

Abstract
The chemical composition of cells at the molecular level determines their growth, differentiation, structure, and function. Probing this composition is a powerful capability as it provides invaluable insight into chemical processes inside cells and, in certain cases, allows one to diagnose diseases based on molecular profiles. However, many techniques analyze fixed cells or lysates of bulk populations, in which information about dynamics and cellular heterogeneity is lost. Recently, nucleic acid-based probes have emerged as a promising platform for the detection of a wide variety of intracellular analytes in live cells with single-cell resolution. This review describes the recent advances in this field and discusses the common strategies for probe design, types of targets that can be identified, current limitations, and future directions.
Keywords: hybridization, aptamer, DNAzyme, theranostics, Diagnostics
Graphical Abstract
Nucleic Acid-Based Structures as Intracellular Probes for Live Cells

1. Introduction
Cells are primarily composed of metal ions, small molecules, proteins, lipids, and nucleic acids.[1] Over the past few decades, numerous methods have been developed for monitoring these analytes and correlating their abundances to fate and health status of the cells. Techniques ranging from microscopy and mass spectrometry to assays such as enzyme-linked immunosorbent assay (ELISA) and reverse transcription polymerase chain reaction (RT-PCR) have vastly improved our understanding of fundamental processes associated with cells.[2–5]
Many techniques for cellular analysis such as northern blotting, western blotting, fluorescence in situ hybridization (FISH), RT-PCR, DNA microarrays, and electron microscopy rely on the fixation or lysis of cells.[5–11] Moreover, as the amount of material from one cell is often insufficient for accurate analysis, a bulk population of cells is used for methods that analyze cell lysates. Information about dynamics of various molecules inside cells and cell-to-cell heterogeneity is often lost in such cases due to ensemble averaging.
The ability to track molecules in live cells is important from a fundamental perspective as it can enable one to determine how the spatial distribution of cellular analytes impacts cellular function, monitor transient processes, and study the evolution of chemical signatures associated with the progression of disease. To highlight this point, Lichtman and Fraser have put forth a useful sports analogy: reconstructing a game of American football and identifying its rules from a series of still snapshots taken at different timepoints from separate games would be a near impossible task.[12]
An indispensable tool for visualizing live cells is light microscopy.[13] However, as methods based on fluorescence typically have greater sensitivity than those based on absorbance, numerous fluorescent tools have been developed for analysis of cellular analytes in live cells.[14] For example, a prominent “zinc spark,” associated with the fertilization of human eggs, could be detected only because of advances in live cell imaging with fluorophore reporters.[15] Conventional light microscopes provide resolution down to 250 nm, while ~50 nm resolution is regularly achieved using super-resolution techniques.[16] These capabilities allow one to not only observe sub-cellular organelles, but also can provide resolution down to the single-molecule level.[17]
Recently, nucleic acid-based probes have gained popularity for visualizing intracellular analytes due to several advantageous properties (listed in section 1.1). Through judicious selection of sequences, a wide variety of targets ranging from nucleic acids to ions, small molecules, and proteins inside living cells can be visualized. In this review, we present an overview of the current state of live cell imaging using nucleic acid-based probes. We begin by delineating why nucleic acid-based probes are attractive. We provide a brief description of the types of nucleic acid-based probes for detecting various analytes (Figure 1) and refer the readers to relevant literature on specific techniques. We then discuss the recent progress in the development of hybridization-based probes, which are primarily used in the detection of nucleic acids. We follow this section by describing how nucleic acids can be utilized to create probes that can detect other analytes, such as proteins, small molecule metabolites, and ions, through the development of aptamers and DNAzymes.[18] We then summarize the various methods used for probe delivery and conclude with a discussion of the current challenges and the areas that need future development.
Figure 1.
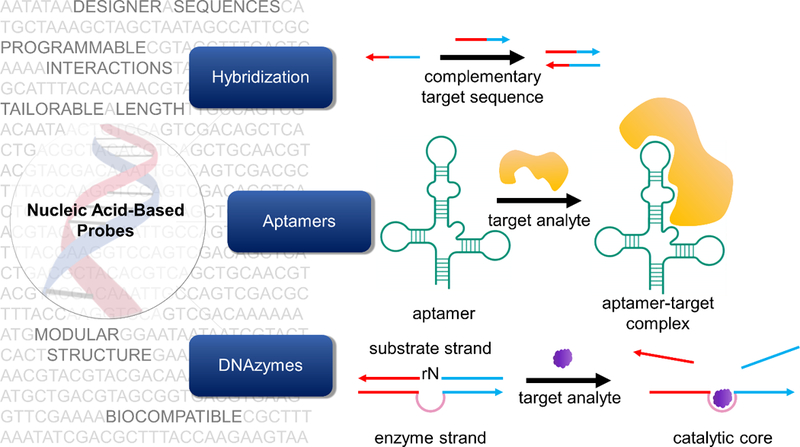
Nucleic acid-based probes can be broadly classified into three types: (i) hybridization-based probes, (ii) aptamers, and (iii) DNAzymes. Hybridization-based probes are single-stranded and recognize complementary targets through Watson-Crick base pairing. For ease of indentification, complementary regions are colored the same in this figure. Aptamers are oligonucleotides evolved through combinatorial selection strategies that can bind to analytes of interest including ions, small molecules, and proteins. Aptamers often adopt complex tertiary structures that enable target recognition. Alternately, target binding can induce conformational changes in their structure. DNAzymes consist of a substrate strand and an enzyme strand (containing a catalytic core) hybridized to each other. The substrate strand contains a single RNA nucleotide which can be catalytically cleaved in the presence of a target.
1.1. Why nucleic acid-based probes?
Nucleic acid-based probes are desirable because of their programmable nature, a property that has led to key advances in several areas of nanotechnology.[19–21] DNA and RNA oligonucleotides (ONTs) can be synthesized across different length scales (1–1,000,000 nt) at relatively low cost, either through chemical or enzymatic processes.[22] Moreover, these probes are amenable to chemical modifications (e.g. with fluorescent tags or custom bases with specific functionalities) at any desired site. Single-stranded probes can be designed to detect other nucleic acids through complementary interactions (hybridization-based probes). They can also be evolved through combinatorial selection techniques such that they bind to any target molecule of interest (aptamers) or catalyze specific reactions (DNAzymes).[23–25] Importantly, these probes, being biopolymers, are biocompatible and non-toxic to cells. Finally, they can be assembled into spherical nucleic acids (SNAs) and taken into cells without the need for transfection agents, which are often toxic.[26] Taken together, these features make nucleic acids particularly attractive as probes.
1.2. The nucleic acid structure
Nucleic acids are anionic polymers composed of nucleotide building blocks. Each nucleotide consists of three subunits: a nitrogenous base, a five-carbon sugar moiety, and a phosphate “backbone” (Figure 2). In this section, we briefly describe the common modifications that are made to nucleotides in the context of generating improved sensors (Figure 2).
Figure 2.
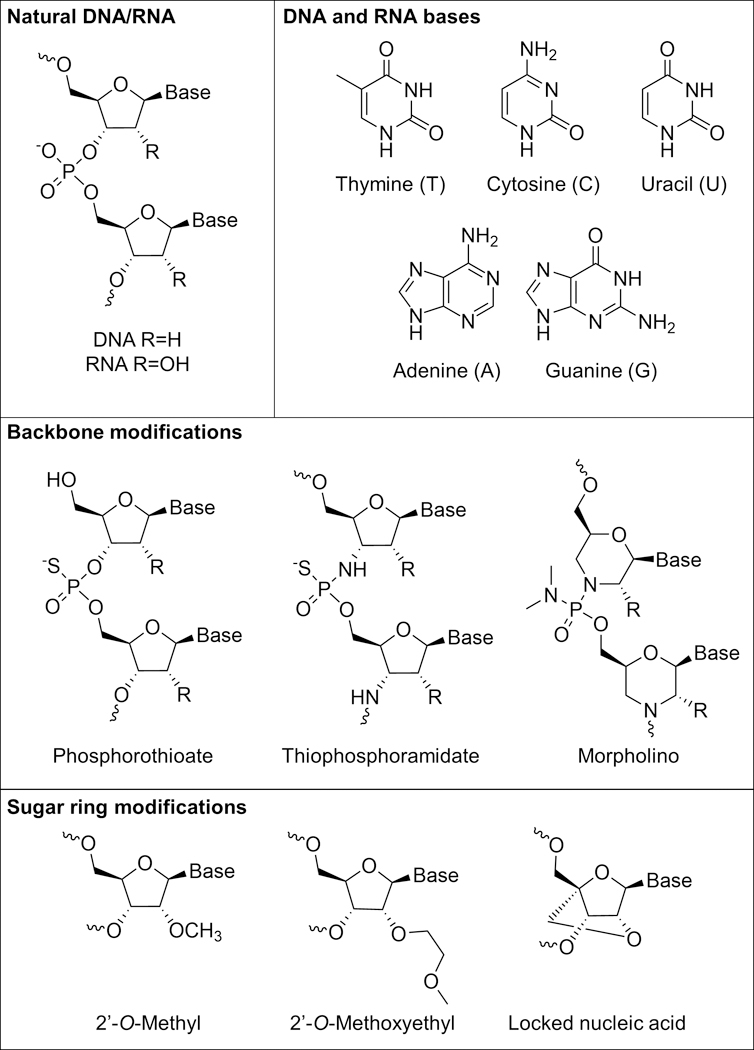
The nucleic acid structure and common modifications to it. Adapted with permission.[27] Copyright 2013, Springer Nature.
Base modifications
Five bases form the foundational alphabet of genetic codes: adenine (A), thymine (T), uracil (U), cytosine (C) and guanine (G). These bases interact with each other through hydrogen bonds, resulting in specific and highly programmable interactions. In classical Watson-Crick base-pairing, A pairs with T while C pairs with G. In RNA, T is substituted by U. Several types of modifications can be made to these standard bases with the goal of increasing stability or functionality. For example, Lannes et al. demonstrated that i-motifs, which have pH-dependent secondary structures, can be engineered to switch their conformation at more acidic or more basic pH by replacing cytosine bases with 5-methylcytosines and 5-bromocytosines, respectively.[28]
Sugar ring modifications
In unmodified RNA, the 2’ position on the sugar is hydroxylated, but in DNA it is not (Figure 2). To impart nuclease resistance, the 2’ position is often modified.[29] Most commonly used modifications include the use of 2’-O-methyl (2’-OMe), 2’-O-methoxyethyl derivatives, and locked nucleic acids (LNAs).[30–32] These modifications can also impact the melting temperature of the nucleic acid duplexes. For example, LNAs have been used in developing probes that bind more strongly to their target nucleic acids.[33]
Backbone modifications
Phosphorothioates (PS), thiophosphoramidates, morpholinos, and peptide nucleic acids (PNAs) are common backbone-modified nucleic acids (Figure 2).[34–37] These modifications influence the thermal and enzymatic stability as well as solubility of the probes. For example, morpholino-based probes for messenger RNA (mRNA) detection afford greater nuclease resistance and higher specificity towards their target, albeit at the cost of reduced solubility.[36,38]
1.3. Types of nucleic acid-based probes
Nucleic acid-based probes can be broadly classified into three groups: (i) hybridization-based probes, (ii) aptamers, and (iii) DNAzymes (Figure 1).
Hybridization-based probes are designed by leveraging Watson-Crick base pairing to detect complementary sequences. Therefore, these probes are used in the detection of nucleic acids including mRNA, microRNA, and non-coding RNA.[39–41] Examples of this type of probe include those used with FISH, molecular beacons (MBs), and NanoFlares.[42–44]
Aptamers are ONT sequences that can be designed to bind to any target of interest. These ONTs are generated through an in vitro process called systematic evolution of ligands by exponential enrichment (SELEX).[23] Aptamers are nucleic acid analogues of antibodies, and recent studies have shown that their performance (in terms of binding affinities, detection limits), in several cases rivals that of antibodies at reduced cost and greater stability.[45] Moreover, aptamers can be evolved using secondary structures alone, without having detailed knowledge of tertiary folding. To date, over 500 aptamers have been generated for more than 100 different targets ranging from ions, small molecules, proteins, to whole cells.[46]
DNAzymes are synthetic DNA structures that can catalyze chemical reactions.[24] So far, no naturally occurring DNAzyme has been identified.[47] However, DNAzymes are obtained through an in vitro combinatorial process by screening a large library of ONTs containing up to 1015 distinct sequences.[48,49] These sequences can be evolved as long single-stranded ONTs to bind specific substrates and subsequently catalyze chemical reactions. The single-stranded ONT is then converted into a two-stranded catalyst. One strand (the substrate strand) consists of a single ribonucleotide and other (the enzyme strand) contains the catalytic core. These two strands are hybridized together through complementary binding arms on either side of the ribonucleotide/catalytic core (Figure 1). The ribonucleotide can be catalytically cleaved, typically, in the presence of specific metal ions. DNAzymes have been used to detect a wide variety of metals,[22] although recent work has focused on detecting other analytes such as RNA.[50]
Combining the advantages of two or more of these classes of probes, hybrid probes can also be generated. Probes termed aptazymes have been designed wherein a DNAzyme is activated only upon aptamer-ligand binding.[51] We further note that the reporting mechanism of a few select nucleic acid-based probes solely take advantage of the nanostructures formed by DNA and cannot be categorized into any of the above mentioned categories. For example, fluorophore-labeled dextran molecules encapsulated within DNA icosahedrons enable the tracking of specific endocytic pathways as the icosahedrons are taken up by the anionic ligand binding receptor pathway.[52] For the purposes of this review, the probes reported in the literature have been classified into one of the three broad categories described above based on our perspective of where the probe design can be most effectively described.
In Table 1, we list selected structures/specific sub-classes of probes that are most commonly used along with example targets that have been visualized using these probes. Additionally, we enumerate the methods of probe delivery, type of signal readout, as well as advantages and disadvantages of each probe type.
Table 1.
Common nucleic acid-based probes
| Probe Type | Target type | Delivery Method | Signaling Method | Advantages and disadvantages |
|---|---|---|---|---|
|
Linear antisense probes A dye-labeled recognition strand binds to its complementary target. |
mRNA,[53] snRNA[54] rRNA,[54] |
1.Microinjection[54] 2. Cell permeabilization[55] 3.Cationic lipids[53] |
Fluorescence |
Advantages 1.Simple probe design Disadvantages 1.High background 2. Unmodified probes prone to degradation 3.Nuclear sequestration possible 4. Requires method for transfection (e.g. Microinjection, Cell Permeabilization, etc.) |
|
Linear FRET probes Hybridization of two different linear antisense probes to adjacent regions of a target sequence occurs, bringing a FRET pair into close proximity. |
mRNA,[56] | 1.Microinjection[56,57] 2.Microporation[58] |
Fluorescence |
Advantages 1.Better selectivity compared to linear antisense probes Disadvantages 1. Unmodified probes prone to degradation 2. Nuclear sequestration possible 3. Requires method for transfection 4. Requires binding to large stretch of RNA for hybridization-based approach |
|
Molecular beacons (MBs)/aptamer beacons/aptamer switch probes Target binding induces opening of a hairpin ONT sequence, separating a fluorophore and quencher. |
mRNA,[59] miRNA,[60] piRNA,[41] small molecules,[61] proteins,[62] temperature[43] |
1. Microinjection[63–67] 2. Cationic lipids[41,60,68–72] 3. Microporation[64,73] 4. Aptamer cell recognition[74,75] |
Fluorescence |
Advantages 1. Better signal to noise ratio compared to linear antisense probes and linear FRET probes 2. Multiplexing commonly done Disadvantages1. Prone to degradation 2. Nuclear sequestration possible 3. Requires method for transfection |
|
Dual FRET beacons Two MBs bind to adjacent regions of a target sequence, whereby MB opening brings a FRET pair near one another resulting in turn-on of FRET signal. |
mRNA[76] | 1. Permeabilization[77] 2. Electroporation[76] 3. Microinjection[78] |
Fluorescence |
Advantages 1. Better signal to noise ratio compared to linear antisense probes, linear FRET probes and MBs 2. Better selectivity compared to MBs Disadvantages 1. Unmodified probes prone to degradation 2. Nuclear sequestration Possible 3. Requires method for transfection 4. Requires binding to large stretch of RNA for hybridization-based approach |
|
FIT probes A duplex sensitive dye of the thiazole orange family acts as a nucleobase surrogate in a recognition sequence such that target binding results in dye fluorescence turn-on. |
mRNA[79] | 1. Permeabilization[80–82] 2. Cationic polymer[79] 3. Microinjection[83] |
Fluorescence |
Advantages 1. Simple probe design (single modification) 2. Lack of false-positive signal due to degradation 3. Highly sensitive to single nucleotide mismatches 4. Multiplexing Possible Disadvantages 1. Nuclear sequestration Possible 2. Unmodified probes prone to degradation 3. Requires method for transfection 4. Less bright than conventional dyes like AlexaFluor488 5. Fluorescence enhancement strongly dependent on dye location in probe |
|
ECHO probes Generally, two thiazole orange fluorophores are covalently attached to one base of a recognition sequence, forming an H-aggregate. Target binding breaks the H-aggregate and turns-on fluorescence. |
mRNA,[84] miRNA,[85] rRNA,[86] small nucleolar RNA[86] |
1. Cationic lipids[85–88] 2. Microinjection[86] |
Fluorescence |
Advantages 1. Lower background signal than FIT probes 2. Multiplexing possible Disadvantages 1. Nuclear sequestration Possible 2. Unmodified probes prone to degradation 3. Requires method for transfection 4. Fluorescence enhancement strongly dependent on sequence of target RNA |
|
SNA-based structures SNAs consist of ONTs covalently functionalized around a spherical nanoparticle core, giving the ONTs a radial arrangement. Examples of nanoparticle cores used include gold,[44] upconverting nanoparticles,[89] micelles,[90] silica,[91] carbon-based,[92] and quantum dot[93] |
mRNA,[44] miRNA,[89] proteins,[94] small molecules,[95] pH,[96] ions[97] |
Active uptake[44,98] | Fluorescence SERS[99] |
Advantages 1. Higher signal to noise ratio than molecular beacons 2. Uptake without transfection reagents 3. Resistant to degradation 4. Biocompatible 5. Multiplexing possible Disadvantages 1. Difficult to quantify endosomal escape 2. Fluorophore/quencher-based structures prone to false-positive signal if degraded |
|
Structures based on ONTs adsorbed to nanoparticle surface ONT probes can be non-covalently associated with various nanoparticle cores for delivery into cells. Examples of nanoparticle cores used include gold,[100] polymer,[101] silica,[102] manganese dioxide,[103] carbon-based,[104] MOFs,[105] iron-oxide,[106] and quantum dot[107] |
mRNA,[108] miRNA,[100] small molecules,[109,110] pH,[111] |
Active Uptake[108] | Fluorescence |
Advantages 1. Uptake without transfection reagents 2. Resistant to degradation 3. Biocompatible 4. Multiplexing possible Disadvantages 1. Difficult to quantify endosomal escape 2. Fluorophore/quencher-based structures prone to false-positive signal if degraded 3. Leaching of oligos from surface possible |
|
Nanoparticles encapsulated with ONTs Nanoparticle cores capable of encapsulating ONTs can be used as a means for carrying probes into cells. Examples of nanoparticle cores used include liposomes[40] and polymers.[112] |
miRNA,[40] mRNA,[112] small molecules[61] |
Active Uptake[112] | Fluorescence |
Advantages 1. Uptake without transfection reagents 2. Resistant to degradation 3. Biocompatible 4. Multiplexing possible Disadvantages 1. Difficult to quantify endosomal escape 2. Fluorophore/quencher-based structures prone to false-positive signal if degraded 3. Strategy limited to use of nanoparticles that can be encapsulated with oligonucleotides |
|
DNA nanomachine/nanostructure-Based DNA can be assembled into 1D, 2D, and 3D nanomachines/nanostructures that act as sensing platforms. |
mRNA,[113] miRNA,[114] pH,[115,116] ions,[117] radicals[118] |
Active uptake[113,119] | Fluorescence, circular dichroism[120] |
Advantages 1. Uptake without transfection reagents 2. Resistant to degradation 3. Multiplexing possible 4. Biocompatible Disadvantages 1. Difficult to quantify endosomal escape 2. Involved design requiring multiple ONT strands 3. Fluorophore/quencher based nanomachines prone to false-positive signal if degraded |
|
Genetically-encoded aptamers RNA aptamers can be expressed in cells such that target binding allows the aptamer to bind a dye that results in a fluorescent signal |
mRNA,[121] small molecules,[122] proteins[123] |
Genetically encoded | Fluorescence |
Advantages 1. Can be expressed in cells, so probe transfection is not necessary 2. Provides a facile way of visualizing small molecules and metabolites which are usually difficult to image 3. Dye has low background 4. Can monitor analytes dynamically in situ 5. Multiplexing possible, in principle Disadvantages 1. Poor folding in vivo 2. Typically requires long exposure times (10-100 ms) for imaging 3. Often requires a tRNA scaffold to promote in cellulo folding 4. Requires genetic modification of cells for imaging |
1.4. Common sensing strategies
Although numerous strategies relying on nucleic acids as probes have been developed for imaging intracellular analytes, most of the techniques employ one or more of the sensing strategies enumerated below:
Single fluorophore label
ONTs labeled with a single fluorophore can be used for detecting intracellular analytes of interest. Examples include probes labeled with commercially available fluorescein isothiocyanate or forced intercalation dyes of the thiazole orange family.[53,124]
Förster resonance energy transfer (FRET)
Nucleic acid probes based on FRET are designed such that the on/off state is dependent upon proximity between a donor and acceptor molecule due to energy transfer between the molecules. Examples include fluorophore/quencher MBs and FRET-based DNAzymes.[125,126]
Nanoparticle-based
Nanoparticles are commonly interfaced with nucleic acids for use in live cell imaging because they often confer advantageous properties over naked nucleic acids. These properties may be the ability to deliver payload into cells without the need of transfection reagents, protect cargo from nuclease degradation, act as a source of fluorescence or as a source of quenching (FRET), exhibit plasmonic properties, and capability to absorb near infrared light for transition to a theranostic platform.[44,106,127–129] Examples of nanomaterials used include gold nanoparticles (e.g. SNAs), graphene oxide nanosheets, quantum dots, MOFs, etc.
Amplification
Incorporating an amplification element into sensing strategies is often used for detecting targets at low abundance. In live cells, target amplification methods like PCR are challenging, and as such, signal amplification methods are the most common route to lowering the limit of detection. Techniques for signal amplification include hybridization chain reaction (HCR), hairpin DNA cascade amplifier (HDCA), and DNA chain reaction (DCR).[72,108,130]
1.5. Design Considerations for Common Application Scenarios
Nucleic-acid based probes may be used in a variety of detection scenarios, broadly classified as: (a) detecting analytes and studying their spatiotemporal behavior, (b) comparing levels of analytes between different cells, (c) sorting rare cells in complex media, and (d) quantifying the amounts of particular analytes present in a cell.
In the simplest case, a nucleic-acid based probe can be used to detect the presence of an analyte of interest in a cell, in certain situations yielding important information about cellular state or the role of an analyte in disease.[131] Moreover, probes can be used to study the spatiotemporal evolution of key analytes, which can provide fundamental insight into the dynamics of these molecules and how these dynamics influence cellular functions.[132,133] For instance, Sticky-flares have been used to show for the first time that β-actin mRNA colocalizes with mitochondria in HeLa cells.[134] Furthermore, ChloropHore has been used for simultaneous tracking of pH and chloride ion concentrations, providing fundamental insight into the relationship between these analytes and Niemann-Pick disease.[117] Microscopy is the primary technique used in studying spatiotemporal behavior of analytes. However, for such an application, the signaling element of the probe (e.g. fluorescent element) must remain associated with the target in the complex cellular environment for it to be possible to study spatiotemporal behavior.
Some researchers are also interested in using nucleic-acid based probes for comparing the levels of analytes between different cell types.[68,135] This may yield fundamental information pertaining to the role of an analyte in diseased versus healthy cells, or allow one to distinguish different cell types based on relative levels of a known intracellular marker. When comparing distinct cell types, differences in probe uptake must be accounted for, preferably through ratiometric analysis. Furthermore, if large numbers of cells are to be compared, flow cytometry will give relatively fast readouts and is thus preferable to microscopy.
Next, sorting and isolating rare cells in complex media based on molecular signatures are important, both in fundamental and applied situations.[136–138] For instance, the isolation of a rare cell may allow for further fundamental study of its genomic profile. On the other hand, knowing that a particular rare cell is present in a sample may allow one to diagnose a disease in a patient— an important application of such a strategy. Again, ratiometric probing is desirable in order to normalize probe uptake across the different cells within a sample. If the isolated cells are used for further study and analysis, the probe used should be biocompatible and not impact cellular viability and function. For example, NanoFlares have been utilized to sort and isolate circulating tumor cells from blood based on their mRNA expession. Moreover, the isolated cells were subjected to mammosphere analysis to verify their retained viability and functionality.[139] These types of sorting studies involving a large population of cells are best suited for fluorescence-activated cell sorting (FACS).
The last broad classification of detection scenarios concerns the ability to quantify targets intracellularly, in contrast to simply detecting or knowing relative amounts. FISH has widely been used for absolute quantification of transcript numbers in fixed cells.[9,140] Methods for quantitative probing in live cells usually incorporate a reference probe/dye as an internal control to which the signal from the targeting probe is normalized. For example, Tan et al. semi-quantitatively measured ATP in live cells using such an approach.[141] However, absolute quantification of intracellular targets in live cells remains a challenge in the field.
For all these detection scenarios, there are more considerations and challenges associated with probe design. These additional factors are discussed in section 6.
2. Hybridization-based probes
2.1. Linear antisense probes
Linear antisense ONTs labeled with a fluorophore can be introduced into live cells as a means of detecting their complementary mRNA transcript (Figure 3A). This method is akin to FISH done with fixed cells, but the washing step to remove unbound probes is not possible with live cells. As such, mRNA detection with linear ONT probes in live cells has an inherently high background signal as bound and unbound probes cannot be distinguished. In spite of its limitations, strategies exist in the literature for mRNA detection using linear ONT probes. One common strategy employed involves adding multiple probes that target the same mRNA sequence, such that binding of the probes results in large local fluorescence intensity that can be distinguished from unhybridized probes.[54] Alternately, mRNA transcripts that are known to localize into specific domains would also result in large local fluorescence intensity from probes and can be visualized.[55] Early studies using linear ONT probes done by Politz et al. targeted the poly A tail to understand how mRNA is trafficked through the nucleus before its eventual release to the cytoplasm.[53] It was found that in large part, mRNA traffics randomly through the nucleus, and therefore, finds nuclear pores by chance before its release into the cytoplasm. In later studies, Molenaar et al. designed probes targeting U1 small nuclear RNA (snRNA), U3 snRNA, 28S ribosomal RNA (rRNA), and poly(A) RNA, and were able to detect these targets in the nucleus upon microinjection.[54] Note that although probes were microinjected into the cytoplasm, all probes were sequestered into the nucleus after approximately 60 seconds, showing the difficulty of the linear ONT approach for imaging targets in the cytoplasm.
Figure 3.
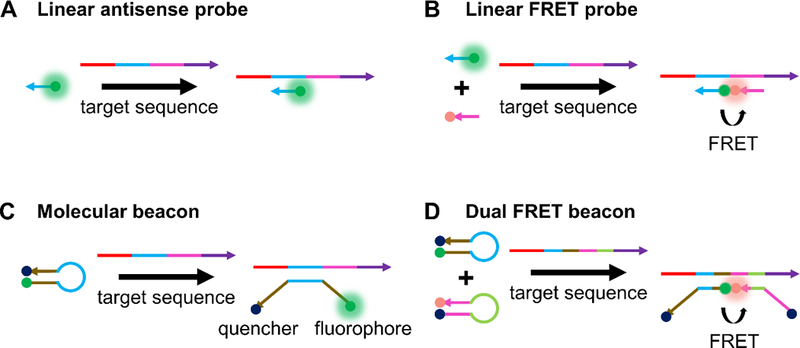
Working principles of common hybridization-based probes. (A) A schematic of linear antisense probes, whereby a dye-labeled recognition sequence binds to its complementary target illustrated in the same color.[9] (B) Linear FRET probes involve hybridization of two different linear antisense probes to adjacent regions of a target sequence, bringing a FRET pair into proximity that results in a fluorescence signal that can be monitored.[56] (C) In the off state, MBs have a duplexed stem region (illustrated in brown) that keeps a fluorophore and quencher in close proximity. Target binding to the loop region (illustrated in light blue) opens the stem region, separates fluorophore and quencher, and turns on fluorescence.[125] (D) Dual FRET beacons incorporate two MBs that can bind to adjacent regions of a target sequence. Similar to linear FRET probes, binding of the MBs to adjacent regions of a target sequence brings a FRET pair near one another, resulting in turn-on of FRET signal.[77]
Taken together, linear antisense probes have largely been limited in their use due to their inherently high background signal and are as such challenging to apply to targets at lower abundance or targets that are more diffuse in cells. Subsequent strategies have sought to create detection strategies with higher signal to noise ratio.
2.2. FRET probes
2.2.1. Linear FRET probes
The use of linear FRET probes is commonly used to reduce the background from linear antisense probes. In this approach, two linear probes that are complementary to the same mRNA target and bind to adjacent regions on the mRNA are synthesized (Figure 3B). The 5’ terminus of one probe and the 3’ terminus of another probe are modified with donor and acceptor dyes, respectively, such that upon binding to mRNA, the FRET pair is brought close together. Proximity results in a decrease in donor fluorescence emission and increase in acceptor fluorescence emission. The first studies done by Tsuji et al. used the FRET signal generated from probe binding to detect cytoplasmic c-fos mRNA in live COS-7 cells.[56] Similar to labeled antisense probes, linear FRET probes are also quickly trafficked to the nucleus after microinjection. As such, cytoplasmic detection required binding the linear FRET probes to a macromolecule (streptavidin) to prevent nuclear sequestration. Okabe et al. also studied c-fos mRNA in COS-7 cells, showing that inducing stress on cells results in the localization of c-fos mRNA in stress granules.[57] Linear FRET probes have also been used for detecting single nucleotide polymorphisms in mRNA. In one example, Dahan et al. designed FRET probes capable of detecting a single nucleotide mutation in the HRAS oncogene.[58]
While the linear FRET approach lowers background stemming from fluorescence from unbound probes, it also confers an additional benefit from the standpoint of selectivity. Because two probes must bind to adjacent regions of the mRNA sequence, it is highly unlikely that aberrant probe binding to mRNA will lead to false positive signal. However, because RNA is associated with proteins and often has complex secondary structure, inaccessibility of stretches of RNA target is a problem common to hybridization probes, especially so for linear FRET probes that often require ~40 bp for targeting.[56] Furthermore, a poor choice of donor/acceptor pairs may lead to a large amount of bleed-through of emission signal from the donor into the acceptor channel.[142]
2.2.2. Molecular beacons
One of the most common methods for visualizing mRNA in live cells is the use of hairpin ONT probes known as molecular beacons, which we refer to as MBs for simplicity. In the “fluorescence off” state of the probe, a stem region is hybridized such that a fluorophore and quencher are close to one another. Target binding to a loop region induces opening of the stem region, resulting in separation of the fluorophore and quencher and introduction of the “fluorescence on” state (Figure 3C). Generally, signal to noise ratios are higher for MBs than for both linear antisense probes and linear FRET probes.[6,125] MBs were introduced by Tyagi et al. in 1996, and first used for live cell imaging in 1998.[125] For instance, Matsuo used MBs to study the distribution of basic fibroblast growth factor mRNA in trabecular cells.[59] In the same year, Sokol et al. leveraged MBs for studying β-actin mRNA and Vav mRNA in K-562 cells.[63]
Since then, numerous papers have been published that take advantage of the utility of MBs. MBs are commonly used for distinguishing different cell types based on mRNA level. In one example, a MB designed for a known oncogene, survivin, fluoresced in breast cancer cells (MDA-MB-435 and MDA-MB-231) but not in normal immortalized mammary epithelial cells (MCF-10A), showing that the beacons could successfully differentiate the cells based on survivin level.[68] Others, like Kang et al., have synthesized conventional MBs to visualize miR-26a and miR-206 simultaneously, two microRNA (miR) that are expressed heavily during myogenic differentiation, in C2C12 cells.[60] Targets need not be limited to miRNA or mRNA, as Park et al. have detected a class of non-coding small RNAs known as piwi-interacting RNAs (piR-36026 and piR-36743) in MCF-7 cells.[41] Others, like Zhao et al.[64] and Chen et al.[73], have claimed to be able to image transcripts at the single molecule level using MBs.
The fluorophore pairs in MBs need not be fluorophore/quencher pairs. Following the work of Zhang et al.,[143] Bohländer et al. described the use of two wavelength shifting MBs for multiplexed miRNA detection.[69] One MB, in the hairpin form, contained a green donor dye and a red acceptor dye in close proximity to one another. In this conformation, exciting the green dye led to emission from the red dye. Upon target binding and opening of the hairpin, exciting the green dye led to emission from the green dye as the two fluorophores were no longer in close proximity. Another MB was designed in the same manner with a blue donor and a yellow emitter. In this way, four colors could be simultaneously monitored for detection of two different targets. The different targets chosen in this paper were miR-21 and miR-31, and they were studied in 293T, SW620, RKO, and WiDr cells.
Yet, one important advantage of fluorophore/quencher MBs is their relative ease of use for multiplexed detection. Namely, FRET is challenging to multiplex because of the difficulty in finding commercially available FRET pairs that fluoresce at distinct wavelengths.[144] Medley et al. showed that the microinjection of multiple fluorophore/quencher MBs containing dyes fluorescing at distinct wavelengths could be used to simultaneously detect mRNA associated with β-actin and manganese superoxide dismutase (MnSOD) mRNA in MDA-MB-231 cells.[65]
A problem with DNA MBs, similar to linear probes, is that they degrade in cells, sometimes within 30 minutes, leading to false positive signals.[66] To help alleviate this issue, modifications such as LNAs, 2’O-Me RNAs, morpholinos, PNAs, or serinol nucleic acids have been introduced into probes to increase biostability.[66,67,73,145–147] Moreover, base modifications can lead to enhanced hybridization kinetics, enhanced specificity, and enhanced target affinity.[148] For instance, Wu et al. introduced LNA into MBs to increase their stability and reduce their propensity for false positive signal due to degradation or protein binding.[66] LNA-enhanced MBs were functional even 24 hours after microinjection into cells, proving useful for long term imaging of MnSOD mRNA in MDA-MB-231 cells. Chen et al. synthesized morpholino molecular beacons (MOR-MBs) for use in vivo.[67] They hypothesized that the superior stability, biocompatibility, and affinity of morpholinos for RNA would lead to advantageous properties. MOR-MBs had no increase in non-specific fluorescence signal in medaka fish embryos over the course of two hours. In contrast, DNA-MB signal increased gradually over the course of 45 minutes, likely due to degradation of the probe by nucleases. MOR-MBs were also able to distinguish between full complement or single base mismatch RNA targets in vivo. A new class of artificial nucleic acids based on a serinol backbone, called serinol nucleic acids, were incorporated into MBs by Murayama et al., and used for their enhanced biostability.[147] While imaging was done in fixed cells, signal to background ratios as high as 930 times were reported in solution, which was about 30 times higher for an equivalent MB synthesized with DNA.
Another issue, similar to other ONT probes already discussed, is that MBs may be sequestered to the nucleus following delivery, which is problematic for imaging targets in the cytoplasm. This sequestration has been prevented by linking MBs to a macromolecule, nanoparticle, or tRNA sequence.[149–151]
For detecting targets of low-abundance, it is sometimes necessary to employ strategies that amplify signal, in turn lowering the limit of detection. Strategies most often employed involve HCR or a slight variation thereof. HCR is an isothermal cascade reaction in which an initiator sequence triggers the hybridization of two complementary sequences trapped in metastable hairpin conformations.[152] Incorporating a donor or acceptor dye (FRET pair) into each of the hairpins is one common way to interface HCR with a readout event for target detection (Figure 4A). Strategies inspired by this approach include cascade hybridization reaction (CHR), developed by Cheglakov et al.[70] and branched HCR (b-HCR), developed by Liu et al.[71]
Figure 4.
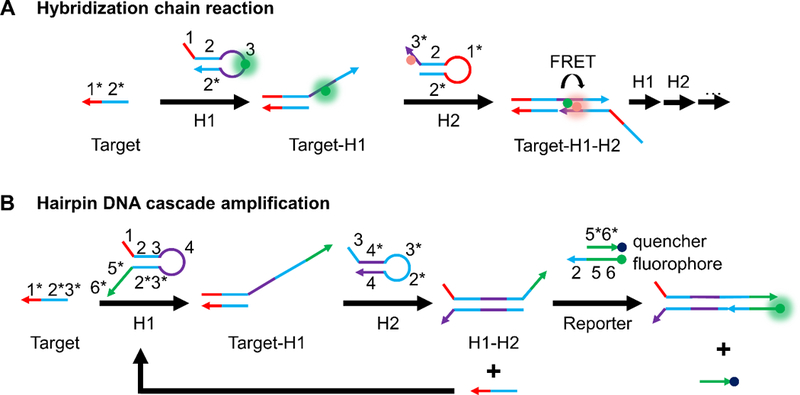
Two common oligonucleotide-based strategies for isothermal amplication of fluorescence signal. (A) Hybridization chain reaction[152] for enabling a FRET-based amplified fluorescence readout. The presence of a target sequence initiates a cascade hybridization reaction between two metastable hairpins (H1 and H2) with extended regions of complementarity. Incorporation of a FRET-pair, one in the loop region of H1 and the other in the toehold region of H2 brings the FRET-pair into close proximity upon HCR. (B) Hairpin DNA cascade amplification.[72] Target binding to H1 exposes a region in H1 that can bind to H2. This releases the target and allows it to bind more H1 strands for amplified signal. The H1-H2 complex can bind to a fluorophore-labeled strand that is pre-hybridized to a quencher-labeled strand, displacing the latter and turning on fluorescence.
Inspired by the work of Yin et al.,[153] another amplification strategy was proposed by the Tan group in 2015, called hairpin DNA cascade amplification (HDCA, Figure 4B).[72] The probe set consists of a total of four distinct strands. When no mRNA target is present, two DNA strands H1 and H2 are each in the form of a hairpin. Additionally, a fluorophore-labeled “reporter” strand is prehybridized to a quencher-labeled strand. When present, mRNA binds to the toehold region of H1, opening the hairpin and exposing bases which can bind to H2, which in turn displaces the mRNA due to stronger binding with H1. This strategy allows the mRNA to participate in more rounds of signal amplification. The H1-H2 complex formed has an overhang that is complementary to the “reporter” strand. The quencher strand is displaced providing a fluorescent readout. In proof-of-concept studies, MnSOD mRNA, associated with tumor proliferation, was detected in MDA-MB-231 cells after transfection of probes with Lipofectamine 3000. Notably, these probes were able to sense MnSOD mRNA even after the mRNA was knocked down using cordycepin, a scenario where a low abundance target could be detected.
One problem with MBs (and linear probes) is that “naked” nucleic acids do not generally enter cells on their own, and thus require transfection strategies like cationic liposomes, conjugation to a cell penetrating peptide, microinjection, or electroporation.[6] Therefore, many recent approaches have sought to use aptamer-based or nanoparticle-based strategies to circumvent the need for these methodologies. While nanoparticle-based strategies will be the subject of a separate section (vide infra), we will briefly discuss strategies for aptamer-based entrance into cells in this section.
The Tan group has designed MBs that start functioning “on demand” and enter cells without transfection reagents (Figure 5).[74] Their carrier probe molecular beacons (CP/MBs) consist of two strands. One strand of the probe is the MB that is complementary to an mRNA transcript of interest. The other strand is an aptamer plus carrier probe (CP) sequence. The aptamer chosen (AS1411) has been found to attach to nucleolin, which is a cell surface marker that is present on many cancer cells. The authors claimed that binding of the CP/MBs to nucleolin allowed for direct cytosolic entry of the probes. To make the probes “on demand,” photocleavable (PC) linkers were incorporated into the CP region. Initially, the CP and MB region are hybridized, not allowing for the MB to bind to complementary mRNA in the cell. Upon UV irradiation, the PC linkers are cleaved and the MB is detached from the CP region, allowing the MB to bind to complementary mRNA. MnSOD was chosen as a model target and imaged in MCF-7 cells. Taken together, this construct allows for targeted delivery to cells, entrance into cells without transfection reagents, and “on demand” activation of the MB allowing for precise spatiotemporally controlled imaging in the cell.
Figure 5.
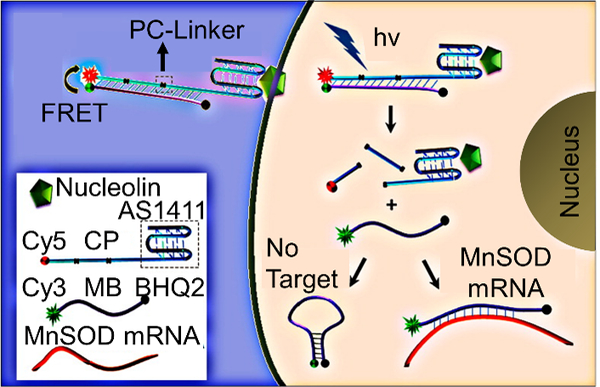
“On-demand” MBs incorporating an aptamer-targeting nucleolin (AS1411) for transfection reagent-free uptake into cells. Irradiation with UV-light activates the MB for use by cleaving the PC linkers and detaching the MB from the CP region. Adapted with permission.[74] Copyright 2013, American Chemical Society.
Furthermore, Kim et al. developed RNA aptamer-based probes for detection of miRNA in live cells.[75] To allow for uptake into cells without the need of transfection reagents, mucin1 (overexpressed in malignant cancer cells)-binding aptamers were hybridized to the probe, resulting in probe endocytosis. As a proof-of-concept, miR-34a was imaged in MCF-7 cells and T47D cells.
2.2.3. Dual FRET beacons
One way to increase the signal to noise ratio of MBs is to employ a dual FRET approach (Figure 3D). In this design, two different MBs are designed that hybridize to adjacent locations on an mRNA transcript of interest. Each probe contains a fluorophore that can form a FRET pair with the fluorophore on the other probe. In the off state, fluorescence of both probes is quenched due to close proximity of a fluorophore and quencher. Upon binding of the MBs to the target, the fluorophores are unquenched and brought in close proximity to one another such that they can form a FRET pair. Importantly, with this strategy, one can differentiate signal due to non-specific separation of fluorophore and quencher from actual signal due to binding by monitoring FRET signal. Santangelo et al. used dual FRET MBs to study the KRAS and survivin oncogenes in live human dermal fibroblasts and MIA PaCa-2 cells.[77] Indeed, they were able to show that signal due to non-specific separation of fluorophore and quencher could be distinguished from signal due to binding, thus reducing false-positive signal. King et al. have shown the utility of dual FRET MBs for detecting Oct4 mRNA and subsequently isolating Oct4 positive pluripotent human embryonic stem cells using flow cytometry.[76]
In spite of the fact that dual FRET beacons can improve signal to noise ratio, they suffer from the same limitations that linear FRET probes suffer. Namely, given the complex secondary structure of RNA and its association with proteins, finding large stretches of RNA that are accessible for probe binding is a challenge. Further, bleed-through of donor emission into the acceptor emission channel may be a problem in certain cases.[142]
2.3. Quencher-free probes
2.3.1. FIT probes
Forced intercalation (FIT) probes were developed in 2005 by the Seitz group as a quencher-free method for nucleic acid detection (Figure 6A).[124] FIT probes consist of a sequence of nucleic acids and a single intercalator dye of the thiazole orange family, whereby the intercalator plays the role of a nucleobase surrogate. Dyes of the thiazole orange family fluoresce when rotation around their methine bridge is restricted. Thus, when a FIT probe binds to its complementary sequence, methine bridge rotation is hindered, which results in fluorescence being turned on. Notably, this strategy results in no false-positive signal as a result of probe degradation, overcoming a key limitation associated with MBs.[83] Furthermore, FIT probes offer fluorescence enhancement, reported to be as high as 195 times upon addition of complementary target.[154] Much of the work in live cell imaging using FIT probes has relied upon the use of peptide nucleic acids (PNA) due to their biostability and both rapid and strong hybridization to complementary ONTs.[79–81,155,156] Kam et al., for example, used PNA FIT probes for detection of KRAS mRNA inside live cells.[79] They showed that FIT probes are sensitive to single nucleotide mismatches directly adjacent to the intercalator, such that single nucleotide polymorphisms (SNPs) in the KRAS gene could be discriminated. Importantly, a conventional fluorophore/quencher MB was unable to discriminate SNPs in the KRAS gene.
Figure 6.
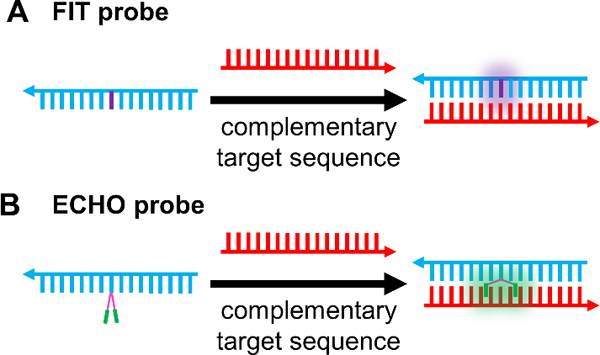
Quencher-free probes. (A) FIT Probes incorporate a viscosity sensitive dye, such that target binding results in turn-on of the dye[124] (B) ECHO probes contain two dyes (e.g. thiazole orange) that are covalently attached to the same base in a probe sequence. In the free probe, the dyes form an H-aggregate resulting in attenuated fluorescence emission. Target binding breaks the H-aggregation, and the dyes intercalate between the bases in the duplex with concomitant fluorescence turn on.[84]
More recent work in the area has looked into enhancing probe brightness, using more accessible nucleobase chemistry, and multiplexing of gene analysis. Imaging in cells and tissues with high sensitivity requires probes that are both responsive and sufficiently bright. Two different approaches have been used to enhance FIT probe brightness. In the first approach, a duplex responsive dye (thiazole orange) is paired to a highly emissive dye (oxazolopyridine analogue, JO).[157] In the single strand, fluorescence is quenched due to dye twisting about the methine bridge and dye-dye contact. Upon binding of complementary target, quenching due to twisting and energy transfer are stopped, leading to fluorescence turn-on. TO is believed to play the role of a light collector that can transfer excitation energy to the highly emissive JO moiety.[157] As proof-of-concept, the TO/JO system was used to image oskar mRNA in the tissue of Drosophila melanogaster using wash-free FISH. Recently, mRNA associated with the mCherry protein was imaged using this system in live FIp-In™ 293 T-Rex cells expressing the gene.[82]
An alternative approach for enhancing the brightness of FIT probes involves the use of locked nucleic acids (LNAs).[83] Hövelmann et al. showed that the introduction of an LNA base directly adjacent to the duplex responsive dye introduces further rigidity in the local environment of the dye, resulting in more restricted rotation around the methine bridge and a concomitant increase in final brightness upon target hybridization. LNA-enhanced FIT probes were then used to track in real time oskar mRNA associated with ribonucleoproteins in Drosophila melanogaster.
Although PNAs impart increased nuclease resistance to the probes, they suffer from several limitations. The neutral backbone of PNAs renders them less soluble compared to conventional phosphate backbone-based ONTs and makes them susceptible to aggregation. Moreover, PNA synthesis is more costly than conventional nucleic acid synthesis and common protocols for nucleic acid transfection are not applicable to them. Therefore, recent work in FIT probes has focused on alternate strategies, for example, by modifying the sugar moieties in ONTs with phosphate backbones.[158] 2’-O-Me RNA and LNAs have been used in FIT probes due to their resistance to nuclease degradation and both rapid and high affinity binding to complementary nucleic acids.[83,154] FIT probes made of DNA have also been used for live cell imaging.[159]
Recently, the Seitz group reported the development of four new dyes for use in FIT probes.[154] Of these, three showed fluorescence enhancement upon addition of complementary target. Importantly, the new dyes fluoresce in distinct wavelength regimes, allowing later studies to do multiplexed live cell imaging of the poly A tail of mRNA and different regions of mCherry mRNA in Flp-In 293 T-REx cells. [159]
2.3.2. ECHO probes
A variant of FIT probes are exciton-controlled hybridization-sensitive fluorescent ONT (ECHO) probes (Figure 6B). When two thiazole orange fluorophores are covalently attached to the same base of a nucleic acid sequence and arranged parallel to one another (H-aggregation), fluorescence is largely attenuated due to excitonic interactions between the two fluorophores. Upon complementary nucleic acid binding, the H-aggregate is broken and each thiazole orange intercalates into the duplex such that fluorescence is turned on.[160] Early studies to assess the feasibility of ECHO probes in live cells consisted of designing probes targeting the poly A tail of RNA. Microinjecting these probes into HeLa cells resulted in fluorescence intensity observable in the cytoplasm and nucleus as a result of probe binding to target.[84]
To aid in multiplexed imaging, Ikeda et al. developed new fluorophores capable of use in ECHO probes.[85] While those fluorophores that were originally developed ranged in emission maxima from 455 nm to 677 nm, more recently near-IR dyes have also been developed for ECHO probes.[87] To show the capability of multiplexed live cell detection, ECHO probes with three different dyes were designed to target three different miRNA sequences. Microinjected probes proved useful for detecting the targeted miRNA in the nucleus of HeLa cells.[85]
Efforts have also been made to introduce base modifications to ECHO probes, as unmodified DNA probes are quickly degraded in cells, making long-term live cell imaging challenging. For instance, a 2’-O-Me-modified ECHO probe targeting the poly-A tail of RNA was used for imaging of HeLa cells.[88] Time-resolved imaging showed that fluorescence was much more intense at the mitotic phase than at the interphase, implying that relatively large quantities of mRNA are expressed as the cell divides. Later studies have shown the capability of ECHO probes to detect other intracellular RNA targets, including 28S rRNA and U3 small nucleolar RNA.[86]
2.3.3. Miscellaneous quencher-free probes
Min et al. took advantage of aggregation-induced emission for detection of miRNA.[161] DNA probes complementary to a target of interest were synthesized with aggregation-induced emission luminogens (AIEgens) on the 5’ end. Upon binding of target, a duplex is formed. Addition of exonuclease III results in successive removal of mononucleotides from the probe sequence and subsequent release of both the RNA target and the AIEgens. The AIEgens are free to aggregate and turn on fluorescence, while the RNA target is free to bind to more probe, thus amplifying signal. The probe was efficacious in detecting miR-21 in HeLa and MCF-7 cells (high expression), and human lung fibroblast cells (HLF) (low expression).
Ro et al. leveraged the unique optical properties of pyrene-linked adenine (PyA) derivatives to detect miRNA in live cells.[162] The probe sequence was designed such that in the presence of the target miRNA, the PyA moieties were brought to close proximity that resulted in a wavelength shift in emission from 455 to 600 nm. A detection limit of 0.63 nm was found in solution, and the strategy was used to image miR-21 in HeLa, MCF-7, and NIH-3T3 cells successfully.
Østergaard et al. have developed quencher-free MBs.[163] 2′-N-(pyren-1-yl)carbonyl-2′-amino LNA monomers are incorporated into the loop region of a MB. Little fluorescence is seen for these LNA monomers when in a single stranded region, but significant fluorescence turn-on is observed when present in a duplexed strand. These so called “glowing LNAs” were used to image mRNA corresponding to component X of pyruvate dehydrogenase complex in 3T3-L1 cells.
2.4. Nanoparticle-based probes
2.4.1. Gold nanoparticle-based
2.4.1.1. NanoFlares
NanoFlares were invented and reported in 2007 by the Mirkin group as a new tool for studying mRNA (Figure 7A).[44] NanoFlares are SNA gold nanoparticle conjugates and consist of a dense shell of duplex DNA on a gold nanoparticle surface. One strand of the duplex, called the recognition strand, has a track (e.g. ~20 base pairs) complementary to an mRNA target of interest. The other strand of the duplex, called the flare strand, consists of a fluorophore and typically has ~8 base pairs of complementary to the recognition strand. When the recognition strand and flare strand are hybridized, the gold nanoparticle quenches fluorescence of the flare strand due to close proximity between the gold and the fluorophore. When the mRNA target of interest is present in the cell at sufficient concentrations, the mRNA binds to the recognition strand and the flare sequence is displaced, resulting in fluorescence turn-on.[164] Owing to the dense packing of DNA on the nanoparticle surface, NanoFlares exhibit high cellular uptake without the need for transfection reagents, display enhanced resistance to nuclease degradation in comparison to free nucleic acid probes, and exhibit little immunogenicity or toxicity.[165] In the first studies, NanoFlares were used to differentiate between different cell lines based on expression of mRNA associated with the oncogenic marker survivin. Specifically, cells that do not express survivin (C166 cells) were differentiated from cells that express large amounts of survivin (SK-BR-3 cells).[44]
Figure 7.
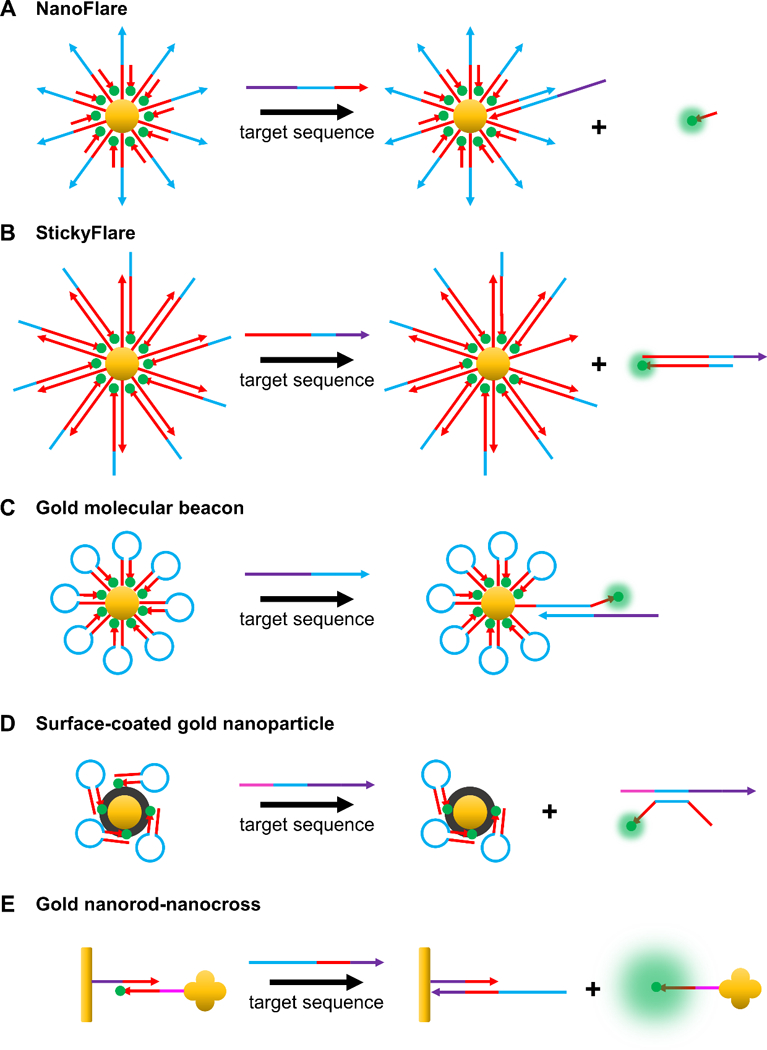
Common gold nanoparticle-based hybridization probes. (A) In the off state of a NanoFlare, a fluorophore-labeled flare strand is hybridized to a gold-bound recognition strand, allowing for gold to quench fluorescence. Target binding to the recognition strand displaces the flare sequence, separates it from gold, and turns on fluorescence.[44] (B) In the StickyFlare construct, the fluorophore-labeled strand is designed to be complementary to the target. Therefore, upon probe-target binding, the target can be monitored, providing valuable spatiotemporal information about its dynamics.[134] (C) Gold MBs consist of fluorophore-labeled hairpin ONTs conjugated to the surface of gold nanoparticles. The principle of action is the same as that of MBs, except the central gold nanoparticle acts as a quencher.[98] (D) As opposed to chemical conjugation, fluorophore-labeled ONTs may also be adsorbed onto surface-coated gold nanoparticles. For example, as depicted in the figure, fluorophore-tagged hairpin ONTs can be adsorbed onto polydopamine (colored black)-coated gold nanoparticles.[167,168] Polydopamine acts as a further source of quenching and can be utilized in photothermal therapy due its ability to absorb NIR light. (E) Anisotropic gold nanoparticles have also been used as cores. For example, a gold nanorod (functionalized with the recognition strand) that acts as a quencher can be attached to a gold nanocross (functionalized with a complementary fluorophore-labeled strand). Upon target binding, the fluorophore-labeled strand attached to the nanocross is released, inducing fluorescence turn on. The presence of the nanocross further enhances the fluorescence due to surface enhanced resonance.[169]
As NanoFlares bind to mRNA, studies in 2009 by Prigodich et al. showed that the platform can be used for the simultaneous detection and regulation of survivin in HeLa cells.[166] Consistent with previous studies, detection of survivin required relatively low concentrations and treatment times of NanoFlares with HeLa cells (0.5 nm and 6 h) while regulation required relatively high concentrations and treatment times (5 nm and 4 days) of NanoFlares with HeLa cells.
Further work has leveraged the capability of NanoFlares to become the first example of a genetic based approach for simultaneous detection and isolation of live circulating tumor cells (CTCs) in and from human blood.[139] Vimentin and fibronectin, both known markers of the epithelial to mesenchymal transition (EMT), were targeted using Nanoflares. A known quantity of mCherry expressing MDA-MB-231 cells was added to samples of whole human blood, followed by incubation with either vimentin, fibronectin, or scramble control NanoFlares. mCherry positive and flare (Cy5) positive cells were isolated using flow cytometry. The NanoFlare was shown to detect as few as 100 CTCs in whole blood samples with an average recovery of cells of around 68%, on par with other techniques for CTC isolation.[170] In a different experiment, a GFP-expressing recurrent cell line known to form mammospheres was spiked into whole human blood. Cells isolated for GFP fluorescence and flare (Cy5) fluorescence were then subjected to a mammosphere analysis. As expected, isolated cells were capable of forming mammospheres indicating that these cells were indeed the GFP-expressing cells originally spiked into human blood and that NanoFlares had no detrimental impact on cell viability.
Recently, NanoFlares have been used for numerous applications ex vivo and in vivo. For instance, Yeo et al. utilized NanoFlares for abnormal scar detection.[135] The connective tissue growth factor (CTGF) mRNA is overexpressed in incidences of hypertrophic and keloidal scars, resulting in overproduction of collagen. In vitro, CTGF-targeting NanoFlares could be used to distinguish between keloidal scar fibroblasts (KSFs), hypertrophic scar fibroblasts (HSFs), and non-diseased dermal fibroblasts (NSFs). Mixing CTGF NanoFlares and HSFs together in matrigel and subsequent subcutaneous injection into mice showed that detection was possible in vivo as well. However, because human skin has more epithelial skin layers than mouse skin, it is in challenging to detect markers of aberrant scar formation in human skin. Using an ex vivo model of human skin, the study showed that NanoFlares infiltrate past the epidermis and distribute in the dermis. Importantly, the penetration depth of NanoFlares was sufficient to detect HSFs that had been injected into the dermal region of an ex vivo skin sample. Taken together, these findings are significant, since the NanoFlare has the potential to change paradigms in skin disease diagnosis from histopotalogy and biopsy to something non-invasive that is more amenable to earlier detection of aberrant scars.
Another example of NanoFlares used for investigating skin wound models comes from Vilela et al.[171] Wound healing involves an epithelial to mesenchymal transition, and as such monitoring of vimentin mRNA using NanoFlares can be used as a handle to monitor the progression of the process. Indeed, the study showed that light-sheet microscopy could be used to study the 3-dimensional distribution of vimentin mRNA in whole tissue samples obtained from mice.
Recently, Moros et al. used gold NanoFlares for detection of mRNA in live Hydra vulgaris organisms.[172] Their study targeted Hymyc1 mRNA, which is a member of the MYC family of proto-oncogenes. After incubating Hydra polyps with NanoFlares targeting Hymyc1, the group found no toxicity to the organism due to the NanoFlares. Furthermore, animals treated with Hymyc 1 targeting NanoFlares showed fluorescence in their body column, but neither in their head nor tentacles, as expected. Scrambled NanoFlares with no target in the organism showed no fluorescence after incubation, while positive control NanoFlares targeting poly A RNA fluoresced throughout the organism, as expected. In another successful use of NanoFlares, Sozer et al. have recently used NanoFlares to detect single nucleotide mutations in Janus kinase 2 mRNA.[173]
Slight variations in the original NanoFlare design have led to NanoFlares with additional capabilities. For example, multiplexed NanoFlares were developed in 2012 to detect multiple mRNA transcripts with a single construct.[39] Recognition sequences targeting both β-actin and survivin were conjugated to the same nanoparticle. By hybridizing flare sequences containing either Cy3 (β-actin) or Cy5 (survivin), both targets could be simultaneously detected. Notably, simultaneous detection allowed for normalizing survivin flare signal to flare signal from a housekeeping gene, narrowing the normally broad distribution of fluorescence values seen due to differences in probe uptake and flare degradation amongst individual cells. The Tang group has also leveraged the strategy of multiplexed detection. In one example, mRNA associated with three different oncogenes was detected with a single construct.[174]
One improvement to the original NanoFlare was introduced by Yang et al. in 2015 in the form of FRET NanoFlares.[175] In this design, the flare strand contains an acceptor dye on the 3’ end and a donor dye on the 5’ end. When mRNA binds to the recognition sequence, the flare strand is displaced. With the proper flare sequence design, the strand folds into a hairpin upon displacement, bringing into close proximity the donor and acceptor dye and allowing for the monitoring of FRET signal as the readout for mRNA binding. This new design holds a key advantage over the original NanoFlare design. Namely, no false-positive signal should be seen due to nuclease degradation or destruction of the gold-sulfur bond. However, this strategy does suffer from the need of having a dual fluorophore-labeled flare strand and necessitates more intricate flare sequence design.
A variant of the NanoFlare design was introduced in 2015 that allowed for simultaneous detection and tracking of mRNA transcripts, termed the Sticky-Flare (Figure 7B).[134] In the Sticky-Flare construct, the flare strand tagged with a fluorophore is complementary to the target mRNA, such that following binding, the mRNA can be tracked in real time. Sticky-Flares designed to target β-actin mRNA allowed for study of the mRNA’s real time dynamics and final localization. β-actin mRNA in HeLa cells was observed to co-localize with the mitochondria, marking the first time this phenomenon has been observed in this cell line.
Other design variations have sought to add signal amplification to NanoFlares. Liang et al. recently developed an entropy driven amplifier in conjunction with conventional NanoFlares, reaching a detection limit of 8 pm in solution and imaging miR-21 in MCF-7, HeLa, HEK293, and MRC-5 cells.[176] Moreover, Li et al. recently interfaced FRET NanoFlares with an amplification element for sensitive miRNA detection.[177] This approach was able to detect miR-let-7a in cells with high expression (MCF-7) and cells with relatively low expression (A549).
Commercially available NanoFlares have also been used successfully by a number of groups for studying genetic content in live cells.[178–181]
2.4.1.2. Gold nanoparticle molecular beacons
Gold nanoparticle molecular beacons (NP-MBs), also variants of SNAs, offer a number of potential key advantages, including resistance to nuclease degradation and entrance into cells without the need of transfection reagents. Gold NP-MBs consist of a dye-labeled hairpin DNA sequence functionalized to a gold nanoparticle (Figure 7C). When the hairpin is closed, the dye is in close proximity to the gold, leading to the fluorescence off state due to gold quenching of fluorescence. When the target mRNA binds the DNA sequence, the hairpin is opened such that the dye is separated from the gold and fluorescence is turned on. Note that this strategy requires careful design of the MB sequence, as hairpin opening must result in sufficient distancing of the fluorophore and gold at risk of observing weak signal. Early work in this area was done by Harry et al., who were able to detect tyrosinase mRNA in melanoma cells.[182] Others, like Xue et al., have detected oncogenes like STAT5B in MCF-7 cells.[183] Uddin et al. have used NP-MB for detection of VCAM-1 mRNA, a marker of inflammation, in retinal microvascular endothelial cells.[184] Work by Qiao et al. has expanded on the original construct by conjugating two different MBs on the gold surface to do multiplexed analysis of survivin and cyclin D1 mRNA.[185] Jayagopal et al. simultaneously imaged GAPDH mRNA and respiratory syncytial virus (RSV) mRNA in Hep-2 cells.[98] In another example from the Tang group, four different oncogenes were detected simultaneously using a gold NP-MB.[186]
Examples of theranostic platforms also exist.[187,188] For example, in 2015, Bao et al. used an in vivo murine model of gastric cancer to show that gold NP-MBs were able to simultaneously detect, target, and knock-down the expression of the oncogenic KRAS gene.[187]
Moreover, Pan et al. have developed constructs capable of simultaneous detection of mRNA and matrix metalloproteipnases (MMPs).[94] MBs specific for TK1 mRNA and GalNAc-T mRNA along with fluorophore-labeled peptides that are cleaved in the presence of MMP-2 and MMP-7 were functionalized to the surface. MB opening and peptide cleavage separated fluorophore from gold, yielding fluorescence. The probe proved useful for the simultaneous monitoring of these cancer associated biomarkers in MCF-7 and MCF-10a cells. Pan et al. have devised a slight variation of the original design to allow multiplexing using monitoring of a FRET signal.[189]
2.4.1.3. Surface-coated gold nanoparticles
Some have coated gold nanoparticles with polydopamine (PDA) as part of their platform. PDA is attractive becaucse it permits immobilization of probes onto the surface through π-π interaction rather than tedious chemical conjugation, acts as another source of fluorescence quenching, and can be used in photothermal therapy because it absorbs light in the near infrared region.[100,190–192]
Choi et al. coated gold nanoparticles with PDA and subsequently adsorbed fluorophore-labeled hairpin probes targeting miRNAs of interest (Figure 7D).[100,167] Duplexing with the miRNA causes release of the probes from the surface and subsequent fluorescence turn-on. The probe proved useful for long term imaging of miR-29b and miR-31, two osteogenic markers, in hMSCs and primary osteoblasts. In particular, a time-dependent fluorescence was seen in primary osteoblasts and hMSCs going through osteogenic differentiation. Importantly, fluorescence signal in these cells was visible for up to 5 days. On the other hand, fluorescence was neither seen in hMSCs that had not differentiated nor in 3T3 fibroblasts, as expected.
Furthermore, Zheng et al. developed a NanoFlare-like construct using PDA-coated gold nanoparticles.[168] Target binding to the recognition strand results in the fluorophore strand’s displacement, leading to fluorescence turn-on. This probe was used to image miR-21 in HeLa cells. Taking advantage of PDA’s ability to absorb near-infrared light and potential as an agent for photothermal therapy, they showed that laser irradiation of HeLa cells treated with the probe resulted in a large decrease in cell viability.
Liu et al. have used PDA-coated gold nanoflowers for miRNA detection.[193] Nanoflowers were used for their high surface area providing access to higher loading densities of ONT probes. Two different hairpins are first adsorbed to the PDA-coated surface, whereby one of the hairpins is fluorophore-labeled. The target of interest opens the fluorophore-labeled hairpin, leading to fluorescence turn-on. Opening of the first hairpin also leads to exposure of a region that can bind to the second hairpin, whereby binding of the second hairpin leads to displacement of the bound target. In this way, the target is recycled to allow for binding to another fluorophore-labeled hairpin, leading to amplified signal. A low detection limit of 400 fm was found in solution. This amplification strategy was used for imaging miR-21 in HeLa cells.
Wu et al. developed the first example of HCR between hairpin probes for live cell mRNA detection.[108] Their construct consists of a gold nanoparticle coated with a layer of cationic peptides that are electrostatically complexed with hairpin DNA. Two different hairpin DNA are associated with the surface, each with a fluorophore that when near the other will form a FRET pair. While associated with the surface, the fluorescence of the dyes is quenched by the gold nanoparticle, leading to the probe “off state.” The target mRNA triggers HCR between the hairpins leading to the “fluorescence on” state in which multiple FRET pairs are formed. The authors claimed a limit of detection of ~0.5 pm in beaker studies. Interestingly, the authors also noted that the constructs bypassed endosomes and entered the cytoplasm directly, enabling them to image survivin mRNA in HeLa cells.
2.4.1.4. Other gold-based approaches
Constructs with gold nanorod cores also have been studied. Interestingly, it has been reported that constructs based on gold nanorods exhibit nearly two fold higher signal to noise ratio than constructs based on gold nanospheres.[194] Furthermore, because gold nanorods absorb near infrared light, they have been widely used in the literature for photothermal therapy, making them candidates for use in theranostic platforms.[195–197]
In a strategy analogous to that used with spherical gold NP-MBs, Wang et al. used gold nanorod MBs for detection of Dll4 mRNA in HUVEC cells.[194] Importantly, they were able to track Dll4 mRNA and study its dynamics in individual cells. In another example, Riahi et al. used gold nanorod MBs for studying β-actin and HSP70 mRNA in human breast adenocarcinoma cells and mice tissues.[198]
Sun et al. developed a dual FRET/surface enhanced resonance nanosensor composed of both gold nanorods and gold nanocrosses (Figure 7E).[169] The recognition strand is conjugated to a gold nanorod, while a fluorophore-labeled complementary strand is conjugated to a gold nanocross. In the case where the two strands are hybridized, the fluorophore is in close proximity to the gold nanorod and fluorescence is quenched. When the target binds the recognition strand, the fluorophore-labeled strand conjugated to the gold nanocross is displaced, resulting in fluorescence turn-on. However, surface enhanced fluorescence is also observed due to the presence of the gold nanocross, resulting in further increase in fluorescence of the dye. The authors used the construct to study miR-21 in HepG2, H9C2, and BRL cells.
Yan et al. electrostatically complexed probe sequences with polyethylenimine (PEI)-modified gold nanorods to achieve miRNA detection with amplified signal.[199] The first element of their system is a quencher containing recognition strand prehybridized with a fluorophore containing strand. Target binding displaces the fluorophore-labeled strand, leading to fluorescence. Adding a fuel strand displaces the bound miRNA target, allowing the target to participate in more fluorophore strand displacement events to amplify signal. The nanosensor was used for detection of miR-21 in MCF-7 tumor bearing mice. Photothermal therapy was also done, as NIR irradiation of the constructs led to reductions in tumor volume.
2.4.2. Upconverting nanoparticle-based
Certain upconverting nanoparticles (UCNPs) have found use in live cell imaging due to their biocompatibility, high photostability, and unique optical properties.[89,120,201,202] These nanoparticles absorb two or more lower energy photons and emit a higher energy photon. In the context of live cell imaging, this property allows probes to be excited with near-IR light and observe emission in the visible range, thereby overcoming issues related to cellular autofluorescence. Li et al. have explored a nanoparticle strategy where a luminescent and circular dichroism (CD) signal can be simultaneously monitored using gold-upconverting nanoparticle (Au-UCNP) pyramids for detection of miRNA in live cells (Figure 8A).[120] In the initial state, DNA linkages between nanoparticles result in the formation of a pyramidal structure. Binding of an miRNA sequence to a recognition region in the pyramid results in structure disassembly and subsequent separation of the gold nanoparticles and UCNPs. This separation is accompanied by an increase in luminescent signal (excitation = 980 nm, emission = 540 nm) from the UCNPs and a decrease in CD signal at 521 nm that can be monitored. To assess the feasibility of this strategy for miRNA detection in live cells, Au-UCNP pyramids were designed and synthesized for miR-21. HeLa cells were either transfected with miR-21 to increase its level or an miR-21 antisense sequence to decrease its level. Indeed, luminescent signal increased and CD signal decreased as the intracellular level of miR-21 increased. However, as the level of miR-21 was decreased in the cell, it was found that monitoring CD signal led to a 4-fold improvement in detection limit when compared to luminescence monitoring. This is important because it may potentially lead to the design of ultra-sensitive probes for intracellular detection based on monitoring CD signal, rather than solely monitoring luminescence signal.
Figure 8.
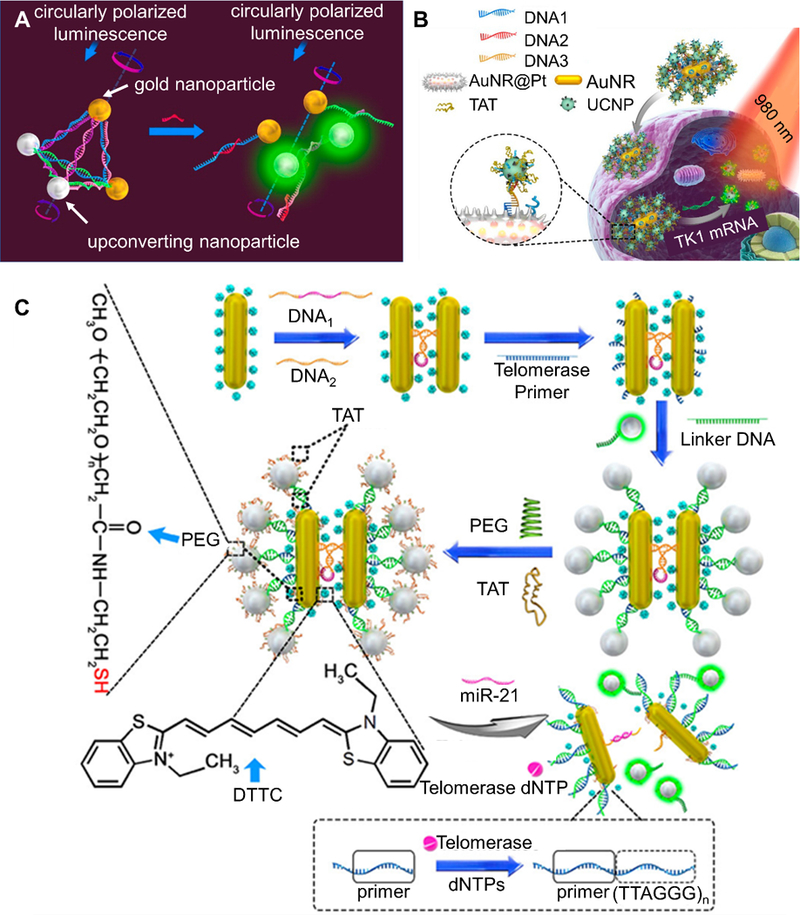
Selected upconverting nanoparticle-based hybridization probes. (A) A dual gold nanoparticle/upconversion system that allows for simultaneous monitoring of luminescent and circular dichroism (CD) signal change upon binding of miRNA. Target binding leads to structural disassembly, giving increased luminescence and decreased CD signal. Adapted with permission.[120] Copyright 2016, American Chemical Society. (B) AuNR/UCNP satellite assembly that is disassembled upon target binding, leading to increase in luminescence due to separation of UCNP and AuNR. A more detailed description of the platform is given in section 2.4.2. Adapted with permission.[89] Copyright 2018, American Chemical Society. (C) A schematic of a AuNR@Pt-UCNPs satellite assembly that can detect both miR-21 and telomerase simultaneously. miRNA binding leads to separation of the nanorods and a change in Raman signal, while telomerase presence leads to separation of gold nanorods and UCNPs and a subsequent luminescence signal. Adapted with permission.[200] Copyright 2017, American Chemical Society.
Gao et al. have used upconverting nanoparticles for mRNA and miRNA detection in live cells (Figure 8B).[89] Gold nanorods are coated with a platinum shell and conjugated with DNA sequence #1, while upconverting nanoparticles are conjugated with DNA sequence #2. To create Au NR@Pt-UCNPs satellite assemblies, DNA sequence #1 and DNA sequence #2 are linked together using DNA sequence #3, which is a recognition sequence for an mRNA of interest. When the mRNA is not present, the satellite assembly is intact and the luminescence of the upconverting nanoparticles is quenched. In the presence of the target mRNA, binding to the recognition sequence results in disassembly of the Au NR@Pt-UCNPs satellites, resulting in separation of the UCNPs from the Au NR@Pt and luminescence turn-on. In solution, the authors found that Au NR@Pt-UCNPs have a detection limit of 1.3 pm. Their efficacy in MCF-7, HeLa, and PCS-460–010 cells was confirmed by detecting TK1 transcripts, with relative luminescence intensities in the cell line corresponding to levels reported q-PCR. Finally, to show the versatility of the platform, miR-21 in HeLa cells was also imaged.
Ma et al. showed the utility of gold nanorod/UCNPs for simultaneous surface-enhanced Raman spectroscopy (SERS)/luminescence-based detection of miR-21 and telemorase in HeLa, MCF-7, and primary uterine fibroblast cells (Figure 8C).[200] A recognition strand (DNA 1) on one nanorod is prehybridized to a different strand (DNA 2) on another nanorod. This hybridization brings the gold nanorods in close proximity. Target binding to the recognition strand dehybridizes the sequences, leading to separation of the nanorods. When the SERS tag 3,3′-diethylthiatricarbocyanine iodide (DTTC) is loaded on the nanoparticle surface, this target binding induced separation leads to a decrease in Raman signal that can be monitored. To detect telomerase, the gold nanorods are further conjugated with a telomerase primer (TP) sequence while upconverting nanoparticles are conjugated with a mismatched sequence. The two components are “glued” using a linker strand, putting the upconverting nanoparticles near the gold nanorod and turning off fluorescence. Telomerase-induced extension of the TP strand results in release of the upconverting nanoparticles, leading to fluorescence turn-on.
2.4.3. Cationic liposome-based
Liposomes are attractive because of their biocompatibility, biodegradeability, ability to enter cells without transfection reagents, and ability to protect cargo from degradation through encapsulation.[40,203]
Kim et al. have used hyaluronic acid-coated liposomes for detection of miR-34a.[40] The hyaluronic acid coating allows for targeting CD44 for endocytic uptake into cells. Liposomes encapsulate within them duplexed strands composed of a fluorophore-labeled recognition strand and a short quencher-labeled strand. Disassembly of the liposome in acidic endosomes is believed to lead to endosomal membrane destabilization and subsequent release of probe into the cytoplasm. Target binding to the recognition strand displaces the quencher strand, recovering fluorescence. Beyond imaging miR-34a in cancerous cells in vitro, the study was also able to show the efficacy of the construct for imaging miR-34a in real time in a mouse breast cancer model.
Han et al. have made use of a similar liposome-based strategy.[204] Unlike Kim et al., no targeting moiety is used on the surface, and uptake is hypothesized to be due to both endocytosis and cell membrane/liposome fusion. Indeed, they were able to show the utility of their sensor for monitoring miRNA associated with adipogenesis. In particular, constructs targeting miR-181a, miR-21, and miR-31 were incubated with bone marrow mesenchymal stromal cells during different differentiation days. As expected, miR-181a and miR-21 expression increased while miR-31 expression decreased as adipogenesis proceeded.
2.4.4. Polymer core-based
Wiraja et al. employed PLGA nanoparticles encapsulating MBs for mRNA detection,[112] using these constructs for studying the dependence of β-actin mRNA in MSCs on culture condition (2D vs. 3D culture).[112] Furthermore, chitosan has been used as a model for a positively charged polymer core for MB delivery.[101] Zhu et al. electrostatically complexed MBs with the chitosan nanoparticles, and used them for detection of MiR-155 in A549, SPC-A1 and PC-3 cells.
Linear FRET probes have been delivered using cationic shell-crosslinked knedel-like nanoparticles (cSCKs).[205] 2 PNA recognition probes labeled respectively with a donor and acceptor fluorophore are each hybridized to short DNA strands to allow for electrostatic complexation with cSCKs. Upon endocytosis, it is believed that cSCKs disrupt the endosomal membrane and release probes into the cytoplasm. Target binding displaces the short DNA complement on both strands. In turn, binding of the fluorophore-labeled recognition strands to adjacent regions in an mRNA of interest can be monitored through FRET signal. A gene associated with inflammation, inducible nitric oxide synthase (iNOS) mRNA, was studied in RAW 264.7 cells.
2.4.5. Micelle-based
Chen et al. have developed MB micelle flares (MBMFs).[90] MBs conjugated to diacyllipids self-assemble into micellular structures with a diacyllipid core and MB shell. Advantages of such a construct include ease of synthesis, biocompatibility, enhanced resistance to nuclease degradation, and uptake into cells without the need of transfection reagents. MBMFs designed to target a known oncogene c-Raf-1 were tested in A549 cells (lung cancer) and HBE135 cells (healthy lung cells). Indeed, MBMFs fluoresced much more strongly in A549 cells than in HBE135 cells. As a negative control, it was shown that non-targeting MBMFs do not fluoresce in A549 cells, indicating the probe was also specific.
Zhang et al. adopted the same structure for use as a theranostic platform.[206] This time doxorubicin, a known cancer drug, was incorporated into the MB stem and in the micelle’s hydrophobic core. Indeed, the constructs proved useful for differentiating cells based on MDR1 mRNA expression, a gene associated with multidrug resistance. Furthermore, their constructs proved useful for knocking down MDR1 through an antisense mechanism. Lastly, release of doxorubicin from the core resulted in high chemotherapeutic efficiency of the theranostic agent in OVCAR8/ADR cells.
2.4.6. Silica core-based
Li et al. delivered probes targeting miR-21 into live MCF-7 cells using silica nanoparticles doped with Ru(bpy)32+ for their strong photostability and biocompatbility.[91] The recognition strand, modified with 2’-O-methyl, contains a carboxyl group for functionalization to amines on the silica nanoparticle surface. The recognition strands also contains a disulfide group and a fluorophore (FAM). A short strand with a quencher is hybridized to the recognition strand, thereby turning off the fluorescence of FAM. An aptamer (AS1411) conjugated to the nanoparticle surface allows for cell specific delivery, whereby the fluorescence of Ru(bpy)32+ is used as a handle for cellular uptake. In the cell, it is claimed that glutathione and acidic conditions cleave the disulfide bond and release the duplexed strand. The target mRNA then binds the recognition strand, releasing the quencher strand and recovering the fluorescence.
Yang et al. have recently developed a novel transfection reagent-free silica@polydopamine-DNA-CeO2 system for simultaneous detection of mi-RNA and H2O2.[102] First, Quasar 670-labeled recognition strands are quenched by PDA due to their adsorption on the silica@PDA surface. Duplex formation upon target hybridization releases the strand, leading to fluorescence recovery. Second, CeO2 nanoparticles adsorbed with FAM-labeled DNA are also decorated around the silica@PDA surface. In a previous study, it has been shown that H2O2 can displace DNA from the CeO2 surface, therefore leading to recovery of initially quenched fluorescence by the CeO2.[207] Taken as a whole, this system could study the interplay between miR-21 and H2O2. For instance, it was found that elevating levels of H2O2 in H9C2 cells led to an increase in expression of miR-21.
Shen et al. reported the use of reverse-transcription helicase-dependent isothermal amplification in conjunction with mesoporous silica nanoparticles (MSNs) for mRNA imaging.[208] As opposed to most strategies which are based on signal amplification, this strategy was based on target amplification, with the authors claiming a detection limit of 4 amol in 40 uL of solution. Constructs were taken up through endocytosis, and the probe was effective in distinguishing A549 (cancerous) and CCC-HPF-1 (healthy) cells based on their relative expression of survivin.
Wang et al. used an HCR-based strategy with MSNs.[209] 2 hairpins able to participate in HCR are electrostatically associated with the MSN surface. One of the hairpins is synthesized with a fluorophore/quencher, such that its opening leads to signal. The authors found a detection limit of 1 pm in solution. Substantiating its efficacy in cells, the probe targeting miR-21 in MCF-7, HeLa and HepG2 cells showed fluorescence bright spots, but showed negligible fluorescence in healthy L-02 cells as expected.
2.4.7. MnO2 nanosheet-based
Probes using manganese dioxide (MnO2) nanosheets have also been constructed. MnO2 offers key advantages, including low cost and ease of preparation, quenching ability, biodegradability, biocompatibility, and efficient delivery of cargo into cells.[103,210,211]
Yang et al. physically adsorbed MBs to the surface of manganese dioxide nanosheets.[103] As mentioned above, manganese dioxide itself also quenches fluorescence, thereby lowering background fluorescence from the case of a free MB alone. Target binding opens the hairpin and releases the probe from the surface, resulting in fluorescence turn-on. Importantly, probes were taken up by cells without the need of transfection reagents. The authors used the system to successfully image miR-21 in HeLa cells.
Li et al. designed a MnO2-based system that incorporated a signal amplification element.[212] Two different fluorophore-labeled hairpins are adsorbed to the MnO2 surface such that they are released upon intracellular degradation of MnO2 in the presence of glutathione. Target binding to one of the hairpins initiates HCR that brings together a FRET pair for signal readout. This strategy proved viable for detecting low abundance miR-21 in HeLa cells. Ou et al. recently used the same strategy, but incorporated two donor fluorophores into one of the DNA strands and an acceptor fluorophore in the other DNA strand.[213] In particular, the two donor/one acceptor (DD-A) system had higher FRET efficiency than the one donor/one acceptor (D-A) system. miR-21 was studied in MCF-7, HepG2, and L02 cells.
2.4.8. Silver nanocluster-based
Silver nanoclusters have also recently found use in live cell RNA detection. Representing a new class of fluorophores, silver nanoclusters are of interest due to their good photostability and low cytotoxicity.[129,214–216] Strategies employing silver nanoclusters take advantage of the high sensitivity of their fluorescence to ONT surroundings. Yeh et al. have reported silver nanoclusters that transition from a dark state to a red emitting state when in close proximity to guanine bases.[129] In this strategy, silver nanoclusters are formed around a 12 bp nucleation strand. Extending off the nucleation strand is a 30 bp recognition sequence that targets a G-rich complement which upon binding turns on fluorescence. However, this original strategy was not applied to live cells.
More recently, Li et al. have extended silver nanoclusters to use in live cells.[214] Again, silver nanoclusters are formed around a nucleic acid nucleation site. However, in this case, hybridization of target to a recognition site close to the silver nanoclusters leads to both fluorescence turn-on and a shift in the silver nanocluster excitation/emission. This “spectrum switching” strategy was used to image TK1 mRNA in HeLa cells, whereby a mutated (scramble) probe was used as a negative control.
2.4.9. Carbon nanomaterial-based
Dong et al. have employed graphene nanoribbons, functionalized with the cationic polymer polyethylenimine (PEI), as a delivery vector for LNA-modified MBs.[217] In particular, a fluorophore/quencher-labeled MB is electrostatically loaded onto PEI-grafted graphene nanoribbons, which is able to enter cells and detect miR-21 in HeLa cells. Because PEI is considered cytotoxic, subsequent strategies have aimed to achieve uptake without such agents.[104]
In this light, graphene oxide (GO) has found use in biosensors due to its ability to quench fluorescence, protect cargo against nuclease degradation, deliver cargo into cells without transfection reagents, and strong affinity for hydrophobic molecules and aromatic rings.[104,218]
For instance, Ryoo et al. have created a biosensor relying on the binding of a fluorophore-labeled peptide nucleic acid with nano-graphene oxide.[104] When bound, the fluorescence of the fluorophore is quenched by the GO. Binding to its complement results in the formation of a duplex and desorption of the probe from the surface, leading to fluorescence turn-on. In their work, Ryoo et al. claimed to reach a detection limit of 1 pm in buffer. The authors could detect miR-21 in live HeLa, MCF-7 cells, and MDA-MB-231 cells; they could also detect miR-29a in Hela cells. In MDA-MB-231 cells, they were able to achieve multiplexed detection of miR-21, miR-125b, and miR-96 using probes with three distinct fluorophores. Pan et al. have recently used a similar strategy for simultaneous detection of mRNA and miRNA.[219] Two different dye-labeled hairpins are adsorbed to the GO surface, complementary for miR-21 and PDCD4 mRNA. The probe was successfully used to image these targets in MCF-7 cells. Lastly, Li et al. have adsorbed hairpins on the GO surface that can participate in an HCR for amplified signal.[220] Two of the hairpins are specific for miR-21 and two other hairpins are specific for miR-let-7a, allowing for multiplexed detection in MCF-7 cells and MCF-10A cells.
A similar strategy based on GO sheets has been proposed by Lu et al.[127] MBs are adsorbed onto the surface of the sheets. The fluorophore on the MB is quenched by both a quencher moiety on the beacon and by the GO sheet. After target binding, the MB is opened and desorbed from the surface, leading to fluorescence turn-on. This strategy was successfully employed to detect survivin mRNA in HeLa cells.
This “double quenching strategy” has also been employed by Yang et al. by using a MB labeled with Cy5 on both ends adsorbed to a GO surface.[221] Quenching from two sources is achieved— self-quenching of the Cy5 molecules and quenching from the GO surface. As such, a low background signal is achieved. Upon opening of the beacon, both Cy5 molecules are turned on, thus resulting in an enhanced signal. Compared to the same system with only one Cy5 molecule, this system had a reported detection limit one order of magnitude lower (30 pm). miR-21 was imaged successfully in MCF-7, HeLa, and HepG2 cells. Furthermore, the system was successfully used for in vivo imaging in three distinct mouse xenograft tumor models.
Li et al. have recently used dye-labeled circular DNA in conjunction with GO for mRNA detection.[222] Release of the circular DNA from the surface after target binding unquenches the dye, leading to signal readout for detection. This strategy was successful in multiplexed detection of c-raf and survivin mRNA in HeLa cells. Compared to ssDNA GO system, circular DNA GO had lower false positive signal, presumably due to the enhanced nuclease resistance of circular DNA compared to ssDNA.
As opposed to physical adsorption of probe sequences onto GO, Yu et al. have devised a different strategy for probe association with GO.[223] In particular, they propose a graft/base pairing strategy, whereby amino-labeled strands pre-duplexed with dye-labeled recognition sequences are covalently attached to the GO surface. In the fluorescence off state, dye-labeled strands are flexible and thus near the GO surface, leading to fluorescence quenching. The formation of a duplex in the recognition region due to target binding rigidifies the strand, leading to separation of fluorophore and surface, giving turn-on of fluorescence. Compared to physical adsorption, which may be prone to probe displacement due to species in cell culture medium or in the cell, chemical conjugation is much more stable. The feasibility of the strategy was tested and confirmed in MCF-7 cells by simultaneous detection of three different miRNA targets— miR-21, miR-let-7a, and miR-125b.
Other carbon nanostructures with strong quenching capabilities have also been used in biosensing applications. For example, Liao et al. have taken advantage of the quenching ability of carbon nanospheres.[224] Initially, a dye-labeled strand is adsorbed on the carbon nanosphere surface. Desorption of the sequence after target binding results in recovery of fluorescence. miR-18a was detected in HepG2 cells using this strategy. In particular, the sensor was used to look at the levels of miR-18a at the S, G2/M, and G1 phases of the cell cycle. Ke et al. have designed a similar system based on polypyrrole nanoparticles.[225] In their system, hairpin DNA labeled with a fluorophore non-covalently associates with the polypyrrole surface, whereby fluorescence is quenched. Upon binding of mRNA target, the hairpin is opened and released, leading to fluorescence turn-on. This polypyrrole based system was used to differentiate cancer cells (MCF-7) from healthy cells (MCF-10A) based on detection of the oncogene c-Myc. Multiplexed detection was also shown by adding a second hairpin to the surface specific for the oncogene TK1, consequently detecting both c-Myc and TK1 simultaneously. One advantage of polypyrrole nanoparticles is their ability to absorb light in the near-IR regime. As such, this paper also showed the utility of polypyrrole nanoparticles to act as agents in photothermal therapy, paving the way for a potential theranostic platform.
Piao et al. have synthesized MBs that use graphite nanoparticles as the quenching moiety.[226] Compared to a conventional dabcyl quencher, using a graphite nanoparticle resulted in an ~11% improvement in quenching efficiency. They also entered cells without transfection reagents. These MBs were used to detect survivin mRNA in MCF-7 cells.
Wu et al. adsorbed MBs to the surface of single-walled carbon nanotubes.[227] The designed constructs were used to detect MnSOD mRNA in MDA-MB-231 cells. Importantly, the use of a carbon nanotube as the delivery vehicle yielded advantageous properties as compared to naked MB delivery, such as an enhanced resistance to enzymatic degradation and transfection reagent free uptake into cells. Moreover, the authors noted that association with the carbon nanotube allows the MB to remain in the cytoplasm, rather than being sequestered to the nucleus.
Noh et al. took advantage of the fluorescence of carbon nanodots to design a system for detecting miRNA.[92] In an SNA type strategy, carbon nanodots were functionalized with a recognition strand prehybridized to a short quencher-labeled strand. When hybridized, the quencher turned off the fluorescence of the carbon nanodot. Upon miRNA induced displacement of the quencher strand, fluorescence was recovered. The feasibility of this strategy in live P19 cells was confirmed by detecting miR-124a.
2.4.10. MOF-based
Metal organic framework (MOF) nanoparticles offer a number of interesting properties, including ease of synthesis, quenching ability, and tailorable chemistry.[228–231]
Wu et al. leveraged the properties of nano-MOFs for detection of miRNA in live cells.[105] A dye-labeled PNA is conjugated to a MOF, whereby metal centers in the MOF function to quench the fluorescence of the dye. Target binding to the PNA results in its release from the MOF and subsequent recovery of fluorescence. In solution, this sensor could detect 10 pm miRNA. The probe was shown to be sensitive to changes in miR-21 level in live MCF-7 cells after addition of an inhibitor. Multiplexed analysis of miR-21, miR-96, and miR-125b was also possible in MDA-MB-231 using three different PNA-MOF complexes.
2.4.11. Plasmonic coupling-based
Although fluorescence-based strategies are most popular for intracellular analyte detection, strategies based on plasmonic coupling have also been developed. For example, Zhou et al used SERS to detect miRNAs of interest.[99] One sequence is conjugated to one gold nanoparticle, and another sequence is conjugated to a second gold nanoparticle. The miRNA of interest hybridizes partially to both sequences, thus bringing the two gold nanoparticles close together. The junctions between the gold nanoparticles form “hot spots,” which can be monitored by SERS if Raman reporters are loaded on the gold nanoparticle surface. In this paper, an alkyne (C≡C) terminated Raman reporter was used for one target of interest, and a nitrile (C≡N) terminated Raman reporter was used for another target of interest. With this ability to multiplex, miR-21 and miR-155 were simultaneously monitored in MCF-7 cells. One advantage of a SERS-based strategy over fluorescence-based methods is potentially greater capability for multiplexing due to sharp spectrally distinct Raman bands. A drawback of a SERS approach is that the equipment required is typically more expensive than that needed for fluorescence-based measurements.[232]
In a similar strategy, Lee et al conjugated a gold nanoparticle with a sequence complementary to an mRNA of interest, while another gold nanoparticle was conjugated with a different sequence complementary to the same mRNA of interest.[128] The sequences are designed such that if the target mRNA is present, the two sequences will bind the mRNA in a manner that will bring the gold nanoparticles in proximity for dimer formation. Due to plasmonic coupling, the formed dimer has a spectral shift that can be detected. In their work, Lee et al. used this system to detect multiple splice variants of BRCA1 mRNA at single-copy number resolution. Because different splice variants resulted in different scattering spectra, the group could effectively distinguish one splice variant from another.
2.4.12. Iron oxide core-based
Lin et al. have pioneered the use of Fe3O4@polydopamine core-shell nanocomposites (Fe3O4@PDA NCs) for use as a theranostic platform.[106] The PDA shell surrounding the Fe3O4 core can adsorb single-stranded nucleic acids that contain a dye and are designed to be complementary to an mRNA target of interest. After mRNA binding to the probe sequence, a duplex is formed that is desorbed from the PDA, resulting in turn on of fluorescence. Lin et al. showed the ability of this platform to differentiate healthy breast cells (MCF-10A) from cancerous breast cells (MCF-7) based on their relative expression of a known oncogene c-Myc. Displaying the versatility of the strategy, they were also able to adsorb multiple probe sequences on the surface with distinct dyes, allowing for simultaneous multiplexed detection of two oncogenes — c-Myc and TK1. These Fe3O4@PDA NCs were also efficacious for photothermal therapy due to PDA’s ability to absorb near IR light and have also shown promise as an MRI contrast agent for imaging cancer cells.
2.4.13. DNA nanostructure-based
Probes based on nanostructures formed from DNA, like the DNA tetrahedron, are of interest in sensing applications because of their biocompatibility, uptake into cells without external agents, and ability to protect cargo from degradation.[113,233]
For instance, a design variation to linear FRET probes was introduced by the Tan group in 2017 with their advent of DNA tetrahedron Nanotweezers (DTNTs) for live cell mRNA detection (Figure 9).[113] DTNTs rely on the self-assembly of four different nucleic acid strands. In the off state, the self-assembly of the four nucleic acid strands keeps a donor and acceptor fluorophore separated from one another. mRNA binding to the DTNT induces a structural change that brings the donor and acceptor fluorophores close to one another, resulting in generation of a FRET signal. Beyond the fact that DTNTs were not prone to false positive signal, they also entered cells without the need of transfection reagents, unlike “naked” nucleic acids. In their initial study, a DTNT was designed targeting TK1 mRNA, which is known to be a marker for tumor growth. Indeed, DTNTs were able to distinguish cancer cells from healthy cells based on the level of TK1 mRNA.
Figure 9.
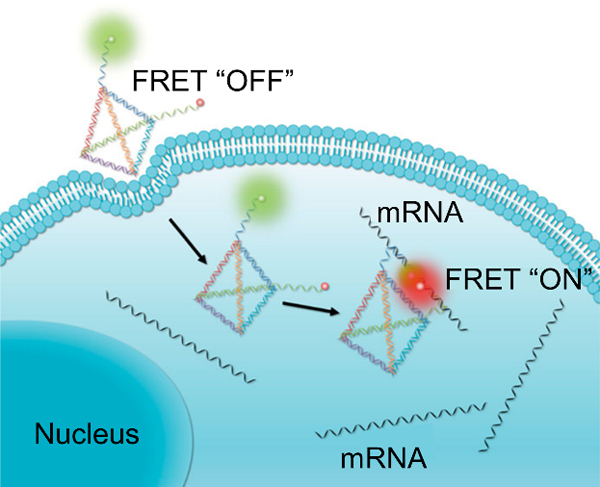
DNA nanotweezers for intracellular oncogenic mRNA detection. Target binding induces a structural change that brings a FRET pair together, resulting in a FRET signal. Adapted with permission.[113] Copyright 2017, American Chemical Society.
Xie et al., on the other hand, described a DNA tetrahedron that incorporated a flurophore and quencher at the end of one of the strands making up the structure, thus putting the two moieties in close proximity when the tetrahedron is in the intact form.[234] Target binding induces a structural change that separates the fluorophore and quencher, turning on fluorescence. The nanosensor had the ability to detect TK1 mRNA in HepG2 cells.
Zhou et al. have expanded on these “tetrahedron” strategies for multiplexed detection of miRNA.[114] Hairpin DNA specific for two different miRNA are incorporated into two of the strands, while a fluorophore and quencher label the termini of two other strands. In the closed state, the intact structure causes the fluorophore and quencher to be near one another. Hairpin opening due to target binding then allows for recovery of fluorescence. Their strategy proved efficacious for imaging miR-155 and miR-21 simultaneously in MDA-MB-231 cells. Wang et al. expanded capabilities to simultaneous detection of three different mRNA targets.[235] c-Myc, TK1 and GalNAc-T mRNA were simultaneously studied in MCF-7 and MCF-10a cells.
Recently, the Tan group has introduced an amplification strategy, entropy-driven 3D DNA amplifier (EDTD), to boost signal from fluorophore/quencher DNA tetrahedron sensors.[236] All properties of tetrahedron probes are retained, including transfection-free cellular uptake and resistance to nuclease degradation. The EDTD probe was used to image TK1 mRNA in both normal and tamofixen (for TK1 knockdown)-treated cells. Conventional DNA tetrahedron molecular beacons (DTMBs) only produced negligible fluorescence in tamofixen treated cells, implying that EDTD tetrahedrons could be used to image mRNAs of low abundance but DTMBs could not.
Tay et al. took inspiration from nature and conventional DNA tetrahedron approaches to design a MB with enhanced resistance to nuclease degradation.[119] They termed their system the nano-snail-inspired nucleic acid locator (nano-SNEL). The first component of their system is a MB that acts as the sensing element. The second component of their system is a DNA tetrahedron. Conjugating the MB to the DNA tetrahedron conferred resistance to nuclease degradation, akin to a snail’s shell affording it protection from environmental conditions. As opposed to naked MBs which do not enter cells, it was also shown that the presence of the DNA nanoshell results in these constructs being taken up by cells without the need of transfection reagents, allowing for detection of GAPDH in live DLD-1 and SW480 cells.
In another recently reported DNA nanostructure system, Ren et al. have developed an amplification strategy that overcomes limitations associated with HCR.[130] In particular, the rate of HCR is reliant on diffusion as each successive hybridizing DNA sequence must be found in a complex 3D milieu. Reactions have been reported to take as long as two hours to go to completion.[108] Ren et al. hypothesized that confining nucleic acids in close proximity to one another would accelerate the kinetics of such DNA chain reactions (DCR). In their design, two different hairpins are alternately hybridized in close proximity along a DNA nanowire. Akin to normal HCR, target binding opens the fluorophore/quencher-labeled hairpin, leading to fluorescence turn-on and a cascade of hybridization between the two hairpins to yield amplified signal. Compared to conventional HCR, the authors noted that this system had a limit of detection ~52 times lower in buffer experiments and required only 15 minutes of incubation time with complementary target. This system was ultimately used to image survivin mRNA in HeLa cells.
2.4.14. Quantum dot-based
Quantum dots (QDs) represent another class of nanoparticles used for live cell RNA detection. Advantages include good quantum yield, size dependent emissive properties, and high photostability.[237,238]
For instance, Dai et al. adsorbed MBs on the surface of molybdenum disulfide quantum dots (MDQDs).[107] MDQDs fluoresce in the blue regime, and can thus be used as a handle for cellular uptake. Transfection reagent-free uptake was achieved and used for imaging of miR-21 in HeLa and HaCaT cells.
Lee et al. employed a slightly different strategy.[93] QDs were covalently conjugated with a cell penetrating peptide for enhanced cellular uptake and a recognition strand prehybridized with a short quencher-labeled sequence. The quencher-labeled sequence, when hybridized, turns off the inherent fluorescence of the QDs. Target binding displaces the quencher sequence, recovering the fluorescence of the QD. Using this design, miR124-a was studied in P19 cells during differentiation.
Dong et al. used graphene QDs (GQDs) covalently conjugated with polyethylene glycol (PEG) and polylactic acid (PLA) as a nanosensor for miRNA detection.[239] PEG and PLA impart stability to the GQD in both aqueous and protein solution. As such, the bioluminescence properties of GQDs are stable over a wide range of pHs. MBs are associated with the GQD surface through π-π interactions. MB opening and release from the surface upon hybridization with target leads to fluorescence turn-on. Importantly, because GQD are photoluminescent themselves, their photoluminescence level in cells can be used as a measure of nanosensor uptake. Intracellularly, the probe was capable of imaging miR-21 in HeLa cells. The probe was also sensitive to downregulation of miR-21, resulting in less nanosensor fluorescence.
Wu et al. designed a two acceptor-one donor theranostic MB based on CdTe/CdS/ZnS QDs.[240] In the closed state, the QD acts as the donor and a BHQ moiety acts as the acceptor. Hairpin opening results in recovery of the QD fluorescence. However, opening of the hairpin also brings the donor QD into close proximity with an acceptor photosensitizer molecule that can produce singlet oxygen to promote cell death. Using MCF-7 cells, which express high level of cyclin D1 mRNA, the study was able to show that the theranostic was efficacious in both detecting the mRNA and inducing cell death.
Lastly, He et al. achieved miRNA detection using gold nanoparticle-QD nano-assemblies.[241] A nucleic acid strand modified with a CdTe QD is prehybridized to a recognition strand that in turn places the QD near the gold nanoparticle. Target binding displaces the QD modified strand, recovering fluorescence. An amplification element was also introduced by incorporating a fuel strand that displaces the target, therefore allowing the target to bind to multiple recognition strands. Shen et al. have used the same strategy, but have made their sensor start functioning “on demand” by pre-functionalizing the entire recognition strand with DNA so as to not allow target binding initially.[242] One of the strands functionalized to the recognition strand contains a photocleavable linker that is cleaved after UV radiation, thus partially exposing the recognition strand and yielding the same system as He et al.
3. Aptamer-based probes
3.1. DNA nanomachine-based
The Krishnan group provided an early demonstration of “DNA nanomachines” that are active in cells.[243] They developed the “I-switch” based on the pH-sensitive i-motif DNA (Figure 10A). The i-motif is a cytosine rich strand that adopts a folded tetraplex conformation at acidic pH held together by C+-H-C linkages and an extended single-stranded conformation at neutral pH. As such, the i-motif can be considered to be an aptamer for proton sensing. The pH ranges sensed by the i-motif can be shifted towards more acidic or basic pH through the incorporation of modified bases or different numbers of cytosine bases.[28,244,245] The I-switch consisted of two DNA duplexes that were linked together through a flexible tether and contained fluorophore-labeled i-motif forming overhangs at both ends (Figure 10A). Under acidic conditions, formation of the i-motif resulted in the “closed” state in which the fluorophores were brought to proximity resulting in a FRET signal. The I-switch could report pH changes in the range of 5.5–6.8. As two fluorophores were used, a ratiometric analysis of uptake and pH change could be performed. Utilizing the I-switch, the authors spatiotemporally mapped the pH changes associated with the endosomal pathway in live cells.
Figure 10.
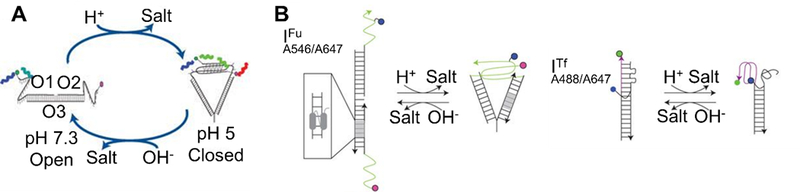
(A) The I –switch nanomachine for monitoring intracellular pH. A structural change from open (low FRET) to closed (high FRET) state occurs as pH is changed from basic to acidic condition. Adapted with permission.[243] Copyright 2009, Springer Nature. (B) By incorporating two sets of FRET pairs as well as two distinct targeting moieties (one on each I-switch), Modi et al. were able to use the I-switch for simultaneous monitoring of the furin retrograde endocytic pathway and the transferrin endocytic/recycling pathway. Adapted with permission.[115] Copyright 2013, Springer Nature.
In a subsequent paper, Krishnan and co-authors demonstrated that pH changes within cellular compartments can be tracked in multicellular organisms.[246,247] In Caenorhabditis elegans worm models, these DNA nanomachines are recognized by scavenger receptors on the surface of scavenger cells called coelomocytes and taken up via receptor-mediated endocytosis. The I-switch has a half-life of ~8 h in cells and reports pH changes over the range 5.3–6.6 by resulting in a five-fold change in FRET signal as it transitions from an open to a closed state.
Modi et al. developed a simultaneous pH mapping technology (SimpHony) using I-switches.[115] They demonstrated that pH changes along two different endocytic pathways (the furin retrograde endocytic pathway and the transferrin endocytic/recycling pathway) can be visualized using two I-switches labeled with two sets of FRET pairs (Figure 10B). A sequence-specific double-stranded DNA binding protein was expressed as a chimera with furin in HeLa cells to allow for monitoring the furin pathway. The I-switch targeting this pathway contained a double-stranded (AT)4 region that could be recognized specifically by the DNA binding protein initiating uptake. The I-switch targeting the transferrin pathway was conjugated to transferrin to allow uptake upon recognition by the transferrin receptor. The use of such DNA nanomachines allowed the sensing of the same analyte in different cellular compartments. Moreover, the modular nature of the I-switch allows custom functionalization with proteins as well as with different FRET pairs. This strategy was later extended for the detection of pH changes associated with the trans-Golgi and cis-Golgi networks, and the endoplasmic reticulum.[244]
Bhatia et al. reported that synthetic DNA constructs can be utilized as hosts to deliver cargo within cells.[52] Fluorescein isothiocyanate-labeled dextran (FD) was encapsulated within a DNA icosahedron. Unlike free FD which cannot be targeted to specific endocytic pathways, encapsulated FD was taken up by the anionic ligand-binding receptor (ALBR) pathway in Drosophila hemocytes, demonstrating that the DNA host altered its targeting properties. Similarly, encapsulated FD was taken up only by ALBR-expressing coelomocytes in Caenorhabditis elegans. The pH-sensitive property of fluorescein enabled monitoring of pH changes associated with endosomal maturation. In a subsequent study, photoresponsive polymers attached to small molecule payloads were loaded into DNA icosahedrons.[248] Light-activated release of the small molecule could be monitored with spatial and temporal precision. Single quantum dots could also be encapsulated within DNA icosahedrons.[249] By modifying the icosahedron with a single endocytic ligand (e.g. folic acid, galectin-3, and the Shiga toxin B-subunit), the endocytic pathway associated with the ligand can be imaged.
In addition to pH, nanomachines that are sensitive to intracellular chloride ion concentrations and that can be targeted to subcellular compartments have also been investigated. Unusual chloride ion concentrations in the lysosome can lead to lysosomal storage disorders. A DNA nanodevice called Clensor can report changes in chloride ion concentrations in a pH-independent manner.[250,251] Clensor consists of three parts: a sensing module containing a 12-base long PNA moiety labeled with a chloride-sensitive dye, 10,10′-bis[3-carboxypropyl]-9,9′-biacridinium dinitrate (BAC), a normalizing module containing a 38-base long Alexa 647-labeled DNA sequence, and a targeting module containing a 26-base long DNA. The probe could be taken up as is through the ALBR pathway due to its negative charge while modification of the targeting moiety with an RNA aptamer that binds to the transferrin receptor enables uptake through the transferrin pathway. Using this probe, chloride ion concentration in the lumen of acidic organelles in Drosophila melanogaster was monitored and it was revealed that the intracellular chloride transporter DmClC-b regulates lysosomal chloride ion concentration.
Recently, combining the advantages of I-switch and Clensor, a probe that can detect pH and chloride ion concentration simultaneously was developed.[117] Using this probe termed as ChloropHore, subpopulations of lysosomes could be chemically resolved based on their chloride and proton content. Importantly, it was shown that lysosomes present in primary skin fibroblasts of healthy individuals could be categorized into two populations, whereas patients with Niemann–Pick disease only had one population. The absent second population could be recovered upon treatment of the disease. Similar strategies have been utilized to develop CalipHluor, a probe for pH-corrected measurement of calcium ions in subcellular compartments[116] and cHOClate, a probe for reporting pH and HOCl simultaneously in phagosomes of macrophages, neutrophils, and monocytes.[118]
One limitation of the early design of I-switch was the relatively long timescale for transforming into the folded state (few minutes). Yang et al. developed the t-switch as a solution to this problem.[252] The t-switch relies on a triplex formation mediated by C+-G-C interactions. Similar to the I-switch, the t-switch reported changes in pH through a FRET-based mechanism, although the response time was only a few seconds. The t-switch was delivered to cells through complexation with the cationic polymer PEI. Compared to the naked t-switch whose dynamic range is pH 5.3 to 6.0, the PEI-t-switch has an extended dynamic range of 4.6 to 7.8. Intracellular pH in HepG2 cells was monitored using this probe.
Pei et al. demonstrated that DNA tetrahedra with dynamic sequences that are responsive to particular targets can be used to develop FRET-based sensors for the target molecules.[253] The authors utilized this construct for sensing ATP in live HeLa cells. Ohtsuka et al. utilized the thrombin binding aptamer (TBA) for detecting K+ in live HeLa cells.[254] The TBA forms a G-quadruplex in the presence of K+ ions. This structural transition was leveraged to create a FRET-based probe. The TBA was biotinylated and conjugated to streptavidin which was further conjugated to biotinylated nuclear export signal peptide (B-NES). The formation of the ternary complex was important as without B-NES the probes were sequestered to the nucleus.
For more information on DNA nanomachine-based probes, the readers are directed to the review by Chakraborty et al.[255]
3.2. Gold nanoparticle-based
In 2009, the Mirkin group introduced aptamer NanoFlares for the detection of intracellular analytes in live cells.[95] Analogous to NanoFlares for mRNA detection, these probes are comprised of a gold nanoparticle core modified with ONT duplexes. One strand of the duplex is the aptamer sequence which is covalently attached to the gold nanoparticle through a gold-sulfur bond. The other strand is a shorter fluorophore-labeled strand whose fluorescence is quenched due to proximity to gold. Upon binding of the target molecule of interest, the aptameric strand folds, the fluorophore-labeled strand is displaced, and its fluorescence is turned on. Using this platform ATP was detected in live HeLa cells. One disadvantage of this strategy is that the binding affinity is reduced by two orders of magnitude compared to the free aptamer in solution, presumably due to partial blocking of the active site of the aptamer by the flare strand[256] and due to steric bulk arising from dense ONT functionalization.
Many groups have used NanoFlares for detecting and measuring other analytes in live cells. For example, by using i-motif forming ONTs (IFOs) labeled with a FRET-pair (rhodamine green and rhodamine red) at the two ends, NanoFlares that measure intracellular pH were developed by Huang et al.[96] Initially, the IFOs are duplexed to cDNA on the gold nanoparticle which results in quenching of the acceptor dye. Lower pH induces a conformational switch such that a quadruplex structure is formed and the two ends of the ONT labeled with dyes are brought to proximity, turning on FRET signal. The presence of two dyes also allows ratiometric analysis of uptake and binding by monitoring the fluorescence ratio of the donor and acceptor. Using this probe, spatial mapping of intracellular pH in HeLa cells, HL-7702 cells, SMMS-7721 cells could be performed. The results revealed that the FRET signal increased with time, indicating that the probes were trafficked from neutral to more acidic environments. Later, using a similar strategy, the group developed aptamer NanoFlares for measuring the concentration of K+ ions by employing ONTs that form a G-quadruplex in the presence of K+ ions.[257]
Wang et al. developed a strategy[258] for plasmonic imaging of intracellular pH gradients in RAW 264.7 cells using two different sizes of gold nanoparticles assembled into core-satellite architectures.[259,260] The core nanoparticle is 50 nm in size and functionalized with G-rich ONT sequences. The satellite nanoparticles are 14 nm in diameter and functionalized with C-rich IFO forming sequences. At higher pH, the nanoparticles are assembled through Watson-Crick base pairing. Under acidic conditions, quadruplex structures are formed from the C-rich i-motifs and the structures are disassembled. The scattering spectra resulting from the assembly (yellow) and disassembly (green) of these structures are different and can be observed using a dark field microscope. As the pH drops along the endocytic pathway from early endosomes to late endosomes/lysosomes, a yellow to green transition in scattering spectra was observed, consistent with disassembly of the nanoparticles at lower pH.
Ma et al. developed a gold nanoparticle-based DNA nanomachine that detects low abundance microRNA through an ATP-fueled amplification strategy.[261] The probe was able to report differences in miR-21 levels between HeLa, HEK-293T, and MRC-5 cell lines.
3.3. Graphene oxide-based
Wang et al. demonstrated that aptamer/GO nanocomposites can be used as probes for monitoring ATP levels in real time in live cells.[109] In this study, a carboxyfluorescein-labeled ATP aptamer was adsorbed onto graphene oxide nanosheets (GO-nS) through π-π stacking interactions. The GO-nS quenched the fluorescence owing to FRET, acted as a transfection vector for the delivery of the ONTs into JB6 cells, and protected the ONT from nuclease degradation. Inside the cells, ATP binding released the aptamer from the GO-nS and turned on fluorescence. This platform was later extended for simultaneous detection of ATP and GTP in MCF-7 cells using two aptamers labeled with spectrally distinct fluorophores.[262] A detailed protocol for this process was later published by Wang et. al.[263] Using a similar strategy, Chen et al demonstrated the first visualization of translocation event of endogenous cytochrome c, a protein mediator of apoptosis, in HeLa cells.[264]
By incorporating a fluorophore-labeled single-stranded DNA (ssDNA) as an internal reference, Tan et al. showed semiquantitative detection of ATP in HeLa cells.[141] The ssDNA is released by non-specific interactions with intracellular proteins and by monitoring its ratio of fluorescence intensity to that of the aptamer MB, the amount of ATP can be determined semiquantitatively.
Intracellular pH has also been probed using DNA-GO-nS complexes. Li et al. engineered fluorophore-labeled ONT probes that transition from a duplex at pH 8 to a triplex at pH 5. The duplex form consists of an ssDNA overhang labeled with rhodamine green. Due to stronger adsorption of ssDNA compared to triplex DNA, the fluorescence is quenched at higher pH. However, acidification induces a conformation shift and causes desorption of the triplex probe resulting in fluorescence turn on. This probe has been used to study apoptosis-induced acidification in Ramos cells.[265]
Shao et al. used IFOs as pH sensors.[111] The IFOs were immobilized on GO through interactions with amine-modified complementary DNA (cDNA). The cDNA was covalently attached to GO through NHS/EDC chemistry. A covalent attachment strategy was used as opposed to the more commonly used physisorption strategy to prevent non-specific release. However, in their study they observed that the interaction of DNA with GO under acidic conditions is too strong to distinguish between open and closed forms. To weaken the interactions, they treated GO with herring sperm DNA. This new probe was capable of detection of pH between the range of 5 and 7.3 and was used to demonstrate pH changes in live HeLa, MCF-7, MDA-MB-231, and A549c cells.
3.4. Upconverting nanoparticle (UCNP)-based
Ma et al. reported lanthanide-doped UCNPs for the detection of cytochrome c.[266] Fluorophore-labeled cytochrome c aptamers are hybridized to cDNA attached to polydopamine-coated UCNPs. The fluorescence is quenched by polydopamine in the absence of cytochrome c and activated when cytochrome c binds to the aptamer strand and displaces it from the surface of the UCNPs. The upconverted luminescence of the nanoparticles serves as an internal reference for probe uptake. The study claimed a detection limit of 20 nm and a dynamic range spanning 50 nm to 10 μm, which includes physiologically relevant concentrations (1–10 μm) of cytosolic cytochrome c found in apoptotic cells. Etoposide induced apoptosis was studied in HepG2 cells using these probes. Low quantum efficiency of the UCNPs and poor DNA loading are two limitations of this strategy.
Zhao et al. developed an NIR-activated probe for imaging ATP in live cells.[267] Their construct was composed of lanthanide-doped UCNPs decorated with photo-cleavable aptamer-based DNA sensors. A fluorophore-labeled aptamer sequence was hybridized to cDNA containing a quencher and a photocleavable group. In the absence of NIR light, the fluorescence was quenched due to FRET and the cDNA prevented ATP-binding. Upon irradiation with 980 nm NIR light, the UCNPs luminesced in the UV and visible blue (peaks at 320, 365, 450, and 475 nm) regime, inducing photolysis of the cDNA into two short fragments. The reduced binding affinity of the generated fragments leads to strand displacement in the presence of ATP. Due to separation of fluorophore and quencher, a fluorescence signal is generated. Using this platform, the authors demonstrated ATP imaging in live HeLa cells as well as in mice bearing HeLa xenograft tumors. The advantage of such a system in which the sensor can be remotely activated by external stimulus is that it adds temporal control to imaging capabilities.
3.5. Polymer core nanoparticle-based
Özalp et al. developed an ATP aptamer that can detect intracellular ATP concentrations in the range between 0.5–8 mm.[61,268,269] The aptamer was converted into a fluorescent sensor using the “aptamer switch probe” strategy developed by Tang et al.[270] An aptamer switch probe consists of three parts: (i) the aptamer sequence, (ii) a short cDNA sequence, and a (iii) PEG linker connecting (i) and (ii). Moreover, the two ends contain a fluorophore and quencher, respectively. Analogous to a MB, in the absence of the target, the probe exists in a hairpin-like conformation that brings the fluorophore-quencher pair in proximity (Figure 11). ATP binding changes the conformation of the probe such that the fluorophore and quencher are separated. The aptamer was encapsulated into polyacrylamide nanparticles for cellular delivery. The porous nanoparticles allow small molecules like ATP to diffuse inside, but prevents nuclease degradation. Real time variation in ATP levels in yeast cells could be monitored using these probes.
Figure 11.
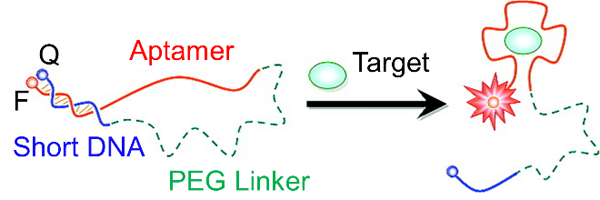
Target analyte detection based on an aptamer switch probe strategy. Target binding to the aptamer induces a structural change that separates a fluorophore and quencher, leading to fluorescence turn-on. Adapted with permission.[270] Copyright 2008, American Chemical Society.
Qiang et al. used polydopamine nanospheres as cores for delivery of ATP-sensing aptamers in cells.[271] Fluorescently-tagged ATP aptamers were adsorbed onto the nanoparticle surface such that the fluorescence was quenched by the core. In the intracellular environment, ATP binding desorbed the strand from the surface, leading to separation of fluorophore and quencher with concomitant increase in fluorescent signal. ATP was sensed in live HeLa cells using this probe.
3.6. Micelle-based
Wu et al. developed switchable aptamer micelle flares for the detection of ATP in live cells.[272] The probe consists of aptamer switch probe–diacyllipid chimeras that self-assemble to form micelles. Target binding causes the conformation of the aptamer to change, separating the fluorophore and the quencher, leading to fluorescence turn on. The fluorescence increases with increasing concentration of ATP (between 0.1–3 mm) in vitro and saturates at ~4-fold enhancement. These probes could be internalized into cells without transfection reagents and colocalization experiments suggested that the majority of the probes are in the cytoplasm as opposed to being in the lysosome. Based on these results, the authors suggested that the probes enter cells mainly via membrane fusion.
3.7. MnO2 nanosheet-based
Zhao developed a bimodal platform for detecting cancer cells based on aptamer-loaded MnO2 nanosheets.[273] MnO2 nanosheets served as a nanocarrier, quencher as well as an MRI contrast agent. The aptamer sgc8 which targets the protein tyrosine kinase 7 (PTK7) was fluorescently tagged and associated with MnO2 nanosheets. Cells expressing PTK7 recognized the aptamer which then dissociated from the MnO2 nanosheets, turning on fluorescence. Upon cellular entry of MnO2 nanosheets, glutathione induced reduction led to the formation of large amounts of Mn2+ which could provide strong MRI contrast.
3.8. MOF-based
Deng et al. developed an upconverting nanoparticle core-MOF-shell based probe for targeted drug delivery and imaging in cancer cells.[274] The MOF surface was functionalized with the AS1411 aptamer that is known to target cancer cells. Following cellular entry, the MOF could release an encapsulated anti-cancer drug. The luminescent UCNP allowed for cellular imaging.
Liu et al. developed an aptamer conjugated MOF-based platform for cellular imaging and photodynamic therapy.[275] The PTK7-targeting aptamer sgc8 was fluorescently tagged and conjugated to the surface of a Zr-based porphyrinic MOF. The MOF acted both as a quencher as well as a photosensitizer for generating reactive oxygen species. Upon binding to PTK7-positive HeLa cells, the fluorescence from the aptamer was turned on allowing cellular imaging. Irradiation with 650 nm induced significant cell death. Thus, this platform acted as a theranostic for cancer.
3.9. Genetically-encoded aptamers
The Jaffrey lab pioneered the development of genetically-encoded aptamers for visualizing intracellular analytes in live cells.[121] Such aptamers are particularly advantageous for tracking of small molecules and metabolites in cells, imaging strategies for which are limited.
Paige et al. developed a 97-nt long aptamer that binds to 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI), a dye resembling the fluorophore in green fluorescent protein (GFP).[122] The photophysical properties of this dye are similar to those of enhanced GFP. The free dye is non-fluorescent whereas upon aptamer binding the dye strongly fluoresces. Due to the characteristic green florescence, the aptamer has been termed “Spinach”. As a first demonstration of the capability of Spinach aptamers in detecting mRNA in live mammalian cells, the authors fused Spinach to the 3′ end of the noncoding RNA 5S, transfected the entity into HEK293T cells, and incubated the cells with DFHBI. The fluorescence distribution from Spinach was similar to that of endogenous 5S, signifying that incorporation of Spinach did not significantly alter the dynamics of the target RNA. In addition to DFHBI, the Jaffrey lab developed a palette of dyes with fluorescence spanning the visible spectrum.[121,276]
Inspired by a strategy proposed by Stojanovic et al.,[277] Paige et al. created modular aptameric sensors for monitoring cellular metabolites (Figure 12). The probe consisted of three parts: (i) Spinach aptamer, (ii) a recognition module, and (iii) a transducer module.[122] When the target binds to the recognition module, the transducer region is duplexed which then leads to DFHBI binding to Spinach resulting in concomitant increase in fluorescence signal. Large fluorescence enhancements up to 32-fold were achieved in solution. Using this strategy, the authors imaged adenosine 5-diphosphate (ADP) and S-adenosylmethionine (SAM) in live E. coli.[122,278] In later studies, Song et al. demonstrated that such a method could be adapted for the imaging of proteins in live bacteria cells.[123] The authors visualized streptavidin as well as the synthesis of MS2 coat protein after infection with MS2 phages in E. coli cells.
Figure 12.
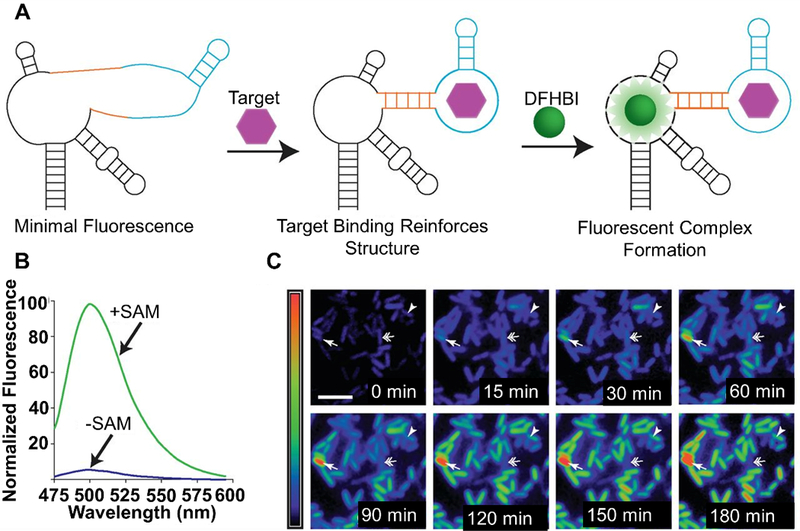
(A) Spinach aptamer composed of Spinach (black), transducer (orange), and aptamer (light blue) for detection of target analytes. Target binding to the aptamer induces a structural change that results in transducer duplexing and concomitant binding of DFHBI to Spinach to give fluorescence turn-on. (B) Fluorescence turn-on of Spinach aptamer in the presence of target SAM. (C) Monitoring of SAM in E. coli cells. Single arrows point to cells with relatively high SAM expression, arrowheads point to cells with increasing levels of SAM over time, while double-arrows point to cells whose SAM content first increases and then decreases over time.[122] Modified with permission from AAAS and authors.
Kellenberger et al. fused the Spinach aptamer to the naturally occurring GEMM-I riboswitch for intracellular detection of cyclic dinucleotides, c-Di-GMP and c-AMP-GMP.[279] Furthermore, enzymatic activity of DncV which catalyzes the synthesis of c-AMP-GMP was monitored. The Jaffrey lab further demonstrated that Spinach-based riboswitches can be utilized for visualizing several metabolites including 5′-pyrophosphate, S-adenosyl-methionine, adenine, and guanine.[280,281] Pothoukalis et al. showed that Spinach aptamer can be used in conjunction with a fluorescent protein to simultaneously monitor both transcription and translation.[282] Ong et al. developed an unstable form of Spinach flanked with bases complementary to an mRNA sequence on either side such that the folded form of Spinach is formed upon binding to a target mRNA. This folded structure can then bind DFHBI and turn on fluorescence.[283]
Despite the strong brightness of Spinach in vitro, in cells, the brightness is significantly reduced due to improper folding of the aptamer. Therefore, ~1 s exposure time is necessary for imaging compared to 10–100 ms exposure times used in imaging GFP-labeled proteins under the same conditions. To address this limitation, Strack et al. developed Spinach2 through systematic mutagenesis of Spinach and demonstrated that it is 2–3-fold brighter than Spinach in both E. coli and HEK293T cells.[284] Using Spinach2, the dynamics and localization of CGG repeat-containing toxic RNAs were investigated.[284,285] Zhang et al. demonstrated by using multiple Spinach aptamers (8–64 repeats), mRNA molecules can be imaged with ~17-fold increased brightness in cells.[286] Importantly, such fusion did not adversely impact mRNA function. Structural minimization of Spinach led to the development of Baby Spinach which retains the functionality of Spinach but consists of 51 nt,[287] which was later utilized for imaging structured cellular RNAs.[288]
A limitation for generating “light up” aptamers like Spinach and Spinach2 is that aptamers are first selected based on their ability to bind fluorophores and then subsequently on their ability to turn on fluorescence. <1% of the aptamers that bind fluorophores lead to fluorescence enhancement.[289] Importantly, function of the isolated aptamers in cellular milieu is often compromised because these aptamers either need ions that are not present intracellularly or are misfolded due to the presence of adjacent sequences introduced for analyte sensing. Therefore, significant in vitro mutagenesis is required to render these probes useful in live cells. Filonov et al. developed an alternate strategy that allowed the selection of fluorescence-enhancing aptamers in cellulo, so that these problems could be mitigated.[289] This strategy led to the selection of the aptamer Broccoli, which binds the dye (Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-2-methyl-1-(2,2,2-trifluoroethyl)-1H-imidazol-5(4H)-one) (DFHBI-1T). Compared to Spinach2, Broccoli has significantly reduced dependence on magnesium for proper folding, resulting in 2-fold increase in fluorescence signal in vivo. Moreover, Broccoli does not require tRNA scaffold which is incorporated into Spinach and Spinach2 to promote folding in cells. The 5S ncRNA was imaged in HEK293T cells using Broccoli. As an alternate strategy, Shu et al. incorporated the pRNA 3WJ motif of the phi29 DNA packaging motor into light up RNA fusions to enable predictable folding.[290] It was subsequently shown that Broccoli could be engineered to give a fluorescence readout in the presence of RNA-modifying enzymes.[291] For example, substitution of specific adenosine bases with N(6)-methyladenosine rendered Broccoli non-fluorescent. Demethylation due to activity of RNA demethylases such as fat mass and obesity-associated protein (FTO) or ALKBH5 generated unmodified Broccoli and restored fluorescence in the presence of DFHBI-1T.
Dolgosheina et al. developed Mango, an aptamer that binds to thiazole orange derivatives and has significantly increased binding affinity and fluorescence efficiency compared to Spinach-type aptamers.[292] These properties could be leveraged to image RNA down to the single molecule level. Later, Autour et al. developed new Mango aptamers for imaging small non-coding RNA such as 5S, U6, and a box C/D scaRNA in fixed and live HEK293T cells.[293] The new aptamers were comparable in brightness to GFP and enhanced GFP. The Jaffrey lab developed Corn, a 28 nt aptamer, which binds to 3,5-difluoro-4-hydroxybenzylidene imidazolinone-2-oxime (DFHO), a mimic of the fluorophore in red fluorescent proteins. Compared to Spinach and Broccoli which lose >50% of fluorescence within 200 ms of constant irradiation, Corn is unaffected up to 10 s. Corn was utilized for quantitative measurement of Pol III transcription in HEK293T cells. DFHO could also bind to Broccoli derivatives yielding Red Broccoli and Orange Broccoli which have red shifted excitation and emission properties; however, these complexes were susceptible to rapid photobleaching.[294,295]
The Nilsen-Hamilton lab developed a FRET-based approach using Intracellular MultiAptamer GEnetic tags (IMAGEtags) for tracking gene expression in vivo. These probes gave higher sensitivity compared to Spinach.[296,297] Jepsen et al. developed a genetically encoded FRET-based strategy for imaging intracellular analytes by incorporating Spinach and Mango aptamers in close proximity in single-stranded RNA origami scaffolds.[298] The validity of this system was demonstrated by imaging SAM in E. Coli cells.
The readers are directed to focused reviews by Ouellet, and You and Jaffrey for further information on genetically encoded-aptamers.[299,300]
3.10. Split aptamers
Split aptamers are comprised of aptameric sequences that have been divided into two parts. In the presence of a target, the two fragments form a ternary complex and this property can be used in designing proximity-dependent sensors. Split aptamers have been developed for detecting adenine, ATP, cocaine, thrombin, 17β-estradiol, D-vasopressin, and theophylline in solution.[301]
Zheng et al. developed a DNA nanoprism-based split-aptamer construct for the detection of ATP in live cells. The aptamer fragments were labeled with a FRET-pair such that in the presence of ATP a FRET signal was obtained.[302] The FRET signal increased linearly in vitro with increasing ATP concentrations between 0.03–2 mm and saturated at a 5-fold enhancement. The probe was also used for reporting changes in ATP concentrations in live HeLa cells.
Recently, Zhang et al. reported the development of a nanoparticle-based split-aptamer sensor for detecting ATP in live cells through HCR (Figure 13).[110] In their work, Zhang et al. used four ONT sequences: (i) two sequences were the aptamer fragments (AP1 and AP2), each fragment consisting of a recognition element that binds to the target and a split initiator element for HCR (Figure 13A). (ii) the remaining sequences were hairpin sequences H1 and H2. Upon binding to ATP, the tail regions of AP1 and AP2 were brought within close proximity, resulting in the formation of an active initiator sequence capable of activating HCR between H1 and H2. H1 was labeled with a fluorophore (TAMRA)-quencher (BHQ) pair such that the fluorescence was turned on by separation of TAMRA from BHQ upon opening of the hairpin due to ATP binding. Moreover, an amplified signal was attained as one ATP binding event results in a cascade of hybridizations. These ONTs were delivered to live cells using amino-functionalized fluorescent block copolymer nanoparticles (BCNs). The positively charged BCNs allowed electrostatic surface adsorption of the negatively charged ONTs as well as efficient transfection. Their inherent fluorescence also allowed the particles to be tracked independently of ATP. These constructs were specific for ATP against CTP, GTP, UTP and TTP, were claimed to give a limit of detection of 30 nm in solution, and reported differences in the relative abundance of ATP inside cells with high signal-to-background ratios.
Figure 13.
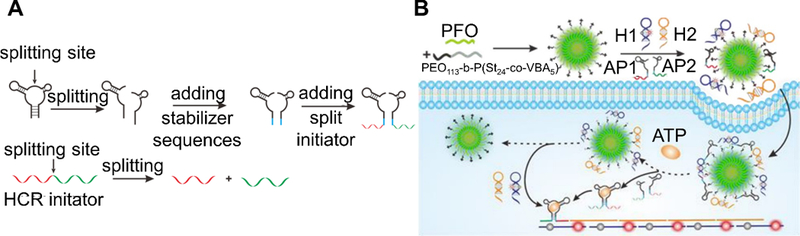
Split aptamers for ATP detection (A) Split aptamer design incorporating HCR for analyte detection. (B) A scheme of a nanoparticle/split aptamer detection strategy for sensing ATP in live cells. Binding of split aptamers AP1 and AP2 to ATP results in their tail regions coming into proximity, acting as an initiator for HCR with H1 and H2. The opening of H1 results in separation of a fluorophore and quencher, giving fluorescence. Adapted with permission.[110] Copyright 2018, American Chemical Society.
Wang et al. developed a “split-broccoli” system for imaging mRNA in live cells.[303] The Broccoli aptamer was split into two fragments and genetically encoded into live mammalian cells. Tandem binding of the aptamer fragments to the target mRNA created a binding pocket where the dye DFHBI could then bind and elicit a fluorescence signal. The authors termed this technique as aptamer-initiated fluorescence complementation (AiFC). The nomenclature was coined by drawing parallels to bimolecular fluorescence complementation (BiFC), a prevalent technique used in imaging protein-protein interactions using split-fluorescent proteins. Using this strategy, CFL1, β-actin, and GAPDH mRNA were imaged in HeLa and HuMSC cells. The authors further demonstrated the versatility of this system by engineering a new iSpinach RNA aptamer for monitoring β-actin. Similarly, Alam et al. also developed a split-broccoli system for monitoring RNA-RNA hybridization in live E. coli cells.[304]
3.11. Other strategies
Analogous to MBs, aptamer beacons were developed by Liang et al. for the imaging of HIV-1 reverse transcriptase in live HeLa and U1 cells.[62] This study addresses the challenge of visualizing endogenous proteins in genetically unmodified cells.
Ke et al. developed a noninvasive technique for the measurement of temperature in live cells using MBs synthesized from L-DNA, the enantiomer of naturally occurring D-DNA.[43] The fluorescence of the MBs is quenched below the melting temperature of the hairpin structure and increases gradually at higher temperatures. The use of L-DNA prevents non-specific binding to cellular proteins and nucleic acids as well as nuclease degradation. Using palladium nanosheets that absorb NIR light (808 nm) to give rise to a photothermal effect, the authors showed that the fluorescence signal could be reversibly tuned over the range of 20–34 °C in live HeLa cells.
Strauss et al. developed slow off-rate modified aptamers (SOMAmers) for monitoring cell surface and intracellular targets using DNA points accumulation in nanoscale topography (DNA-PAINT).[305] DNA-PAINT is a single molecule imaging technique that allows ~ 5 nm spatial resolution.[306] The method involves the binding of dye-labeled DNA strands to their complements transiently. Unlike traditional aptamers, SOMAmers contain modified bases with hydrophobic residues allowing increased range of protein targets with high-affinity ligands. 7 different SOMAmers that can quantitatively image proteins in different cellular compartments such as the cell membrane, nucleus, lysosomal membrane, peroxisomes, and mitochondria, were identified. These proteins included EGFR, GFP-labeled Nup107, catalase proteins, ErbB2, HSP90, LIMP-2, and HSP60.
Several strategies aimed at identifying and isolating whole live cells based on aptamers or studying proteins on cell membranes have been published.[307–315] However, most of these aptamers target cell surface markers as opposed to intracellular markers and are, therefore, beyond the scope of this review.
4. DNAzyme-based probes
4.1. Gold nanoparticle-based
The Lu group pioneered the development of DNAzyme-based probes for imaging metal ions in live cells (Figure 14).[97] The 39E DNAzyme was chosen as a model as it detects its target uranyl ions with excellent selectivity (>1 million-fold) and sensitivity (45 pm). A NanoFlare-like construct was used, consisting of a 13 nm gold core and a dense 39E DNAzyme shell. The enzyme strand of the DNAzyme was covalently attached to the gold core through a short thiolated poly A region at the 3’ end. The substrate strand, labeled with Cy3 at the 5’ end, was hybridized to the enzyme strand, resulting in quenched fluorescence due to proximity to the gold surface. The purpose of adding the poly A region was to ensure that steric bulk due to dense functionalization did not impede hybridization with the substrate strand. However, it was observed that this also led to decreased quenching efficiency. Therefore, the substrate strand was further modified with a quencher, BHQ, at the 3’ end. In the presence of uranyl ions, the substrate strand was cleaved at the ribonucleotide site, resulting in a shorter strand with much lower melting temperature (21 °C) compared to the original intact strand (60 °C). Therefore, at physiological temperature (37 °C), the Cy3 containing short strand separated from the nanoparticle and fluorescence could be observed. Using this strategy, it was demonstrated that uranyl ions can be imaged in live HeLa cells pretreated with uranyl citrate. Moreover, using Lysotracker it was shown that the probes were mostly localized in the lysosomes. Li et al. later extended this strategy for the simultaneous imaging of Zn2+ and Cu2+ using two distinct fluorophores (FAM for Zn2+ and Cy5 for Cu2+) in HepG2 cells.[316] In this context, we mention that a recent review by Zhou et al. provides a comprehensive overview of the state of metal sensing by DNA.[22]
Figure 14.
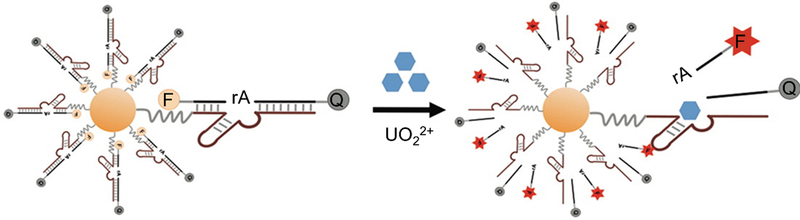
DNAzymes immobilized on the surface of gold nanoparticles for detection of uranyl ions. A fluorophore on the substrate strand is quenched by both gold and BHQ. Cleavage of the substrate strand in the presence of uranyl ions leads to separation of the fluorophore from both quenching elements and a subsequent fluorescence turn on. Adapted with permission.[97] Copyright 2013, American Chemical Society.
The Wang group has developed several DNAzyme functionalized gold nanoparticle-based probes for live cell imaging. For example, Yang et al. reported the first aptazyme-based gold nanoparticle probe for the detection of ATP in live cells (Figure 15).[317] A 13 nm gold nanoparticle was densely functionalized with 3’-TAMRA-labeled substrate strands to which 5’-BHQ-2-labeled aptazymes specific for ATP were hybridized. The number of aptazyme strands were lower than the number of substrate strands. The aptazyme could not form a stable and active structure without ATP. However, in the presence of ATP and Mg2+ (a cofactor), a stable structure could be formed, leading to ATP-dependent cleavage of the substrate strand, separating the fluorophore from the quencher and eliciting a fluorescence signal. As one aptazyme can bind to several substrate strands in succession, a small number of ATP molecules could lead to a large fluorescence readout allowing a reported detection limit as low as 200 nm. The aptazyme-gold nanoparticle construct was used to image ATP in live HeLa cells. They later developed hairpin-locked DNAzyme probes[318] and split-DNAzyme-based probes for imaging microRNA in live cells.[319]
Figure 15.
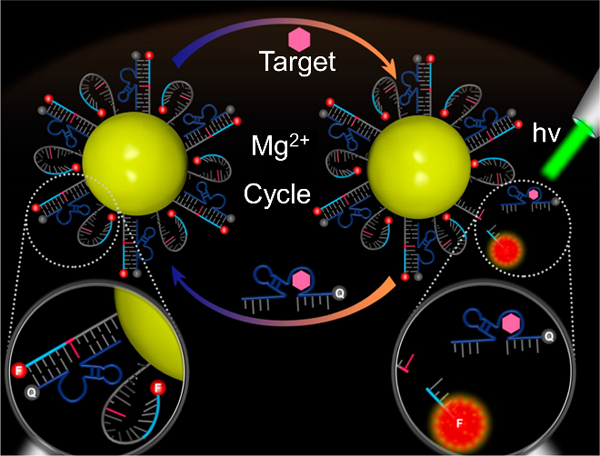
Workflow of aptazyme/gold nanoparticle-based method for detection of ATP molecules. Fluorophore-labeled substrate strands are conjugated to gold nanoparticles and prehybridized with a quencher-labeled aptazyme strand. The aptazyme is activated in the presence of ATP, leading to cleavage of the substrate strand and separation of fluorophore and quencher to yield fluorescence. Active aptazymes can bind to multiple substrate strands in succession, leading to amplified signal. Adapted with permission.[317] Copyright 2016, American Chemical Society.
Using a slightly modified strategy employing two DNA hairpin strands (including a Zn2+-specific DNAzyme), Liu et al. developed a probe for detecting microRNA.[320] Similarly, Peng et al. developed a “DNAzyme motor” probe, also for detecting microRNA.[50]
One limitation of DNAzymes is metal-dependent degradation prior to intracellular entry. As a potential solution to this problem, Wang et al. developed NIR activated DNAzymes.[321] Gold nanoshells were functionalized with a three-stranded DNAzyme precursor. Upon irradiation, local changes in temperature resulted in release of the DNAzyme from the gold surface, which could then form the active structure. In this study, Zn2+ was imaged in HeLa cells.
4.2. Graphene oxide-based
Kim et al. developed a theranostic platform for detection and regulation of mRNA based on DNAzyme-graphene oxide nanosheet (nGO) complexes.[322] nGO protected the DNAzyme against nuclease degradation and allowed its delivery into cells without the use of transfection reagents. They demonstrated that using this construct, the Hepatitis C virus non-structural gene (NS3) can be detected and down-regulated in Huh-7-rep liver cells. The DNAzyme sequence used consisted of a catalytic domain flanked on both sides by bases complementary to the NS3 mRNA and a fluorophore label (FAM) at the 5’ end. The fluorescence of FAM was quenched by 95% upon complexation of the DNAzyme with nGO and could be recovered upon separation of the DNAzyme from the nGO via binding to the target mRNA. Post mRNA binding, the DNAzyme elicited sequence specific mRNA cleavage resulting in changes in gene expression both at the mRNA and protein levels. The DNAzyme-nGO complex reduced mRNA and protein levels down to 32.6% and 26%, respectively, outperforming lipofectamine-transfected DNAzyme which led to reduction to 57.3% and 30.2%, respectively.
4.3. Polymer core nanoparticle-based
Zhang et al. developed dendritic polyethylene–cationic poly(p-phenylene ethynylene) nanoparticles for the delivery of DNAzymes into live cells.[323] The 8–17 DNAzyme that undergoes catalytic degradation in the presence of Pb2+ was used in this study. The DNAzyme was bound to the polymer core through electrostatic interactions. The 5’ end of the substrate strand was labeled with Cy5.5 while the 3’ end of the enzyme strand was labeled with BHQ-3, resulting in quenched fluorescence in the absence of Pb2+. In buffer, addition of increasing concentrations of Pb2+ (up to 10 μM) led to increase in fluorescence signal (~10-fold) due to degradation of the substrate strand resulting in separation of the fluorophore and the quencher. The nanocomposite was internalized into HepG2 cells without transfection reagents. The cells were then treated with 0 to 725 mm Pb2+. The blue-green fluorescence of the nanoparticle probe enabled monitoring uptake whereas the far-red fluorescence of the Cy5.5 allowed monitoring of Pb2+ concentration. Using confocal imaging and flow cytometry, the authors demonstrated that the probe responded to changing concentrations of Pb2+ in live cells.
4.4. Dendrimer-based
The presence of inorganic cores used in delivering DNA-based probes has been shown to induce toxicity at high concentrations in some cases. To overcome this limitation, Meng et al. developed a nanocarrier composed entirely of DNA using a dendrimer-based strategy.[324] The sensing moiety of the dendrimer was based on a DNAzyme for detecting L-histidine and consisted of 4 strands as shown in Figure 16. The strand Y1b was labeled with a quencher, BHQ-1, while the substrate strand X was labeled with the fluorophore FAM. In the presence of L-histidine, the substrate strand was cleaved, resulting in a shorter strand with a much lower melting temperature (12.7 °C) compared to the original full-length strand (44.3 °C). At physiological temperature, the strand dehybridized, resulting in separation of FAM and BHQ-1 leading to fluorescence enhancement. In buffer, the dynamic range was established to be 5 μm −10 mm, with a reported detection limit of 500 nm. Importantly, the dendrimer-based architecture imparted resistance to nuclease degradation as well as stability against degradation due to non-specific protein binding. In cell lysate, similar titration curves were obtained, with a dynamic range of 5–100 μm. To test whether these constructs function in live cells, MCF-7 (human breast cancer) cells were used as a model. Cells treated with the dendrimer nanocarrier, followed by 100 mm L-histidine and 150 mm K+ (a cofactor in the catalytic reaction) showed significantly increased fluorescence compared to cells treated without one or more of the components. The authors also developed a sensor for detecting ATP using an ATP aptamer instead of the DNAzyme. Taken together, these results represent an advance towards developing more biocompatible nucleic acid-based probes.
Figure 16.
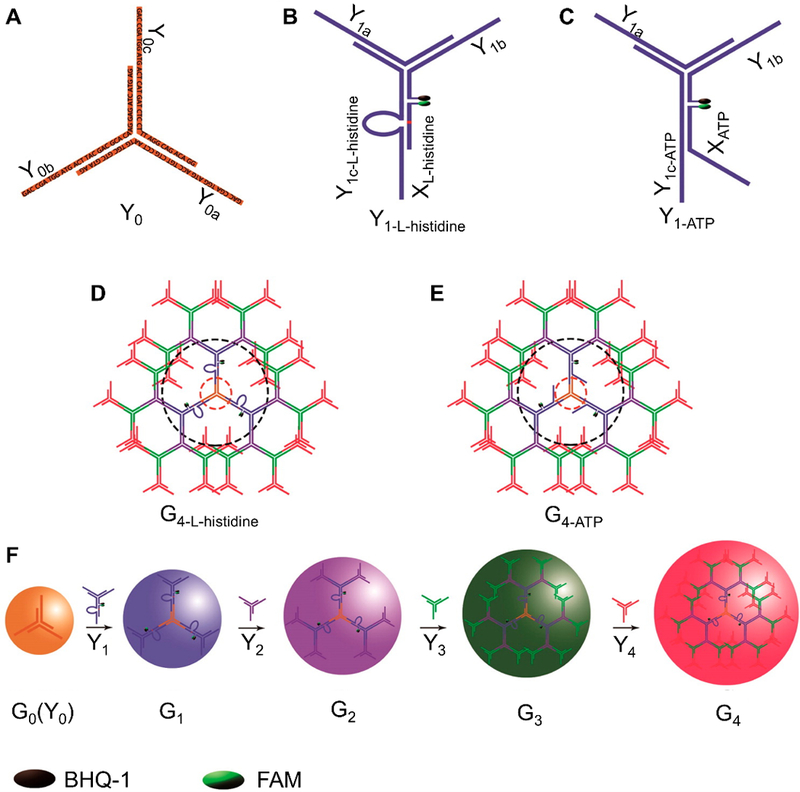
Design of dendrimer-based nanoparticles for detection of ATP and histidine. (A) The assembly of Y0 containing a 3-way junction made from single-stranded components Y0a, Y0b, and Y0c. (B) The assembly of Y1-L-histidine from single-stranded components Y1a, BHQ1-Y1b, Y1c-L-histidine and FAM-XL-histidine. (C) The assembly of Y1-ATP from single-stranded components Y1a, BHQ1-Y1b, Y1c-ATP and FAM-XATP. (D) The assembly of G4-L-histidine from components Y0, Y1-L-histidine, Y2, Y3, and Y4. (E) The assembly of G4-ATP from components Y0, Y1-ATP, Y2, Y3, and Y4. (F) Layer-by-layer development of generations 1–4 of DNA dendrimers.[324] Adapted with permission. Copyright 2014, American Chemical Society.
4.5. MOF-based
Yi et al. developed a MOF-based probe for ratiometric detection of intracellular microRNA.[325] The 8–17 DNAzyme labeled with a FRET-pair was complexed with a zeolitic imidazolate framework-8 (ZIF-8). This nanostructure was taken up by cells. In the endosome, acidic digestion of ZIF-8 released Zn2+, which acts as a cofactor for the DNAzyme. In the presence of the target microRNA, a change in FRET signal is observed due to DNAzyme activity. This strategy was utilized in imaging miR-21 in MCF-7, HeLa, and L02 cells. Chen et al. developed MOF nanoparticles capped with DNAzymes and aptazymes as a therapeutic platform for treating cancer cells.[326] Doxorubicin was incorporated into the porous MOF structures and the DNAzymes/aptazymes acted as a cap. In the presence of a target metal ion (Mg2+ or Pb2+) or target ligand for the aptamer (ATP), DNAzyme/aptazyme activity was observed, leading to drug release.
4.6. FRET-based
Hwang et al. developed photocaged DNAzymes for monitoring metal ions in live cells with spatial as well as temporal precision.[327] One challenge of using DNAzymes is premature cleavage of the substrate strand before the probes are completely internalized. As the 2’ position of the ribonucleotide adenosine plays an important role in the catalytic activity of the DNAzyme, the authors modified the site with a photocleavable 2′‐O‐nitrobenzyl protection group. The 8–17 DNAzyme was chosen to image Zn2+ in live HeLa cells. The substrate strand was modified with fluorescein on the 5’ end and BHQ-1 on the 3’ end while the enzyme strand was modified with dabcyl on the 5’ end. In the absence of the target, the two strands were hybridized, and the fluorescence was quenched. Notably, incubation of the probes with 50 μM Zn2+ in buffer and human serum for long periods (up to 7 days) did not result in significant increase in fluorescence indicating that cleavage was minimum in the absence of light. In contrast, the unprotected adenosine containing DNAzyme degraded within an hour. These probes were transfected into HeLa cells using Lipofectamine 2000, and upon irradiation with 365 nm light, the nitrobenzyl protection group was cleaved, thereby leading to Zn2+-dependent cleavage of the substrate strand with concomitant increase in fluorescence signal from fluorescein. Nuclear localization of the probes was observed, consistent with previous reports suggesting ONTs delivered using Lipofectamine are sequestered into the nucleus. One advantage of using this system is that it is highly modular i.e. by using the same substrate strand, but by replacing the catalytic core of the enzyme strand with a core sensitive to another metal ion, a new probe can be developed. The authors demonstrated this capability by using GR-5 as the catalytic core for detecting Pb2+.
As an alternate strategy (but similar fluorescence reporting mechanism) to prevent false positive signal, Cui et al. developed L-DNAzymes to image Cu2+ in live HeLa cells.[328] Their rationale for choosing L-DNAzymes was to prevent false positive signal that may arise from digestion of probes by nucleases, non-specific protein or off-target binding of the probes. The L-DNAzyme probe, being an enantiomer of D-DNAzyme, retained its catalytic activity in the presence of the achiral metal ion, while imparting enhanced biostability to the probe. Compared to D-DNAzymes, these probes were resistant to nuclease degradation and resisted binding to single-stranded DNA protein. These probes were transfected into the cells using Lipofectamine and upon treatment of the cells with Cu2+, fluorescence signal could be observed.
Torabi et al. developed the first Na+-specific DNAzyme by in vitro selection and converted it into a fluorescent sensor for imaging Na+ in live cells.[329] The DNAzyme was characterized by fast catalytic rate (observed rate constant ~0.1 min−1), high selectivity (>10–000 fold) for Na+ over competing ions (K+, Li+, etc.), and a dynamic range (0.135–50 mm) encompassing physiological concentrations of Na+. By attaching a quencher (Iowa Black) to the 3’ ends of the enzyme and substrate strands as well as a fluorophore (FAM) to the 5’ end of the substrate strand, a fluorescent sensor for Na+ was developed. The ribonucleotide adenosine site in the substrate strand was protected by photocleavable 2′‐O‐nitrobenzyl protection group to enable controlled activation of the DNAzyme after probe internalization. Later, Wu et al. developed a catalytic hairpin assembly-based strategy for amplification of signal coming from Na+-specific activity of the NaA43 DNAzyme.[330]
4.7. Other strategies
Zhang et al. developed a theranostic platform through which low-abundance microRNAs can be visualized using an in situ amplification strategy and drugs can be released in the presence of target microRNAs.[331] DNAzymes were attached to multifunctional nanoparticles. Two microRNAs, miR-21 and miR-145, which are up- and down- regulated in many types of tumor cells, respectively, were chosen as targets. Fluorophore-quencher-labeled multicomponent DNAzymes were attached to mesoporous silica-coated gold nanorods via azide-alkyne click chemistry. These constructs had a dynamic range between 2.5–250 pm and a reported detection limit of ~1 pm. They could be internalized into cells (HeLa, NIH 3T3, and HL-60) and in the presence of their targets, cleavage of the fluorophore-labeled substrate strands led to increased fluorescence. Using calibration curves, the authors quantified the number of transcripts per cell. Upon NIR irradiation, the gold nanorod could cause local increase in temperature, modulating cleavage activity. Moreover, doxorubicin could be incorporated within the mesoporous silica and the DNAzymes acted as a “gatekeeper,” releasing the encapsulated drug only in the presence of the target microRNAs.
He et al. developed probes employing a dual amplification strategy for the detection of mRNA in live cells.[332] These probes consisted of a ZnO core onto which four hairpin DNA strands were adsorbed. This architecture allowed the probes to be taken up inside cells without the need for transfection reagents and impeded nuclease degradation. In the endosome, it is believed that acid-triggered dissolution of the ZnO core resulted in cytosolic delivery of the nucleic acids. In the presence of the target mRNA, active DNAzymes were formed by self-assembly of the hairpin strands via HCR, which lead to a fluorescence readout. This strategy yielded a reported limit of detection as low as 0.1 fm. Using this method, the survivin mRNA, often overexpressed in cancer cells, was imaged in a variety of cells, including HeLa, L02, C166, MCF-10A, HepG2, SMCC-7721, and MCF-7 cells.
Some other recent developments in the area of DNAzyme-based detection involve the use of MnO2 nanosheet carriers[333] and magnetic field-activated DNAzymes for imaging mRNA,[334] enzyme-initiated DNAzyme nanodevices,[335] as well as upconverting nanoparticles-based probes.[336]
5. Methods for Probe Delivery
The cell membrane is impervious to ONTs, making intracellular delivery of nucleic acid-based probes challenging. Moreover, for imaging, it is important to consider not only the probe uptake, but also the quantity of free probe available for binding to the desired targets. For example, a probe that targets a molecule in the cytoplasm must be able to reach the cytoplasm and should not be confined within cellular compartments. Some methods which have been widely utilized to deliver nucleic acid-based imaging probes include the use of microinjection, transfection reagents, and electroporation.[6] Recently, several nanoparticle-based platforms as well as aptamer-based strategies have been developed as transfection reagent-free strategies for intracellular delivery of probes via receptor-mediated internalization.[44,74]
Microinjection involves direct introduction of imaging probes into the cytoplasm of the cells. While structures of various sizes can be delivered effectively, the throughput is low. Moreover, microinjection can damage the cell and impede normal cell functions.
As an alternative strategy, various transfection reagents have emerged as popular vectors for introducing imaging probes inside cells. One class of transfection reagents involves toxin-based cell membrane permeabilization. For example, streptolysin O (SLO), is a bacterial protein that oligomerizes on the cell-surface in a cholesterol-dependent manner to form reversible pores with diameters of 25–30 nm. These pores facilitate the transfer of nucleic-acid based probes from extracellular space indirectly into the cytoplasm;[337] the cell membrane can be restored after SLO treatment by placing the cells in normal cell culture medium. Cell penetrating peptides (CPPs) (e.g. HIV-1 Tat peptide and its derivatives) are another class of transfection reagents. The positive charge associated with the cationic amino acids aids in cellular transfection.[338] Cationic liposome-based transfection reagents such as Lipofectamine have also been developed.[339] A major limitation of transfection reagents is induction of toxicity. Moreover, in some cases, transfection reagents can also alter gene expression. Some transfection reagents are susceptible to endosomal entrapment. Probes that enter through endosomal pathways are prone to rapid degradation by nucleases. Therefore, even when the probes do escape, only a small fraction of the probes remain functional.
To overcome challenges associated with endosomal entrapment, electroporation has been used to deliver imaging probes to cells with over 90% transfection efficiency.[150,340] Electroporation entails the application of strong electric fields to transiently destabilize cell membranes so that external reagents may be introduced within cells. Typically, low cell viability is often associated with electroporation. Although recent advances such as microporation have minimized cell mortality (>85% cell viability[150]), optimization is necessary to determine the electrical parameters for efficient cellular delivery. The parameters depend on electrode design, cell type as well as identity of molecules to be introduced within cells.
Many nanomaterial-based nucleic acid imaging probes do not require transfection as they are taken up by cells through receptor-mediated endocytosis. For example, the spherical nucleic acid architecture which involves a dense DNA shell grafted to a nanoparticle core allows NanoFlares and structurally similar probes to be taken up by endocytosis.[139] Graphene-oxide based probes are also taken up spontaneously by cells.[263] A small fraction of probes being released from the endosome results in stoichiometrically meaningful quantities for detection. However, it is not known quantitatively what percentage of these probes escape from the endosome and remain functional. Inability to detect desired targets in the cytosol may arise from lack of careful optimization of incubation conditions, given that probe uptake and endosomal escape may be cell line and preparation dependent. It should be noted that in fluorophore-quencher based systems, separation of the fluorophore and the quencher due to nuclease degradation in the endosomes can be a significant contributor to the incidence of false positive signals.
Certain aptamer sequences can also enable probe internalization within cells without the need of transfection reagents. For example, the AS1411 aptamer that targets nucleolin, a protein often overexpressed on the surface of various cancer cell lines, has been used to deliver probes preferentially to cancer cells.[74] Similarly, the transferrin aptamer has been utilized to image the transport of chloride ions along the transferrin pathway using DNA nanomachines.[250] Importantly, the use of aptamers allows probes to be delivered along different pathways so that chemical changes along these pathways can be individually imaged.[115,250]
6. Challenges
6.1. Target accessibility
Probes must be able to bind to their targets to elicit a detectable signal. However, not all targets are easily accessible for binding to the probes. For example, parts of an mRNA sequence might be bound to proteins or may have secondary structures, rendering it difficult for a complementary probe to bind. Therefore, while designing new probes, several sequences may have to be designed and tested to find the optimal probe. Studies have suggested that the starting and terminating codon regions of mRNAs fare as better targets.[341] Yet, probes must be carefully designed as a shift of only a few bases either towards the 3’ or 5’ can lead to an ineffective probe. Target analytes may also be localized within membrane-bound organelles and inability of probes to enter these organelles will impede detection.
6.2. Selection of fluorophores and quenchers
To establish quantitative imaging of intracellular analytes with high signal-to-background ratios, fluorophores and quenchers must be carefully chosen. For example, cellular autofluorescence in the green channel contributes to significant background. These effects can be mitigated by using dyes with red-shifted fluorsecence or by developing brighter dyes with greater quantum yields. Alternately, lanthanide-based dyes which have longer fluorescence lifetimes can be used for imaging to overcome interference from autofluorescence. A limitation of such an approach is the need for more complex instrumentation. Another important consideration is the photostability of the dye. Long-term cellular imaging necessitates the use of dyes that are resistant to photobleaching. Designing fluorophores with desired brightness, photostability, and biocompatibility is still an active field of research. A major limitation of fluorescence-based approaches in general is that multiplexed imaging of more than 5 or 6 analytes is challenging due to the limited numbers of spectrally distinct fluorophores available. Moreover, the fluorescence intensity of fluorophores can also be impacted by local pH or interaction with other molecules, which can complicate the interpretation of results.
6.3. Sensitivity and selectivity of probes
The most challenging hurdle in developing fluorescent probes for imaging analytes in living cells is sensitivity of detection. Low-abundance molecules are particularly difficult to observe, although several amplification strategies such as HCR have been developed to enhance signal. Moreover, the binding affinity of the probes play an important role in determining the sensitivity with which targets can be detected. This is particularly important for aptamer-based probes which are typically evolved in vitro and therefore do not behave as expected inside cells and often show poor folding. Also important are the kinetics of probe-target binding. If the dynamics of the analyte of interest occur faster than the target binds, it will be impossible to follow the analyte with spatiotemporal precision. Importantly, probes should be carefully designed to be selective for their targets to avoid false positive signals.
6.4. Probe delivery and stability
As mentioned above, there are numerous challenges associated with the delivery of probes in live cells. Each technique has its own advantage and disadvantages. Probes must be able to reach their targets to be able to detect them. Therefore, probe stability is of paramount importance. This is particularly important for monitoring long-term dynamics. Nucleic acid-based probes are inherently susceptible to enzymatic degradation. However, their stability can be increased via modification of the nucleic acid structure or association with nanoparticles. Also important for long-term monitoring of live cells is the impact of the probes themselves on the cells. For example, binding of antisense/DNAzyme probes to mRNA can induce down-regulation or degradation of mRNA over long treatment times.[166] Some of these challenges can be mitigated by using low concentrations of probes, while ensuring the concentration is high enough to elicit a signal, or shorter treatment times.
7. Conclusions and outlook
Nucleic acid-based probes are revolutionizing the field of live cell imaging, offering a simple and versatile way of detecting important intracellular analytes in real time. We summarize the commonly recurring themes in the literature we have reviewed here to give the reader a sense for where the field of nucleic acid sensors is and where the field is going.
The majority of literature employs fluorescence-based techniques for monitoring analytes in live cells. Similarly, technologies that have been commercialized, such as molecular beacons and NanoFlares, generally employ fluorescence-based readout. Moreover, because biological systems involve the interplay of multiple analytes, many papers seek to achieve multiplexed detection with their sensor platform. Another common theme is a push to find more red-shifted dyes to overcome background noise from autofluorescence of cells and tissues. Examples include the development of quinoline blue for use in FIT probes and the development of the Red Broccoli aptamer as an alternative for the original Spinach aptamer.[154,294] Beyond this, structures that permit long-term imaging of cellular analytes are commonly sought after. This is usually achieved by increasing nucleic acid-based probe stability (i.e. introducing base/backbone/sugar modifications) and using fluorescent reporters that have robust photostability (i.e. quantum dots).[66,237,238]
Furthermore, the discovery of SNA based NanoFlares began a marked push in the field towards the development of nanoparticle-based platforms. As previously mentioned, such platforms confer a variety of interesting properties including the ability to deliver payload into cells without the need of transfection reagents, protect cargo from nuclease degradation, and act as a source of fluorescence/quenching. Nanoparticles have also helped advance the development of theranostics, another recurring area of interest in the works presented here.[106,166,342] Examples include theranostics based on DNAzymes and NIR-absorbing nanoparticles interfaced with MBs.[225,343]
A final theme encountered has been the push for developing and incorporating amplification strategies into biosensors. This is motivated by the importance of being able to detect low abundance targets in live cells.
The majority of current work has focused on cells in 2D culture, while less focus has been given to investigating tissues or processes in vivo. Developing robust sensors that function in more challenging environments requires many design considerations. First, probes must be resistant to degradation, which can be achieved through the introduction of base, sugar ring and backbone modifications on the nucleic acids or their association with nanoparticles. Robust sensing in tissues and in vivo is ideally done with dyes with high brightness and wavelength emission profiles in the “optically transparent” NIR window (800–950 nm).[344] The development of stable and biocompatible dyes that fit these criteria is an active area of research, and we expect their development to greatly advance the capabilities of nucleic acid-based sensors. Moreover, conventional delivery techniques like microinjection or commercial transfection reagents are generally not possible for use in vivo. As such, we expect that nanoparticle-based delivery methods will be continually developed, with an emphasis on designing systems with biocompatible and biodegradable nanoparticles that allow targeted delivery to specific regions of interest without sequestration into the liver. Taken together, these advances will not only provide invaluable information about cellular processes with impressive resolution but also usher in a new era of in vivo bioimaging, enabling fluorescence-guided surgery based on different levels of expression of intracellular analytes. Moreover, since many of these probes can also regulate target expression levels they can be potentially translated into a therapeutic platform, giving rise to a new class of nucleic acid-based theranostics.
Nucleic acid-based sensors such as hybridization-based probes, aptamers, and DNAzymes have emerged as key tools for studying analytes in live cells. This review describes the current state of intracellular nucleic acid-based probes and discusses the common strategies for probe design, what targets can be detected, and how current limitations may be overcome in the future.
Acknowledgements
D.S. and S.B.E. contributed equally to this work. This material is based on research sponsored by Air Force Research Laboratory under agreement number FA8650–15-2–5518. The U.S. Government is authorized to reproduce and distribute reprints for Governmental purposes notwithstanding any copyright notation thereon. The views and conclusions contained herein are those of the authors and should not be interpreted as necessarily representing the official policies or endorsements, either expressed or implied, of Air Force Research Laboratory or the U.S. Government. It was also supported by the Air Force Office of Scientific Research award FA9550–17-1–0348, the Vannevar Bush Faculty Fellowship program sponsored by the Basic Research Office of the Assistant Secretary of Defense for Research and Engineering and funded by the Office of Naval Research award N00014–15-1–0043, and the National Cancer Institute of the National Institutes of Health under award U54CA199091. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Biographies

Devleena Samanta graduated from Stanford University in 2017 with a PhD in Chemistry. Advised by Richard N. Zare, she gained a broad research experience in nanomaterial synthesis, drug delivery, and microdroplet chemistry. Currently, she is a postdoctoral fellow in Professor Mirkin’s group developing nanoparticle-based biodiagnostic tools. She is interested in exploring how rationally designed nanomaterials can be exploited for biological applications.

Sasha B. Ebrahimi graduated summa cum laude in 2016 from The University of Illinois at Urbana-Champaign with a B.S. in chemical engineering. He is currently a doctoral candidate at Northwestern University advised by Professor Mirkin. His research interests lie at the interface of nanotechnology and diagnostics. His current research focuses on developing novel nanoparticle-based sensing platforms for detecting intracellular analytes implicated in various forms of cancer.

Chad A. Mirkin is a chemist and nanoscience expert, known for his invention and development of spherical nucleic acids, dip-pen nanolithography and related cantilever-free, scanning probe-based nanopatterning methodologies, and contributions to supramolecular chemistry and nanoparticle synthesis. He has authored over 730 manuscripts and over 1,100 patent applications worldwide (over 330 issued) and founded multiple companies (e.g., AuraSense, Nanosphere, Exicure, TERA-print, and CDJ Technologies). He has been recognized with over 130 awards and elected into all three US National Academies. Mirkin was an advisor to President Obama for eight years, and he is an Associate Editor of JACS.
Footnotes
Conflict of interest
C.A.M. has financial interests in Exicure, Inc. which could potentially benefit from the outcomes of this research.
Contributor Information
Devleena Samanta, Department of Chemistry and International Institute for Nanotechnology, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA.
Sasha B. Ebrahimi, Department of Chemical and Biological Engineering, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA.
Chad A. Mirkin, Department of Chemistry and International Institute for Nanotechnology, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
References
- [1].Cooper GGM, Hausman RE, The Cell: A Molecular Approach, Sinauer Associates, Sunderland, Massachusetts; 2004. [Google Scholar]
- [2].Adams SR, ackey MRM, Ramachandra R, Palida Lemieux SF, Steinbach P, Bushong EA, Butko MT, Giepmans BNG, Ellisman MH, Tsien RY, Cell Chem. Biol. 2016, 23, 1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Han X, Aslanian A, Yates JR, Curr. Opin. Chem. Biol. 2008, 12, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Aydin S, Peptides 2015, 7, 4. [DOI] [PubMed] [Google Scholar]
- [5].Deepak S, Kottapalli K, Rakwal R, Oros G, Rangappa K, Iwahashi H, Masuo Y, Agrawal G, Curr. Genomics 2007, 8, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bao G, Rhee WJ, Tsourkas A, Annu. Rev. Biomed. Eng. 2009, 11, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Farrell RE, RNA Methodologies, Elsevier Inc., 2010. [Google Scholar]
- [8].Mahmood T, Yang PC, Am N J. Med. Sci. 2012, 4, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cui C, Shu W, Li P, Front. Cell Dev. Biol. 2016, 4, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Harrington CA, Rosenow C, Retief J, Curr. Opin. Microbiol. 2000, 3, 285. [DOI] [PubMed] [Google Scholar]
- [11].Graham L, Orenstein JM, Nat. Protoc. 2007, 2, 2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lichtman JW, Fraser SE, Nat. Neurosci. 2001, 4, 1215. [DOI] [PubMed] [Google Scholar]
- [13].Stephens DJ, Allan VJ, Science 2003, 300, 82. [DOI] [PubMed] [Google Scholar]
- [14].Specht EA, Braselmann E, Palmer AE, Annu. Rev. Physiol. 2017, 79, 93. [DOI] [PubMed] [Google Scholar]
- [15].Duncan FE, Que EL, Zhang N, Feinberg EC, O’Halloran TV, Woodruff TK, Sci. Rep. 2016, 6, 24737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Godin AG, Lounis B, Cognet L, Biophys. J. 2014, 107, 1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sydor AM, Czymmek KJ, Puchner EM, Mennella V, Trends Cell Biol. 2015, 25, 730. [DOI] [PubMed] [Google Scholar]
- [18].Liu J, Cao Z, Lu Y, Chem. Rev. 2009, 109, 1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ, Nature 1996, 382, 607. [DOI] [PubMed] [Google Scholar]
- [20].Jones MR, Seeman NC, Mirkin CA, Science 2015, 347, 1260901. [DOI] [PubMed] [Google Scholar]
- [21].Jones MR, Mirkin CA, Nature 2012, 491, 42. [DOI] [PubMed] [Google Scholar]
- [22].Zhou W, Saran R, Liu J, Chem. Rev. 2017, 117, 8272. [DOI] [PubMed] [Google Scholar]
- [23].Stoltenburg R, Reinemann C, Strehlitz B, Biomol. Eng. 2007, 24, 381. [DOI] [PubMed] [Google Scholar]
- [24].Hollenstein M, Molecules 2015, 20, 20777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ellington AD, Szostak JW, Nature 1990, 346, 818. [DOI] [PubMed] [Google Scholar]
- [26].Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AKR, Han MS, Mirkin CA, Science 2006, 312, 1027. [DOI] [PubMed] [Google Scholar]
- [27].DeVos SL, Miller TM, Neurotherapeutics 2013, 10, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lannes L, Halder S, Krishnan Y, Schwalbe H, ChemBioChem 2015, 16, 1647. [DOI] [PubMed] [Google Scholar]
- [29].Prakash TP, Chem. Biodivers. 2011, 8, 1616. [DOI] [PubMed] [Google Scholar]
- [30].Verma S, Eckstein F, Annu. Rev. Biochem. 1998, 67, 99. [DOI] [PubMed] [Google Scholar]
- [31].Grünweller A, Hartmann RK, BioDrugs 2007, 21, 235. [DOI] [PubMed] [Google Scholar]
- [32].Teplova M, Minasov G, Tereshko V, Inamati GB, Cook PD, Manoharan M, Egli M, Nat. Struct. Biol. 1999, 6, 535. [DOI] [PubMed] [Google Scholar]
- [33].Vester B, Wengel J, Biochemistry 2004, 43, 13233. [DOI] [PubMed] [Google Scholar]
- [34].Eckstein F, Nucleic Acid Ther 2014, 24, 374. [DOI] [PubMed] [Google Scholar]
- [35].Farschtschi N, Gorenstein DG, Tetrahedron Lett. 1988, 29, 6843. [Google Scholar]
- [36].Corey DR, Abrams JM, Genome Biol. 2001, 2, 1015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pellestor F, Paulasova P, Eur. J. Hum. Genet. 2004, 12, 694. [DOI] [PubMed] [Google Scholar]
- [38].Garg A, Wesolowski D, Alonso D, Deitsch KW, Ben Mamoun C, Altman S, Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Prigodich AE, Randeria PS, Briley WE, Kim NJ, Daniel WL, Giljohann DA, Mirkin CA, Anal. Chem. 2012, 84, 2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim E, Yang J, Park J, Kim S, Kim NH, Yook JI, Suh JS, Haam S, Huh YM, ACS Nano 2012, 6, 8525. [DOI] [PubMed] [Google Scholar]
- [41].Park YK, Jung WY, Park MG, Song SK, Lee YS, Heo H, Kim S, Medchemcomm 2017, 8, 2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Levsky JM, Singer RH, Cell Sci J 2003, 116, 2833. [DOI] [PubMed] [Google Scholar]
- [43].Ke G, Wang C, Ge Y, Zheng N, Zhu Z, Yang CJ, J. Am. Chem. Soc. 2012, 134, 18908. [DOI] [PubMed] [Google Scholar]
- [44].Seferos DS, Giljohann DA, Hill HD, Prigodich AE, Mirkin CA, J. Am. Chem. Soc. 2007, 129, 15477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bunka DHJ, Stockley PG, Nat. Rev. Microbiol. 2006, 4, 588. [DOI] [PubMed] [Google Scholar]
- [46].“Apta-Index™ (Aptamer Database),” can be found under https://www.aptagen.com/
- [47].Hwang K, Hosseinzadeh P, Lu Y, Inorganica Chim. Acta 2016, 452, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Breaker RR, Joyce GF, Chem. Biol. 1994, 1, 223. [DOI] [PubMed] [Google Scholar]
- [49].Joyce GF, Annu. Rev. Biochem. 2004, 73, 791. [DOI] [PubMed] [Google Scholar]
- [50].Peng H, Li XF, Zhang H, Le XC, Nat. Commun. 2017, 8, 14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Famulok M, Hartig JS, Mayer G, Chem. Rev. 2007, 107, 3715. [DOI] [PubMed] [Google Scholar]
- [52].Bhatia D, Surana S, Chakraborty S, Koushika SP, Krishnan Y, Nat. Commun. 2011, 2, 339. [DOI] [PubMed] [Google Scholar]
- [53].Politz JC, Browne ES, Wolf DE, Pederson T, Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Molenaar C, Marras SA, Slats JC, Truffert JC, Lemaitre M, Raap AK, Dirks RW, Tanke HJ, Nucleic Acids Res. 2001, 29, E89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Paillasson S, Van De Corput M, Dirks RW, Tanke HJ, Robert-Nicoud M, Ronot X, Exp. Cell Res. 1997, 231, 226. [DOI] [PubMed] [Google Scholar]
- [56].Tsuji A, Koshimoto H, Sato Y, Hirano M, Sei-Iida Y, Kondo S, Ishibashi K, Biophys. J. 2000, 78, 3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Okabe K, Harada Y, Zhang J, Tadakuma H, Tani T, Funatsu T, Nucleic Acids Res. 2011, 39, e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dahan L, Huang L, Kedmi R, Behlke MA, Peer D, PLoS One 2013, 8, e72389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Matsuo T, Biochim. Biophys. Acta - Gen. Subj. 1998, 1379, 178. [DOI] [PubMed] [Google Scholar]
- [60].Kang WJ, Cho YL, Chae JR, Lee JD, Choi KJ, Kim S, Biomaterials 2011, 32, 1915. [DOI] [PubMed] [Google Scholar]
- [61].Nielsen LJ, Olsen LF, Ozalp VC, ACS Nano 2010, 4, 4361. [DOI] [PubMed] [Google Scholar]
- [62].Liang Y, Zhang Z, Wei H, Hu Q, Deng J, Guo D, Cui Z, Zhang XE, Biosens. Bioelectron. 2011, 28, 270. [DOI] [PubMed] [Google Scholar]
- [63].Sokol DL, Zhang X, Lu P, Gewirtz AM, Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhao D, Yang Y, Qu N, Chen M, Ma Z, Krueger CJ, Behlke MA, Chen AK, Biomaterials 2016, 100, 172. [DOI] [PubMed] [Google Scholar]
- [65].Medley CD, Drake TJ, Tomasini JM, Rogers RJ, Tan W, Anal. Chem. 2005, 77, 4713. [DOI] [PubMed] [Google Scholar]
- [66].Wu Y, Yang CJ, Moroz LL, Tan W, Anal. Chem. 2008, 80, 3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chen J, Wu J, Hong Y, Chem. Commun. 2016, 52, 3191. [DOI] [PubMed] [Google Scholar]
- [68].Peng XH, Cao ZH, Xia JT, Carlson GW, Lewis MM, Wood WC, Yang L, Cancer Res. 2005, 65, 1909. [DOI] [PubMed] [Google Scholar]
- [69].Bohländer PR, Abba ML, Bestvater F, Allgayer H, Wagenknecht HA, Org. Biomol. Chem. 2016, 14, 5001. [DOI] [PubMed] [Google Scholar]
- [70].Cheglakov Z, Cronin TM, He C, Weizmann Y, J. Am. Chem. Soc. 2015, 137, 6116. [DOI] [PubMed] [Google Scholar]
- [71].Liu L, Liu JW, Wu H, Wang XN, Yu RQ, Jiang JH, Anal. Chem. 2018, 90, 1502. [DOI] [PubMed] [Google Scholar]
- [72].Wu C, Cansiz S, Zhang L, Teng IT, Qiu L, Li J, Liu Y, Zhou C, Hu R, Zhang T, Cui C, Cui L, Tan W, J. Am. Chem. Soc. 2015, 137, 4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chen M, Ma Z, Wu X, Mao S, Yang Y, Tan J, Krueger CJ, Chen AK, Sci. Rep. 2017, 7, 1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Qiu L, Wu C, You M, Han D, Chen T, Zhu G, Jiang J, Yu R, Tan W, J. Am. Chem. Soc. 2013, 135, 12952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kim J, Lee E, Kang YY, Mok H, Chem. Commun. 2015, 51, 9038. [DOI] [PubMed] [Google Scholar]
- [76].King FW, Liszewski W, Ritner C, Bernstein HS, Stem Cells Dev. 2011, 20, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Santangelo PJ, Nix B, Tsourkas A, Bao G, Nucleic Acids Res. 2004, 32, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bratu DP, Cha B-J, Mhlanga MM, Kramer FR, Tyagi S, Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kam Y, Rubinstein A, Nissan A, Halle D, Yavin E, Mol. Pharm. 2012, 9, 685. [DOI] [PubMed] [Google Scholar]
- [80].Kummer S, Knoll A, Socher E, Bethge L, Herrmann A, Seitz O, Bioconjugate Chem. 2012, 23, 2051. [DOI] [PubMed] [Google Scholar]
- [81].Kummer S, Knoll A, Socher E, Bethge L, Herrmann A, Seitz O, Angew. Chem. Int. Ed. 2011, 50, 1931. [DOI] [PubMed] [Google Scholar]
- [82].Fang GM, Chamiolo J, Kankowski S, Hövelmann F, Friedrich D, Löwer A, Meier JC, Seitz O, Chem. Sci. 2018, 9, 4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hövelmann F, Gaspar I, Loibl S, Ermilov EA, Röder B, Wengel J, Ephrussi A, Seitz O, Angew. Chem. Int. Ed. 2014, 53, 11370. [DOI] [PubMed] [Google Scholar]
- [84].Kubota T, Ikeda S, Okamoto A, Bull. Chem. Soc. Jpn. 2009, 82, 110. [Google Scholar]
- [85].Ikeda S, Kubota T, Yuki M, Okamoto A, Angew. Chem. Int. Ed. 2009, 48, 6480. [DOI] [PubMed] [Google Scholar]
- [86].Oomoto I, Suzuki-Hirano A, Umeshima H, Han YW, Yanagisawa H, Carlton P, Harada Y, Kengaku M, Okamoto A, Shimogori T, Wang DO, Nucleic Acids Res. 2015, 43, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ikeda S, Yanagisawa H, Nakamura A, Wang DO, Yuki M, Okamoto A, Org. Biomol. Chem. 2011, 9, 4199. [DOI] [PubMed] [Google Scholar]
- [88].Kubota T, Ikeda S, Yanagisawa H, Yuki M, Okamoto A, Bioconjugate Chem. 2009, 20, 1256. [DOI] [PubMed] [Google Scholar]
- [89].Gao R, Hao C, Xu L, Xu C, Kuang H, Anal. Chem. 2018, 90, 5414. [DOI] [PubMed] [Google Scholar]
- [90].Chen T, Wu CS, Jimenez E, Zhu Z, Dajac JG, You M, Han D, Zhang X, Tan W, Angew. Chem. Int. Ed. 2013, 52, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Li H, Mu Y, Lu J, Wei W, Wan Y, Liu S, Anal. Chem. 2014, 86, 3602. [DOI] [PubMed] [Google Scholar]
- [92].Noh EH, Ko HY, Lee CH, Jeong MS, Chang YW, Kim S, J. Mater. Chem. B 2013, 1, 4438. [DOI] [PubMed] [Google Scholar]
- [93].Lee J, Moon S, Lee Y, Ali B, Al-Khedhairy A, Ali D, Ahmed J, Al Salem A, Kim S, Sensors 2015, 15, 12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Pan W, Yang H, Li N, Yang L, Tang B, Chem. - Eur. J. 2015, 21, 6070. [DOI] [PubMed] [Google Scholar]
- [95].Zheng D, Seferos DS, Giljohann DA, Patel PC, Mirkin CA, Nano Lett. 2009, 9, 3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Huang J, Ying L, Yang X, Yang Y, Quan K, Wang H, Xie N, Ou M, Zhou Q, Wang K, Anal. Chem. 2015, 87, 8724. [DOI] [PubMed] [Google Scholar]
- [97].Wu P, Hwang K, Lan T, Lu Y, J. Am. Chem. Soc. 2013, 135, 5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Jayagopal A, Halfpenny KC, Perez JW, Wright DW, J. Am. Chem. Soc. 2010, 132, 9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Zhou W, Li Q, Liu H, Yang J, Liu D, ACS Nano 2017, 11, 3532. [DOI] [PubMed] [Google Scholar]
- [100].Choi CKK, Choi CHJ, Bian L, Methods Mol. Biol. 2017, 1570, 155. [DOI] [PubMed] [Google Scholar]
- [101].Zhu HZ, An JH, Yao Q, Han J, Li XT, Jiang FL, Chen GP, Peng LN, Li YS, Sun JG, Chen ZT, Molecules 2014, 19, 14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Yang L, Ren Y, Pan W, Yu Z, Tong L, Li N, Tang B, Anal. Chem. 2016, 88, 11886. [DOI] [PubMed] [Google Scholar]
- [103].Yang K, Zeng M, Fu X, Li J, Ma N, Tao L, RSC Adv. 2015, 5, 104245. [Google Scholar]
- [104].Ryoo SR, Lee J, Yeo J, Na HK, Kim YK, Jang H, Lee JH, Han SW, Lee Y, Kim VN, Min DH, ACS Nano 2013, 7, 5882. [DOI] [PubMed] [Google Scholar]
- [105].Wu Y, Han J, Xue P, Xu R, Kang Y, Nanoscale 2015, 7, 1753. [DOI] [PubMed] [Google Scholar]
- [106].Lin L-S, Cong Z-X, Cao J-B, Ke K-M, Peng Q-L, Gao J, Yang H-H, Liu G, Chen X, ACS Nano 2014, 8, 3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Dai W, Dong H, Fugetsu B, Cao Y, Lu H, Ma X, Zhang X, Small 2015, 11, 4158. [DOI] [PubMed] [Google Scholar]
- [108].Wu Z, Liu GQ, Yang XL, Jiang JH, J. Am. Chem. Soc. 2015, 137, 6829. [DOI] [PubMed] [Google Scholar]
- [109].Wang Y, Li Z, Hu D, Lin CT, Li J, Lin Y, J. Am. Chem. Soc. 2010, 132, 9274. [DOI] [PubMed] [Google Scholar]
- [110].Zhang C-H, Wang H, Liu J-W, Sheng Y-Y, Chen J, Zhang P, Jiang J-H, ACS Sens. 2018, 3, 2526. [DOI] [PubMed] [Google Scholar]
- [111].Shao C, Liang J, He S, Luan T, Yu J, Zhao H, Xu J, Tian L, Anal. Chem. 2017, 89, 5445. [DOI] [PubMed] [Google Scholar]
- [112].Wiraja C, Yeo DC, Chew SY, Xu C, J. Mater. Chem. B 2015, 3, 6148. [DOI] [PubMed] [Google Scholar]
- [113].He L, Lu DQ, Liang H, Xie S, Luo C, Hu M, Xu L, Zhang X, Tan W, ACS Nano 2017, 11, 4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Zhou W, Li D, Xiong C, Yuan R, Xiang Y, ACS Appl. Mater. Interfaces 2016, 8, 13303. [DOI] [PubMed] [Google Scholar]
- [115].Modi S, Nizak C, Surana S, Halder S, Krishnan Y, Nat. Nanotechnol. 2013, 8, 459. [DOI] [PubMed] [Google Scholar]
- [116].Narayanaswamy N, Chakraborty K, Saminathan A, Zeichner E, Leung K, Devany J, Krishnan Y, Nat. Methods 2019, 16, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Leung K, Chakraborty K, Saminathan A, Krishnan Y, Nat. Nanotechnol. 2019, 14, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Thekkan S, Jani MS, Cui C, Dan K, Zhou G, Becker L, Krishnan Y, Nat. Chem. Biol. 2018, DOI 10.1038/s41589-018-0176-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Tay CY, Yuan L, Leong DT, ACS Nano 2015, 9, 5609. [DOI] [PubMed] [Google Scholar]
- [120].Li S, Xu L, Ma W, Wu X, Sun M, Kuang H, Wang L, Kotov NA, Xu C, J. Am. Chem. Soc. 2016, 138, 306. [DOI] [PubMed] [Google Scholar]
- [121].Paige JS, Wu KY, Jaffrey SR, Science 2011, 333, 6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Paige JS, Nguyen-Duc T, Song W, Jaffrey SR, Science 2012, 335, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Song W, Strack RL, Jaffrey SR, Nat. Methods 2013, 10, 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Köhler O, Jarikote DV, Seitz O, ChemBioChem 2005, 6, 69. [DOI] [PubMed] [Google Scholar]
- [125].Tyagi S, Kramer FR, Nat. Biotechnol. 1996, 14, 303. [DOI] [PubMed] [Google Scholar]
- [126].Zhou W, Ding J, Liu J, Org. Biomol. Chem. 2017, 15, 6959. [DOI] [PubMed] [Google Scholar]
- [127].Lu CH, Zhu CL, Li J, Liu JJ, Chen X, Yang HH, Chem. Commun. 2010, 46, 3116. [DOI] [PubMed] [Google Scholar]
- [128].Lee K, Cui Y, Lee LP, Irudayaraj J, Nat. Nanotechnol. 2014, 9, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Yeh HC, Sharma J, Han JJ, Martinez JS, Werner JH, Nano Lett. 2010, 10, 3106. [DOI] [PubMed] [Google Scholar]
- [130].Ren K, Xu Y, Liu Y, Yang M, Ju H, ACS Nano 2018, 12, 263. [DOI] [PubMed] [Google Scholar]
- [131].Su X, Xiao X, Zhang C, Zhao M, Appl. Spectrosc. 2012, 66, 1249. [DOI] [PubMed] [Google Scholar]
- [132].Chin LK, Lee CH, Chen BC, Lab Chip 2016, 16, 2014. [DOI] [PubMed] [Google Scholar]
- [133].Voss S, Krüger DM, Koch O, Wu Y-W, Proc. Natl. Acad. Sci. 2016, 113, 14348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Briley WE, Bondy MH, Randeria PS, Dupper TJ, Mirkin CA, Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Yeo DC, Wiraja C, Paller AS, Mirkin CA, Xu C, Nat. Biomed. Eng. 2018, 2, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Yu M, Stott S, Toner M, Maheswaran S, Haber DA, Cell Biol J 2011, 192, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Balasubramanian S, Kagan D, Jack Hu CM, Campuzano S, Lobo-Castañon MJ, Lim N, Kang DY, Zimmerman M, Zhang L, Wang J, Angew. Chemie - Int. Ed. 2011, 50, 4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Chen Y, Li P, Huang PH, Xie Y, Mai JD, Wang L, Nguyen NT, Huang TJ, Lab Chip 2014, 14, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Halo TL, McMahon KM, Angeloni NL, Xu Y, Wang W, Chinen AB, Malin D, Strekalova E, Cryns VL, Cheng C, Mirkin CA, Thaxton CS, Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Battich N, Stoeger T, Pelkmans L, Nat. Methods 2013, 10, 1127. [DOI] [PubMed] [Google Scholar]
- [141].Tan X, Chen T, Xiong X, Mao Y, Zhu G, Yasun E, Li C, Zhu Z, Tan W, Anal. Chem. 2012, 84, 8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Sanderson J, Understanding Light Microscopy, Wiley, 2019. [Google Scholar]
- [143].Zhang P, Beck T, Tan W, Angew. Chemie - Int. Ed. 2001, 40, 402. [PubMed] [Google Scholar]
- [144].Bunt G, Wouters FS, Biophys. Rev. 2017, 9, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Socher E, Bethge L, Knoll A, Jungnick N, Herrmann A, Seitz O, Angew. Chem. Int. Ed. 2008, 47, 9555. [DOI] [PubMed] [Google Scholar]
- [146].Kuhn H, V Demidov V, Coull JM, Fiandaca MJ, Gildea BD, Frank-Kamenetskii MD, J. Am. Chem. Soc. 2002, 124, 1097. [DOI] [PubMed] [Google Scholar]
- [147].Murayama K, Kamiya Y, Kashida H, Asanuma H, ChemBioChem 2015, 16, 1298. [DOI] [PubMed] [Google Scholar]
- [148].Wang K, Tang Z, Yang CJ, Kim Y, Fang X, Li W, Wu Y, Medley CD, Cao Z, Li J, Colon P, Lin H, Tan W, Angew. Chem. Int. Ed. 2009, 48, 856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Chen AK, Behlke MA, Tsourkas A, Nucleic Acids Res. 2007, 35, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Chen AK, Behlke MA, Tsourkas A, Nucleic Acids Res. 2008, 36, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Mhlanga MM, Vargas DY, Fung CW, Kramer FR, Tyagi S, Nucleic Acids Res. 2005, 33, 1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Dirks RM, Pierce NA, Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Yin P, Choi HMT, Calvert CR, Pierce NA, Nature 2008, 451, 318. [DOI] [PubMed] [Google Scholar]
- [154].Hövelmann F, Gaspar I, Chamiolo J, Kasper M, Steffen J, Ephrussi A, Seitz O, Chem. Sci. 2016, 7, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Kummer S, Knoll A, Herrmann A, Seitz O, Methods Mol. Biol. 2013, 1039, 291. [DOI] [PubMed] [Google Scholar]
- [156].Kolevzon N, Hashoul D, Naik S, Rubinstein A, Yavin E, Chem. Commun. 2016, 52, 2405. [DOI] [PubMed] [Google Scholar]
- [157].Hövelmann F, Gaspar I, Ephrussi A, Seitz O, J. Am. Chem. Soc. 2013, 135, 19025. [DOI] [PubMed] [Google Scholar]
- [158].Hövelmann F, Bethge L, Seitz O, ChemBioChem 2012, 13, 2072. [DOI] [PubMed] [Google Scholar]
- [159].Chamiolo J, Fang G, Hövelmann F, Friedrich D, Knoll A, Loewer A, Seitz O, ChemBioChem 2018, 20, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Okamoto A, Chem. Soc. Rev. 2011, 40, 5815. [DOI] [PubMed] [Google Scholar]
- [161].Min X, Zhang M, Huang F, Lou X, Xia F, ACS Appl. Mater. Interfaces 2016, 8, 8998. [DOI] [PubMed] [Google Scholar]
- [162].Ro JJ, Lee HJ, Kim BH, Chem. Commun. 2018, 54, 7471. [DOI] [PubMed] [Google Scholar]
- [163].Østergaard ME, Cheguru P, Papasani MR, Hill RA, Hrdlicka PJ, J. Am. Chem. Soc. 2010, 132, 14221. [DOI] [PubMed] [Google Scholar]
- [164].Chinen AB, Guan CM, Ferrer JR, Barnaby SN, Merkel TJ, Mirkin CA, Chem. Rev. 2015, 115, 10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Cutler JI, Auyeung E, Mirkin CA, J. Am. Chem. Soc. 2012, 134, 1376. [DOI] [PubMed] [Google Scholar]
- [166].Prigodich AE, Seferos DS, Massich MD, Giljohann DA, Lane BC, Mirkin CA, ACS Nano 2009, 3, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Choi CKK, Li J, Wei K, Xu YJ, Ho LWC, Zhu M, To KKW, Choi CHJ, Bian L, J. Am. Chem. Soc. 2015, 137, 7337. [DOI] [PubMed] [Google Scholar]
- [168].Zheng A, Zhang X, Huang Y, Cai Z, Liu X, Liu J, RSC Adv. 2018, 8, 6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Sun J, Pi F, Ji J, Lei H, Gao Z, Zhang Y, Habimana JDD, Li Z, Sun X, Anal. Chem. 2018, 90, 3099. [DOI] [PubMed] [Google Scholar]
- [170].Noble PB, Cutts JH, Can. Vet. J. 1967, 8, 110. [PMC free article] [PubMed] [Google Scholar]
- [171].Vilela P, Heuer-Jungemann A, El-Sagheer A, Brown T, Muskens OL, Smyth NR, Kanaras AG, Small 2018, 14, e1703489. [DOI] [PubMed] [Google Scholar]
- [172].Moros M, Kyriazi M-E, El-Sagheer AH, Brown T, Tortiglione C, Kanaras AG, ACS Appl. Mater. Interfaces 2018, 11, 13905. [DOI] [PubMed] [Google Scholar]
- [173].Sozer S, Aptullahoglu E, Shivarov V, Yavuz AS, Int. J. Lab. Hematol. 2019, DOI 10.1111/ijlh.12991. [DOI] [PubMed] [Google Scholar]
- [174].Li N, Chang C, Pan W, Tang B, Angew. Chem. Int. Ed. 2012, 51, 7426. [DOI] [PubMed] [Google Scholar]
- [175].Yang Y, Huang J, Yang X, Quan K, Wang H, Ying L, Xie N, Ou M, Wang K, J. Am. Chem. Soc. 2015, 137, 8340. [DOI] [PubMed] [Google Scholar]
- [176].Liang CP, Ma PQ, Liu H, Guo X, Yin BC, Ye BC, Angew. Chem. Int. Ed. 2017, 56, 9077. [DOI] [PubMed] [Google Scholar]
- [177].Li D, Zhou W, Yuan R, Xiang Y, Anal. Chem. 2017, 89, 9934. [DOI] [PubMed] [Google Scholar]
- [178].Da Sacco S, Thornton ME, Petrosyan A, Lavarreda-Pearce M, Sedrakyan S, Grubbs BH, De Filippo RE, Perin L, Stem Cells Transl. Med. 2017, 6, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Zakiryanova GK, Kustova E, Urazalieva NT, Amirbekov A, Baimuchametov ET, Nakisbekov NN, Shurin MR, Immunity, Inflamm. Dis. 2017, 5, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Heun Y, Pogoda K, Anton M, Pircher J, Pfeifer A, Woernle M, Ribeiro A, Kameritsch P, Mykhaylyk O, Plank C, Kroetz F, Pohl U, Mannell H, Mol. Ther. 2017, 25, 1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Amatya VJ, Mawas AS, Kushitani K, El-Din MMM, Takeshima Y, Int. J. Oncol. 2016, 48, 1599. [DOI] [PubMed] [Google Scholar]
- [182].Harry SR, Hicks DJ, Amiri KI, Wright DW, Chem. Commun. 2010, 46, 5557. [DOI] [PubMed] [Google Scholar]
- [183].Xue J, Shan L, Chen H, Li Y, Zhu H, Deng D, Qian Z, Achilefu S, Gu Y, Biosens. Bioelectron. 2013, 41, 71. [DOI] [PubMed] [Google Scholar]
- [184].Uddin MDI, Jayagopal A, Wong A, McCollum GW, Wright DW, Penn JS, Nanomedicine 2018, 14, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Qiao G, Gao Y, Li N, Yu Z, Zhuo L, Tang B, Chem. - Eur. J. 2011, 17, 11210. [DOI] [PubMed] [Google Scholar]
- [186].Pan W, Zhang T, Yang H, Diao W, Li N, Tang B, Anal. Chem. 2013, 85, 10581. [DOI] [PubMed] [Google Scholar]
- [187].Bao C, Conde J, Curtin J, Artzi N, Tian F, Cui D, Sci. Rep. 2015, 5, 12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Conde J, Rosa J, de la Fuente JM, V Baptista P, Biomaterials 2013, 34, 2516. [DOI] [PubMed] [Google Scholar]
- [189].Pan W, Li Y, Wang M, Yang H, Li N, Tang B, Chem. Commun. 2016, 52, 4569. [DOI] [PubMed] [Google Scholar]
- [190].Qiang W, Li W, Li X, Chen X, Xu D, Chem. Sci. 2014, 5, 3018. [Google Scholar]
- [191].Cheng Y, Zhang S, Kang N, Huang J, Lv X, Wen K, Ye S, Chen Z, Zhou X, Ren L, ACS Appl. Mater. Interfaces 2017, 9, 19296. [DOI] [PubMed] [Google Scholar]
- [192].Nam J, Son S, Ochyl LJ, Kuai R, Schwendeman A, Moon JJ, Nat. Commun. 2018, 9, 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Liu J, Du P, Zhang J, Shen H, Lei J, Chem. Commun. 2018, 54, 2550. [DOI] [PubMed] [Google Scholar]
- [194].Wang S, Riahi R, Li N, Zhang DD, Wong PK, Adv. Mater. 2015, 27, 6034. [DOI] [PubMed] [Google Scholar]
- [195].Ali MRK, Wu Y, Tang Y, Xiao H, Chen K, Han T, Fang N, Wu R, El-Sayed MA, Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Wu Y, Ali MRK, Dong B, Han T, Chen K, Chen J, Tang Y, Fang N, Wang F, El-Sayed MA, ACS Nano 2018, 12, 9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Yin D, Li X, Ma Y, Liu Z, Chem. Commun. 2017, 53, 6716. [DOI] [PubMed] [Google Scholar]
- [198].Riahi R, Wang S, Long M, Li N, Chiou PY, Zhang DD, Wong PK, ACS Nano 2014, 8, 3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Yan N, Wang X, Lin L, Song T, Sun P, Tian H, Liang H, Chen X, Adv. Funct. Mater. 2018, 28, 1800490. [Google Scholar]
- [200].Ma W, Fu P, Sun M, Xu L, Kuang H, Xu C, J. Am. Chem. Soc. 2017, 139, 11752. [DOI] [PubMed] [Google Scholar]
- [201].Jo HL, Song YH, Park J, Jo EJ, Goh Y, Shin K, Kim MG, Lee KT, Nanoscale 2015, 7, 19397. [DOI] [PubMed] [Google Scholar]
- [202].Wu S, Butt HJ, Adv. Mater. 2016, 28, 1208. [DOI] [PubMed] [Google Scholar]
- [203].Alavi M, Karimi N, Safaei M, Adv. Pharm. Bull. 2017, 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Han S, Kang B, Jang E, Ki J, Kim E, Jeong MY, Huh YM, Son HY, Haam S, Adv. Healthc. Mater. 2018, 7, 1701019. [DOI] [PubMed] [Google Scholar]
- [205].Wang Z, Zhang K, Shen Y, Smith J, Bloch S, Achilefu S, Wooley KL, Taylor JS, Org. Biomol. Chem. 2013, 11, 3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Zhang R, Gao S, Wang Z, Han D, Liu L, Ma Q, Tan W, Tian J, Chen X, Adv. Funct. Mater. 2017, 27, 1701027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Gao W, Wei X, Wang X, Cui G, Liu Z, Tang B, Chem. Commun. 2016, 52, 3643. [DOI] [PubMed] [Google Scholar]
- [208].Shen X, Wang Y, Zhang Y, Ouyang J, Na N, Adv. Funct. Mater. 2018, 28, 1803286. [Google Scholar]
- [209].Wang Y, Yu Z, Zhang Z, Ren R, Zhang S, Analyst 2016, 141, 2861. [DOI] [PubMed] [Google Scholar]
- [210].Yuan Y, Wu S, Shu F, Liu Z, Chem. Commun. 2014, 50, 1095. [DOI] [PubMed] [Google Scholar]
- [211].Zhang XL, Zheng C, Guo SS, Li J, Yang HH, Chen G, Anal. Chem. 2014, 86, 3426. [DOI] [PubMed] [Google Scholar]
- [212].Li J, Li D, Yuan R, Xiang Y, ACS Appl. Mater. Interfaces 2017, 9, 5717. [DOI] [PubMed] [Google Scholar]
- [213].Ou M, Huang J, Yang X, He X, Quan K, Yang Y, Xie N, Li J, Wang K, ChemBioChem 2018, 19, 147. [DOI] [PubMed] [Google Scholar]
- [214].Li J, You J, Zhuang Y, Han C, Hu J, Wang A, Xu K, Zhu JJ, Chem. Commun. 2014, 50, 7107. [DOI] [PubMed] [Google Scholar]
- [215].Kunwar P, Hassinen J, Bautista G, Ras RHA, Toivonen J, ACS Nano 2014, 8, 11165. [DOI] [PubMed] [Google Scholar]
- [216].Díez I, Ras RHA, Nanoscale 2011, 3, 1963. [DOI] [PubMed] [Google Scholar]
- [217].Dong H, Ding L, Yan F, Ji H, Ju H, Biomaterials 2011, 32, 3875. [DOI] [PubMed] [Google Scholar]
- [218].Chung C, Kim YK, Shin D, Ryoo SR, Hong BH, Min DH, Acc. Chem. Res. 2013, 46, 2211. [DOI] [PubMed] [Google Scholar]
- [219].Pan W, Liu B, Gao X, Yu Z, Liu X, Li N, Tang B, Nanoscale 2018, 10, 14264. [DOI] [PubMed] [Google Scholar]
- [220].Li L, Feng J, Liu H, Li Q, Tong L, Tang B, Chem. Sci. 2016, 7, 1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Yang L, Liu B, Wang M, Li J, Pan W, Gao X, Li N, Tang B, ACS Appl. Mater. Interfaces 2018, 10, 6982. [DOI] [PubMed] [Google Scholar]
- [222].Li J, Li J, Zhou J, Liu T, Chen S, Yang H, Chem. Commun. 2018, 54, 896. [DOI] [PubMed] [Google Scholar]
- [223].Yu J, He S, Shao C, Zhao H, Li J, Tian L, Nanoscale 2018, 10, 7067. [DOI] [PubMed] [Google Scholar]
- [224].Liao X, Ju H, Chem. Commun. 2015, 51, 2141. [DOI] [PubMed] [Google Scholar]
- [225].Ke K, Lin L, Liang H, Chen X, Han C, Li J, Yang HH, Chem. Commun. 2015, 51, 6800. [DOI] [PubMed] [Google Scholar]
- [226].Piao Y, Liu F, Seo TS, ACS Appl. Mater. Interfaces 2012, 4, 6785. [DOI] [PubMed] [Google Scholar]
- [227].Wu Y, Phillips JA, Liu H, Yang R, Tan W, ACS Nano 2008, 2, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Zhu X, Zheng H, Wei X, Lin Z, Guo L, Qiu B, Chen G, Chem. Commun. 2013, 49, 1276. [DOI] [PubMed] [Google Scholar]
- [229].Chen L, Zheng H, Zhu X, Lin Z, Guo L, Qiu B, Chen G, Chen ZN, Analyst 2013, 138, 3490. [DOI] [PubMed] [Google Scholar]
- [230].Li H, Eddaoudi M, O’Keeffe M, Yaghi OM, Nature 1999, 402, 276. [Google Scholar]
- [231].Rowsell JLC, Yaghi OM, Microporous Mesoporous Mater. 2004, 73, 3. [Google Scholar]
- [232].Chen K, Ong YH, Yuen C, Liu Q, in Imaging in Dermatology, (Eds: Hamblin MR, Avci P, Gupta GK), Academic Press, 2016, pp. 141–154. [Google Scholar]
- [233].Xie N, Liu S, Yang X, He X, Huang J, Wang K, Analyst 2017, 142, 3322. [DOI] [PubMed] [Google Scholar]
- [234].Xie N, Huang J, Yang X, Yang Y, Quan K, Wang H, Ying L, Ou M, Wang K, Chem. Commun. 2016, 52, 2346. [DOI] [PubMed] [Google Scholar]
- [235].Wang S, Xia M, Liu J, Zhang S, Zhang X, ACS Sens. 2017, 2, 735. [DOI] [PubMed] [Google Scholar]
- [236].He L, Lu D, Liang H, Xie S, Zhang X, Liu Q, Yuan Q, Tan W, J. Am. Chem. Soc. 2018, 140, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S, Science 2005, 307, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Gao X, Yang L, Petros JA, Marshall FF, Simons JW, Nie S, Curr. Opin. Biotechnol. 2005, 16, 63. [DOI] [PubMed] [Google Scholar]
- [239].Dong H, Dai W, Ju H, Lu H, Wang S, Xu L, Zhou SF, Zhang Y, Zhang X, ACS Appl. Mater. Interfaces 2015, 7, 11015. [DOI] [PubMed] [Google Scholar]
- [240].Wu D, Song G, Li Z, Zhang T, Wei W, Chen M, He X, Ma N, Chem. Sci. 2015, 6, 3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].He X, Zeng T, Li Z, Wang G, Ma N, Angew. Chem. Int. Ed. 2016, 55, 3073. [DOI] [PubMed] [Google Scholar]
- [242].Shen Y, Li Z, Wang G, Ma N, ACS Sens. 2018, 3, 494. [DOI] [PubMed] [Google Scholar]
- [243].Modi S, Swetha MG, Goswami D, Gupta GD, Mayor S, Krishnan Y, Nat. Nanotechnol. 2009, 4, 325. [DOI] [PubMed] [Google Scholar]
- [244].Modi S, Halder S, Nizak C, Krishnan Y, Nanoscale 2014, 6, 1144. [DOI] [PubMed] [Google Scholar]
- [245].Halder S, Krishnan Y, Nanoscale 2015, 7, 10008. [DOI] [PubMed] [Google Scholar]
- [246].Surana S, Bhat JM, Koushika SP, Krishnan Y, Nat. Commun. 2011, 2, 340. [DOI] [PubMed] [Google Scholar]
- [247].Surana S, Krishnan Y, Methods Mol. Biol. 2013, DOI 10.1007/978-1-62703-336-7_2. [DOI] [PubMed] [Google Scholar]
- [248].Veetil AT, Chakraborty K, Xiao K, Minter MR, Sisodia SS, Krishnan Y, Nat. Nanotechnol. 2017, 12, 1183. [DOI] [PubMed] [Google Scholar]
- [249].Bhatia D, Arumugam S, Nasilowski M, Joshi H, Wunder C, Chambon V, Prakash V, Grazon C, Nadal B, Maiti PK, Johannes L, Dubertret B, Krishnan Y, Nat. Nanotechnol. 2016, 11, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Saha S, Prakash V, Halder S, Chakraborty K, Krishnan Y, Nat. Nanotechnol. 2015, 10, 645. [DOI] [PubMed] [Google Scholar]
- [251].Prakash V, Saha S, Chakraborty K, Krishnan Y, Chem. Sci. 2016, 7, 1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Yang M, Zhang X, Liu H, Kang H, Zhu Z, Yang W, Tan W, Anal. Chem. 2015, 87, 5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Pei H, Liang L, Yao G, Li J, Huang Q, Fan C, Angew. Chem. Int. Ed. 2012, 51, 9020. [DOI] [PubMed] [Google Scholar]
- [254].Ohtsuka K, Sato S, Sato Y, Sota K, Ohzawa S, Matsuda T, Takemoto K, Takamune N, Juskowiak B, Nagai T, Takenaka S, Chem. Commun. 2012, 48, 4740. [DOI] [PubMed] [Google Scholar]
- [255].Chakraborty K, Veetil A, Jaffrey SR, Krishnan Y, Annu. Rev. Biochem. 2016, 85, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Nutiu R, Li Y, J. Am. Chem. Soc. 2003, 125, 4771. [DOI] [PubMed] [Google Scholar]
- [257].Yang Y, Huang J, Yang X, Quan K, Xie N, Ou M, Tang J, Wang K, Chem. Commun. 2016, 52, 11386. [DOI] [PubMed] [Google Scholar]
- [258].Wang C, Du Y, Wu Q, Xuan S, Zhou J, Song J, Shao F, Duan H, Chem. Commun. 2013, 49, 5739. [DOI] [PubMed] [Google Scholar]
- [259].Xu X, Rosi NL, Wang Y, Huo F, Mirkin CA, J. Am. Chem. Soc. 2006, 128, 9286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Huo F, Lytton-Jean AKR, Mirkin CA, Adv. Mater. 2006, 18, 2304. [Google Scholar]
- [261].Ma PQ, Liang CP, Zhang HH, Yin BC, Ye BC, Chem. Sci. 2018, 9, 3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Wang Y, Li Z, Weber TJ, Hu D, Lin CT, Li J, Lin Y, Anal. Chem. 2013, 85, 6755. [DOI] [PubMed] [Google Scholar]
- [263].Wang Y, Tang L, Li Z, Lin Y, Li J, Nat. Protoc. 2014, 9, 1944. [DOI] [PubMed] [Google Scholar]
- [264].Chen TT, Tian X, Liu CL, Ge J, Chu X, Li Y, J. Am. Chem. Soc. 2015, 137, 982. [DOI] [PubMed] [Google Scholar]
- [265].Li XM, Song J, Cheng T, Fu PY, Anal. Bioanal. Chem, 2013, 405, 5993. [DOI] [PubMed] [Google Scholar]
- [266].Ma L, Liu F, Lei Z, Wang Z, Biosens. Bioelectron. 2017, 87, 638. [DOI] [PubMed] [Google Scholar]
- [267].Zhao J, Gao J, Xue W, Di Z, Xing H, Lu Y, Li L, J. Am. Chem. Soc. 2018, 140, 578. [DOI] [PubMed] [Google Scholar]
- [268].Özalp VC, Nielsen LJ, Olsen LF, ChemBioChem 2010, 11, 2538. [DOI] [PubMed] [Google Scholar]
- [269].Özalp VC, Pedersen TR, Nielsen LJ, Olsen LF, J. Biol. Chem. 2010, 285, 37579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Tang Z, Mallikaratchy P, Yang R, Kim Y, Zhu Z, Wang H, Tan W, J. Am. Chem. Soc. 2008, 130, 11268. [DOI] [PubMed] [Google Scholar]
- [271].Qiang W, Hu H, Sun L, Li H, Xu D, Anal. Chem. 2015, 87, 12190. [DOI] [PubMed] [Google Scholar]
- [272].Wu C, Chen T, Han D, You M, Peng L, Cansiz S, Zhu G, Li C, Xiong X, Jimenez E, Yang CJ, Tan W, ACS Nano 2013, 7, 5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Zhao Z, Fan H, Zhou G, Bai H, Liang H, Wang R, Zhang X, Tan W, J. Am. Chem. Soc. 2014, 136, 11220. [DOI] [PubMed] [Google Scholar]
- [274].Deng K, Hou Z, Li X, Li C, Zhang Y, Deng X, Cheng Z, Lin J, Sci. Rep. 2015, 5, 7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Liu Y, Hou W, Xia L, Cui C, Wan S, Jiang Y, Yang Y, Wu Q, Qiu L, Tan W, Chem. Sci. 2018, 9, 7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Song W, Strack RL, Svensen N, Jaffrey SR, J. Am. Chem. Soc. 2014, 136, 1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Stojanovic MN, Kolpashchikov DM, J. Am. Chem. Soc. 2004, 126, 9266. [DOI] [PubMed] [Google Scholar]
- [278].Strack RL, Song W, Jaffrey SR, Nat. Protoc. 2014, 9, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Kellenberger CA, Wilson SC, Sales-Lee J, Hammond MC, J. Am. Chem. Soc. 2013, 135, 4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].You M, Litke JL, Jaffrey SR, Proc. Natl. Acad. Sci. U.S.A. 2015, 112, E2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Litke JL, You M, Jaffrey SR, Methods Enzymol. 2016, 572, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Pothoulakis G, Ceroni F, Reeve B, Ellis T, ACS Synth. Biol. 2014, 3, 182. [DOI] [PubMed] [Google Scholar]
- [283].Ong WQ, Citron YR, Sekine S, Huang B, ACS Chem. Biol. 2017, 12, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Strack RL, Disney MD, Jaffrey SR, Nat. Methods 2013, 10, 1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Strack RL, Jaffrey SR, Methods Enzymol. 2015, 550, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Zhang J, Fei J, Leslie BJ, Han KY, Kuhlman TE, Ha T, Sci. Rep. 2015, 5, 17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Warner KD, Chen MC, Song W, Strack RL, Thorn A, Jaffrey SR, Ferré-D’Amaré AR, Nat. Struct. Mol. Biol. 2014, 21, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Okuda M, Fourmy D, Yoshizawa S, Nucleic Acids Res. 2017, 45, 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Filonov GS, Moon JD, Svensen N, Jaffrey SR, J. Am. Chem. Soc. 2014, 136, 16299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Shu D, Khisamutdinov EF, Zhang L, Guo P, Nucleic Acids Res. 2014, 42, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Svensen N, Jaffrey SR, Cell Chem. Biol. 2016, 23, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Dolgosheina EV, Jeng SCY, Panchapakesan SSS, Cojocaru R, Chen PSK, Wilson PD, Hawkins N, Wiggins PA, Unrau PJ, ACS Chem. Biol. 2014, 9, 2412. [DOI] [PubMed] [Google Scholar]
- [293].Autour A, Jeng SCY, Cawte AD, Abdolahzadeh A, Galli A, Panchapakesan SSS, Rueda D, Ryckelynck M, Unrau PJ, Nat. Commun. 2018, 9, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Song W, Filonov GS, Kim H, Hirsch M, Li X, Moon JD, Jaffrey SR, Nat. Chem. Biol. 2017, 13, 1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Warner KD, Sjekloća L, Song W, Filonov GS, Jaffrey SR, Ferré-D’Amaré AR, Nat. Chem. Biol. 2017, 13, 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Shin I, Ray J, Gupta V, Ilgu M, Beasley J, Bendickson L, Mehanovic S, Kraus GA, Nilsen-Hamilton M, Nucleic Acids Res. 2014, 42, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Ilgu M, Ray J, Bendickson L, Wang T, Geraskin IM, Kraus GA, Nilsen-Hamilton M, Methods 2016, 98, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Jepsen MDE, Sparvath SM, Nielsen TB, Langvad AH, Grossi G, Gothelf KV, Andersen ES, Nat. Commun. 2018, 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].You M, Jaffrey SR, Annu. Rev. Biophys. 2015, 44, 187. [DOI] [PubMed] [Google Scholar]
- [300].Ouellet J, Front. Chem. 2016, 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Chen A, Yan M, Yang S, Trends Anal. Chem. 2016, 80, 581. [Google Scholar]
- [302].Zheng X, Peng R, Jiang X, Wang Y, Xu S, Ke G, Fu T, Liu Q, Huan S, Zhang X, Anal. Chem. 2017, 89, 10941. [DOI] [PubMed] [Google Scholar]
- [303].Wang Z, Luo Y, Xie X, Hu X, Song H, Zhao Y, Shi J, Wang L, Glinsky G, Chen N, Lal R, Fan C, Angew. Chem. Int. Ed. 2018, 57, 972. [DOI] [PubMed] [Google Scholar]
- [304].Alam KK, Tawiah KD, Lichte MF, Porciani D, Burke DH, ACS Synth. Biol. 2017, 6, 1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Strauss S, Nickels PC, Strauss MT, Jimenez Sabinina V, Ellenberg J, Carter JD, Gupta S, Janjic N, Jungmann R, Nat. Methods 2018, 15, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Schnitzbauer J, Strauss MT, Schlichthaerle T, Schueder F, Jungmann R, Nat. Protoc. 2017, 12, 1198. [DOI] [PubMed] [Google Scholar]
- [307].Medley CD, Smith JE, Tang Z, Wu Y, Bamrungsap S, Tan W, Anal. Chem. 2008, 80, 1067. [DOI] [PubMed] [Google Scholar]
- [308].Wang K, Fan D, Liu Y, Wang E, Biosens. Bioelectron. 2015, 73, 1. [DOI] [PubMed] [Google Scholar]
- [309].Liu G, Mao X, Phillips JA, Xu H, Tan W, Zeng L, Anal. Chem. 2009, 81, 10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Jo H, Her J, Ban C, Biosens. Bioelectron. 2015, 71, 129. [DOI] [PubMed] [Google Scholar]
- [311].Borghei YS, Hosseini M, Dadmehr M, Hosseinkhani S, Ganjali MR, Sheikhnejad R, Anal. Chim. Acta 2016, 904, 92. [DOI] [PubMed] [Google Scholar]
- [312].Zhang X, Xiao K, Cheng L, Chen H, Liu B, Zhang S, Kong J, Anal. Chem. 2014, 86, 5567. [DOI] [PubMed] [Google Scholar]
- [313].Sheng W, Chen T, Kamath R, Xiong X, Tan W, Fan ZH, Anal. Chem. 2012, 84, 4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Sheng W, Chen T, Tan W, Fan ZH, ACS Nano 2013, 7, 7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Liang H, Chen S, Li P, Wang L, Le J, Li J, Yang HH, Tan W, J. Am. Chem. Soc. 2018, 140, 4186. [DOI] [PubMed] [Google Scholar]
- [316].Li L, Feng J, Fan Y, Tang B, Anal. Chem. 2015, 87, 4829. [DOI] [PubMed] [Google Scholar]
- [317].Yang Y, Huang J, Yang X, Quan K, Wang H, Ying L, Xie N, Ou M, Wang K, Anal. Chem. 2016, 88, 5981. [DOI] [PubMed] [Google Scholar]
- [318].Yang Y, Huang J, Yang X, He X, Quan K, Xie N, Ou M, Wang K, Anal. Chem. 2017, 89, 5850. [DOI] [PubMed] [Google Scholar]
- [319].Wu Y, Huang J, Yang X, Yang Y, Quan K, Xie N, Li J, Ma C, Wang K, Anal. Chem. 2017, 89, 8377. [DOI] [PubMed] [Google Scholar]
- [320].Liu J, Cui M, Zhou H, Yang W, ACS Sens. 2017, 2, 1847. [DOI] [PubMed] [Google Scholar]
- [321].Wang W, Satyavolu NSR, Wu Z, Zhang JR, Zhu JJ, Lu Y, Angew. Chem. Int. Ed. 2017, 56, 6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Kim S, Ryoo SR, Na HK, Kim YK, Choi BS, Lee Y, Kim DE, Min DH, Chem. Commun. 2013, 49, 8241. [DOI] [PubMed] [Google Scholar]
- [323].Zhang L, Huang H, Xu N, Yin Q, J. Mater. Chem. B 2014, 2, 4935. [DOI] [PubMed] [Google Scholar]
- [324].Meng HM, Zhang X, Lv Y, Zhao Z, Wang NN, Fu T, Fan H, Liang H, Qiu L, Zhu G, Tan W, ACS Nano 2014, 8, 6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Yi JT, Chen TT, Huo J, Chu X, Anal. Chem. 2017, 89, 12351. [DOI] [PubMed] [Google Scholar]
- [326].Chen WH, Yu X, Cecconello A, Sohn YS, Nechushtai R, Willner I, Chem. Sci. 2017, 8, 5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Hwang K, Wu P, Kim T, Lei L, Tian S, Wang Y, Lu Y, Angew. Chem. Int. Ed. 2014, 53, 13798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].Cui L, Peng R, Fu T, Zhang X, Wu C, Chen H, Liang H, Yang CJ, Tan W, Anal. Chem. 2016, 88, 1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Torabi S-F, Wu P, McGhee CE, Chen L, Hwang K, Zheng N, Cheng J, Lu Y, Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Wu Z, Fan H, Satyavolu NSR, Wang WJ, Lake R, Jiang JH, Lu Y, Angew. Chem. Int. Ed. 2017, 56, 8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Zhang P, He Z, Wang C, Chen J, Zhao J, Zhu X, Li CZ, Min Q, Zhu JJ, ACS Nano 2015, 9, 789. [DOI] [PubMed] [Google Scholar]
- [332].He D, He X, Yang X, Li HW, Chem. Sci. 2017, 8, 2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [333].Chen F, Bai M, Zhao Y, Cao K, Cao X, Zhao Y, Anal. Chem. 2018, 90, 2271. [DOI] [PubMed] [Google Scholar]
- [334].Bakshi SF, Guz N, Zakharchenko A, Deng H, Tumanov AV, Woodworth CD, Minko S, Kolpashchikov DM, Katz E, J. Am. Chem. Soc. 2017, 139, 12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [335].Chen F, Bai M, Cao K, Zhao Y, Cao X, Wei J, Wu N, Li J, Wang L, Fan C, Zhao Y, ACS Nano 2017, 11, 11908. [DOI] [PubMed] [Google Scholar]
- [336].Yang Z, Loh KY, Chu Y-T, Feng R, Satyavolu NSR, Xiong M, Nakamata Huynh SM, Hwang K, Li L, Xing H, Zhang X, Chemla YR, Gruebele M, Lu Y, J. Am. Chem. Soc. 2018, 140, 17656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Giles RV, Spiller DG, Grzybowski J, Clark RE, Nicklin P, Tidd DM, Nucleic Acids Res. 1998, 26, 1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [338].Becker-Hapak M, McAllister SS, Dowdy SF, Methods 2001, 24, 247. [DOI] [PubMed] [Google Scholar]
- [339].Dalby B, Cates S, Harris A, Ohki EC, Tilkins ML, Price PJ, Ciccarone VC, Methods 2004, 33, 95. [DOI] [PubMed] [Google Scholar]
- [340].Young JL, Dean DA, Adv. Genet. 2015, 89, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [341].Rhee WJ, Santangelo PJ, Jo H, Bao G, Nucleic Acids Res. 2008, 36, e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [342].Jani MS, Veetil AT, Krishnan Y, Nat. Rev. Mater. 2019, DOI 10.1038/s41578-019-0105-4. [DOI] [Google Scholar]
- [343].Zhou W, Ding J, Liu J, Theranostics 2017, 7, 1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].Lei Z, Li X, Luo X, He H, Zheng J, Qian X, Yang Y, Angew. Chem. Int. Ed. 2017, 56, 2979. [DOI] [PubMed] [Google Scholar]