

Graphical Abstarct
Procedure (Note 1)
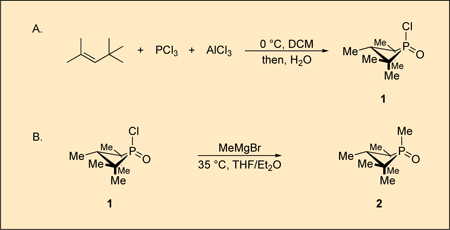
A. 1-Chloro-2,2,3,4,4-pentamethylphosphetane 1-oxide (1).
To a flame-dried 500 mL round-bottomed flask (cooled under vacuum) equipped with a magnetic stir bar (1.25 in, egg-shaped) is added aluminum chloride (28.0 g, 210 mmol, 1.0 equiv) and the flask is sealed with rubber septum (Note 2). After evacuating the flask and back-filling with nitrogen three times, dry dichloromethane (125 mL, 1.7 M) is added via syringe and the heterogeneous mixture is cooled to 0–2 °C with an ice bath while stirring under positive nitrogen pressure (Notes 3 and 4) (Figure 1A). Phosphorus trichloride (18.3 mL, 210 mmol, 1.0 equiv) is added via syringe and stirred for 5 min (Note 5). Lastly, 2,4,4-trimethyl-2-pentene (32.7 mL, 210 mmol, 1.0 equiv) is added via syringe over a period of five min and stirring is continued at 0–2 °C for 2 h (Notes 6, 7, and 8) (Figure 1B).
Figure 1.

A) Reaction set-up/aluminum chloride stirring in CH2Cl2 at 0 °C. B) Five min after the addition of 2,4,4-trimethyl-2-pentene, white precipitate is observed. C) Quench set-up. (Photos provided by the checkers)
The reaction is quenched by the slow addition of distilled water (125 mL) via a 250-mL pressure-equalized dropping funnel over 45 min while maintaining the temperature at 0–2 °C (Note 9) (Figure 1C). The reaction mixture is then transferred to a 500-mL separatory funnel and shaken. The organic phase is separated and the aqueous layer is extracted with one 125-mL portion of dichloromethane. The combined organic layers are washed with saturated NaCl solution (100 mL) and the brine solution is back-extracted with dichloromethane (125 mL). The combined organic phases are dried over anhydrous sodium sulfate (50 g, 15 min), filtered, and concentrated via rotary evaporation (ca. 100 mmHg, 23 °C) (Notes 10 and 11).
The crude residue is further dried in vacuo (ca. 0.2 mmHg, 23 °C) for a period of 45 min, backfilling the flask with nitrogen and breaking up the solids into smaller pieces every ten min with a spatula before placing the flask back under vacuum. Crude product 1, a white crystalline solid, is transferred to a tared 100-mL round-bottom flask and weighed to give 36.7 g (90% crude), and to this solid is added hexanes (55 mL, ca. 1.5 mL/1 g of 1). The mixture is rotated and heated to ca. 65 °C using a rotary evaporator in a fume hood, at atmospheric pressure and with a cold finger, until the solids are completely dissolved (Note 12). The hot vessel is wrapped and covered with aluminum foil and cooled slowly over 45 min to 23 °C, then placed in a freezer at –20 °C for 60 min. The resulting solids are collected on a coarse-porosity fritted funnel, then covered by a watch glass and dried by suction. The solid is broken up with a spatula to give phosphetane oxide 1 (29.3 g, 72%, 97:3 anti:syn) (Notes 13 and 14) (Figure 2).
Figure 2.

Recrystallized phosphetane oxide 1. Crystals are thick needles and tend to clump together during the recrystallization. (Photo provided by the submitters)
B. anti-1,2,2,3,4,4-Hexamethylphosphetane 1-oxide (2).
To a flame-dried 500 mL round-bottomed flask (cooled under vacuum) equipped with a magnetic stir bar (1.25 in, egg-shaped) is added phosphetane oxide 1 (15.0 g, 77 mmol, 1.0 equiv) and the flask is sealed with a rubber septum. After evacuating the vessel and back-filling with nitrogen three times, dry tetrahydrofuran (52 mL) is added via syringe and the homogeneous mixture is cooled to 0–2 °C with an ice bath while stirring (Note 15). A solution of methylmagnesium bromide (3 M in diethyl ether, 28 mL, 84 mmol, 1.1 equiv) is added over a period of 10 min with stirring (Note 16). After the addition, the mixture is warmed to 23 °C and then heated at 35 °C in a water bath for 2 h and then returned to 23 °C (Note 17) (Figure 3A).
Figure 3.

A) Reaction set-up/reaction mixture heating at 35 °C after Grignard addition with precipitate formation. B) Reaction mixture after quenching with saturated ammonium chloride. (Photo provided by the checkers)
The reaction vessel is immersed in an ice bath and the reaction is quenched by addition of saturated ammonium chloride (20 mL) (Note 18). Additional distilled water (60 mL) is added and the mixture is vigorously stirred until all solids went into solution. The reaction mixture is poured into a 0.5-L separatory funnel and the organic and aqueous layers are partitioned. The aqueous layer is separated and set aside temporarily. The organic layer is transferred directly into a 500 mL round-bottomed flask. The ethereal solvents are removed via rotary evaporation (ca. 100 mmHg, 23 °C) (Note 19) and the resulting residue is redissolved in dichloromethane (15 mL). The aqueous layer is reintroduced to the separatory and extracted with dichloromethane (two portions, 80 mL then 50 mL) (Note 20). The combined dichloromethane phases are then transferred back into the separatory funnel and washed with brine (50 mL). The aqueous brine phase is back-extracted with dichloromethane (50 mL) and all of the aqueous layers are combined and back-extracted with one last portion of dichloromethane (75 mL). The combined dichloromethane phases are dried over anhydrous sodium sulfate (25 g, 15 min), filtered through a plug of cotton, and concentrated by rotary evaporation (ca 100 mmHg, 23 °C). The product is further dried in vacuo (ca. 0.2 mmHg, 23 °C) giving crude phosphetane oxide 2 as a white amorphous solid. To purify, crude 2 is placed inside a 150 mL beaker and then diethyl ether (90 mL) is added at 23 °C. The resulting slurry is stirred by hand with a spatula thoroughly for 3 min and then cooled in an ice bath. After chilling for 10 min, the solids are collected in a medium-porosity fritted funnel via vacuum filtration and the product is rinsed with additional diethyl ether (15 mL) that is cooled in an ice bath (~2 mL portions via pipette). Thereafter, the fritted funnel is covered with a watch glass and compound 2 is dried via vacuum suction filtration for 10 min before transferring to an appropriately sized tared vial (11.7 g, 87%, single isomer) (Notes 21, 22 and 23) (Figure 4).
Figure 4.

Product 2 after Et2O slurry and vacuum filtration. (Photo provided by the submitters)
Working with Hazardous Chemicals
The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as “Prudent Practices in the Laboratory” (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one’s own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
Discussion
Trivalent phosphorus compounds are good O-atom acceptors for synthetic chemistry. Typically, the resulting phosphine oxide is generated as terminal stoichiometric waste. Rendering the phosphine oxide susceptible to reductive turnover opens possibilities for catalytic O-atom transfer chemistry (Scheme 1).
Scheme 1.

A general representation of PIII/PV=O redox cycling in which starting material A is converted to product B and generalized reductant [R] is employed
It is known that small-ring phosphine oxides undergo rapid rates of deoxygenation when exposed to hydrosilane reductants.2 Our group has demonstrated that four-member phosphacycles (i.e. phosphetanes) are good candidates for catalysis involving interconversion of R3PIII and R3PV=O compounds.3 Phosphetanes are most conveniently prepared by the McBride reaction, which is initiated by phosphenium addition to a suitable unsaturated hydrocarbon, followed by cationic rearrangement and ring formation.4 The procedure detailed above is a representative example of such a McBride synthesis that is adaptable with minor modification to a range of phosphetane targets.
As an illustration of catalytic PIII/PV=O redox applications, our group disclosed in 2017 that phosphetane 2, in conjunction with a terminal hydrosilane reductant, catalyzes the reductive N–N bond forming Cadogan cyclization of o-nitrobenzaldimines, o-nitroazobenzenes, and related substrates.5 This catalytic route to 2H-indazoles, 2H-benzotriazoles, and related heterocycles serves as a catalytic complement to the traditional stoichiometric Cadogan method, which is typically conducted by exhaustive deoxygenation with an excess of a tricoordinate phosphine reagent to provide heterocyclic products.6 Despite longstanding utility, one practical limitation of (super)stoichiometric phosphine-mediated Cadogan cyclizations concerns efficiency and ease of isolation; two equivalents of phosphine oxide are generated per cyclization reaction and excess equivalents of phosphine reagent remain in the reaction mixture. Also, phosphine reagents are often air-sensitive and require special handling and storage in order to ensure their quality. By contrast, the developed catalytic protocol employs the solid, bench-stable, cyclic phosphine oxide 2, which can be handled and manipulated on scale. While many methods exist for the preparation of each of these heterocycles, the generality of the Cadogan protocol has made this transformation distinctive and useful in synthesis.7
Subsequently, our group demonstrated in 2018 an extension of the catalytic Cadogan reaction to C–N bond forming substrate classes. Use of 20 mol% of 1,2,2,3,4,4-hexamethylphosphetane 1-oxide (2) with phenylsilane as terminal reductant converts o-nitrobiphenyl and o-nitrostyrenes into carbazole and indole products, respectively (Table 1).8 The method is robust, scalable, and operationally simple. In the case of carbazole formation, the products precipitate from solution following cooling and pure material can be obtained after rinsing the solid products with dichloromethane. One limitation of the reaction is that substrates bearing multiple nitro moieties undergo unselective functionalization, as the PIII phosphetane apparently does not discriminate between the o-nitro group and others present in the molecule. Furthermore, a small amount of unproductive reduction of the nitroarene to aniline was observed in electron-withdrawn o-nitrobiaryl and -styrenyl derivatives. In almost all cases, this byproduct could be easily removed during purification. Overall, a diverse set of heterocyclic products (>50 examples published to date) can be synthesized by using a substoichiometric amount of anti-1,2,2,3,4,4-hexamethylphosphetane 1-oxide (2) in combination with phenylsilane as terminal reductant.
Table 1.
Scope of phosphetane catalyzed Cadogan cyclizations
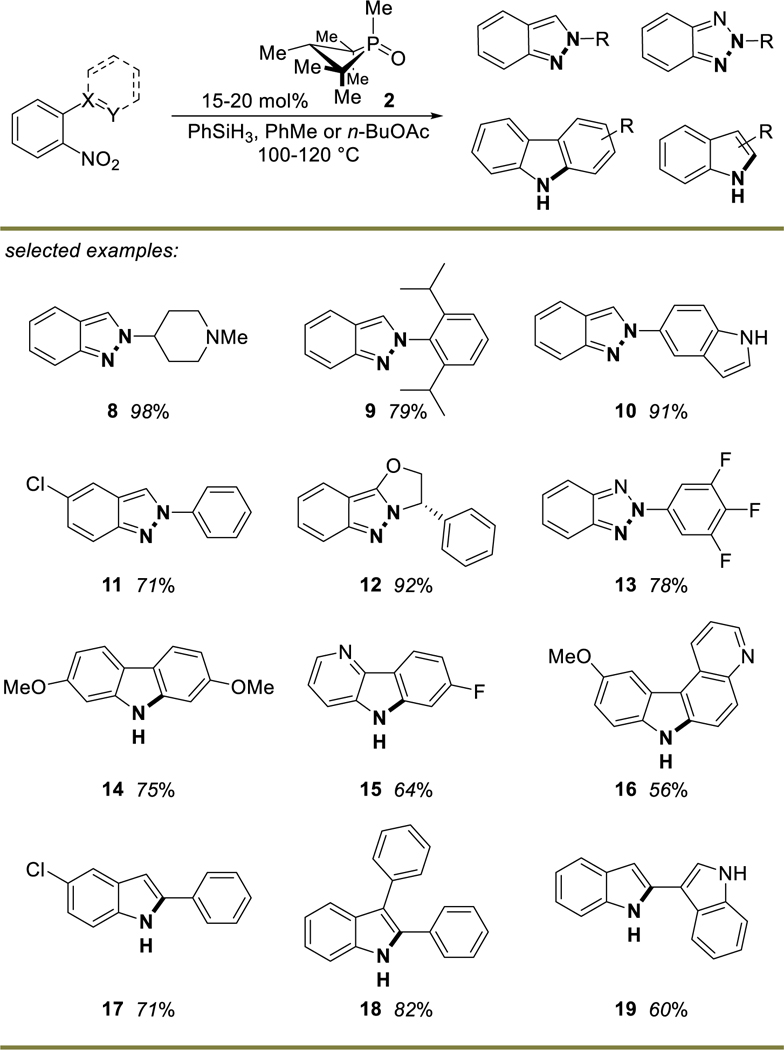 |
Yields shown are for pure isolated material. Products 8–13 were conducted on 0.5 mmol scale (15 mol% 2, 100 °C, 1 M toluene) and 14–19 were conducted on 1-gram scale (20 mol% 2, 120 °C, 1 M butyl acetate). 2 equiv of phenylsilane in all reactions.
Beyond intramolecular reactions, our group reported in 2018 the intermolecular reductive C–N cross coupling of nitroarenes and boronic acids via PIII/PV=O catalysis using 15 mol% of 1,2,2,3,4,4-hexamethylphosphetane 1-oxide (2) and phenylsilane.9 This organophosphorus-catalyzed synthesis of (hetero)arylamines represents a chemically orthogonal complement to transition metal catalyzed methods such as Buchwald-Hartwig, Ullmann, and Chan-Lam couplings.10 A diverse set of (hetero)arylamine products (>30 examples published to date) have been synthesized by the developed PIII/PV=O method (Table 2).
Table 2.
Scope of phosphetane catalyzed C-N coupling
 |
Yields shown are for pure isolated material, except for 33 which was determined by quantitative NMR. Reactions were conducted on 1 mmol scale in m-xylene (0.5 M), except as noted. Compound 22 (3 mmol); 28–30 (1 gram); 33 (0.5 mmol).
In summary, using a substoichiometric amount of anti-1,2,2,3,4,4-hexamethylphosphetane 1-oxide (2) in the presence of phenylsilane represents a general O-atom transfer system to drive the deoxygenative transformations of nitroarene substrates, including reductive cyclization and reductive C–N coupling. The optimized synthetic preparation of phosphetane 2 demonstrated in the above procedure provides access to decagram quantities of this useful compound.
Biography

Trevor Nykaza is from Oak Lawn, IL and attended Lake Forest College where he received his B.A. (2014). In 2014, he joined the Radosevich research group to investigate the design and application of geometrically constrained organophosphorus compounds in redox-driven synthetic methodology. He is now a senior graduate student focused on the application of phosphetane based PIII/PV=O redox-cycling as a general deoxygenation platform, with a specific interest in the manipulation of nitro compounds.

Julian Cooper is from Houston, TX and attended Rice University where he received his B.S. (2014). Now a senior graduate student in the Radosevich Lab, Julian has focused on the development of new transformations that take advantage of the biphilic reactivity of geometrically constrained phosphorus compounds.

Alex Radosevich is originally from Waukegan, IL. He received his B.S. degree from the Univ. of Notre Dame (2002) and his Ph.D. degree from the Univ. of California, Berkeley with F. Dean Toste (2007). After an NIH postdoctoral fellowship stay with Daniel G. Nocera at MIT, he joined the faculty at the Pennsylvania State University in 2010. In August of 2016, he returned to MIT where he is Assoc. Professor.

Robert-Cristian Raclea is originally from Galati, Romania. Currently, he is a senior year undergraduate student in the chemistry department of Imperial College London, and his research interests include natural product synthesis and the development of new synthetic methodologies. In 2019, he completed a research internship in the laboratory of Prof. Mohammad Movassaghi at the Massachusetts Institute of Technology as part of the MIT-Imperial College London exchange program.

Kyan D’Angelo was born in Toronto, ON. He received his B.Sc. and M.Sc. degrees from the University of Toronto where he conducted research in the laboratory of Professor Mark Taylor on catalytic stereoselective glycosylation reactions. In 2016, he began his Ph.D. studies under the supervision of Professor Mohammad Movassaghi at the Massachusetts Institute of Technology. His current research focuses on the total synthesis of alkaloid natural products.
Appendix. Chemical Abstracts Nomenclature (Registry Number)
Aluminum chloride: Aluminum chloride; (7446-70-0)
Dichloromethane: Methane, dichloro-; (75-09-2)
Phosphorus trichloride: Phosphorous trichloride; (7719-12-2)
2,4,4-Trimethyl-2-pentene: 2-Pentene, 2,4,4-trimethyl-; (107-40-4)
Sodium chloride: Sodium chloride (NaCl); (7647-14-5)
Sodium sulfate: Sulfuric acid sodium salt (1:2); (7757-82-6)
1-Chloro-2,2,3,4,4-pentamethylphosphetane 1-oxide: Phosphetane, 1-chloro-2,2,3,4,4-pentamethyl-, 1-oxide; (29276-11-7)
Tetrahydrofuran: Furan, tetrahydro-; (109-99-9)
Methylmagnesium bromide: Magnesium, bromomethyl-; (75-16-1)
Ammonium chloride: Ammonium chloride ((NH4)Cl); (12125-02-9)
Notes
Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of “Prudent Practices in the Laboratory” (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also “Identifying and Evaluating Hazards in Research Laboratories” (American Chemical Society, 2015) which is available via the associated website “Hazard Assessment in Research Laboratories” at https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with aluminum chloride, phosphorus trichloride, 2,4,4-trimethyl-2-pentene, methylmagnesium bromide, dichloromethane, tetrahydrofuran, diethyl ether, hexanes, ammonium chloride, sodium sulfate, and sodium chloride.
Aluminum chloride (99.9%) was purchased from Strem Chemicals (Catalog: 93–1387, Lot: 25835400) and used as received. Prior to its use, the reagent was stored in a desiccator using CaSO4 as desiccant (Drierite®).
Dichloromethane was purchased from Fischer Scientific (D37–4) and degassed by bubbling with argon for 1 h before passing through a Glass Contour Solvent Purification System. The solvent was transferred in three portions using a 60 mL syringe (50, 50, 25 mL), washing any aluminum chloride on the upper vessel wall down to the bottom of the flask in the process.
The mixture was stirred at 300 rpm throughout the reaction.
Phosphorus trichloride (99%, ReagentPlus®) was purchased from Sigma-Aldrich as a Sure/Seal™ bottle and used as received. The material was added in one portion over 1 min.
2,4,4-Trimethyl-2-pentene (98%) was purchased from Acros Organics (Code: 162631000, Lot: A0367167) and used as received.
Concurrent with the addition of 2,4,4-trimethyl-2-pentene (6.5 mL/min) the aluminum chloride dissolved. A white precipitate was observed after 5 min. Bubbling may be observed if addition proceeds too quickly. If 2,4,4-trimethyl-2-pentene is added more slowly than described it is possible to see precipitation during the addition.
The poor solubility of the cationic reaction intermediates and products in CH2Cl2 make quantitation of reaction progress difficult. The consumption of PCl3 (31P NMR (162 MHz, chloroform-d) δ 220 ppm) can be monitored by analyzing reaction aliquots (0.1 mL reaction mixture, in 0.4 mL chloroform-d).
With stirring, the rubber septum was removed and quickly replaced with a 250-mL pressure-equalized dropping funnel to which an adaptor connected to Tygon tubing was previously attached. The other end of the tubing was directed into a scrubbing solution of saturated aqueous sodium bicarbonate (500 mL). Caution: water should be added slowly at first (0.5 mL/min over 20 min, maintaining 0 °C), as evolution of large quantities of gas (HCl) is observed during the addition. Bubbling can become quite vigorous. Addition rate can be increased gradually, and care should be taken to ensure that the reaction mixture is stirring. Formation of a white precipitate will intensify with increased addition of water and then dissolve with vigorous continued stirring. If all solids do not dissolve following the initial water addition, then the liquid phase can be decanted from the remaining solids and additional water may be added to the flask with stirring until the solids are dissolved. All aqueous layers should then be combined in the separatory funnel.
The dried solution was filtered into a 1 L pear-shaped round-bottomed flask using a funnel plugged with cotton.
Concentration via rotary evaporation may give 1 as an oil. Scratching this oil with a glass stir rod will induce the formation of a solid.
Compound 1 is recrystallized to improve isomeric ratio and to increase the purity of the compound. Additionally, using crude 1 directly appears to result in decreased yield of 2. The submitters added boiling hexanes (69 °C) to a 150 mL beaker containing the crude and then hand-stirred and heated to dissolve solids. This was a performed successfully by the checkers on smaller scale, but the modified protocol was preferred as the use of a rotary evaporator enabled safer handling of flammable vapors, as well as providing efficient agitation.
Compound 1 will slowly hydrolyze in the presence of moisture; it may be stored in a sealed container at –20 °C for an extended period. Purity of recrystallized compound 1 was assessed using quantitative NMR (16.8 mg 1,3,5-trimethoxybenzene (>99.9%), 19.5 mg of 1 dissolved in 1 mL chloroform-d, purity 97%). 1H NMR (400 MHz, chloroform-d) δ: 0.87 (dd, J = 7.1, 1.6 Hz, 3H), 1.23 – 1.35 (m, 12H), 1.73 (qd, J = 7.1, 3.9 Hz, 1H). 13C NMR (101 MHz, chloroform-d) δ: 7.2 (d, J = 30.7 Hz), 18.1 (d, J = 3.6 Hz), 25.8 (d, J = 6.8 Hz), 42.2, 56.8 (d, J = 57.1 Hz). Proton coupled 31P NMR (162 MHz, chloroform-d) δ: 80.78 – 82.25 (m). IR (diamond ATR, neat) 2979, 2961, 2912, 2863, 1454, 1386, 1375, 1366, 1337, 1274, 1248, 1212, 1170, 1074, 1054, 1021, 990, 933, 903, 752, 661, 634, 558, 502 cm-1. mp (flame-sealed capillary): 66–69 °C. Data were collected rapidly after the preparation of 1 and the compound was used within 1 h in the second step.
The preparation of 1 was duplicated on half-scale (105 mmol) yielding 18.8 g crude 1 (97 mmol, 92%) and 14.6 g recrystallized 1 (75 mmol, 71%, ≥97:3 anti:syn, purity ≥97%).
Tetrahydrofuran (THF) was purchased from Fisher Scientific (T397) and degassed by bubbling argon for 1 h before passing through a Glass Contour Solvent Purification System.
A new bottle of methylmagnesium bromide (3.0 M in Et2O) (Catalog: 189898–100ML; Lot: SHBJ6161) was purchased from Sigma-Aldrich as a Sure/Seal™ bottle and used as received. Slight excess of the Grignard reagent is necessary as compound 1 is hygroscopic and may contain small quantities of water. MeMgBr was added via a 30 mL syringe (two portions, 14 mL x 2 is suggested). Note that precipitate formation may set in quickly after heating the reaction mixture. The submitter’s procedure involved heating in an oil bath, which was successfully tested by the checkers, but changing to a water bath was deemed more practical.
Reaction progress was monitored by analyzing reaction aliquots (0.3 mL of reaction mixture) for the consumption of compound 1 (31P NMR (162 MHz, THF/Et2O) δ: 85.1 ppm). 31P chemical shift variance is solvent dependent – the sample was not locked. The checkers found monitoring of the reaction to not be helpful, as yields were reproducibly high when the reaction is simply allowed to run for the indicated time.
During the addition of saturated ammonium chloride (3.5 mL/min), a large quantity of white solids precipitate initially and stirring may become difficult.
Concentration and redissolution of the initial ether/THF layer with CH2Cl2 is necessary to minimize emulsion formation during the work up.
Compound 2 is water soluble and copious CH2Cl2 is necessary for maximum recovery. Excessive shaking of the separatory funnel may result in the formation of an emulsion which will separate slowly (~20 min). The dried solution was filtered through a funnel plugged with cotton.
Purity of compound 2 was assessed using quantitative NMR (16.8 mg 1,3,5-trimethoxybenzene (>99.9%), 17.4 mg of 2 dissolved in 1.0 mL chloroform-d, purity = 97%). Characterization data for 2: 1H NMR (400 MHz, chloroform-d) δ: 0.83 (dd, J = 7.1, 1.7 Hz, 3H), 1.05 – 1.26 (m, 12H), 1.43 – 1.53 (m, 4H). 13C NMR (101 MHz, chloroform-d) δ: 7.0 (d, J = 23.1 Hz), 9.9 (d, J = 41.1 Hz), 17.5 (d, J = 4.3 Hz), 24.8 (d, J = 3.7 Hz), 42.7 (d, J = 6.0 Hz), 45.5 (d, J = 59.4 Hz). Proton coupled 31P NMR (162 MHz, chloroform-d) δ: 58.4 – 59.7 (m). IR (diamond ATR, neat) 2974, 2946, 2908, 2863, 1460, 1414, 1371, 1307, 1237, 1182, 1154, 1073, 1056, 1021, 989, 929, 885, 643, 609, 559, 491, 443, 418 cm-1. mp: 167–169 °C.
Phosphetane oxide 2 is bench-stable and does not require special storage or handling.
The preparation of 2 was duplicated on half-scale (7.5 g, 39 mmol) yielding 5.8 g 2 (87%, single isomer, purity = 97%).
References
- 1.Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States, radosevich@mit.edu We thank NIH NIGMS (GM114547) and MIT for funding support. ORCID (ATR): 0000-0002-5373-7343
- 2.(a) Marsi KL J. Org. Chem 1974, 39, 265–267. [Google Scholar]; (b) O’Brien CJ; Tellez JL; Nixon ZS; Kang LJ; Carter AL; Kunkel SR; Przeworski KC; Chass GA Angew. Chem. Int. Ed 2009, 48, 6836–6839. [DOI] [PubMed] [Google Scholar]; (c) van Kalkeren HA; Leenders SHAM; Hommersom CRA; Rutjes FPJT; van Delft FL Chem. Eur. J 2011, 17, 11290–11295. [DOI] [PubMed] [Google Scholar]
- 3.(a) Zhao W; Yan PK; Radosevich AT J. Am. Chem. Soc 2015, 137, 616–619. [DOI] [PubMed] [Google Scholar]; (b) Reichl KD; Dunn NL; Fastuca NJ; Radosevich AT J. Am. Chem. Soc 2015, 137, 5292–5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Jungermann E; McBride JJ; Clutter R; Mais AJ Org. Chem 1962, 27, 606–610. [Google Scholar]; (b) McBride JJ; Jungermann E; Killheffer JV; Clutter RJ J. Org. Chem 1962, 27, 1833–1836. [Google Scholar]
- 5.Nykaza TV; Harrison TS; Ghosh A; Putnik RA; Radosevich AT J. Am. Chem. Soc 2017, 139, 6839–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li JJ; Name Reactions for Heterocyclic Chemistry II; Wiley, New York, 2011; pp. 112–124. [Google Scholar]
- 7.(a) Preparation of 2H-indazoles:Schmidt A; Beutler A; Snovydovych B Eur. J. Org. Chem 2008, 2008, 4073–4095 and citing literature. [Google Scholar]; (b) Preparation of 2H-benzotriazoles:Kim BH; Kim SK; Lee YS; Jun YM; Baik W; Lee BM Tetrahedron Lett 1997, 38, 8303–8306. [Google Scholar]; (c) Jo J; Lee HY; Liu W; Olasz A; Chen C-H; Lee DJ Am. Chem. Soc 2012, 134, 16000–16007. [DOI] [PubMed] [Google Scholar]; (d) Dong J; Jin B; Sun P Org. Lett 2014, 16, 4540–4542. [DOI] [PubMed] [Google Scholar]; (e) Ryu T; Min J; Choi W; Jeon WH; Lee PH Org. Lett 2014, 16, 2810–2813. [DOI] [PubMed] [Google Scholar]; (f) Shang X; Zhao S; Chen W; Chen C; Qiu H Chem. - Eur. J 2014, 20, 1825–1828. [DOI] [PubMed] [Google Scholar]; (g) Khatun N; Modi A; Ali W; Patel BK J. Org. Chem 2015, 80, 9662–9670. [DOI] [PubMed] [Google Scholar]; (h) Preparation of indoles:Humphrey GR; Kuethe JT Chem. Rev 2006, 106, 2875–2911. [DOI] [PubMed] [Google Scholar]; (i) Preparation of carbazoles:Schmidt AW; Reddy KR; Knölker H-J Chem. Rev 2012, 112, 3193–3328. [DOI] [PubMed] [Google Scholar]
- 8.Nykaza TV; Ramirez A; Harrison TS; Luzung MR; Radosevich AT J. Am. Chem. Soc 2018, 140, 3103–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nykaza TV; Cooper JC; Li G; Mahieu N; Ramirez A; Luzung MR; Radosevich AT J. Am. Chem. Soc 2018, 140, 15200–15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Hartwig JF Angew. Chem. Int. Ed 1998, 37, 2046–2067. [DOI] [PubMed] [Google Scholar]; (b) Wolfe JP; Buchwald SL Angew. Chem. Int. Ed 1999, 38, 2413–2416. [DOI] [PubMed] [Google Scholar]; (c) Jiang L; Buchwald SL Metal-Catalyzed Cross-Coupling Reactions De Meijere A; Diderich F Eds.; Wiley-Blackwell: Hoboken, NJ, 2008; ed. 2, pp. 699–760. [Google Scholar]; (d) Jiang Y; Ma D Catalysis without Precious Metals Bullock RM Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2010; pp. 213–233. [Google Scholar]; (e) Bariwal J; Eycken E. V. der. Chem. Soc. Rev 2013, 42, 9283–9303. [DOI] [PubMed] [Google Scholar]; (f) Corcoran EB; Pirnot MT; Lin S; Dreher SD; DiRocco DA; Davies IW; Buchwald SL; MacMillan DWC Science 2016, 353, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Schlummer B; Scholz U Adv. Synth. Catal 2004, 346, 1599–1626. [Google Scholar]; (h) Torborg C; Beller M Adv. Synth. Catal 2009, 351, 3027–3043. [Google Scholar]; (i) Yin J Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, Crawley ML; Trost BM Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2012; pp. 97–163. [Google Scholar]; (j) Ruiz-Castillo P; Buchwald SL Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]