with rtracklayer 1.40.6 (Lawrence et al. 2009). The genome was divided into 25-bp bins and the average enrichment value per bin was calculated using the tileGenome and binnedAverage functions of GenomicRanges 1.32.7 (Lawrence et al. 2013). Gene annotations were imported from PomBase (Lock et al. 2019) and converted to genomic coordinates with the makeTxDbFromGFF function from GenomicFeatures 1.32.3 (Lawrence et al. 2013). Finally, mean and confidence interval per each genotype were generated during signal track plotting using the DataTrack command from Gviz 1.24.0 (Hahne and Ivanek 2016). For the P-value track, reads for H3K9me2 ChIP-seq in each isolate of each strain were extended to 200 bp and counted into sliding 150-bp windows beginning every 30 bp using the windowCounts function from R package csaw 1.18.0 (Lun and Smyth 2016). Global background was determined from 5-kb bins and a filter of 1.7 times the global average was applied with the filterWindows function and subsetting. Composition bias was corrected using the TMM method via the normFactors and asDGEList functions and then dispersion was calculated via estimateDisp function before a generalized linear model based on genotype was fit with glmQLfit. P-values result from testing a contrast between Δepe1Δset1 and Δepe1 based on the fitted model and summarizing the per window P-values over 300-bp or 1200-bp bins. P-values were interpreted as colors based on the specified ranges and added to the signal track plots with the AnnotationTrack command from Gviz. Peaks were called with epic2 0.0.14 (Stovner and Saetrom 2019) with the following flags: --effective-genome-fraction 0.999968 -bin 200 -g 3 -fs 200 -fdr 0.05. Regions of known heterochromatin formation were imported from a previously curated list (Parsa et al. 2018). Regions were extended by 10 kb on each side to account for differences in coordinates that may exist for different genome assemblies, as well as variable spreading. Peaks and known regions were plotted using Gviz (Hahne and Ivanek 2016). For the global analysis comparison between Δepe1Δset1 and Δepe1 genotypes, the average value per 300-bp window for the input normalized H3K9me2 ChIP-seq was computed using deeptools2 3.1.3 (Ramírez et al. 2016) function multiBigWigSummary. The counts per bin output file was read into R 3.6.0 and the mean value for each genotype was computed per bin. The log2 ratio of the Δepe1Δset1 genotype average over the Δepe1 genotype average was computed for each bin that had H3K9me2 signal above a threshold of 1.5 times the global average calculated in the same manner as for P-value track.
Clr4 purification
The chromodomain of Clr4 (residues 6–64, Clr4-CD) and SET domain (residues 192–490, Clr4-SET) were each cloned into MacroLab vector 14C containing N-terminal 6xHis and maltose-binding protein (MBP) tags. Full-length Clr4 was expressed from a previously described vector (Al-Sady et al. 2013). Proteins were expressed as described (Al-Sady et al. 2013) except that for Clr4-SET and full-length Clr4 LB was substituted for 2XYT medium supplemented with 10 μM ZnSO4. Lysis and Talon affinity resin purification (Takara Bio) and size exclusion chromatography were essentially as described (Al-Sady et al. 2013). Lysis buffer was 100 mM HEPES (pH 7.5), 300 mM NaCl, 10% glycerol, 7.5 mM imidazole, 0.5% Triton-X100, 1 mM β-mercaptoethanol, and protease inhibitors. For Clr4-SET and full-length Clr4, Triton was substituted for 0.01% Igepal NP-40. After final size exclusion chromatography, Clr4-CD was eluted into FP storage buffer (20 mM HEPES at pH 7.5, 100 mM KCl, 10% glycerol, 5 mM β-mercaptoethanol). Clr4-SET and full-length Clr4 were eluted into Clr4 storage buffer (100 mM Tris at pH 8.5, 100 mM KCl, 10% glycerol, 1 mM MgCl2, 20 µM ZnSO4, 10 mM β-mercaptoethanol). All proteins were flash-frozen and stored at −80°C. Protein concentrations were determined by Sypro Ruby (Bio-Rad) gel staining against a BSA standard curve and for Clr4-CD and Clr4-SET were verified by UV absorption at 280 nm using the theoretical extinction coefficient (ExPasy ProtParam) 88,810 cm−1M−1 and 98,210 cm−1M−1 for Clr4-CD and Clr4-SET, respectively.
Fluorescence polarization assay
Fluorescence polarization assay for binding of Clr4-CD or full-length Clr4 to H3 tail peptides was performed as described (Canzio et al. 2013). Ten nanomolar H3 tail peptide with K4me0K9me0 (unmodified), K4me0K9me3, or K4me3K9me3 modifications (GenScript) was used as probe. Reactions were performed in FP buffer (20 mM HEPES at pH 7.5, 100 mM KCl, 10% glycerol, 0.01%–0.1% NP-40 substitute) and incubated for 20 min at room temperature prior to measurement. Fluorescence polarization measurements and data analysis including fitting of curves were performed as described (Canzio et al. 2013).
Histone methyltransferase assay
Multiple turnover kinetic assays were performed as described (Al-Sady et al. 2013) with the following modifications. Reactions contained 100 µM cold SAM (disulfate tosylate; Abcam) and 10–15 µM 3HSAM tracer (55–75 Ci/mmol; PerkinElmer) and were incubated with 1 µM Suv39/Clr4-SET or full-length and varying amounts of biotinylated H3(1–20) peptide with K4me0 (unmodified), K4me2, or K4me3 (GenScript). Reactions were performed at 30°C in Clr4 reaction buffer (100–120 mM Tris at pH 8.5, 100 mM KCl, 10% glycerol, 1 mM MgCl2, 20 µM ZnSO4, 10 mM β-cmercaptoethanol).
Licor Western blot
For Western blot lysate method 2, whole-cell total protein extracts were prepared as described (Al-Sady et al. 2016). For anti-Flag Western blot lysate method 1, pellets from 1 mL of saturated overnight cultures were flash-frozen, then resuspended in 10% trichloro-acetic acid, mixed by vortexing, and then incubated for 10 min on ice. The precipitate was washed once with cold acetone and the pellet was air-dried and then resuspended in 40 µL of Tris/HCl (pH 8) with 200 µL of 2× Laemmli sample buffer. Four-hundred microliters of 0.5-mm glass beads was added per tube, and each sample was mixed in a platform vortexer twice for 60 sec. The bottom of the tube was then pierced with a 26G needle and the supernatant was recovered into another tube by centrifugation. Prior to loading the gel all samples were boiled for 10 min and then centrifuged at >10,000g for 2 min to remove insoluble material. Western blot was performed as described (Al-Sady et al. 2016) and the following primary antibodies were used H3K4me2 (Active Motif, 39141), H3K4me3 (Active Motif, 39159), anti-GAPDH (Thermo Scientific, MA5-15738) and anti-Flag M2 (Sigma). H3K4me2/3 blots were coincubated with anti-GAPDH antisera and followed by both secondary antibodies as described (Al-Sady et al. 2016). For the anti-Flag Western blot, given the size difference between GAPDH and Set1, the membrane was cut between the 50- and 75-kDa bands on the ladder. The larger half was incubated with anti-Flag 1° and then antimouse 2° while the smaller half was incubated separately with anti-GAPDH 1° and then antimouse 2°.
Data sets
All available sequencing data sets were deposited in the Gene Expression Omnibus with the superseries accession number GSE140067.
Supplementary Material
Acknowledgments
We thank Hiten D Madhani for the generous gifts of strains and reagents and use of laboratory equipment. Additionally, we thank Sandra Catania, Michael McManus, Sy Redding, and Kieran Mace for helpful discussions on data acquisition, analysis, and interpretation. This work was supported by grants from the National Institutes of Health (NIH; DP2GM123484) and the University of California at San Francisco Program for Breakthrough Biomedical Research (partially funded by the Sandler Foundation) to B.A.-S., and the ARCS Foundation Scholarship and Hooper Graduate Fellowship to R.A.G. R.A.G. was additionally supported by an NIH grant to attend the Cold Spring Harbor Laboratory course Statistical Methods for Functional Genomics. We acknowledge the course instructors for their helpful comments on this work, in particular Sean Davis for insightful suggestions on statistical analysis of the screen hits. This work was supported by grants awarded to S.B. from the German Research Foundation (BR 3511/2-1) and the European Union Network of Excellence EpiGeneSys (HEALTH-2010-257082). S.B. is Member of the Collaborative Research Center 1064 funded by the German Research Foundation and acknowledges infrastructure support. J.E.B. is a Natural Sciences and Engineering Research Council of Canada postdoctoral fellow. Flow cytometry and FACS data were generated in the University of California at San Francisco Parnassus Flow Cytometry Core, which is supported by Diabetes Research Center (DRC) grants NIH P30 DK063720 and NIHS10 1S10OD021822-01.
Author contributions: R.A.G., B.A.-S., R.R.B., and S.B. designed experiments. R.A.G, B.A.-S., and R.R.B. performed all experiments. R.A.G., B.A.-S., R.R.B., N.A.S., and J.E.B. analyzed and discussed the data. R.A.G. and B.A.-S. prepared the manuscript draft. All authors revised the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.328468.119.
References
- Al-Sady B, Madhani HD, Narlikar GJ. 2013. Division of labor between the chromodomains of HP1 and Suv39 methylase enables coordination of heterochromatin spread. Mol Cell 51: 80–91. 10.1016/j.molcel.2013.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sady B, Greenstein RA, El-Samad HJ, Braun S, Madhani HD. 2016. Sensitive and quantitative three-color protein imaging in fission yeast using spectrally diverse, recoded fluorescent proteins with experimentally-characterized in vivo maturation kinetics. PloS One 11: e0159292 10.1371/journal.pone.0159292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausio J, Van Holde KE. 1986. Histone hyperacetylation: its effects on nucleosome conformation and stability. Biochemistry 25: 1421–1428. 10.1021/bi00354a035 [DOI] [PubMed] [Google Scholar]
- Aygün O, Mehta S, Grewal SI. 2013. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat Struct Mol Biol 20: 547–554. 10.1038/nsmb.2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub N, Goldshmidt I, Lyakhovetsky R, Cohen A. 2000. A fission yeast repression element cooperates with centromere-like sequences and defines a mat silent domain boundary. Genetics 156: 983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub N, Noma K, Isaac S, Kahan T, Grewal SIS, Cohen A. 2003. A novel jmjC domain protein modulates heterochromatization in fission yeast. Mol Cell Biol 23: 4356–4370. 10.1128/MCB.23.12.4356-4370.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrales RR, Forn M, Georgescu PR, Sarkadi Z, Braun S. 2016. Control of heterochromatin localization and silencing by the nuclear membrane protein Lem2. Genes Dev 30: 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AC, Felsenfeld G. 1999. Stopped at the border: boundaries and insulators. Curr Opin Genet Dev 9: 191–198. 10.1016/S0959-437X(99)80029-X [DOI] [PubMed] [Google Scholar]
- Binda O, LeRoy G, Bua DJ, Garcia BA, Gozani O, Richard S. 2010. Trimethylation of histone H3 lysine 4 impairs methylation of histone H3 lysine 9: regulation of lysine methyltransferases by physical interaction with their substrates. Epigenetics 5: 767–775. 10.4161/epi.5.8.13278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, Garcia JF, Rowley M, Rougemaille M, Shankar S, Madhani HD. 2011. The Cul4-Ddb1Cdt2 ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell 144: 41–54. 10.1016/j.cell.2010.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower-Toland B, Wacker DA, Fulbright RM, Lis JT, Kraus WL, Wang MD. 2005. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J Mol Biol 346: 135–146. 10.1016/j.jmb.2004.11.056 [DOI] [PubMed] [Google Scholar]
- Bühler M, Verdel A, Moazed D. 2006. Tethering RITS to a nascent transcript initiates RNAi- and heterochromatin-dependent gene silencing. Cell 125: 873–886. 10.1016/j.cell.2006.04.025 [DOI] [PubMed] [Google Scholar]
- Bühler M, Spies N, Bartel DP, Moazed D. 2008. TRAMP-mediated RNA surveillance prevents spurious entry of RNAs into the Schizosaccharomyces pombe siRNA pathway. Nat Struct Mol Biol 15: 1015–1023. 10.1038/nsmb.1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S, Kim T. 2010. The role of cotranscriptional histone methylations. Cold Spring Harb Symp Quant Biol 75: 95–102. 10.1101/sqb.2010.75.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, Grewal SIS. 2005. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat Genet 37: 809–819. 10.1038/ng1602 [DOI] [PubMed] [Google Scholar]
- Canzio D, Liao M, Naber N, Pate E, Larson A, Wu S, Marina DB, Garcia JF, Madhani HD, Cooke R, et al. 2013. A conformational switch in HP1 releases auto-inhibition to drive heterochromatin assembly. Nature 496: 377–381. 10.1038/nature12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferré F, et al. 2011. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 471: 513–517. 10.1038/nature09806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JQ, Li Y, Pan X, Lei BK, Chang C, Liu ZX, Lu H. 2010. The fission yeast inhibitor of growth (ING) protein Png1p functions in response to DNA damage. J Biol Chem 285: 15786–15793. 10.1074/jbc.M110.101832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin HG, Pradhan M, Estève PO, Patnaik D, Evans TC Jr., Pradhan S. 2005. Sequence specificity and role of proximal amino acids of the histone H3 tail on catalysis of murine G9A lysine 9 histone H3 methyltransferase. Biochemistry 44: 12998–13006. 10.1021/bi0509907 [DOI] [PubMed] [Google Scholar]
- Collazo E, Couture JF, Bulfer S, Trievel RC. 2005. A coupled fluorescent assay for histone methyltransferases. Anal Biochem 342: 86–92. 10.1016/j.ab.2005.04.007 [DOI] [PubMed] [Google Scholar]
- Dirk LM, Flynn EM, Dietzel K, Couture JF, Trievel RC, Houtz RL. 2007. Kinetic manifestation of processivity during multiple methylations catalyzed by SET domain protein methyltransferases. Biochemistry 46: 3905–3915. 10.1021/bi6023644 [DOI] [PubMed] [Google Scholar]
- D'Urso A, Takahashi YH, Xiong B, Marone J, Coukos R, Randise-Hinchliff C, Wang JP, Shilatifard A, Brickner JH. 2016. Set1/COMPASS and Mediator are repurposed to promote epigenetic transcriptional memory. Elife 5: e16691 10.7554/eLife.16691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan ED, Braun CR, Gygi SP, Moazed D. 2014. Post-transcriptional regulation of meiotic genes by a nuclear RNA silencing complex. RNA 20: 867–881. 10.1261/rna.044479.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, Liu XS. 2012. Identifying ChIP-seq enrichment using MACS. Nat Protoc 7: 1728–1740. 10.1038/nprot.2012.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flury V, Georgescu PR, Iesmantavicius V, Shimada Y, Kuzdere T, Braun S, Bühler M. 2017. The histone acetyltransferase Mst2 protects active chromatin from epigenetic silencing by acetylating the ubiquitin ligase Brl1. Mol Cell 67: 294–307.e9. 10.1016/j.molcel.2017.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PS, Larkin M, Thillainadesan G, Dhakshnamoorthy J, Balachandran V, Xiao H, Wellman C, Chatterjee R, Wheeler D, Grewal SIS. 2018. Iron homeostasis regulates facultative heterochromatin assembly in adaptive genome control. Nat Struct Mol Biol 25: 372–383. 10.1038/s41594-018-0056-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JF, Dumesic PA, Hartley PD, El-Samad H, Madhani HD. 2010. Combinatorial, site-specific requirement for heterochromatic silencing factors in the elimination of nucleosome-free regions. Genes Dev 24: 1758–1771. 10.1101/gad.1946410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JF, Al-Sady B, Madhani HD. 2015. Intrinsic toxicity of unchecked heterochromatin spread is suppressed by redundant chromatin boundary functions in Schizosacchromyces pombe. G3 (Bethesda) 5: 1453–1461. 10.1534/g3.115.018663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg DS, Anlembom TE, Wang J, Patel SR, Li B, Hinnebusch AG. 2014. NuA4 links methylation of histone H3 lysines 4 and 36 to acetylation of histones H4 and H3. J Biol Chem 289: 32656–32670. 10.1074/jbc.M114.585588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez EB, Espinosa JM, Forsburg SL. 2005. Schizosaccharomyces pombe mst2+ encodes a MYST family histone acetyltransferase that negatively regulates telomere silencing. Mol Cell Biol 25: 8887–8903. 10.1128/MCB.25.20.8887-8903.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein RA, Jones SK, Spivey EC, Rybarski JR, Finkelstein IJ, Al-Sady B. 2018. Noncoding RNA-nucleated heterochromatin spreading is intrinsically labile and requires accessory elements for epigenetic stability. Elife 7: e32948 10.7554/eLife.32948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SIS, Bonaduce MJ, Klar AJS. 1998. Histone deacetylase homologs regulate epigenetic inheritance of transcriptional silencing and chromosome segregation in fission yeast. Genetics 150: 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. 2008. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948–951. 10.1038/nature06947 [DOI] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. 2007. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130: 77–88. 10.1016/j.cell.2007.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, Ivanek R. 2016. Visualizing genomic data using Gviz and bioconductor. Methods Mol Biol 1418: 335–351. 10.1007/978-1-4939-3578-9_16 [DOI] [PubMed] [Google Scholar]
- Hall IM, Shankaranarayana GD, Noma KI, Ayoub N, Cohen A, Grewal SIS. 2002. Establishment and maintenance of a heterochromatin domain. Science 297: 2232–2237. 10.1126/science.1076466 [DOI] [PubMed] [Google Scholar]
- Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, Crabtree GR. 2012. Dynamics and memory of heterochromatin in living cells. Cell 149: 1447–1460. 10.1016/j.cell.2012.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias N, Currie MA, Jih G, Paulo JA, Siuti N, Kalocsay M, Gygi SP, Moazed D. 2018. Automethylation-induced conformational switch in Clr4 (Suv39h) maintains epigenetic stability. Nature 560: 504–508. 10.1038/s41586-018-0398-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada M, Nichols RJ, Parsa JY, Homer CM, Benn RA, Hoxie RS, Madhani HD, Shuman S, Schwer B, Pleiss JA. 2016. Phospho-site mutants of the RNA Polymerase II C-terminal domain alter subtelomeric gene expression and chromatin modification state in fission yeast. Nucleic Acids Res 44: 9180–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S, Noma K, Grewal SI. 2004. RNAi-independent heterochromatin nucleation by the stress-activated ATF/CREB family proteins. Science 304: 1971–1976. 10.1126/science.1099035 [DOI] [PubMed] [Google Scholar]
- Kagansky A, Folco HD, Almeida R, Pidoux AL, Boukaba A, Simmer F, Urano T, Hamilton GL, Allshire RC. 2009. Synthetic heterochromatin bypasses RNAi and centromeric repeats to establish functional centromeres. Science 324: 1716–1719. 10.1126/science.1172026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmizis A, Santos-Rosa H, Penkett CJ, Singer MA, Vermeulen M, Mann M, Bähler J, Green RD, Kouzarides T. 2007. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature 449: 928–932. 10.1038/nature06160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalik KM, Shimada Y, Flury V, Stadler MB, Batki J, Bühler M. 2015. The Paf1 complex represses small-RNA-mediated epigenetic gene silencing. Nature 520: 248–252. 10.1038/nature14337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurukuti S, Tiwari VK, Tavoosidana G, Pugacheva E, Murrell A, Zhao Z, Lobanenkov V, Reik W, Ohlsson R. 2006. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci 103: 10684–10689. 10.1073/pnas.0600326103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusevic D, Kudithipudi S, Iglesias N, Moazed D, Jeltsch A. 2017. Clr4 specificity and catalytic activity beyond H3K9 methylation. Biochimie 135: 83–88. 10.1016/j.biochi.2017.01.013 [DOI] [PubMed] [Google Scholar]
- Lan F, Zaratiegui M, Villén J, Vaughn MW, Verdel A, Huarte M, Shi Y, Gygi SP, Moazed D, Martienssen RA, et al. 2007. S. pombe LSD1 homologs regulate heterochromatin propagation and euchromatic gene transcription. Mol Cell 26: 89–101. 10.1016/j.molcel.2007.02.023 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantermann AB, Straub T, Strålfors A, Yuan GC, Ekwall K, Korber P. 2010. Schizosaccharomyces pombe genome-wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae. Nat Struct Mol Biol 17: 251–U215. 10.1038/nsmb.1741 [DOI] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, Carey V. 2009. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25: 1841–1842. 10.1093/bioinformatics/btp328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. 2013. Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NN, Chalamcharla VR, Reyes-Turcu F, Mehta S, Zofall M, Balachandran V, Dhakshnamoorthy J, Taneja N, Yamanaka S, Zhou M, et al. 2013. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155: 1061–1074. 10.1016/j.cell.2013.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. 2006. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442: 91–95. 10.1038/nature04802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Huarte M, Zaratiegui M, Vaughn MW, Shi Y, Martienssen R, Cande WZ. 2008. Lid2 is required for coordinating H3K4 and H3K9 methylation of heterochromatin and euchromatin. Cell 135: 272–283. 10.1016/j.cell.2008.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Motamedi MR, Yip CK, Wang Z, Walz T, Patel DJ, Moazed D. 2009. An α motif at Tas3 C terminus mediates RITS cis spreading and promotes heterochromatic gene silencing. Mol Cell 34: 155–167. 10.1016/j.molcel.2009.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litt MD, Simpson M, Gaszner M, Allis CD, Felsenfeld G. 2001. Correlation between histone lysine methylation and developmental changes at the chicken β-globin locus. Science 293: 2453–2455. 10.1126/science.1064413 [DOI] [PubMed] [Google Scholar]
- Lock A, Rutherford K, Harris MA, Hayles J, Oliver SG, Bähler J, Wood V. 2019. PomBase 2018: user-driven reimplementation of the fission yeast database provides rapid and intuitive access to diverse, interconnected information. Nucleic Acids Res 47: D821–D827. 10.1093/nar/gky961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz DR, Meyer LF, Grady PJ, Meyer MM, Cam HP. 2014. Heterochromatin assembly and transcriptome repression by Set1 in coordination with a class II histone deacetylase. Elife 3: e04506 10.7554/eLife.04506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, Smyth GK. 2016. csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res 44: e45 10.1093/nar/gkv1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ III, Voigt P, Martin SR, Taylor WR, De Marco V, et al. 2009. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461: 762–767. 10.1038/nature08398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina DB, Shankar S, Natarajan P, Finn KJ, Madhani HD. 2013. A conserved ncRNA-binding protein recruits silencing factors to heterochromatin through an RNAi-independent mechanism. Genes Dev 27: 1851–1856. 10.1101/gad.226019.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. 2011. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol 18: 867–874. 10.1038/nsmb.2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheyeva IV, Grady PJ, Tamburini FB, Lorenz DR, Cam HP. 2014. Multifaceted genome control by Set1 Dependent and Independent of H3K4 methylation and the Set1C/COMPASS complex. PLoS Genet 10: e1004740 10.1371/journal.pgen.1004740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A. 2001. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci 98: 12902–12907. 10.1073/pnas.231473398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller MM, Fierz B, Bittova L, Liszczak G, Muir TW. 2016. A two-state activation mechanism controls the histone methyltransferase Suv39h1. Nat Chem Biol 12: 188–193. 10.1038/nchembio.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J, Klar AJ, Grewal SI. 2000. A chromodomain protein, Swi6, performs imprinting functions in fission yeast during mitosis and meiosis. Cell 101: 307–317. 10.1016/S0092-8674(00)80840-5 [DOI] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. 2001. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292: 110–113. 10.1126/science.1060118 [DOI] [PubMed] [Google Scholar]
- Nielsen AL, Oulad-Abdelghani M, Ortiz JA, Remboutsika E, Chambon P, Losson R. 2001. Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol Cell 7: 729–739. 10.1016/S1097-2765(01)00218-0 [DOI] [PubMed] [Google Scholar]
- Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D. 2002. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev 16: 479–489. 10.1101/gad.967202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Grewal SIS. 2002. Histone H3 lysine 4 methylation is mediated by Set1 and promotes maintenance of active chromatin states in fission yeast. Proc Natl Acad Sci 99: 16438–16445. 10.1073/pnas.182436399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Allis CD, Grewal SI. 2001. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293: 1150–1155. 10.1126/science.1064150 [DOI] [PubMed] [Google Scholar]
- Noma K, Cam HP, Maraia RJ, Grewal SI. 2006. A role for TFIIIC transcription factor complex in genome organization. Cell 125: 859–872. 10.1016/j.cell.2006.04.028 [DOI] [PubMed] [Google Scholar]
- Oki M, Kamakaka RT. 2005. Barrier function at HMR. Mol Cell 19: 707–716. 10.1016/j.molcel.2005.07.022 [DOI] [PubMed] [Google Scholar]
- Parsa JY, Boudoukha S, Burke J, Homer C, Madhani HD. 2018. Polymerase pausing induced by sequence-specific RNA-binding protein drives heterochromatin assembly. Genes Dev 32: 953–964. 10.1101/gad.310136.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, et al. 2005. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122: 517–527. 10.1016/j.cell.2005.06.026 [DOI] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. 2016. deepTools2: a next generation Web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165. 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, et al. 2000. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599. 10.1038/35020506 [DOI] [PubMed] [Google Scholar]
- Reinke H, Hörz W. 2003. Histones are first hyperacetylated and then lose contact with the activated PHO5 promoter. Mol Cell 11: 1599–1607. 10.1016/S1097-2765(03)00186-2 [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Zhang K, Zofall M, Chen E, Grewal SI. 2011. Defects in RNA quality control factors reveal RNAi-independent nucleation of heterochromatin. Nat Struct Mol Biol 18: 1132–1138. 10.1038/nsmb.2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguev A, Schaft D, Shevchenko A, Aasland R, Shevchenko A, Stewart AF. 2003. High conservation of the Set1/Rad6 axis of histone 3 lysine 4 methylation in budding and fission yeasts. J Biol Chem 278: 8487–8493. 10.1074/jbc.M209562200 [DOI] [PubMed] [Google Scholar]
- Sadeghi L, Prasad P, Ekwall K, Cohen A, Svensson JP. 2015. The Paf1 complex factors Leo1 and Paf1 promote local histone turnover to modulate chromatin states in fission yeast. EMBO Rep 16: 1673–1687. 10.15252/embr.201541214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. 2002. Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411. 10.1038/nature01080 [DOI] [PubMed] [Google Scholar]
- Schlichter A, Cairns BR. 2005. Histone trimethylation by Set1 is coordinated by the RRM, autoinhibitory, and catalytic domains. EMBO J 24: 1222–1231. 10.1038/sj.emboj.7600607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott KC, Merrett SL, Willard HF. 2006. A heterochromatin barrier partitions the fission yeast centromere into discrete chromatin domains. Curr Biol 16: 119–129. 10.1016/j.cub.2005.11.065 [DOI] [PubMed] [Google Scholar]
- Shankaranarayana GD, Motamedi MR, Moazed D, Grewal SIS. 2003. Sir2 regulates histone H3 lysine 9 methylation and heterochromatin assembly in fission yeast. Curr Biol 13: 1240–1246. 10.1016/S0960-9822(03)00489-5 [DOI] [PubMed] [Google Scholar]
- Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81: 65–95. 10.1146/annurev-biochem-051710-134100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stovner EB, Saetrom P. 2019. epic2 efficiently finds diffuse domains in ChIP-seq data. Bioinformatics 35: 4392–4393. 10.1093/bioinformatics/btz232 [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Cam HP, Sugiyama R, Noma K-i, Zofall M, Kobayashi R, Grewal SIS. 2007. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell 128: 491–504. 10.1016/j.cell.2006.12.035 [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Thillainadesan G, Chalamcharla VR, Meng Z, Balachandran V, Dhakshnamoorthy J, Zhou M, Grewal SIS. 2016. Enhancer of rudimentary cooperates with conserved RNA-processing factors to promote meiotic mRNA decay and facultative heterochromatin assembly. Mol Cell 61: 747–759. 10.1016/j.molcel.2016.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja N, Zofall M, Balachandran V, Thillainadesan G, Sugiyama T, Wheeler D, Zhou M, Grewal SI. 2017. SNF2 family protein Fft3 suppresses nucleosome turnover to promote epigenetic inheritance and proper replication. Mol Cell 66: 50–62.e6. 10.1016/j.molcel.2017.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Ilin S, Rogers RS, Tanny JC, Lavender H, Li H, Baker L, Boyle J, Blair LP, Chait BT, et al. 2006. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol Cell 24: 785–796. 10.1016/j.molcel.2006.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong K, Keller T, Hoffman CS, Annunziato AT. 2012. Schizosaccharomyces pombe Hat1 (Kat1) is associated with Mis16 and is required for telomeric silencing. Eukaryot Cell 11: 1095–1103. 10.1128/EC.00123-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trewick SC, Minc E, Antonelli R, Urano T, Allshire RC. 2007. The JmjC domain protein Epe1 prevents unregulated assembly and disassembly of heterochromatin. EMBO J 26: 4670–4682. 10.1038/sj.emboj.7601892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Belmont AS. 2017. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169: 780–791. 10.1016/j.cell.2017.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatasubrahmanyam S, Hwang WW, Meneghini MD, Tong AH, Madhani HD. 2007. Genome-wide, as opposed to local, antisilencing is mediated redundantly by the euchromatic factors Set1 and H2A.Z. Proc Natl Acad Sci 104: 16609–16614. 10.1073/pnas.0700914104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrier L, Taglini F, Barrales RR, Webb S, Urano T, Braun S, Bayne EH. 2015. Global regulation of heterochromatin spreading by Leo1. Open Biol 5: 150045 10.1098/rsob.150045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Zhang Y. 2001. Mi2, an auto-antigen for dermatomyositis, is an ATP-dependent nucleosome remodeling factor. Nucleic Acids Res 29: 2517–2521. 10.1093/nar/29.12.2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tadeo X, Hou H, Tu PG, Thompson J, Yates JR III, Jia S. 2013. Epe1 recruits BET family bromodomain protein Bdf2 to establish heterochromatin boundaries. Genes Dev 27: 1886–1902. 10.1101/gad.221010.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lawry ST, Cohen AL, Jia S. 2014. Chromosome boundary elements and regulation of heterochromatin spreading. Cell Mol Life Sci 71: 4841–4852. 10.1007/s00018-014-1725-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Reddy BD, Jia S. 2015. Rapid epigenetic adaptation to uncontrolled heterochromatin spreading. Elife 4: e06179 10.7554/eLife.06179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. 2009. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet 41: 246–250. 10.1038/ng.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirén M, Silverstein RA, Sinha I, Walfridsson J, Lee H-M, Laurenson P, Pillus L, Robyr D, Grunstein M, Ekwall K. 2005. Genomewide analysis of nucleosome density histone acetylation and HDAC function in fission yeast. EMBO J 24: 2906–2918. 10.1038/sj.emboj.7600758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo H, Dam Ha S, Lee SB, Buratowski S, Kim T. 2017. Modulation of gene expression dynamics by co-transcriptional histone methylations. Exp Mol Med 49: e326 10.1038/emm.2017.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, et al. 2002. The genome sequence of Schizosaccharomyces pombe. Nature 415: 871–880. 10.1038/nature724 [DOI] [PubMed] [Google Scholar]
- Xhemalce B, Kouzarides T. 2010. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev 24: 647–652. 10.1101/gad.1881710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Fischle W, Sugiyama T, Allis CD, Grewal SIS. 2005. The nucleation and maintenance of heterochromatin by a histone deacetylase in fission yeast. Mol Cell 20: 173–185. 10.1016/j.molcel.2005.10.002 [DOI] [PubMed] [Google Scholar]
- Yamane K, Mizuguchi T, Cui B, Zofall M, Noma K, Grewal SI. 2011. Asf1/HIRA facilitate global histone deacetylation and associate with HP1 to promote nucleosome occupancy at heterochromatic loci. Mol Cell 41: 56–66. 10.1016/j.molcel.2010.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Mosch K, Fischle W, Grewal SIS. 2008a. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat Struct Mol Biol 15: 381–388. 10.1038/nsmb.1406 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008b. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, et al. 2013. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152: 642–654. 10.1016/j.cell.2012.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Grewal SI. 2006. Swi6/HP1 recruits a JmjC domain protein to facilitate transcription of heterochromatic repeats. Mol Cell 22: 681–692. 10.1016/j.molcel.2006.05.010 [DOI] [PubMed] [Google Scholar]
- Zofall M, Yamanaka S, Reyes-Turcu FE, Zhang K, Rubin C, Grewal SI. 2012. RNA elimination machinery targeting meiotic mRNAs promotes facultative heterochromatin formation. Science 335: 96–100. 10.1126/science.1211651 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.