

Abstract
FUN14 domain-containing protein 1 (Fundc1)-dependent mitophagy, mainly activated by ischemic/hypoxic preconditioning, benefits acute myocardial reperfusion injury and chronic metabolic syndrome via sustaining mitochondrial homeostasis. Mitochondrial fission plays a pathogenic role in ischemic acute kidney injury (AKI) through perturbation of mitochondrial quality and activation of mitochondrial apoptosis. The aim of our study was to explore the role of Fundc1 mitophagy in ischemia preconditioning (IPC)-mediated renoprotection. Proximal tubule-specific Fundc1 knockout (Fundc1PTKO) mice were subjected to ischemia reperfusion injury (IRI) and IPC prior to assessment of renal function, mitophagy, mitochondrial quality control, and Drp1-related mitochondrial fission. Following exposure to IPC, Fundc1 mitophagy was activated through post-transcriptional phosphorylation at Ser17. Interestingly, IRI-mediated renal injury, inflammation, and tubule cell death were mitigated by IPC whereas proximal tubule-specific Fundc1 knockout (Fundc1PTKO) mice abolished IPC-offered renoprotection. Mechanistically, IRI-evoked mitochondrial damage was improved by IPC whereas Fundc1 deficiency provoked mitochondrial abnormality, manifested by impaired mitochondrial quality and hyperactivated Drp1-dependent mitochondrial fission. Interestingly, Fundc1 deficiency-associated mitochondrial dysfunction was reversed by pharmacological inhibition of mitochondrial fission. In vivo, Fundc1 deletion-caused renal injury, severe pro-inflammatory response, and tubule cell death could be nullified by way of knockout Drp1 on Fundc1PTKO background. Finally, we also revealed that IPC triggered Fundc1 mitophagy activation through UNC-51-like kinase 1 (Ulk1) and Ulk1 ablation interrupted IPC-mediated Fundc1 activation and thus attenuated IPC-induced renoprotection. Fundc1 mitophagy, primarily driven by IPC, confers resistance to AKI through reconciliation of mitochondrial fission, implicating the therapeutic potential of targeting mitochondrial homeostasis for AKI.
Keywords: Fundc1, Mitophagy, Ulk1, IPC, AKI, Mitochondrial fission, Drp1
Graphical abstract
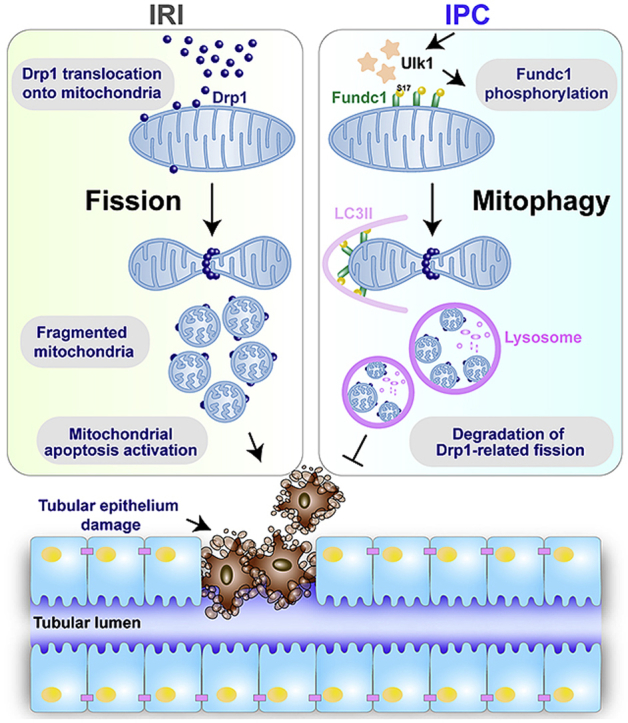
1. Introduction
Acute kidney injury (AKI) often develops in renal ischemia-reperfusion injury (IRI), sepsis and nephrotoxicity. Ample evidence has indicated that overt renal tubular cell injury predisposes AKI via intrinsic and/or extrinsic programmed cell death (PCD) [1], which is tightly associated with changes in mitochondrial function [2]. One of the key adaptive responses of mitochondria to various stress insults is the activation of mitophagy, a cellular process that promptly and selectively clears long-lived or damaged mitochondria in an autophagosome-dependent manner [3]. Molecular examination reveals that mitophagy is mainly under the regulation of receptor-independent [4] or -dependent mechanisms [5]. The former is well known as PINK1/Parkin-induced mitophagy with its beneficial role in renoprotective action documented recently [6]. FUN14 domain-containing protein 1 (Fundc1) is a novel mitophagy receptor and governs mitochondrial turnover through direct interaction with LC3 [7]. Fundc1 mitophagy is activated by hypoxia preconditioning through post-transcriptional phosphorylation at Ser17 [8,9]. Our previous studies indicated that proper activation of Fundc1 mitophagy is vital for not only maintenance of mitochondrial performance but also cell/organ homeostasis in myocardial IRI [4], microvascular reperfusion defects [10], and platelet activation/aggregation [11]. In addition, beneficial effects of Fundc1-dependent mitophagy on mitochondrial quality control have also been documented in sepsis-induced liver injury [12], cancer [13] and neuronal dysfunction [9] although little is known with regards to the role of Fundc1-mediated mitophagy in AKI.
Mitophagy is typically turned on by renal ischemic preconditioning (IPC) that serves as a beneficial and practical avenue to lower the incidence of AKI [14]. Thus, it is plausible to speculate that Fundc1-induced mitophagy may contribute to IPC-offered renoprotection against AKI, if any. It is now recognized that mitochondrial fission is an early molecular event preceding AKI whereas pharmacological intervention and/or genetic ablation of gene related to fission effectively attenuate AKI-related renal injury [15]. In fact, mitochondrial quality control, ROS oxidative stress, and mitochondrial apoptosis are all closely controlled by mitochondrial fission in AKI [16]. Although a tie between mitophagy activation and fission inhibition has been reported in several disease models [17,18], the precise nature through which IPC-activated mitophagy attenuates AKI in particular if any mitochondrial fission involvement has not been fully elucidated. The aim of our study is to explore whether Fundc1 mitophagy is required for IPC-mediated renoprotection in AKI via attenuating mitochondrial fission.
2. Materials and methods
2.1. Animal
Fundc1fl/fl mice have been described by our previous studies [4,11]. KapCre mice (Stock No: 008781) and Ulk1fl/fl mice (Stock No: 017976) were obtained from the Jackson Laboratory. Drp1fl/fl mice were generated as previously described [15]. The renal proximal tubule-conditional Fundc1 knockout (Fundc1PTKO), Drp1 knockout (Drp1PTKO) and Ulk1 knockout (Ulk1PTKO) mice were created by breeding KapCre mice with Fundc1fl/fl, Drp1fl/fl mice, and Ulk1fl/fl mice, respectively. Besides, Fundc1PTKO mice crossed with Drp1fl/fl mice to obtain renal proximal tubule-conditional Fundc1-Drp1 double knockout mice (Fundc1-Drp1PTKO mice; Fundc1fl/fl, Drp1fl/fl, KapCre). All mice were crossed on a C57BL/6 background for at least three generations. All animal experiments were conducted in accordance with a protocol approved by the Animal Institutional Care and Use Committees at the University of Wyoming and Chinese PLA General Hospital.
2.2. Renal IRI and IPC models in vivo
Renal ischemia AKI was induced using the ischemia reperfusion injury (IRI) model (eight-week-old male mice, n = 6/group). In brief, following treatment with buprenorphine (0.075 mg/kg body wt) and tribromoethanol (250 mg/kg body wt), renal pedicles were exposed by flank incisions for bilateral clamping to induce 30 min ischemia followed by 24 h reperfusion. Sham-operated group received the same operation with the exception of vessel clamping. To induce IPC, mice were anesthetized with 1%–3% isoflurane through inhalation (Baxter, Deerfield, IL) and then bilateral renal vessels were cross-clamped for 15-min to produce ischemia, followed by 1-h reperfusion to induce renal IPC model according to the previous studies [2,6]. After 30 min of recovery, IPC-treated mice were subjected to IRI. A graphic explanation of the surgery procedure is illustrated in Supplemental Fig. 1A. Renal tissues and blood samples were collected 1 h after IRI. BUN ELISA kit (MBS751125) and Creatinine (Cr) ELISA Kit (MBS2540563), purchased from MyBioSource, Inc., were used to detect the levels of BUN and Cr after IRI. Cell-permeable Z-IETD-FMK (Abcam; ab141382, 100 μM), Z-ATAD-FMK (Abcam; ab141383, 10 μM), Z-LEHD-FMK (Selleck Chemicals, Houston, TX, USA; No. S7313, 50 μM), and Z-VAD-FMK (Selleck Chemicals, Houston, TX, USA; No. S7023, 20 μM) were first dissolved in DMSO and then diluted in PBS (the final concentration of DMSO in the solution injected into animals was <1%). To block the activity of caspase-8, caspase-12, and caspase-9, mice were intraperitoneally injected with Z-IETD-FMK, Z-ATAD-FMK, and Z-LEHD-FMK, respectively, 24 h before IRI, according to previously reported protocols [19,20].
2.3. Histopathology and immunohistochemistry
Following IRI procedures, mice were humanely euthanized, and samples of mouse kidneys were collected. Tissues were fixed with formalin and were then embedded in paraffin. Sections were prepared for hematoxylin and eosin (H&E) staining and tubular injury index was determined as previously reported. In addition, sections (3–4 μm thickness) were deparaffinized, rehydrated by serial immersions in ethanol, blocked with a peroxidase blocking solution for 5 min [21]. Primary antibody (Fundc1, 1:250, #ab224722, Abcam) was used to incubate with sections followed by subsequent staining with a biotin-linked secondary antibody prior to streptavidin (Dako Cytomation, K3461) treatment. Slides were then treated with an AEC solution for 10 min at room temperature and images were acquired using an Axioskop 40 microscope (Zeiss, Oberkochen, Germany).
2.4. Cellular isolation and mimicked IRI model in vitro
Primary tubule cells were isolated from the above gene-knockout mice according to the previous studies [22]. Then, these “normal” tubule cells were cultured in complete DMEM medium with 10% FBS for 1–3 days and then subjected to mimicked IPC (mIPC) and/or mimicked IRI (mIRI) in vitro. The mimicked IRI (mIRI) was induced through incubating isolated normal tubule cells with 10 mM rotenone in glucose-free DMEM for 3-h followed by 3-h full culture medium incubation with 10% FBS at 37 °C/5% CO2. The mIPC was induced via 30-min rotenone treatment followed by 30-min recovery in fresh culture medium with 10% FBS at 37 °C/5% CO2. To inhibit the activity of lysosome-mediated protein degradation and ubiquitin-proteasome system, tubule cells were pre-treated with bafilomycin A1 (Selleck Chemicals, Houston, TX, USA; No. S1413, 0.1 μM) and MG132 (Selleck Chemicals, Houston, TX, USA; No. S2619, 30 μM) 4 h before mIRI in vitro. Besides, Mdivi-1 (Selleck Chemicals, Houston, TX, USA; No. S7162, 5 μM), an inhibitor of mitochondrial division, was used to incubated with tubule cells 2 h before mIRI to suppress excessive mitochondrial fission [23]. To block the activity of caspase-3, caspase-8, caspase-12, and caspase-9, tubule cells were treated with Z-VAD-FMK (Selleck Chemicals, Houston, TX, USA; No. S7023, 20 μM), Z-IETD-FMK (Abcam; ab141382, 100 μM), Z-ATAD-FMK (Abcam; ab141383, 10 μM), and Z-LEHD-FMK (Selleck Chemicals, Houston, TX, USA; No. S7313, 50 μM), respectively, for 2 h prior to mIRI in vitro.
2.5. TUNEL staining and cell viability assay
Paraffin-embedded sections were stained using the In Situ Cell Death Detection Kit (Sigma-Aldrich) following the manufacturer's instructions. Briefly, 4-μm kidney sections were deparaffinized and rehydrated. Then, sections were incubated with a TUNEL reagent mixture for 30 min at room temperature, and were rinsed with PBS three times, 5 min each. Nuclei were stained with DAPI [24]. Sections were visualized under a Leica TCS SP5 II microscope and apoptotic areas were quantified in 15 independent fields. Percentage of stained area was calculated using the Image J software. Besides, cellular viability was determined using the MTT and LDH release assays (Beyotime, China), as our previously described [25].
2.6. Caspase activity assay
To assess caspase activity, caspase commercial kits (Beyotime Institute of Biotechnology, China) were used according to the manufacturer's protocol. In brief, for caspase-9 activity assay, 5 μl of Ac-LEHD-pNA (Beyotime, #P9728, 200 μM) was added to samples for 45 min at 37 °C. For caspase-8 activity assay, 5 μl of Ac-IETD-pNA (Beyotime, #P9723, 0.2 mM) was used to incubate with samples for 1 h at 37 °C/5% CO2 in the dark. Caspase-3 activity was determined through incubation of Ac-DEVD-pNA (Beyotime, #P9710, 0.2 mM) with samples for 1 h at 37 °C/5% CO2 in the dark. Then, absorbance was recorded at 405 nm to reflect caspase-3/8/9 activities [26]. Caspase-12 activity was determined using a Caspase-12 Fluorometric Assay Kit (BioVision, #K139). In brief, 5 μl of the ATAD-AFC substrate (50 μM final concentration) was incubated with samples for 1 h at 37 °C/5% CO2 in the dark. Then, absorbance was evaluated at 505 nm using a microplate reader (Epoch 2; BioTek Instruments, Inc., Winooski, VT, USA).
2.7. Immunoblot analysis and co-immunoprecipitation
Kidney cortex and primary tubule cells were harvested and lysed with a RIPA lysis buffer (Beyotime) including phosphatase and protease cocktail (Biotool). Protein lysates were prepared and centrifuged at 12,000 rpm at 4 °C for 10 min to remove insoluble materials. Protein concentration was determined using a BCA protein assay kit (Bestbio) [27]. An equivalent quantity of protein was mixed with an SDS-PAGE loading buffer (Beyotime) and was subjected to SDS-PAGE. The separated proteins were transferred onto PVDF membranes and probed with primary antibody and an appropriate peroxidase-conjugated secondary antibody. Bands were visualized using a Western-Light chemiluminescent detection system (Image Station 4000 MM Pro, XLS180, Kodak, USA). p-FUNDC1 (1:500) and t-FUNDC1 (1:1000) poly-clonal antibodies were produced by immunizing rabbits with synthesized and purified phosphorylated and nonphosphorylated peptides from FUNDC1 (Abgent, SuZhou, China) according to our previous studies [4]. The primary antibodies used in the present study was as follows: Ulk1 (Abcam, #ab167139, 1:1000), Drp1 (Abcam, #ab184247, 1:1000), Tom20 (Abcam, #ab186734, 1:1000), VDAC1 (Abcam, #ab15895, 1:1000), GAPDH (Abcam, #ab181602, 1:1000), LC3II (Cell Signaling Technology, #12513, 1:1000), p62 (Cell Signaling Technology, #5114, 1:1000), LC3I/II (Cell Signaling Technology, #13118, 1:1000), Kim1 (MyBioSource, # MBS2006453, 1:1000), Tim23 (Santa Cruz Biotechnology, #sc-514463, 1:1000), Parkin, (Santa Cruz Biotechnology, #sc-32282, 1:1000), Bnip3 (Abcam, #ab10433, 1:1000), p-Drp1S616 (MyBioSource, #MBS9386761, 1:1000).
Protein interaction was estimated using a Co-Immunoprecipitation Kit (Pierce, 26149) as our previously described [25,28]. Briefly, proteins from samples were cross-linked in 1% paraformaldehyde followed by rinsing in PBS containing 100 mmol/L glycine. Samples were then lysed by sonication in PBS with 1% Triton X-100 and were incubated with respective antibodies and protein A/G agarose. Bands were visualized using a Western-Light chemiluminescent detection system (Image Station 4000 MM Pro, XLS180, Kodak, USA) [29].
2.8. qPCR for gene expression
RNA was isolated using a RNeasy Mini Kit (Qiagen #74104) according to the manufacturer's instructions. Reverse transcription was performed with the Verso cDNA synthesis kit (Thermo Fisher Scientific/Invitrogen Life Sciences) per instruction from the supplier [30]. Quantitative PCR was performed using SYBR Green supermix (Bio-Rad #1725120). mRNA levels were measured using real-time quantitative PCR (qPCR) with an ABI 7900 HT cycler (Thermo Fisher, MA, US). The qPCR primers were as follows: Ccl2, forward primer 5′-GTTGGCTCAGCCAGATGCA-3′ and reverse primer 5′-AGCCTACTCATTGGGATCATCTTG-3′; IL6, forward primer 5′-TGGCTAAGGACCAAGACCATCCAA -3′ and reverse primer 5′-AACGCACTAGGTTTGCCGAGTAGA-3′; Drp1, forward primer 5′-TAGTGGGCAGGGACCTTCTT-3′ and reverse primer 5′-TGCTTCAACTCCATTTTCTTCTCC-3′; Mff, forward primer 5′-AAGTGGCTCTCACCCTAGCA-3′ and reverse primer 5′-TGCCCCACTCACCAAATGT-3′; Fis1, forward primer 5′-CAAGGAACTGGAGCGGCTCATTA-3′ and reverse primer 5′- GGACACAGCAAGTCCGATGAGT-3′; Mid49, forward primer 5′-TGTGGTGGACTTCCTCTTGGC-3′ and reverse primer 5′-GAGAATGAATGGGCGTGGG-3′; Mid51, forward primer 5′-AGGATGACAATGGCATTGGC-3′ and reverse primer 5′-CCGATCGTACATCCGCTTAAC-3′; GAPDH, forward primer 5′-ACGGCAAATTCAACGGCACAGTCA-3′ and reverse primer 5′-TGGGGGCATCGGCAGAAGG-3′.
2.9. Fractionation of mitochondria
Samples were harvested, rinsed with PBS, and were suspended in an isolation buffer (3 mM Hepes-KOH (pH 7.4), 0.21 M mannitol, 0.07 M sucrose, 0.2 mM EGTA) on ice. Then, homogenates were loaded on 0.34 M sucrose followed by centrifugation at 500×g. These steps were repeated for three times before centrifugation of supernatants at 10,000×g to extract mitochondrial fraction [31]. VDAC was used as the loading control for mitochondrial western blots.
2.10. mtDNA strand breaks detection
mtDNA strand breaks were measured based on our previous study [32]. In brief, mitochondrial suspension, isolated from treated cells, was centrifuged at 15 000 g at 4 °C for 30 min. Then, sediment was incubated with 0.25 mmol/L inositol, 10 mmol/L Na3PO4, and 1 mmol/L MgCl2 at 4 °C for 30 min (pH 7.2). Fluorometric analysis of DNA unwinding methods were reported by Birnboim and Jevcak [33].
2.11. siRNA knockdown assay
Mouse Parkin siRNA was purchased from Santa Cruz Biotechnology. To knockdown Parkin expression, tubule cells were washed and incubated with 20 nM siRNA in an OptiMEM media (Life Technologies #31985070) supplemented with 1:50 Oligofectamine (Life Technologies #12252011) for 5 h. Cells were washed with PBS and were then incubated overnight with complete DMEM medium with 10% FBS [34]. The next day, cells were washed with PBS and were collected for experimentation. Western blot was used to verify the knockdown efficiency.
2.12. Adenovirus-mediated Drp1 overexpression
Construction of adenovirus vectors containing Drp1 was generated as our previously described [18,35]. In brief, plasmids of pDC316-mCMV-Drp1 were designed and produced by the Shanghai GenePharma Co., Ltd. (Shanghai, China). Plasmids were transfected into 293 T cells using lipofectamine 2000. After transfection for 48 h, viral supernatant was collected and was filtered through a 0.45-lm filter to obtain adenovirus-Drp1 (Ad-Drp1). Thereafter, tubule cells were infected with Ad-Drp1 for 6 h at 37 °C/5% CO2. The media were then replaced with fresh culture medium [36]. After 24-h culture, cells were washed with PBS and collected for experimentation. Western blot was used to evaluate the overexpression efficiency.
2.13. Mitochondrial potential and ROS staining
MitoSOX red mitochondrial superoxide indicator (M36008) and CellROX™ Green Reagent (C10444), purchased from Invitrogen, Inc., were used to stain mitochondrial ROS (mito-ROS) and cytoplasmic ROS (cyto-ROS), respectively. In brief, cells were stained with the MitoSOX red mitochondrial superoxide indicator for 30 min at 37 °C/5% CO2 in the dark. After rinsing with PBS, cells were labelled with a CellROX™ Green Reagent for 15 min at 37 °C/5% CO2 in the dark. Samples were subsequently washed with PBS to remove free probes. Nuclei were stained with DAPI. ROS quantification was performed through the fluorescence intensity of mito-ROS and cyto-ROS, based on our previous studies [37,38]. Mitochondrial membrane potential was detected using the JC-1 assay (Invitrogen™, T3168) according to manufacturer's protocol [39,40]. In brief, cells were washed with PBS and were then stained with JC-1 probe for 30 min at 37 °C/5% CO2 in the dark. Subsequently, PBS was used to remove free probes and images were captured under a Leica TCS SP5 II confocal spectral microscope. The red-to-green fluorescence ratio was employed to evaluate the changes in mitochondrial membrane potential. To determine immunofluorescence, the red/green immunosignals were converted into an average grayscale intensity which was subsequently analyzed using Image-Pro Plus 6.0 software.
2.14. Mitophagy detection and mPTP opening assay
Mt-Keima is a ratiometric pH-sensitive fluorescent probe that is targeted into the mitochondrial matrix. A low-ratio mt-Keima derived fluorescence (543/458 nm) reports neutral environment, whereas a high-ratio fluorescence reports acidic pH. Thus, mt-Keima enables differential imaging of mitochondria in the cytoplasm and mitochondria in acidic lysosomes. Mt-Keima probe (pMT-mKeima-Red, #AM-V-251, MBL Medical & Biological Laboratories Co., ltd. Woburn, MA) was transfected to cells at 37 °C/5% CO2 following the manufacturer's instructions [41]. Fluorescent images were captured using Leica TCS SP5 II confocal spectral microscope. Ratio (543/458 nm) of mt-Keima emission light were calculated as a value of mitophagy. mPTP was determined using an Image-IT™ LIVE Mitochondrial Transition Pore Assay Kit (Invitrogen™, Catalog #I35103). In brief, cells were washed with PBS and were then stained using Calcein at 37 °C for 15 min in the dark. Subsequently, PBS was used to remove residual dyes to minimize background. Then, absorbance was recorded at 494 nm using a microplate reader (Epoch 2; BioTek Instruments, Inc., Winooski, VT, USA).
2.15. Confocal microscopy
For confocal microscopy, samples were fixed through 4% paraformaldehyde, permeabilized using 0.5% Triton X-100, blocked with 5% bovine serum albumin, and were then incubated with primary antibodies: Tom20, (mitochondrial antibody, 1:500, Abcam, #ab186734) and Ki67 (a proliferation marker, 1:100, Abcam, # ab15580), at 4 °C overnight, followed by incubation with the appropriate secondary antibody. Nuclei were stained with DAPI prior to mounting. Confocal fluorescence images were captured using a Leica TCS SP5 II confocal spectral microscope. At least 200 mitochondria were used to evaluate the average mitochondrial length based on our previous studies [4,10]. Besides, at least 100 cells were recorded to calculate the number of cells with fragmented mitochondria.
2.16. Pulse-chase analysis
Cells were radiolabeled with [35 S]-Met (100 mCi) for the indicated durations in normal culture condition. Following rinses with PBS, cells were chased in a complete DMEM medium with 10% FBS for the indicated durations [42]. Whole-cell lysates were immunoprecipitated with Protein G plus beads coated in the indicated antibodies. Precipitated proteins were eluted with a SDS-PAGE loading buffer and were analyzed through western blots.
2.17. Mitochondrial respiration and ETCs activity detection
Mitochondrial respiration was measured via analyzing the mitochondrial oxygen consumption rates (OCR), as our previously described [28]. In brief, cells were seeded at 40,000 cells/well on 96- well XFe96 cell culture microplates and were cultured for 48 h under an XFe96 extracellular flux analyzer (Agilent Technologies). For respiration assays, cells were incubated in a CO2-free environment for 1 h, and OCR was measured every 3 min over 90 min. First, OCR was quantified in basal conditions (20 mmol/L glucose), then 1 μmol/L oligomycin (ATP synthase inhibitor), next with 0.125 μmol/L FCCP (mitochondrial respiration uncoupler), and finally with 1 μmol/L rotenone/antimycin A (complex I and III inhibitors, respectively). Complex I Activity Assay Kit (ab109721) and Complex II Activity Assay Kit (ab109908), purchased from Abcam, were used to determine the activity of ETCs activity per manufacturer's protocol [43].
2.18. Statistical analysis
Data are presented as means ± standard error (S.E.M). Using GraphPad Prism 5.0 software. Quantitative data were analyzed by t-test for the comparison between two groups and by ANOVA followed by Tukey post-test for comparison between multiple groups. P < 0.05 was considered statistically significant.
3. Results
3.1. Fundc1 mitophagy is induced by IPC via post-transcriptional modification at Ser17
To discern the role of Fundc1-mediated mitophagy in ischemic AKI, western blots were used to observe the alterations of p-Fundc1S17 over a 24-h post-IRI time frame. Moderate level of p-Fundc1S17 was found in renal cortex lysates at baseline (Fig. 1A). After IRI exposure, p-Fundc1S17 was overtly suppressed and its level was no longer detectable 6-h post-IRI, compared with the baseline levels (Fig. 1A). Interestingly, upon IPC treatment, p-Fundc1S17 was significantly increased from the baseline levels. Importantly, IPC substantially mitigated the inhibitory effect of IRI on p-Fundc1S17 and maintained p-Fundc1S17 level at near-baseline levels throughout IRI periods (Fig. 1A). In comparison, other mitophagy-related protein, such as Parkin, was abundant at baseline (Fig. 1A). IRI caused a similar decline in Parkin, the level of which was almost undetectable 6-h post-IRI. IPC seemed to elicit a mild action on Parkin expression with no statistical difference reached in Parkin expression between IPC treatment and baseline condition (Fig. 1A). In addition, IPC failed to sustain Parkin during the IRI period. These results indicate that IRI stress significantly represses mitophagy signals including Fundc1 and Parkin whereas IPC primarily activates Fundc1. Other autophagy parameters such as LC3II was predictably downregulated in response to IRI stress (Fig. 1A), an effect that was followed by an accumulation of mitochondrial membrane protein (Tim23). In comparison with IRI group, IPC also maintained LC3II expression and reduced Tim23 to near-normal levels (Fig. 1A). These data indicate that Fundc1-related mitophagy is inhibited at the stage of IRI and is stabilized by IPC.
Fig. 1.

IPC primarily activates Fundc1-related mitophagy in IRI kidneys. (A) Fundc1f/f mice were subjected to IRI (30-min ischemia and 24-h reperfusion) with or without IPC. Then, proteins were isolated from renal cortex and western blots were used to evaluate the level of mitophagy parameters. (B) Proteins were isolated from renal cortex in Fundc1f/f and proximal tubule-specific Fundc1 knockout (Fundc1PTKO) mice. Then, level of Fundc1 was determined using western blots. (C) Fundc1f/f and Fundc1PTKO mice were subjected to IRI (30-min ischemia and 24-h reperfusion) with or without IPC treatment. Then, mitophagy-related parameters such as Fundc1, Parkin, LC3 and Tim23 were analyzed using Western blot. (D–E) Primary tubule cells were isolated from Fundc1f/f and Fundc1PTKO mice and received mimicked IPC (mIPC) and/or mimicked IRI (mIRI) using rotenone-mediated nutrient deprivation. Then, mt-Kemia assay was employed to assess mitophagy in vitro. The fluorescence intensity ratio of 534/458 nm was used to quantify mitophagy index. Experiments were repeated for at least three times and data are shown as mean ± SEM (n = 6 mice or 3 independent cell isolations per group). Fundc1f/f mice in sham group or Fundc1f/f tubule cells in control group were used as the normalizer for all the conditions. *P < 0.05.
To better clarify the causal role of Fundc1 in IPC-sustained mitophagy, proximal tubule-specific Fundc1 knockout (Fundc1PTKO) and littermate control (Fundc1f/f) mice were used. At the physiological condition, Fundc1 expression is abundant in tubule whereas it is sparse in glomeruli (Supplemental Fig. 1B). The knockdown efficiency of Fundc1PTKO was confirmed using Western blot in whole kidney tissues (Supplemental Fig. 1C) and cortex lysates (Fig. 1B). Similar to the above findings, IRI drastically suppressed p-Fundc1 and Parkin, leading to LC3II reduction and Tim23 accumulation (Fig. 1C). IPC could primarily upregulate p-Fundc1 level rather than Parkin, an effect that was coincidentally followed by increased LC3II and decreased Tim23 (Fig. 1C). Deletion of Fundc1 abolished IPC-stabilized p-Fundc1 level, resulting in LC3II downregulation and Tim23 accumulation (Fig. 1C). In line with these observations, mitophagy index was also detected through mt-Keima assay in vitro using isolated primary tubule epithelial cells from Fundc1f/f and Fundc1PTKO mice in the absence or presence of the lysosomal inhibitor Baf-A1. As shown in Fig. 1D–E, although mitophagy was attenuated by mIRI, IPC could restore mitophagy activity in Fundc1f/f cells rather than Fundc1PTKO cells. These data indicate that Fundc1, mainly preserved by IPC, contributes to mitophagy preservation under renal IRI.
Notably, baseline deletion of Fundc1 had no influence of LC3II and Tim23 (Fig. 1C). Interestingly, a compensatory increase in Parkin expression (Fig. 1C) rather than Bnip3/Nix (Supplemental Fig. 1D) was induced in response to baseline Fundc1 deficiency. Once silencing of Parkin in Fundc1-depleted cells, basal mitophagy was largely impaired (Supplemental Fig. 1E), as evidenced by decreased LC3II and increased Tim23. These data suggest that baseline Fundc1 deletion activates Parkin and the latter compensates mitophagy activity. However, under IRI, both p-Fundc1 and Parkin were suppressed and thus mitophagy compensatory signal was absent in vivo (Fig. 1C) and in vitro (Supplemental Fig. 1E). In contrast to IRI, IPC predominantly sustained p-Fundc1 to near-physiological levels and failed to interrupt IRI-mediated Parkin downregulation. Under this condition, Parkin-mediated compensatory mechanism was inactive and concurrent Fundc1 deletion blunted IPC-induced mitophagy, as evidenced by decreased LC3II and elevated Tim23 (Fig. 1C and Supplemental Fig. 1E). This finding explains that Fundc1 deficiency primarily affects IPC-mediated mitophagy in kidney IRI.
3.2. IPC-preserved renal function is aggravated by Fundc1 deficiency
Next, contribution of Fundc1 mitophagy to renoprotection rendered by IPC was evaluated. Levels of BUN and creatinine (Cr) were significantly higher in IRI group when compared with the sham group (Fig. 2A–B). Along the same line, IRI overtly increased the expression of kidney injury molecule 1 (Kim1), the effects of which were mitigated by IPC treatment (Supplemental Figs. 2A–B). These beneficial effects of IPC were attenuated in Fundc1PTKO mice. Besides, HE staining exhibited that IRI triggered tubular injury (Fig. 2C–D) and these structural damages were correlated with more TUNEL+ apoptotic cells (Fig. 2E–F) as well as increased Ki67+ proliferative cells (Supplemental Figs. 2C–D). Interestingly, the IPC offered beneficial effects on IRI-mediated histopathological changes, tubular death and tubule cell proliferative recovery in Fundc1f/f mice but not Fundc1PTKO mice. This pro-survival action of IPC on tubule cells was also validated in vitro using MTT and LDH release assays (Supplemental Figs. 2E–F).
Fig. 2.

Fundc1 deletion attenuates IPC-mediated renoprotection in IRI kidneys. (A–B) Following IRI, levels of BUN and Cr were determined in various groups. (C–D) H&E staining for reperfused kidneys. Tubular injury index was determined. (E–F) TUNEL assay for IRI kidneys. The number of apoptotic cells was evaluated. (G–H) qPCR was used to observe pro-inflammation factors. Experiments were repeated at least three times and data are shown as mean ± SEM (n = 6 mice per group). Fundc1f/f mice in sham group were used as the normalizer for all the conditions. *P < 0.05.
It has been reported that inflammation response is associated with the severity of AKI [16]. Levels of inflammation factors such as Ccl2 and IL-6 were significantly elevated by IRI compared to that in the sham group, as evaluated through qPCR (Fig. 2G–H). However, IPC treatment inhibited the upregulation of inflammation factors; the effect of which was abolished in Fundc1PTKO mice. These data indicate that AKI-related renal injury and inflammation response are relieved by IPC in a Fundc1 mitophagy-dependent fashion.
3.3. Fundc1 ablation compromises IPC-sustained mitochondrial quality control
After mIRI, the content of double-strand mitochondrial DNA (mtDNA) was reduced whereas mIPC maintained mtDNA integrity in Fundc1f/f but not Fundc1PTKO tubule cells (Fig. 3A). Structurally, mtDNA copy and transcription are fine-tuned by mtDNA genomic stability. However, mtDNA replication (Supplemental Fig. 3A) and translation (Supplemental Figs. 3B–C) were significantly reduced by mIRI, but was mostly normalized by mIPC in Fundc1f/f cells rather than Fundc1PTKO cells. Functionally, mtDNA plays a pivotal role in encoding mitochondrial electron transport chain complexes (ETC). mIRI exposure inactivated ETC whereas mIPC reversed ETC activity in Fundc1f/f cells but to a lesser extent, in Fundc1PTKO cells (Supplemental Figs. 3D–E). Due to ETC dysfunction, ATP production (Fig. 3B) and mitochondrial respiration (Fig. 3C–D) were inhibited whereas mitochondrial ROS (mito-ROS) and cytoplasmic ROS (cyto-ROS) were boosted in response to mIRI (Fig. 3E). mIPC substantially restored ATP synthesis and effectively neutralized mito- and cyto-ROS overload, the effects of which were weakened by Fundc1 deletion. Other than energy metabolism, mitochondrial biogenesis, as assessed via the transcriptional levels of PGC1α and Sirt3, was significantly inhibited by mIRI and improved by mIPC in Fundc1f/f rather than Fundc1PTKO cells (Fig. 3F–G). These data support that mitochondrial quality control is likely handled by IPC through Fundc1-mediated mitophagy.
Fig. 3.

IPC-mediated mitochondrial protection was abolished in response to Fundc1 depletion. (A) Primary tubule cells were isolated from Fundc1f/f and Fundc1PTKO mice. mtDNA double strand breaks were determined in vitro. (B–D) Cellular ATP production and mitochondrial OCR rate were determined using an XFe96 extracellular flux analyzer. (E) Mitochondrial ROS and cytoplasmic ROS production were determined using the MitoSOX red indicator and CellROX™ green reagent, respectively. The immunosignals of mito-ROS and cyto-ROS were measured using Image-Pro Plus 6.0 software. (F–G) In vitro, RNA was isolated from cells and then the transcriptions of PGC1α and Sirt3 were measured using qPCR. Experiments were repeated at least three times and data are shown as mean ± SEM (n = 3 independent cell isolations per group). Fundc1f/f tubule cells in control group were used as the normalizer for all the conditions. *P < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.4. IPC-inhibited mitochondrial apoptosis is amplified by Fundc1 deficiency
Mitochondria damage triggers programmed cell death. Data from in vitro study revealed that mIRI provoked TUNEL apoptosis; the effect of which was mitigated by mIPC in Fundc1f/f cells but not Fundc1PTKO cells (Fig. 4A). Interestingly, Z-VAD-FMK, an irreversible pan-caspase inhibitor, suppressed TUNEL apoptosis and largely “reversed” mIPC-exerted anti-apoptotic effect in Fundc1PTKO cells (Fig. 4A). Besides, results of mitochondrial potential dissipation (an early event of mitochondrial apoptosis, Fig. 4B–C) and mPTP opening (a late feature of mitochondrial death, Fig. 4D) also supported that Fundc1 mitophagy was required for mIPC-mediated reconciliation of mitochondrial apoptosis.
Fig. 4.

IPC-mediated tubule survival is attenuated by Fundc1 deficiency due to activated mitochondrial apoptosis. (A) Primary tubule cells were isolated from Fundc1f/f and Fundc1PTKO mice. Cell death was evaluated using TUNEL assay. Z-VAD-FMK, an inhibitor of caspase-3, was incubated with tubule cells prior to mIRI procedure. (B–C) Mitochondrial potential was stained using JC-1 probe. Then, red-to-green fluorescence ratio was used to quantify mitochondrial potential. (D) mPTP opening rate was determined in response to Fundc1 deletion and/or mIPC treatment. (E–F) In vivo, kidney tissues were collected and the caspase-12/8 activities were measured via ELISA. (G) In vivo, Z-ATAD-FMK (an inhibitor of caspase-12), Z-IETD-FMK (a caspase-8 blocker), and Z-LEHD-FMK (an inhibitor of caspase-9) were injected into mice 24-h before IRI. Then, kidney tissues were isolated and activity of caspase-3 was measured using ELISA. (H) In vitro, cell death was measured using TUNEL staining. Z-ATAD-FMK, Z-IETD-FMK, and Z-LEHD-FMK were used to inhibit the activation of caspase-12, caspase-8 and caspase-9, respectively. Experiments were repeated at least three times and data are shown as mean ± SEM (n = 6 mice or 3 independent cell isolations per group). Fundc1f/f mice in sham group or Fundc1f/f tubule cells in control group were used as the normalizer for all the conditions. *P < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To clarify whether non-mitochondrial apoptosis pathways, such as caspase-12-related endoplasmic reticulum apoptosis or caspase-8-invovled Fas death signal, were affected by Fundc1, we analyzed the activities of caspase-12/8. In Fig. 4E–F, caspase-12/8 was activated by IRI and inhibited by IPC in vivo whereas Fundc1 deficiency cannot interrupt IPC-mediated caspase-12/8 inhibition, suggesting non-mitochondrial apoptosis is unlikely handled by Fundc1. Besides, neither Z-ATAD-FMK (an inhibitor of caspase-12) nor Z-IETD-FMK (a caspase-8 blocker) could reverse IPC-mediated anti-apoptotic effect in Fundc1-deleted cells (Fig. 4G). However, Z-LEHD-FMK, a caspase-9 inhibitor, restored mIPC-induced pro-survival action in Fundc1-null cells (Fig. 4G). Similar observations were made in vitro through TUNEL staining (Fig. 4H). Thus, IPC is capable of blocking mitochondria-dependent or -independent apoptosis induced by renal IRI. Among the wide array of apoptotic signal, mitochondria-initiated death rather than non-mitochondria apoptosis seems to be regulated by IPC-induced Fundc1 mitophagy.
3.5. Hyperactivation of Drp1-related mitochondrial fission accounts for mitochondrial dysfunction induced by Fundc1 deficiency
Drp1-related mitochondrial fission is noted as an initial step to aggravate renal IRI [15]. Fundc1 mitophagy has been reported to suppress fission [4,10]. Thus, we speculated that IPC-activated Fundc1 mitophagy regulates mitochondrial quality control and apoptosis through counteracting Drp1-related fission. In vitro, mitochondrial morphology staining demonstrated that mIRI resulted in the cleavage of mitochondria from an elongated network into small spheres or short rods (Fig. 5A); this effect of which could be inhibited by mIPC in Fundc1f/f rather than Fundc1PTKO cells. In vivo molecular investigation illustrated that both total Drp1 (t-Drp1) (Fig. 5B) and mitochondrial Drp1 (mito-Drp1) (Fig. 5C) were upregulated and reduced by IRI and IPC, respectively. Interestingly, Fundc1 deletion provoked an accumulation of t-Drp1 and mito-Drp1 in IPC-treated kidney (Fig. 5B–C). Of note, Drp1 transcript was analyzed and the result showed that, among the tested genes related to mitochondrial fission (including Drp1, Mff, Fis1, Mid49, Mid51), Drp1 transcript was upregulated to ~ two-fold in response to IRI (Supplemental Fig. 4). Nevertheless, IPC and/or Fundc1 deletion could not affect IRI-augmented Drp1 transcription, suggesting that IPC and Fundc1 might modulate Drp1 expression at the post-transcriptional level. Protein stability analysis using a pulse-chase assay exhibited that degradation rate of t-Drp1 was prolonged by mIRI and accelerated by mIPC (Fig. 5D); this effect was absent in Fundc1-deleted cells, suggesting that IPC-mediated Drp1 downregulation was due to its rapid degradation. Given a cardinal role of Fundc1 in promoting mitochondrial degradation, we questioned whether Fundc1-activated mitophagic degradation affected Drp1 stability. To this end, cells were treated with MG132 and BafA1 to inhibit proteasome and lysosome, respectively. In Fig. 5E, t-Drp1/mito-Drp1 downregulation was induced by mIPC and this effect could be reversed by BafA1 rather than MG132 in Fundc1f/f cells, hinting that mitochondria-located Drp1 and fragmented mitochondria are eliminated by IPC through the Fundc1-mediated mitophagy-lysosomal degradation system.
Fig. 5.

IPC requires Fundc1 to degrade Drp1 and to inhibit fatal mitochondrial division. (A) Immunofluorescence for mitochondria. At least 200 mitochondria were used to calculate the average mitochondrial length. Meanwhile, the number of cells with fragmented mitochondria was recorded. (B–C) Proteins were isolated and the levels of total Drp1 (t-Drp1) and mitochondrial Drp1 (mito-Drp1) were detected in various groups. (D) The chase time of t-Drp1 in tubule cells with or without Fundc1 deletion. (E) BafA1 and MG132 were used to inhibit the mitophagic degradation and ubiquitin-proteasome system, respectively. Then, levels of t-Drp1 and mito-Drp1 were measured. (F–G) JC-1 probe was used to evaluate mitochondrial potential. Mdivi-1 was employed to inhibit the Drp1 mitochondrial fission. Experiments were repeated at least three times and data are shown as mean ± SEM (n = 6 mice or 3 independent cell isolations per group). Fundc1f/f mice in sham group or Fundc1f/f tubule cells in control group were used as the normalizer for all the conditions. *P < 0.05.
To test whether Drp1 inhibition may offset Fundc1 deficiency-induced mitochondrial quality defects, Fundc1-deleted cells were incubated with Mdivi-1. Mdivi-1 treatment had no regulatory effect on mitophagy activity under normal, mIRI and mIPC conditions (Supplemental Figs. 5A–B), excluding the possibility that Drp1-related fission is required for Fundc1-related mitophagy. However, Mdivi-1 treatment alone attenuated mIRI-mediated mitochondrial damage (Fig. 5F–G) and cell death (Supplemental Fig. 5C). Importantly, Fundc1 deficiency-mediated mitochondrial membrane potential reduction (Fig. 5F–G) and mitochondrial apoptosis re-activation (Supplemental Fig. 5C) could be also reversed by Mdivi-1. In contrast, adenovirus transfection-mediated Drp1 overexpression (Supplemental Figs. 5D–E) abolished the protective effects exerted by mIPC on mitochondrial potential (Supplemental Figs. 5F–G) and tubule cell viability (Supplemental Figs. 5H–I). Taken together, these results demonstrate that Fundc1 deficiency disrupts IPC-exerted mitochondrial protection possibly through hyper-activation of Drp1-related mitochondrial fission.
3.6. Ablation Drp1 reveres renal dysfunction in Fundc1PTKO mice
To discern if hyper-activation of Drp1-related fission is involved in mitochondrial dysfunction and renal injury in Fundc1-deleted mice, we crossed Fundc1PTKO mice with Drp1f/f mice to generate proximal tubule-specific Fundc1/Drp1 double knockout (Fundc1/Drp1PTKO) mice. The Drp1 knockout in proximal tubule was confirmed using western blots (Supplemental Figs. 6A–B). Knockout Drp1 attenuated IRI-mediated renal dysfunction in vivo (Supplemental Figs. 6C–F) and tubule cell damage in vitro (Supplemental Figs. 6G–H). Compared to the IRI group, IPC maintained renal function (Fig. 6A–B), attenuated proximal tubule damage (Fig. 6C–D), reduced tubule cells death (Fig. 6E–F) and repressed inflammation response (Fig. 6G–H). However, IPC-mediated renoprotections were nullified in Fundc1PTKO mice and were restored via knock-outing Drp1 in Fundc1PTKO mice (Fig. 6A–H). Altogether, our results highlighted that Fundc1 deficiency abrogates renoprotective action of IPC on IRI kidney whereas knock-outing Drp1 in the Fundc1PTKO background could correct IPC-afforded renal protection.
Fig. 6.

Deletion of Drp1 restores IPC-mediated renoprotection in Fundc1-depleted mice. (A–B) Fundc1PTKO mice crossed with Drp1fl/fl mice to obtain renal proximal tubule-conditional Fundc1-Drp1 double knockout (Fundc1-Drp1PTKO) mice. After IRI, levels of BUN and Cr were determined. (C–D) HE staining for reperfused kidneys. Tubular injury index was determined. (E–F). TUNEL assay for IRI kidneys. The number of apoptotic was detected. (G–H) qPCR was used to observe the alterations of inflammation factors upregulation. Experiments were repeated at least three times and data are shown as mean ± SEM (n = 6 mice per group). Fundc1f/f mice in sham group were used as the normalizer for all the conditions. *P < 0.05.
3.7. IPC activates Fundc1 through Ulk1 and Ulk1 ablation abolishes IPC-mediated renoprotection
Lastly, to identify the signaling pathways responsible for the upregulation of Fundc1 mitophagy induced by IPC, we examined Ulk1 which was recently identified as an initial stimulus for Fundc1 phosphorylation under hypoxia exposure [8,9]. Protein analysis illustrated that IRI repressed levels of Ulk1 and p-Fundc1S17 whereas IPC could upregulate their levels (Fig. 7A). Interestingly, genetic ablation of Ulk1 inhibited IPC-mediated improvement of p-Fundc1S17 (Fig. 7A), attributing an essential role for Ulk1 on Fundc1 phosphorylation especially under hypoxia condition. Further, Ulk1 deletion had no effect on basal mitophagy level whereas it impaired mIPC-mediated mitochondrial degradation, as assessed through the ratio of mt-Keima assay (Fig. 7B). Similarly, baseline deletion of Ulk1 had no adverse effect on renal function, as assessed by histological staining (Fig. 7C) and BUN/Cr levels (Fig. 7D–E). However, IPC-mediated renal protection was ineffective in Ulk1PTKO mice. Molecular scrutiny demonstrated that IPC-ameliorated mitochondrial apoptosis (Supplemental Fig. 7A) and Drp1 fission (Supplemental Figs. 7B–C) were re-activated in Ulk1PTKO mice as a possible consequence of Fundc1 mitophagy inactivation. Finally, the molecular basis by which Ulk1 modulated Fundc1 was explored. Proteins interaction assay illustrated that Ulk1 was able to interact with Fundc1 in vitro (Supplemental Fig. 7D) and in vivo (Supplemental Fig. 7E), which was in accordance with the previous findings [8,9]. In sum, our data uncover that IPC improves Fundc1 mitophagy via Ulk1, a core component for IPC-rendered renoprotection.
Fig. 7.

Ulk1 is an upstream regulator for Fundc1 mitophagy in response to IPC. (A) Ulk1f/f mice and proximal tubule-specific Ulk1 knockout (Ulk1PTKO) mice were subjected to IRI model (30-min ischemia and 24-h reperfusion) with or without IPC pretreatment. Then, proteins were isolated from kidneys and western blots were used to analyze the expression of Ulk1 and Fundc1. (B) Primary tubule cells were isolated from Ulk1f/f and Ulk1PTKO mice and were subject to mimicked IPC (mIPC) and/or mimicked IRI (mIRI) using rotenone-mediated nutrient deprivation. mt-Kemia assay was used to evaluate mitophagy index via analyzing the ratio of 534/458 nm. (C) HE staining for reperfused kidneys. (D–E) After IRI, levels of BUN and Cr were determined. (F) Schematic diagram depicting IRI-induced renal damage and the role of Fundc1 mitophagy in IPC-mediated renoprotection through reducing Drp1-related mitochondrial fission. Experiments were repeated at least three times and data are shown as mean ± SEM (n = 6 mice or 3 independent cell isolations per group). Ulk1f/f mice in sham group or Ulk1f/f tubule cells in control group were used as the normalizer for all the conditions. *P < 0.05.
4. Discussion
Fundc1-mediated mitophagy was originally reported in hypoxic environment [44]. Later work unveiled critical roles of Fundc1 mitophagy in the rapid and precise reconstruction of mitochondrial quality and integrity [45]. In this study, we offered novel insights into a role for Fundc1 mitophagy in kidney ischemic preconditioning using an ischemic AKI model. Our results revealed for the first time that (1) Fundc1 mitophagy is activated in IPC-treated kidneys and loss of Fundc1 “inevitably” abolishes IPC-conferred kidney protection, denoting the obligatory role of Fundc1 mitophagy in IPC-associated renoprotection; (2) mechanistically, IPC requires Fundc1-activated mitophagy to degrade the mitochondria-localized Drp1, thereby eliminating IRI-activated fatal mitochondrial fission; (3) Fundc1 deficiency-mediated mitochondrial damage and therapeutic ineffectiveness of IPC could be reversed through simultaneous genetic ablation of Drp1, indicating the relationship between mitophagy and fission in kidney IPC; (4) finally, Ulk1 is required for IPC-mediated Fundc1 mitophagy and Ulk1 deletion hampers IPC-mediated preservation of mitochondrial and renal function (Fig. 7F). This work has consolidated a pivotal role for Fundc1 mitophagy as the key molecular messenger for IPC-mediated kidney protection and the therapeutic potential of targeting the IPC-Ulk1-Fundc1-Drp1 axis in the clinical management of AKI.
Mitophagy is a complex process and regulated by receptor-dependent or -independent pathways. The former comprises of Fundc1 or Bnip3 whereas the latter is managed by Parkin. Based on our results, baseline Fundc1 deletion was associated with a compensatory upregulation of Parkin, rather than Bnip3 or Nix. Accordingly, mitophagy homeostasis was unaffected by Fundc1 deficiency unless both of Fundc1 and Parkin were removed simultaneously. This compensatory mechanism was previously proposed and verified by several in-depth studies [[46], [47], [48]]. For example, germ-line Parkin ablation in mice has proven not to be an ideal experimental model for mitophagy depletion, in part due to compensation from Mul1-mediated mitophagy under physiological states [46]. Besides, Bcl2 like 13 (BCL2L13) is the mammalian homologue of Atg32, the primary mitophagy receptor found in yeasts. BCL2L13 expression may somewhat compensate the basal mitophagy activity in Atg32-null yeasts [47]. Moreover, lack of mitophagy in Mfn2-knockout mice could be compensated by activation of nonselective autophagy under normal condition [48]. In fact, autophagy-related compensatory mechanism was first described in Ulk1-knockout mice [49,50]. Ulk1/2 serve as the key upstream inducers of autophagy through regulation of ATGs. However, mice lacking either Ulk1 or Ulk2 globally were viable without any notable defect in autophagy or development [51]. In comparison, mice lacking both Ulk1 and Ulk2 could not survive beyond 24 h of birth [49,50]. Here, our data have provided convincing evidence that baseline mitophagy is governed by a complex molecular machinery and Fundc1 loss is backed by Parkin upregulation to sustain physiological mitophagy activity. Therefore, baseline Fundc1 deletion seems to display little impact on kidney function. This point receives supports from earlier work that baseline deletion of Fundc1 exhibited little pathological role in organ development and function including liver [52], heart [4], adipose [53], and muscle [54].
In response to IRI, data from our present study suggested that mitophagy parameters (e.g., Fundc1 and Parkin) were dramatically repressed. Although several studies argue that mitophagy (autophagy) is induced by IRI, a recent study using novel autophagy-reporter mice demonstrated that autophagy is inhibited within 24-h post-IRI with an augmented autophagy around 1-3d post-IRI [55]. This concept has been supported by recent reports that mitophagy is in fact inhibited in the setting of renal IRI [56,57]. Several mechanisms may potentially contribute to the discrepancies in mitophagy (autophagy) activation/deactivation under IRI, including animal age [58], species or strains [58], and oxidative stress status [59].
IPC has been proposed to be an inducer for mitophagy [2,16]. In our hands, IPC required Ulk1 to turn on Fundc1, a phenomenon closely in line with the previous findings [8,9] where hypoxia/ischemia preconditioning promoted the interaction between Ulk1 and Fundc1, accounting for IPC-induced Fundc1 phosphorylation and mitophagy upregulation. These data should explain the requirement of the Ulk1/Fundc1 axis in regulating IPC-induced mitophagy although baseline deletion of Ulk1 or Fundc1 offers little effect on mitophagy. With pretreatment with IPC prior to IRI challenge, p-Fundc1 levels were sustained at near-normal levels whereas Parkin expression was reduced to undetectable levels. Under this setting, Parkin-mediated compensatory mechanism is inactive while Fundc1 is required for mitophagy preservation. These observations explain that: 1) baseline Fundc1 deletion has no obvious inhibitory action on mitophagy activity due to the existence of Parkin-mediated compensatory mechanism; 2) IPC primarily maintains p-Fundc1 expression and fails to interrupt IRI-mediated Parkin loss, and therefore, Fundc1 deletion abolishes IPC-mediated mitophagy activation.
Hyperactive or fatal fission is usually accompanied with mitochondrial fragmentation, respiratory defect, and mitochondrial apoptosis [60]. Besides, pharmacological inhibition of fission or genetic ablation of Drp1 could attenuate ischemic AKI [61]. Defects in mitophagy are known to prompt aberrant mitochondrial fragmentation throughout cytoplasm in acute spinal cord injury [62], Parkinson's disease [63], and heart ischemia burden [64]. Here, we found that mitochondria-localized Drp1 and fragmented mitochondria were eliminated through Fundc1-elicted mitophagic degradation. This is well exemplified by a recent study that Fundc1-HSC70 interaction promotes the mitochondrial translocation of the unfolded cytosolic proteins for degradation via lysosome [65]. Besides, other groups also found the critical role of mitophagy-lysosome degradative system in removing other mitochondria-located proteins such as Bnip3 [66] and Fis1 [65]. In contrast, several careful examinations also found degradation of mitochondrial fission-related proteins such as mitochondrial elongation factor 1 (MIEF1) [67] and mitofusin 1 (Mfn1) [68] through ubiquitin-proteasome system (UPS). In fact, a set of complex interactions between mitophagic degradation and UPS have been extensively discussed [69,70] and thus degradation pathway choice may be dependent on disease condition and specific pathological process.
Finally, our results revealed the role for Ulk1 as an upstream regulator for Fundc1 mitophagy in the face of IPC, in line with previous studies [8,9]. Unlike autophagy, the molecular regulatory network of mitophagy remains elusive. To-date, three possible regulators have been identified for Fundc1 namely CK2α [4], PGAM5 [45] and Ulk1 [8]. While Ischemia/hypoxia prefers to activate Fundc1 through Ulk1 signal, reperfusion/reoxygenation trends to suppress Fundc1 via CK2α. Additional experiments are necessary to better explain whether Ulk1 downregulation or CK2α upregulation would participate in the IRI-mediated Fundc1 inactivation and mitophagy inhibition. Meanwhile, other phosphorylation sites do exist for Fundc1 including Ser13 and Tyr18 while the precise roles of these post-transcriptional modification modalities remain to be elucidated for Fundc1 in AKI-induced pathology.
Several experimental limitations should be considered for the present study. First and foremost, cell death was found to be partially attenuated by IPC and/or Z-VAD-FMK, indicating contribution from non-apoptotic pathways such as necroptosis in IRI kidney. Nonetheless, the casual relationship between Fundc1 and necroptosis remains futile. Second, further attempts using various autophagy-monitored mice should be desirable to obtain a more complete picture with regards to alterations and roles of mitophagy in our experimental setting of IRI kidney. In conclusion, data from our present study have convincingly demonstrated that IPC-mediated renoprotection is managed by Fundc1 mitophagy, a compensatory mechanism to prevent fission execution, and sustain mitochondrial homeostasis, ultimately conferring renoprotective effect.
Data availability
The datasets generated and/or analyzed during the study are available from the corresponding author with reasonable request.
Contribution statement
HZ and JW involved in conception and design, performance of experiments, data analysis and interpretation, and manuscript preparation; JW and PJZ involved in the development of methodology, HZ and RBL involved in data acquisition, HZ and JR involved in financial support, study supervision and final approval of manuscript.
Funding
This work was supported in part by National Key R&D Program of China (2017YFA0506000), and the NSFC (81900252, 81900254, 81870249, 81870249 and 91749128).
Declaration of competing interest
All authors declare that they have no conflict of interest.
Acknowledgements
None.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101415.
Contributor Information
Jun Ren, Email: jren@uwyo.edu.
Hao Zhou, Email: zhouhao@plagh.org.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Yang Y., Song M., Liu Y., Liu H., Sun L., Peng Y., Liu F., Venkatachalam M.A., Dong Z. Renoprotective approaches and strategies in acute kidney injury. Pharmacol. Ther. 2016;163:58–73. doi: 10.1016/j.pharmthera.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Livingston M.J., Wang J., Zhou J., Wu G., Ganley I.G., Hill J.A., Yin X.M., Dong Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy. 2019;15(12):2142–2162. doi: 10.1080/15548627.2019.1615822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y., Whaley-Connell A.T., Sowers J.R., Ren J. Autophagy as an emerging target in cardiorenal metabolic disease: from pathophysiology to management. Pharmacol. Ther. 2018;191:1–22. doi: 10.1016/j.pharmthera.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou H., Zhu P., Wang J., Zhu H., Ren J., Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018;25(6):1080–1093. doi: 10.1038/s41418-018-0086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rakovic A., Ziegler J., Martensson C.U., Prasuhn J., Shurkewitsch K., Konig P., Paulson H.L., Klein C. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 2018;26(8):1428–1441. doi: 10.1038/s41418-018-0219-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang C., Han H., Yan M., Zhu S., Liu J., Liu Z., He L., Tan J., Liu Y., Liu H., Sun L., Duan S., Peng Y., Liu F., Yin X.M., Zhang Z., Dong Z. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. 2018;14(5):880–897. doi: 10.1080/15548627.2017.1405880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuang Y., Ma K., Zhou C., Ding P., Zhu Y., Chen Q., Xia B. Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy. 2016;12(12):2363–2373. doi: 10.1080/15548627.2016.1238552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu W., Tian W., Hu Z., Chen G., Huang L., Li W., Zhang X., Xue P., Zhou C., Liu L., Zhu Y., Zhang X., Li L., Zhang L., Sui S., Zhao B., Feng D. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15(5):566–575. doi: 10.1002/embr.201438501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L., Wang P., Dong H., Wang S., Chu H., Yan W., Zhang X. Ulk1/FUNDC1 prevents nerve cells from hypoxia-induced apoptosis by promoting cell autophagy. Neurochem. Res. 2018;43(8):1539–1548. doi: 10.1007/s11064-018-2568-x. [DOI] [PubMed] [Google Scholar]
- 10.Zhou H., Wang J., Zhu P., Zhu H., Toan S., Hu S., Ren J., Chen Y. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2alpha. Basic Res. Cardiol. 2018;113(4):23. doi: 10.1007/s00395-018-0682-1. [DOI] [PubMed] [Google Scholar]
- 11.Zhou H., Li D., Zhu P., Hu S., Hu N., Ma S., Zhang Y., Han T., Ren J., Cao F., Chen Y. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J. Pineal Res. 2017;63(4) doi: 10.1111/jpi.12438. [DOI] [PubMed] [Google Scholar]
- 12.Yan M., Yu Y., Mao X., Feng J., Wang Y., Chen H., Xie K., Yu Y. Hydrogen gas inhalation attenuates sepsis-induced liver injury in a FUNDC1-dependent manner. Int. Immunopharmacol. 2019;71:61–67. doi: 10.1016/j.intimp.2019.03.021. [DOI] [PubMed] [Google Scholar]
- 13.Wu L., Zhang D., Zhou L., Pei Y., Zhuang Y., Cui W., Chen J. FUN14 domain-containing 1 promotes breast cancer proliferation and migration by activating calcium-NFATC1-BMI1 axis. EBioMedicine. 2019;41:384–394. doi: 10.1016/j.ebiom.2019.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Z., Gong R. Remote ischemic preconditioning for kidney protection: GSK3beta-centric insights into the mechanism of action. Am. J. Kidney Dis. 2015;66(5):846–856. doi: 10.1053/j.ajkd.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perry H.M., Huang L., Wilson R.J., Bajwa A., Sesaki H., Yan Z., Rosin D.L., Kashatus D.F., Okusa M.D. Dynamin-related protein 1 deficiency promotes recovery from AKI. J. Am. Soc. Nephrol. 2018;29(1):194–206. doi: 10.1681/ASN.2017060659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhan M., Brooks C., Liu F., Sun L., Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83(4):568–581. doi: 10.1038/ki.2012.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin Q., Li R., Hu N., Xin T., Zhu P., Hu S., Ma S., Zhu H., Ren J., Zhou H. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018;14:576–587. doi: 10.1016/j.redox.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H., Li D., Zhu P., Ma Q., Toan S., Wang J., Hu S., Chen Y., Zhang Y. Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia-reperfusion injury. J. Pineal Res. 2018;65(3) doi: 10.1111/jpi.12503. [DOI] [PubMed] [Google Scholar]
- 19.Equils O., Moffatt-Blue C., Ishikawa T.O., Simmons C.F., Ilievski V., Hirsch E. Pretreatment with pancaspase inhibitor (Z-VAD-FMK) delays but does not prevent intraperitoneal heat-killed group B Streptococcus-induced preterm delivery in a pregnant mouse model. Infect. Dis. Obstet. Gynecol. 2009;2009 doi: 10.1155/2009/749432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terlizzi M., Di Crescenzo V.G., Perillo G., Galderisi A., Pinto A., Sorrentino R. Pharmacological inhibition of caspase-8 limits lung tumour outgrowth. Br. J. Pharmacol. 2015;172(15):3917–3928. doi: 10.1111/bph.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou L., Zhang H., Davies K.J.A., Forman H.J. Aging-related decline in the induction of Nrf2-regulated antioxidant genes in human bronchial epithelial cells. Redox Biol. 2018;14:35–40. doi: 10.1016/j.redox.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terryn S., Jouret F., Vandenabeele F., Smolders I., Moreels M., Devuyst O., Steels P., Van Kerkhove E. A primary culture of mouse proximal tubular cells, established on collagen-coated membranes. Am. J. Physiol. Renal. Physiol. 2007;293(2):F476–F485. doi: 10.1152/ajprenal.00363.2006. [DOI] [PubMed] [Google Scholar]
- 23.Kim S.Y., Nair D.M., Romero M., Serna V.A., Koleske A.J., Woodruff T.K., Kurita T. Transient inhibition of p53 homologs protects ovarian function from two distinct apoptotic pathways triggered by anticancer therapies. Cell Death Differ. 2019;26(3):502–515. doi: 10.1038/s41418-018-0151-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krause J., Loser A., Lemoine M.D., Christ T., Scherschel K., Meyer C., Blankenberg S., Zeller T., Eschenhagen T., Stenzig J. Rat atrial engineered heart tissue: a new in vitro model to study atrial biology. Basic Res. Cardiol. 2018;113(5):41. doi: 10.1007/s00395-018-0701-2. [DOI] [PubMed] [Google Scholar]
- 25.Zhou H., Zhu P., Guo J., Hu N., Wang S., Li D., Hu S., Ren J., Cao F., Chen Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507. doi: 10.1016/j.redox.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karwi Q.G., Bice J.S., Baxter G.F. Pre- and postconditioning the heart with hydrogen sulfide (H2S) against ischemia/reperfusion injury in vivo: a systematic review and meta-analysis. Basic Res. Cardiol. 2018;113(1):6. doi: 10.1007/s00395-017-0664-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hao L., Sun Q., Zhong W., Zhang W., Sun X., Zhou Z. Mitochondria-targeted ubiquinone (MitoQ) enhances acetaldehyde clearance by reversing alcohol-induced posttranslational modification of aldehyde dehydrogenase 2: a molecular mechanism of protection against alcoholic liver disease. Redox Biol. 2018;14:626–636. doi: 10.1016/j.redox.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou H., Du W., Li Y., Shi C., Hu N., Ma S., Wang W., Ren J. Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J. Pineal Res. 2018;64(1) doi: 10.1111/jpi.12450. [DOI] [PubMed] [Google Scholar]
- 29.Ibanez-Cabellos J.S., Perez-Machado G., Seco-Cervera M., Berenguer-Pascual E., Garcia-Gimenez J.L., Pallardo F.V. Acute telomerase components depletion triggers oxidative stress as an early event previous to telomeric shortening. Redox Biol. 2018;14:398–408. doi: 10.1016/j.redox.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frank D., Vince J.E. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019;26(1):99–114. doi: 10.1038/s41418-018-0212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beckendorf J., Van Den Hoogenhof M.M.G., Backs J. Physiological and unappreciated roles of CaMKII in the heart. Basic Res. Cardiol. 2018;113(4):29. doi: 10.1007/s00395-018-0688-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou H., Hu S., Jin Q., Shi C., Zhang Y., Zhu P., Ma Q., Tian F., Chen Y. Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc. 2017;6(3) doi: 10.1161/JAHA.116.005328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Birnboim H.C., Jevcak J.J. Fluorometric method for rapid detection of DNA strand breaks in human white blood cells produced by low doses of radiation. Cancer Res. 1981;41(5):1889–1892. [PubMed] [Google Scholar]
- 34.Basalay M.V., Davidson S.M., Gourine A.V., Yellon D.M. Neural mechanisms in remote ischaemic conditioning in the heart and brain: mechanistic and translational aspects. Basic Res. Cardiol. 2018;113(4):25. doi: 10.1007/s00395-018-0684-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou H., Zhang Y., Hu S., Shi C., Zhu P., Ma Q., Jin Q., Cao F., Tian F., Chen Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J. Pineal Res. 2017;63(1) doi: 10.1111/jpi.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bacmeister L., Schwarzl M., Warnke S., Stoffers B., Blankenberg S., Westermann D., Lindner D. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol. 2019;114(3):19. doi: 10.1007/s00395-019-0722-5. [DOI] [PubMed] [Google Scholar]
- 37.Zhou H., Wang S., Zhu P., Hu S., Chen Y., Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335–346. doi: 10.1016/j.redox.2017.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou H., Shi C., Hu S., Zhu H., Ren J., Chen Y. BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis. 2018;21(3):599–615. doi: 10.1007/s10456-018-9611-z. [DOI] [PubMed] [Google Scholar]
- 39.Audia J.P., Yang X.M., Crockett E.S., Housley N., Haq E.U., O'donnell K., Cohen M.V., Downey J.M., Alvarez D.F. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y12 receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Res. Cardiol. 2018;113(5):32. doi: 10.1007/s00395-018-0692-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Staudacher V., Trujillo M., Diederichs T., Dick T.P., Radi R., Morgan B., Deponte M. Redox-sensitive GFP fusions for monitoring the catalytic mechanism and inactivation of peroxiredoxins in living cells. Redox Biol. 2018;14:549–556. doi: 10.1016/j.redox.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amanakis G., Kleinbongard P., Heusch G., Skyschally A. Attenuation of ST-segment elevation after ischemic conditioning maneuvers reflects cardioprotection online. Basic Res. Cardiol. 2019;114(3):22. doi: 10.1007/s00395-019-0732-3. [DOI] [PubMed] [Google Scholar]
- 42.Aluja D., Inserte J., Penela P., Ramos P., Ribas C., Iniguez M.A., Mayor F., Jr., Garcia-Dorado D. Calpains mediate isoproterenol-induced hypertrophy through modulation of GRK2. Basic Res. Cardiol. 2019;114(3):21. doi: 10.1007/s00395-019-0730-5. [DOI] [PubMed] [Google Scholar]
- 43.Abukar Y., Ramchandra R., Hood S.G., Mckinley M.J., Booth L.C., Yao S.T., May C.N. Increased cardiac sympathetic nerve activity in ovine heart failure is reduced by lesion of the area postrema, but not lamina terminalis. Basic Res. Cardiol. 2018;113(5):35. doi: 10.1007/s00395-018-0695-9. [DOI] [PubMed] [Google Scholar]
- 44.Liu L., Feng D., Chen G., Chen M., Zheng Q., Song P., Ma Q., Zhu C., Wang R., Qi W., Huang L., Xue P., Li B., Wang X., Jin H., Wang J., Yang F., Liu P., Zhu Y., Sui S., Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012;14(2):177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 45.Chen G., Han Z., Feng D., Chen Y., Chen L., Wu H., Huang L., Zhou C., Cai X., Fu C., Duan L., Wang X., Liu L., Liu X., Shen Y., Zhu Y., Chen Q. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell. 2014;54(3):362–377. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 46.Yun J., Puri R., Yang H., Lizzio M.A., Wu C., Sheng Z.H., Guo M. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife. 2014;3 doi: 10.7554/eLife.01958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao K., Wang K., Zhao M., Xu T., Klionsky D.J. Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J. Cell Biol. 2011;193(4):755–767. doi: 10.1083/jcb.201102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song M., Chen Y., Gong G., Murphy E., Rabinovitch P.S., Dorn G.W. 2nd, Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ. Res. 2014;115(3):348–353. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee E.J., Tournier C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy. 2011;7(7):689–695. doi: 10.4161/auto.7.7.15450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheong H., Lindsten T., Wu J., Lu C., Thompson C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. U. S. A. 2011;108(27):11121–11126. doi: 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kundu M., Lindsten T., Yang C.Y., Wu J., Zhao F., Zhang J., Selak M.A., Ney P.A., Thompson C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112(4):1493–1502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li W., Li Y., Siraj S., Jin H., Fan Y., Yang X., Huang X., Wang X., Wang J., Liu L., Du L., Chen Q. FUN14 domain-containing 1-mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatology. 2019;69(2):604–621. doi: 10.1002/hep.30191. [DOI] [PubMed] [Google Scholar]
- 53.Wu H., Wang Y., Li W., Chen H., Du L., Liu D., Wang X., Xu T., Liu L., Chen Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy. 2019:1–17. doi: 10.1080/15548627.2019.1596482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu T., Xu Z., Liu L., Guo Q., Wu H., Liang X., Zhou D., Xiao L., Liu L., Liu Y., Zhu M.S., Chen Q., Gan Z. Mitophagy directs muscle-adipose crosstalk to alleviate dietary obesity. Cell Rep. 2018;23(5):1357–1372. doi: 10.1016/j.celrep.2018.03.127. [DOI] [PubMed] [Google Scholar]
- 55.Li L., Wang Z.V., Hill J.A., Lin F. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J. Am. Soc. Nephrol. 2014;25(2):305–315. doi: 10.1681/ASN.2013040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ji J., Zhou X., Xu P., Li Y., Shi H., Chen D., Li R., Shi H. Deficiency of apoptosis-stimulating protein two of p53 ameliorates acute kidney injury induced by ischemia reperfusion in mice through upregulation of autophagy. J. Cell Mol. Med. 2019;23(4):2457–2467. doi: 10.1111/jcmm.14094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng J., Li H., Zhang Y., Wang Q., Zhao S., Meng P., Li J. Mammalian STE20-like kinase 1 deletion alleviates renal ischaemia-reperfusion injury via modulating mitophagy and the AMPK-YAP signalling pathway. Cell. Physiol. Biochem. 2018;51(5):2359–2376. doi: 10.1159/000495896. [DOI] [PubMed] [Google Scholar]
- 58.Jankauskas S.S., Pevzner I.B., Andrianova N.V., Zorova L.D., Popkov V.A., Silachev D.N., Kolosova N.G., Plotnikov E.Y., Zorov D.B. The age-associated loss of ischemic preconditioning in the kidney is accompanied by mitochondrial dysfunction, increased protein acetylation and decreased autophagy. Sci. Rep. 2017;7 doi: 10.1038/srep44430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ishihara M., Urushido M., Hamada K., Matsumoto T., Shimamura Y., Ogata K., Inoue K., Taniguchi Y., Horino T., Fujieda M., Fujimoto S., Terada Y. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Renal. Physiol. 2013;305(4):F495–F509. doi: 10.1152/ajprenal.00642.2012. [DOI] [PubMed] [Google Scholar]
- 60.Kapitsinou P.P., Haase V.H. Molecular mechanisms of ischemic preconditioning in the kidney. Am. J. Physiol. Renal. Physiol. 2015;309(10):F821–F834. doi: 10.1152/ajprenal.00224.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin J.L., Gruszczyk A.V., Beach T.E., Murphy M.P., Saeb-Parsy K. Mitochondrial mechanisms and therapeutics in ischaemia reperfusion injury. Pediatr. Nephrol. 2019;34(7):1167–1174. doi: 10.1007/s00467-018-3984-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kullmann A.F., Truschel S.T., Wolf-Johnston A.S., Mcdonnell B.M., Lynn A.M., Kanai A.J., Kessler T.M., Apodaca G., Birder L.A. Neurourol Urodyn; 2019. Acute Spinal Cord Injury Is Associated with Mitochondrial Dysfunction in Mouse Urothelium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song C., Zhang J., Qi S., Liu Z., Zhang X., Zheng Y., Andersen J.P., Zhang W., Strong R., Martinez P.A., Musi N., Nie J., Shi Y. Cardiolipin remodeling by ALCAT1 links mitochondrial dysfunction to Parkinson's diseases. Aging Cell. 2019;18(3) doi: 10.1111/acel.12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saito T., Nah J., Oka S.I., Mukai R., Monden Y., Maejima Y., Ikeda Y., Sciarretta S., Liu T., Li H., Baljinnyam E., Fraidenraich D., Fritzky L., Zhai P., Ichinose S., Isobe M., Hsu C.P., Kundu M., Sadoshima J. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Investig. 2019;129(2):802–819. doi: 10.1172/JCI122035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li Y., Xue Y., Xu X., Wang G., Liu Y., Wu H., Li W., Wang Y., Chen Z., Zhang W., Zhu Y., Ji W., Xu T., Liu L., Chen Q. A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 2019;38(3) doi: 10.15252/embj.201798786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park C.W., Hong S.M., Kim E.S., Kwon J.H., Kim K.T., Nam H.G., Choi K.Y. BNIP3 is degraded by ULK1-dependent autophagy via MTORC1 and AMPK. Autophagy. 2013;9(3):345–360. doi: 10.4161/auto.23072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xian H., Liou Y.C. Loss of MIEF1/MiD51 confers susceptibility to BAX-mediated cell death and PINK1-PRKN-dependent mitophagy. Autophagy. 2019:1–19. doi: 10.1080/15548627.2019.1596494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou Q., Li H., Li Y., Tan M., Fan S., Cao C., Meng F., Zhu L., Zhao L., Guan M.X., Jin H., Sun Y. Inhibiting neddylation modification alters mitochondrial morphology and reprograms energy metabolism in cancer cells. JCI Insight. 2019;4(4) doi: 10.1172/jci.insight.121582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saito T., Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ. Res. 2015;116(8):1477–1490. doi: 10.1161/CIRCRESAHA.116.303790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoshii S.R., Kishi C., Ishihara N., Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011;286(22):19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the study are available from the corresponding author with reasonable request.