

Abstract
The major cellular antioxidant glutathione (GSH) protects cancer cells from oxidative damage that can lead to the induction of ferroptosis, an iron‐dependent form of cell death triggered by the aberrant accumulation of lipid peroxides. Inhibitors of the cystine‐glutamate antiporter subunit xCT, which mediates the uptake of extracellular cystine and thereby promotes GSH synthesis, are thus potential anticancer agents. However, the efficacy of xCT‐targeted therapy has been found to be diminished by metabolic reprogramming that affects redox status in cancer cells. Identification of drugs for combination with xCT inhibitors that are able to overcome resistance to xCT‐targeted therapy might thus provide the basis for effective cancer treatment. We have now identified the vasodilator oxyfedrine (OXY) as a sensitizer of cancer cells to GSH‐depleting agents including the xCT inhibitor sulfasalazine (SSZ). Oxyfedrine contains a structural motif required for covalent inhibition of aldehyde dehydrogenase (ALDH) enzymes, and combined treatment with OXY and SSZ was found to induce accumulation of the cytotoxic aldehyde 4‐hydroxynonenal and cell death in SSZ‐resistant cancer cells both in vitro and in vivo. Microarray analysis of tumor xenograft tissue showed cyclooxygenase‐2 expression as a potential biomarker for the efficacy of such combination therapy. Furthermore, OXY‐mediated ALDH inhibition was found to sensitize cancer cells to GSH depletion induced by radiation therapy in vitro. Our findings thus establish a rationale for repurposing of OXY as a sensitizing drug for cancer treatment with agents that induce GSH depletion.
Keywords: aldehyde dehydrogenase (ALDH), drug repurposing, ferroptosis, glutathione (GSH), xCT
In the present study, we showed that the vasodilator oxyfedrine is a powerful sensitizer of cancer cells to xCT‐targeted therapy.

1. INTRODUCTION
Ferroptosis is a type of cell death induced by oxidative stress and consequent lipid peroxidation in an iron‐dependent method.1, 2 The cystine‐glutamate antiporter subunit xCT mediates the uptake of extracellular cystine, which contributes to synthesis of the major cellular antioxidant glutathione (GSH). Given that depletion of GSH induces ferroptosis, xCT is considered a promising target for cancer treatment.3, 4 However, cellular sensitivity to inducers of ferroptosis including xCT inhibitors has been found to be affected by cellular metabolic status5, 6 and the nuclear factor erythroid 2 (NF‐E2)‐related factor 2 (Nrf2)‐dependent antioxidant signaling pathway in cancer cells.7 Existence of tumor cell heterogeneity with regard to ferroptosis sensitivity might thus be a barrier to the clinical application of xCT inhibitors in cancer therapy.
We recently carried out a synthetic lethal screen of a drug library to identify agents that might reverse the resistance of cancer cells to xCT inhibitors. This screen showed that the oral anesthetic dyclonine (DYC) sensitized cancer cells to the cytotoxic effects of inhibition either of xCT activity or of GSH synthesis.8 Dyclonine has a structural motif responsible for the covalent inhibition of aldehyde dehydrogenase (ALDH) enzymes. In the presence of the xCT inhibitor sulfasalazine (SSZ), DYC induced intracellular accumulation of the cytotoxic aldehyde 4‐hydroxynonenal (4‐HNE) in SSZ‐resistant cancer cells and thereby induced the necrotic death of these cells.8 However, given that DYC is a local anesthetic, it might not be suitable for systemic use in combination with SSZ in the clinical setting.
We have now found that the existing drug oxyfedrine (OXY), which is a structural analog of DYC, also triggers the intracellular accumulation of 4‐HNE in cooperation either with GSH‐depleting agents such as SSZ and buthionine sulfoximine (BSO) or with radiation treatment, and that it thereby induces severe oxidative damage in cancer cells.
2. MATERIALS AND METHODS
2.1. Reagents and antibodies
Sulfasalazine and BSO were from Santa Cruz Biotechnology, DYC was from Tokyo Chemical Industry and OXY was from Alfa Chemistry. For immunoblot analysis, human xCT was detected with rabbit monoclonal antibodies (#12691, 1:1000 dilution; Cell Signaling Technology), Nrf2 with rabbit monoclonal antibodies (ab62352, 1:500 dilution; Abcam) and β‐actin with mouse monoclonal antibodies (sc‐47778, 1:500; Santa Cruz Biotechnology). For immunocytofluorescence analysis, 4‐HNE was detected with mouse monoclonal antibodies (MAB3249, 1:100; R&D Systems). For immunohistochemical or immunohistofluorescence analysis, 4‐HNE was detected with mouse monoclonal antibodies (HNEJ‐2, 1:100; JaICA) and cyclooxygenase‐2 (COX‐2) was detected with rabbit polyclonal antibodies (HPA001335, 1:100; Atlas Antibodies).
2.2. Cell culture
DMS114 and HCT116 cells were obtained from ATCC; SBC3 cells were from Juntendo University; HSC‐2, HSC‐3, and HSC‐4 cells were from RIKEN Cell Bank; OSC19 cells were from Kanazawa University as previously described8; SCC25 cells were from DS Pharma Biochemical; and A549 cells were from the Riken Bioresource Center, as previously described.9 OSC19 SSZR cells were established as previously described.8 HCT116, SBC3, HSC‐2, HSC‐3, HSC‐4, OSC19, OSC19‐SSZR, and SCC25 cells were cultured in DMEM supplemented with 10% FBS, and DMS114 and A549 cells were cultured in RPMI 1640 medium supplemented with 10% FBS. All cells were maintained under 5% CO2 at 37°C, were used for experiments within 6‐12 months after receipt, and were characterized by short tandem repeat (STR) analysis before use.
2.3. In vitro cell viability assay
Cells were seeded in 96‐well plates (4000 cells/well), cultured overnight, and exposed to test agents in complete medium. Cell survival was subsequently analyzed with the use of a Cell Titer‐Glo 2.0 luminescence‐based cell viability kit (Promega).
2.4. In vitro GSH assay
Cells were seeded in six‐well plates (140 000 cells/well), cultured overnight, and exposed to test agents in complete medium, after which intracellular GSH level was determined with the use of a GSH‐Glo glutathione assay kit (Promega).
2.5. Measurement of intracellular reactive oxygen species
Cells were seeded in six‐well plates (140 000 cells/well), cultured overnight, exposed to test agents in complete medium for 48 hours, washed twice with PBS, incubated for 15 minutes at 37°C with 2.5 µmol/L chloromethyl‐dihydrodichlorofluorescein diacetate (CM‐H2DCF‐DA; Life Technologies) in serum‐free medium, and washed again before measurement of intracellular reactive oxygen species (ROS) by flow cytometry.
2.6. Flow cytometry
Flow cytometric analysis was carried out with an Attune Acoustic Focusing Cytometer (Life Technologies). Cells were passed through a 40‐μm nylon mesh to yield a single‐cell suspension, and apoptotic cells were excluded on the basis of positive staining with propidium iodide. The ALDEFLUOR assay (Stemcell Technologies) was carried out according to the manufacturer's protocol.
2.7. RNA interference
A549 cells were transfected with annealed siRNAs (chimeric RNA‐DNA duplexes from Japan Bioservice) for 48 hours with the use of the Lipofectamine RNAi MAX reagent (Invitrogen). The siRNA sequences are as follows (overhang DNA sequences are shown in lowercase): control siRNA, 5′‐UUCUCCGAACGUGUCACGUtt‐3′ and 5′‐ACGUGACACGUUCGGAGAAtt‐3′; Nrf2 siRNA, 5′‐UCCUACUGUGAUGUGAAAUtt‐3′ and 5′‐AUUUCACAUCACAGUAGGAgc‐3′.
2.8. Immunoblot analysis
Cells were washed with PBS and lysed in Laemmli sample buffer. Equal amounts of lysate protein were subjected to SDS‐PAGE, after which the separated proteins were transferred to a PVDF membrane and exposed to primary antibodies. Immune complexes were detected with HRP‐conjugated secondary antibodies and Chemiluminescence Reagent Plus (Perkin‐Elmer Japan).
2.9. Immunocytofluorescence analysis
Cells were fixed with 4% paraformaldehyde, treated with 0.2% Triton X‐100 in PBS for 30 minutes, washed with PBS, incubated for 1 hour first with 3% BSA in PBS and then with primary antibodies to 4‐HNE diluted in PBS, washed three times with PBS, and then exposed for 1 hour to Alexa Fluor 488‐conjugated secondary antibodies (Life Technologies) diluted in PBS. All incubations were carried out at room temperature. Nuclei were stained with Hoechst 33 342 (Invitrogen) at 5 μg/mL DAPI, and the cells were then observed with a BZ‐X800 fluorescence microscope (Keyence).
2.10. In vivo drug treatment
HCT116 cells (2 × 106 cells/site) were implanted s.c. in the flank of athymic nude mice (CLEA Japan). The mice were then injected i.p. with physiological saline, SSZ (350 mg/kg per day), OXY (10 mg/kg per day), or both drugs. All animal experiments were carried out in accordance with protocols approved by the Ethics Committee of Keio University. Volumes of tumors were measured at days 1, 7, and 14.
2.11. Immunostaining of tumor tissue
For immunohistochemical analysis, tissue was fixed with 4% paraformaldehyde, embedded in paraffin, and sectioned at a thickness of 4 μm. The sections were depleted of paraffin, rehydrated in a graded series of ethanol solutions, and washed with PBS before exposure to microwave radiation (500 W) for 10 minutes in citrate buffer (pH 6.0). They were then treated with 0.2% Triton X‐100 in PBS for 30 minutes, washed with PBS, incubated for 1 hour at room temperature first with 3% BSA in PBS and then with primary antibodies diluted in PBS, and washed again three times with PBS. Immune complexes were detected with the use of a Vectastain Elite Kit and ImmPACT DAB Peroxidase Substrate (Vector Laboratories), and the sections were counterstained with hematoxylin and viewed with an FV10i confocal microscope (Olympus).
2.12. Microarray analysis
Samples were processed for microarray analysis at the Core Instrumentation Facility of Keio University School of Medicine. Total RNA was extracted from tumor tissue with the use of the Trizol reagent (Invitrogen). cRNA probes were synthesized from the total RNA and subjected to hybridization with a Clariom S Pico array (Affymetrix). Raw intensity data for each experiment were analyzed with the use of Transcriptome Analysis Console (TAC) software (Applied Biosystems). Microarray data are available in the GEO database under the accession number http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137952.
2.13. In vitro radiation treatment
HCT116 and HSC‐4 cells were exposed to 4, 6, and 10 Gy of ionizing radiation in a day with the use of an X‐ray irradiator (MBR‐1520R‐4; Hitachi) at settings of 150 kV and 20 mA.
2.14. Statistical analysis
Data are presented as means ± SD and were analyzed with the unpaired Student's t test with the use of SPSS v25 software (IBM). P < .05 was considered statistically significant.
3. RESULTS
3.1. Dyclonine analog OXY functions as an ALDH inhibitor in cancer cells
To examine the impact of DYC as a sensitizer to GSH‐depleting drugs, we assessed the effects of the combination of DYC and either SSZ or the GSH synthesis inhibitor BSO in multiple cancer cell lines including non‐small cell lung cancer (A549), small cell lung cancer (DMS114 and SBC3), head and neck squamous cell carcinoma (HSC‐2, HSC‐3, HSC‐4, OSC19 and its SSZ‐resistant clone OSC19‐SSZR,8 and SCC25), and colon cancer (HCT116) cells. Consistent with our previous observations,8 the combination of DYC and either SSZ or BSO greatly reduced the viability of all the tested cell lines, with the effect of each drug combination being greater than that of the corresponding single agents (Figure 1A). DYC thus reversed the acquired resistance of OSC19‐SSZR cells to SSZ (Figure 1A). Although these results show the efficacy of DYC as a sensitizer to the cytotoxic action of xCT‐targeted drugs, the fact that DYC is an oral anesthetic would likely be problematic with regard to its systemic administration. We therefore examined whether the vasodilator OXY, which has a structure similar to that of DYC10 (Figure 1B), might mimic this sensitizing action of DYC. Given that the synthetic lethal effect of DYC and GSH‐depleting agents relies on ALDH inhibition by DYC, we first investigated whether OXY also inhibits ALDH activity. Similar to our previous findings with DYC,8 OXY suppressed ALDH activity in HCT116 and HSC‐4 cells (Figure 1C, Figure S1), suggesting that OXY might also have the ability to sensitize cancer cells to GSH‐depleting agents.
Figure 1.
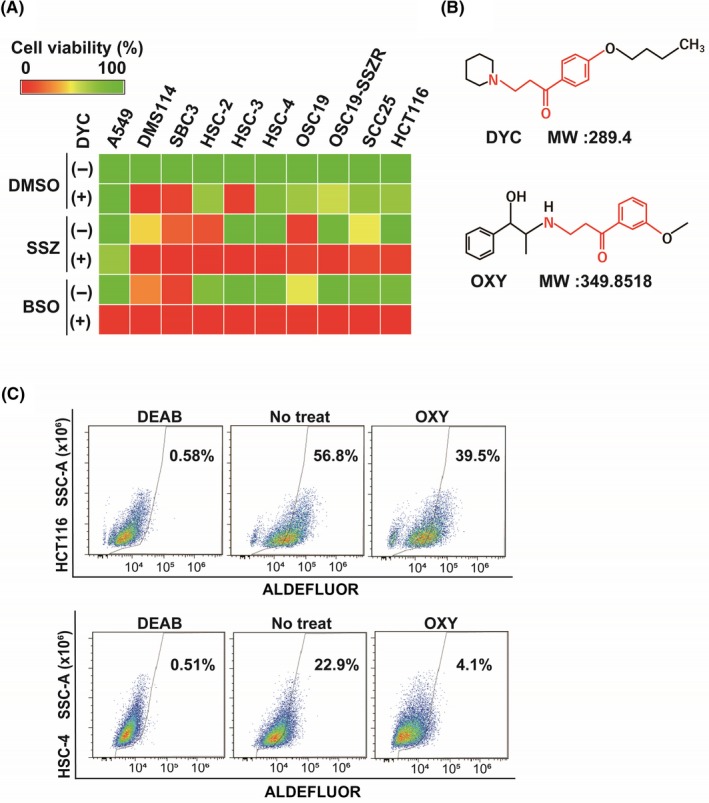
Oxyfedrine (OXY) is a structural analog of dyclonine (DYC) and functions as an aldehyde dehydrogenase (ALDH) inhibitor in cancer cells. A, The indicated cancer cell lines were cultured for 48 h with DMSO vehicle, sulfasalazine (SSZ; 400 µmol/L), buthionine sulfoximine (BSO; 100 µmol/L), or DYC (50 µmol/L), as indicated, and were then assayed for viability. Data are means from three independent experiments and are presented as a heat map. B, Chemical structure of DYC and OXY. The structural motif responsible for covalent inhibition of ALDH is shown in red. MW, molecular weight. C, HCT116 and HSC‐4 cells were cultured with or without OXY (50 µmol/L) for 48 h, stained with ALDEFLUOR (Stemcell Technologies) in the absence or presence of the ALDH inhibitor diethylaminobenzaldehyde (DEAB; 15 µmol/L), and then analyzed by flow cytometry for ALDH activity. SSC‐A, side scatter area
3.2. Combined treatment with OXY and either SSZ or BSO induces cell death as a result of the accumulation of 4‐HNE
To examine whether OXY might influence the cytotoxicity of GSH‐depleting agents, we studied HCT116 and HSC‐4 cells, both of which manifest resistance to SSZ alone.8 Treatment with BSO alone and the combination with OXY and SSZ or BSO induced marked depletion of GSH in both cell lines, whereas SSZ had a smaller (but still significant) effect (Figure 2A), indicating that xCT inhibition by SSZ alone is not sufficient to deprive the cells of most of their GSH. We next examined the effect of single or combined treatment with OXY and each of the two GSH‐depleting agents on cell viability. Treatment with OXY, SSZ, or BSO alone had no significant effect on the viability of HCT116 or HSC‐4 cells, whereas combined treatment with OXY and either SSZ or BSO greatly reduced the viability of both cell lines (Figure 2B). These results thus suggested that, like DYC, OXY acts as a sensitizer for GSH‐depleting agents, with the drug combinations inducing cell death in HCT116 and HSC‐4 cells.
Figure 2.

Oxyfedrine (OXY) sensitizes cancer cells to the effects of glutathione (GSH) depletion by sulfasalazine (SSZ) and buthionine sulfoximine (BSO). A, Intracellular content of GSH in HCT116 and HSC‐4 cells incubated for 48 h in the absence or presence of SSZ (400 µmol/L), BSO (100 µmol/L), or OXY (50 µmol/L) as indicated. Data are expressed relative to the corresponding value for control cells and are means ± SD from three independent experiments. *P < .05, **P < .01 (Student's t test). B, HCT116 and HSC‐4 cells were cultured for 48 h as in (A) and were then assayed for cell viability. Data are means ± SD from three independent experiments. **P < .01 (Student's t test). C, HCT116 and HSC‐4 cells cultured as in (A) for 24 h were subjected to immunofluorescence analysis of 4‐HNE (green). Nuclei were also stained with DAPI (blue). Scale bars, 100 µm. D, HCT116 and HSC4 cells cultured as in (A) for 48 h were assayed for reactive oxygen species by flow cytometric analysis after loading with chloromethyl‐dihydrodichlorofluorescein diacetate (CM‐H2DCF‐DA; Life Technologies)
We next tested the effect of combined treatment with OXY and GSH‐depleting agents on the abundance of the cytotoxic aldehyde 4‐HNE, a major end product of lipid peroxidation. Whereas SSZ, BSO, or OXY alone had little effect on 4‐HNE abundance, combination of OXY with either SSZ or BSO induced marked intracellular accumulation of 4‐HNE in HCT116 and HSC‐4 cells (Figure 2C), suggesting that inhibition of both GSH synthesis and ALDH activity allows accumulation of the cytotoxic aldehyde and leads to cell death.
Reaction of 4‐HNE with various thiol‐containing proteins that participate in redox signaling can result in the generation of ROS.11, 12 We therefore next examined the impact of the combination of OXY with SSZ or BSO on ROS levels with the use of the fluorescent probe CM‐H2DCF‐DA. Treatment with BSO alone, which largely depleted the cells of GSH (Figure 2A), increased the intracellular ROS level in both HCT116 and HSC‐4 cells, whereas SSZ alone had little such effect (Figure 2D). These results indicated that monotherapy with SSZ is not sufficient to deplete GSH to a level that allows ROS accumulation in these cells. However, combined treatment with OXY and SSZ was found to increase intracellular ROS levels in both HCT116 and HSC‐4 cells (Figure 2D), suggesting that simultaneous inhibition of xCT and ALDH might give rise to a vicious cycle of cytotoxic aldehyde generation and ROS accumulation in cancer cells.
3.3. Nrf2 activation reduces the efficacy of combination therapy with OXY and SSZ
Given that activation of the transcription factor Nrf2 results in upregulation of xCT expression and thereby protects cancer cells against ferroptosis,13 we next studied A549 cells, which harbor a mutation in the gene for Kelch‐like ECH‐associated protein 1 (Keap1) that gives rise to the constitutive expression of Nrf214 and the resistance to ferroptosis induced by sulfasalazine (Figure 1A). Amounts of Nrf2 and its downstream target xCT were markedly higher in A549 cells than in HCT116 or HSC‐4 cells (Figure 3A), suggesting that constitutive Nrf2 expression results in a high level of xCT expression in A549 cells. To determine whether activation of Nrf2 signaling affects the efficacy of combined treatment with OXY and either SSZ or BSO, we examined the effects of these drug combinations in A549 cells. Induction of cell death by combined treatment with OXY and SSZ was less pronounced in A549 cells than in HCT116 or HSC‐4 cells, whereas combined treatment with OXY and BSO reduced cell viability in A549 cells to an extent similar to that apparent in HCT116 or HSC‐4 cells (Figure 2B, Figure 3B). These results suggested that SSZ is less effective than BSO in inducing cell death in combination with OXY in cancer cells that manifest constitutive Nrf2 activation.
Figure 3.
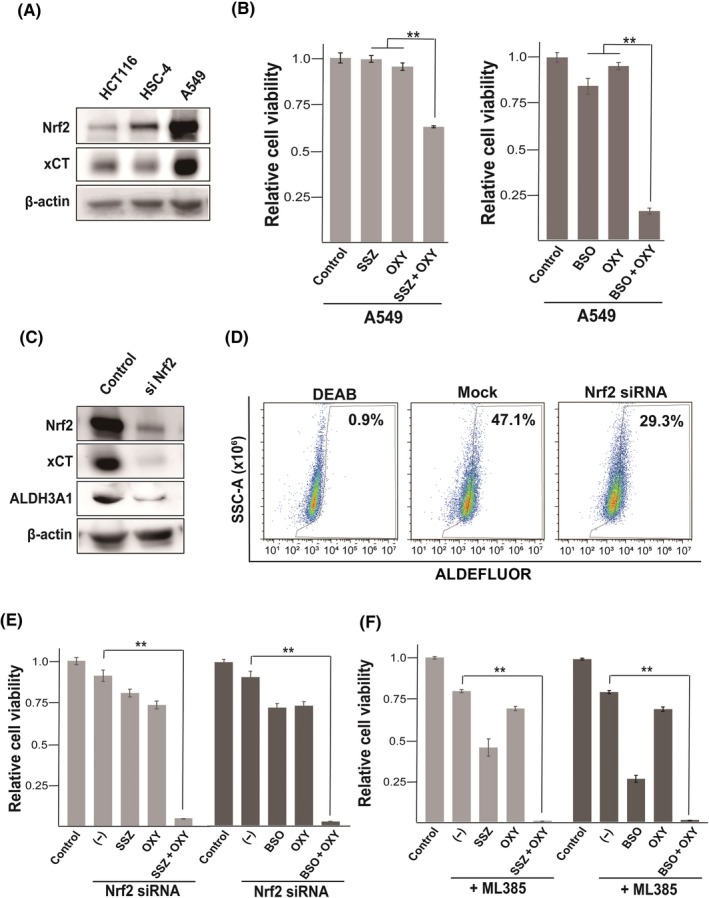
Nuclear factor erythroid 2 (NF‐E2)‐related factor 2 (Nrf2) signaling limits cancer cell sensitivity to combination therapy with sulfasalazine (SSZ) and oxyfedrine (OXY). A, Immunoblot analysis of Nrf2, xCT, and β‐actin (loading control) in HCT116, HSC‐4, and A549 cells. B, A549 cells were cultured for 48 h in the absence or presence of SSZ (400 µmol/L), buthionine sulfoximine (BSO; 100 µmol/L), or OXY (50 µmol/L), as indicated, and were then assayed for cell viability. Data are means ± SD from three independent experiments. **P < .01 (Student's t test). C, Immunoblot analysis of Nrf2, xCT and ALDH3A1 in A549 cells transfected with a control of Nrf2 siRNA. D, A549 and A549 transfected with siNrf2 cells were cultured with or without OXY (50 µmol/L) for 48 h, stained with ALDEFLUOR (Stemcell Technologies) in the absence or presence of the aldehyde dehydrogenase (ALDH) inhibitor diethylaminobenzaldehyde (DEAB; 15 µmol/L), and then analyzed by flow cytometry for ALDH activity. SSC‐A, side scatter area. E, A549 cells transfected with control or Nrf2 siRNAs were cultured for 48 h as in (B) and then assayed for cell viability. Data are means ± SD from three independent experiments. **P < .01 (Student's t test). F, A549 cells were cultured for 48 h in the absence or presence of SSZ (400 µmol/L), BSO (100 µmol/L), OXY (50 µmol/L), or ML385 (5 µmol/L) as indicated, and were then assayed for cell viability. Data are means ± SD from three independent experiments. **P < .01 (Student's t test)
To examine further the relevance of Nrf2 signaling to the reduced efficacy of combined treatment with OXY and SSZ in A549 cells, we depleted the cells of Nrf2 with a specific siRNA. Transfection of A549 cells with the Nrf2 siRNA resulted in marked depletion of both Nrf2 and xCT (Figure 3C), indicating that xCT is indeed upregulated as a result of constitutive Nrf2 activation in A549 cells. Furthermore, Nrf2 siRNA markedly decreased the expression of ALDH3A1 that plays a role in the resistance to xCT inhibitor in cancer cells,8 and leads to the reduction of ALDH activity (Figure 3C,D). Together, the constitutive Nrf2 activation might confer an ability to resist combination therapy with OXY and SSZ in cancer cells through upregulation of xCT and ALDH3A1. We thus examined the consequence of Nrf2 depletion on the efficacy of combination therapy and found that Nrf2 siRNA resulted in marked enhancement of the cytotoxic effect of combined treatment with OXY and either SSZ or BSO in A549 cells (Figure 3E), suggesting that activation of Nrf2 signaling protects cancer cells from oxidative damage induced by simultaneous inhibition of ALDH and either xCT activity or GSH synthesis. We also found that the specific Nrf2 inhibitor ML385 greatly enhanced the cytotoxic effect of combined treatment with OXY and either SSZ or BSO in A549 cells (Figure 3F). Together, these results suggested that the efficacy of combination therapy with OXY and either SSZ or BSO may be limited by Nrf2 activity and that the simultaneous use of a Nrf2 inhibitor may enhance the effect of such combination therapy in cancer cells that harbor an Nrf2 or Keap1 mutation.
3.4. Combination therapy with OXY and SSZ induces 4‐HNE accumulation in tumor cells and suppression of tumor growth
To examine the impact of combination therapy with OXY and SSZ in vivo, we studied tumors formed by HCT116 cells, which are resistant to SSZ monotherapy. The combination therapy greatly limited tumor growth as determined by measurement of tumor volume and weight, whereas OXY or SSZ alone had no significant effect (Figure 4A,B). These results thus suggested that simultaneous inhibition of ALDH and xCT by OXY and SSZ, respectively, might be effective for the treatment of tumors that manifest resistance to SSZ monotherapy.
Figure 4.
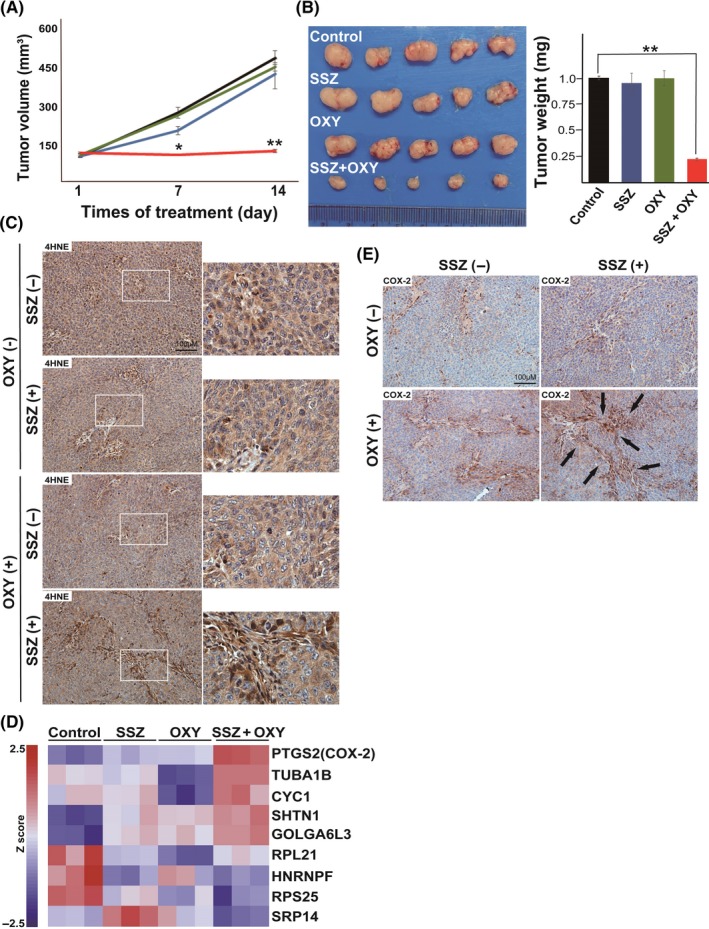
Synthetic lethality of combination therapy with oxyfedrine (OXY) and sulfasalazine (SSZ) in vivo. A, Volume of tumors formed after s.c. injection of HCT116 cells and treatment with saline (control), SSZ (350 mg/kg per day), OXY (10 mg/kg per day), or the combination of both drugs in nude mice. Data are means ± SD for four or five mice per group. *P < .05, **P < .01 versus the corresponding control value (Student's t test). B (Left) HCT116 tumors isolated from mice after treatment for 16 days as in (A). (Right) Weight of HCT116 tumors after treatment for 16 d as in (A). Data are means ± SD for five mice per group. **P < .01 (Student's t test). C, Immunohistochemical staining of 4‐hydroxynonenal (4‐HNE; brown) in HCT116 tumors after treatment for 16 d as in (A). Boxed regions in left images are shown at higher magnification on the right. Scale bar, 100 µm. D, Microarray analysis for the differentially expressed genes in HCT116 tumors after treatment for 16 days as in (A). Data are expressed as Z score with triplicates per group and are presented as a heat map. E, Immunohistochemical staining for COX‐2 (brown) in HCT116 tumors after treatment for 16 d as in (A). Arrows indicate areas of prominent staining. Scale bar, 100 µm
We carried out immunohistochemical analysis to determine whether combination therapy with OXY and SSZ induces 4‐HNE accumulation in HCT116 tumors. Abundance of 4‐HNE was markedly higher in tumors treated with both OXY and SSZ compared with those treated with either agent alone or control tumors (Figure 4C), indicating that combination therapy with OXY and SSZ limits the ability of cancer cells to prevent 4‐HNE accumulation in vivo.
To identify biomarkers that might reflect the efficacy of combination therapy with OXY and SSZ, we carried out microarray analysis of HCT116 tumors. Such analysis showed that the abundance of COX‐2 mRNA was markedly increased in tumors treated with the combination therapy (Figure 4D). Given that 4‐HNE has been shown to induce COX‐2 expression,15, 16 we next carried out immunohistochemical analysis of COX‐2 in tumor samples. COX‐2 expression was markedly increased in tumors subjected to combination therapy with OXY and SSZ (Figure 4E). Of note, the expression of COX‐2 was detected predominantly in tumor cells around the stroma, and the COX‐2–positive area appeared similar to that positive for 4‐HNE (Figure 4C,E). The expression of COX‐2 as well as the abundance of 4‐HNE are thus potential biomarkers for monitoring the efficacy of combination therapy with OXY and SSZ (Figure 5D).
Figure 5.
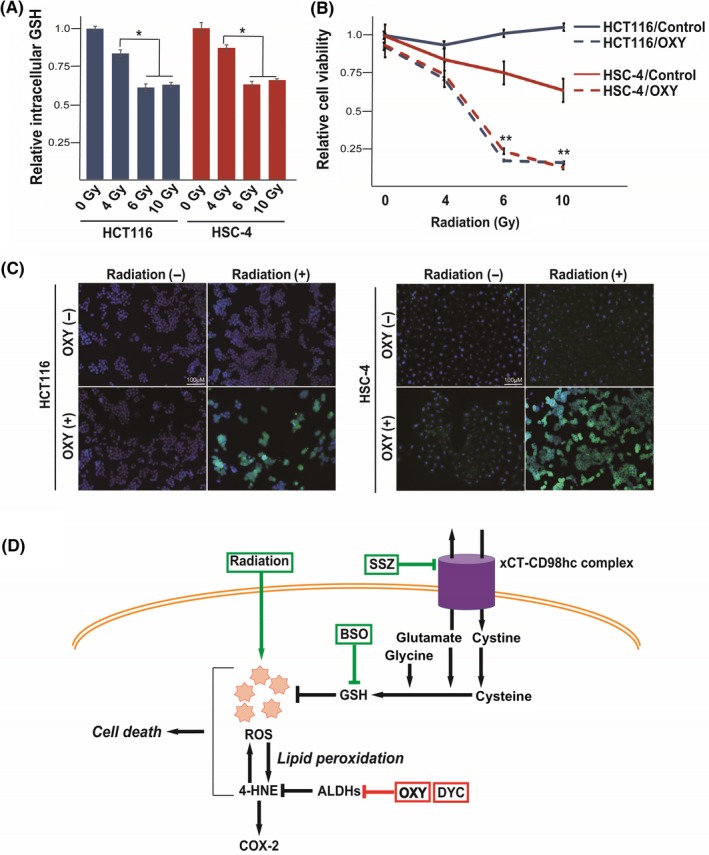
Oxyhedrine (OXY) sensitizes cancer cells to radiation treatment. A, Intracellular content of glutathione (GSH) in HCT116 and HSC‐4 cells at 24 h after onset of irradiation with 0, 4, 6, or 10 Gy. Data are expressed relative to the corresponding value for control cells and are means ± SD from three independent experiments. *P < .05. (Student's t test). B, Viability of HCT116 and HSC‐4 cells at 24 h after onset of irradiation with 0, 4, 6, or 10 Gy in the absence (control) or presence of OXY (50 µmol/L). Data are means ± SD from three independent experiments. **P < .01 versus the corresponding control value (Student's t test). C, Immunofluorescence staining of 4‐hydroxynonenal (4‐HNE; green) in HCT116 and HSC‐4 cells at 24 h after the onset of irradiation with 0 or 6 Gy in the absence or presence of OXY (50 µmol/L). Nuclei were also stained with DAPI (blue). Scale bars, 100 µm. D, Proposed mechanism for the action of combination therapy with OXY and either a GSH‐depleting drug (sulfasalazine [SSZ] or buthionine sulfoximine [BSO]) or radiation. Simultaneous inhibition of aldehyde dehydrogenase (ALDH) and depletion of GSH leads to accumulation of the cytotoxic aldehyde 4‐HNE and consequent cell death. COX‐2 gene expression is upregulated by 4‐HNE and may serve as a biomarker for such combination therapy. ROS, reactive oxygen species
3.5. Oxyfedrine sensitizes cancer cells to GSH depletion induced by radiation treatment
Given that radiation treatment induces ROS generation and depletion of GSH17, 18 (Figure 5A), we next examined whether OXY might sensitize cancer cells to radiation. Whereas HCT116 and HSC‐4 cells manifested resistance to irradiation alone, the combination of OXY with 6 or 10 Gy of radiation exerted a cytotoxic effect in both cell lines (Figure 5B). These results thus suggested that irradiation might be applied to deplete intracellular GSH and thereby exert a synergistic antiproliferative effect with OXY in cancer cells locally. Immunocytofluorescence analysis showed that the combination of OXY and radiation induced marked intracellular accumulation of 4‐HNE in HCT116 and HSC‐4 cells, whereas OXY or radiation alone had little effect (Figure 5C). Together, these results thus indicated that OXY treatment is effective for sensitizing cancer cells not only to GSH‐depleting drugs but also to radiation therapy.
4. DISCUSSION
Ferroptosis is a form of regulated but nonapoptotic cell death that results from iron‐dependent lipid peroxidation, and various inducers of ferroptosis have recently been identified.6 The cystine‐glutamate antiporter subunit xCT plays a key role in suppression of ferroptosis in cancer cells and is considered a promising target for cancer therapy.4, 19 Recent clinical studies with the xCT inhibitor SSZ in patients with advanced gastric or lung cancer20, 21, 22 have shown that the antitumor effect of SSZ monotherapy is limited to a subpopulation of tumor cells that express variant isoforms of CD44 and whose viability is dependent on xCT activity. Interferon‐γ released from CD8+ T cells was recently shown to suppress xCT expression and thereby sensitize cancer cells to ferroptosis.23 The efficacy of therapy with an xCT inhibitor might thus depend on the simultaneous use of either a cytotoxic drug or an agent that further sensitizes cancer cells to ferroptosis. On the basis of this premise, we recently carried out a drug screen and identified DYC as a sensitizer for GSH‐depleting agents in cancer cells.8
Oxyfedrine and DYC share a common motif that consists of an aromatic ketone and amine and which undergoes β‐elimination and thereby generates a vinyl ketone intermediate that interacts covalently with ALDH and inhibits its enzymatic activity.10 We have now shown that, like DYC, OXY has the ability to inhibit ALDH activity, with such inhibition giving rise to a synergistic cytotoxic effect with GSH‐depleting agents in cancer cells. ALDH‐mediated 4‐HNE detoxification is thus a candidate therapeutic target for enhancement of the anticancer action of GSH‐depleting drugs or radiation therapy.
The transcription factor Nrf2 regulates the transcription of components of antioxidant systems, detoxification, NADPH regeneration, lipid metabolism and heme metabolism.24 Activation of Nrf2 has been shown to upregulate xCT expression and thereby protects cancer cells against ferroptosis.13 In this study, we showed that the constitutive activation of Nrf2 results in high expression of xCT and ALDH3A1, and Nrf2 depletion sensitizes Nrf2‐overexpressing cancer cells to combination therapy with OXY and SSZ. Together, the gene mutation status of KEAP1 and Nrf2 (NFE2L2) is a critical determinant for the efficacy of combination therapy with OXY and SSZ.
4‐Hydroxynonenal is a highly reactive lipid peroxidation product and preferentially forms adducts with cysteine residues of thiol‐containing proteins that function in redox signaling and antioxidant systems.25 Capacity for 4‐HNE detoxification may contribute to resistance to xCT‐targeted therapy and ferroptosis induction in cancer cells. Simultaneous inhibition of ALDH activity and GSH function thus leads to the accumulation of 4‐HNE, which induces further accumulation of ROS, resulting in consequent cell death (Figure 5D). Furthermore, we found that the abundance of COX‐2 mRNA, which is increased by 4‐HNE,26 was upregulated in tumors subjected to combination therapy with OXY and SSZ. Expression of COX‐2 might thus serve as a biomarker for the effectiveness of such combination therapy.
An engineered form of human cyst(e)inase that can degrade both cystine and cysteine systemically was recently developed.27 Clinical use of this modified enzyme or xCT inhibitors is expected to effectively deplete GSH in tumor tissue. Identification of drugs that act synergistically with GSH‐depleting agents to induce cell death in cancer cells may therefore provide the basis for a potential novel type of cancer therapy. Our results have now established a rationale for repurposing OXY to target 4‐HNE metabolism by ALDH in cancer cells that manifest resistance to xCT‐targeted monotherapy.
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This study was supported by grants (to O.N.) from the Project for Cancer Research and Therapeutic Evolution (P‐CREATE) of the Japan Agency for Medical Research and Development (AMED). We thank I. Ishimatsu for technical assistance, M. Kobori for help with preparation of the manuscript, and R. Suzuki (Core Instrumentation Facility, Keio University School of Medicine) for assistance with microarray analysis.
Otsuki Y, Yamasaki J, Suina K, et al. Vasodilator oxyfedrine inhibits aldehyde metabolism and thereby sensitizes cancer cells to xCT‐targeted therapy. Cancer Sci. 2020;111:127–136. 10.1111/cas.14224
REFERENCES
- 1. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149:1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)‐ cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633‐1640. [DOI] [PubMed] [Google Scholar]
- 4. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell. 2011;19:387‐400. [DOI] [PubMed] [Google Scholar]
- 5. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun X, Ou Z, Chen R, et al. Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Okazaki S, Shintani S, Hirata Y, et al. Synthetic lethality of the ALDH3A1 inhibitor dyclonine and xCT inhibitors in glutathione deficiency‐resistant cancer cells. Oncotarget. 2018;9:33832‐33843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Minakata K, Takahashi F, Nara T, et al. Hypoxia induces gefitinib resistance in non‐small‐cell lung cancer with both mutant and wild‐type epidermal growth factor receptors. Cancer Sci. 2012;103:1946‐1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khanna M, Chen CH, Kimble‐Hill A, et al. Discovery of a novel class of covalent inhibitor for aldehyde dehydrogenases. J Biol Chem. 2011;286:43486‐43494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhong H, Yin H. Role of lipid peroxidation derived 4‐hydroxynonenal (4‐HNE) in cancer: focusing on mitochondria. Redox Biol. 2015;4:193‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ansari SA, Pendurthi UR, Rao LVM. The lipid peroxidation product 4‐ hydroxy‐2‐nonenal induces tissue factor decryption via ROS generation and the thioredoxin system. Blood Adv. 2017;1:2399‐2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fan Z, Wirth AK, Chen D, et al. Nrf2‐Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. 2017;6:e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Singh A, Bodas M, Wakabayashi N, Bunz F, Biswal S. Gain of Nrf2 function in non‐small‐cell lung cancer cells confers radioresistance. Antioxid Redox Signal. 2010;13:1627‐1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zarrouki B, Soares AF, Guichardant M, Lagarde M, Geloen A. The lipid peroxidation end‐product 4‐HNE induces COX‐2 expression through p38MAPK activation in 3T3‐L1 adipose cell. FEBS Lett. 2007;581:2394‐2400. [DOI] [PubMed] [Google Scholar]
- 16. Uchida K. HNE as an inducer of COX‐2. Free Radic Biol Med. 2017;111:169‐172. [DOI] [PubMed] [Google Scholar]
- 17. Ashwood‐Smith MJ. The effect of whole‐body x‐irradiation on the glutathione content of rat thymus. Int J Radiat Biol Relat Stud Phys Chem Med. 1961;3:125‐132. [DOI] [PubMed] [Google Scholar]
- 18. Fierro S, Yoshikawa M, Nagano O, Yoshimi K, Saya H, Einaga Y. In vivo assessment of cancerous tumors using boron doped diamond microelectrode. Sci Rep. 2012;2:901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nagano O, Okazaki S, Saya H. Redox regulation in stem‐like cancer cells by CD44 variant isoforms. Oncogene. 2013;32:5191‐5198. [DOI] [PubMed] [Google Scholar]
- 20. Shitara K, Doi T, Nagano O, et al. Dose‐escalation study for the targeting of CD44v(+) cancer stem cells by sulfasalazine in patients with advanced gastric cancer (EPOC1205). Gastric Cancer. 2017;20:341‐349. [DOI] [PubMed] [Google Scholar]
- 21. Shitara K, Doi T, Nagano O, et al. Phase 1 study of sulfasalazine and cisplatin for patients with CD44v‐positive gastric cancer refractory to cisplatin (EPOC1407). Gastric Cancer. 2017;20:1004‐1009. [DOI] [PubMed] [Google Scholar]
- 22. Otsubo K, Nosaki K, Imamura CK, et al. Phase I study of salazosulfapyridine in combination with cisplatin and pemetrexed for advanced non‐small‐cell lung cancer. Cancer Sci. 2017;108:1843‐1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang W, Green M, Choi JE, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hayes JD, Dinkova‐Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199‐218. [DOI] [PubMed] [Google Scholar]
- 25. Breitzig M, Bhimineni C, Lockey R, Kolliputi N. 4‐Hydroxy‐2‐nonenal: a critical target in oxidative stress? Am J Physiol Cell Physiol. 2016;311:C537‐C543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumagai T, Kawamoto Y, Nakamura Y, et al. 4‐Hydroxy‐2‐nonenal, the end product of lipid peroxidation, is a specific inducer of cyclooxygenase‐2 gene expression. Biochem Biophys Res Commun. 2000;273:437‐441. [DOI] [PubMed] [Google Scholar]
- 27. Cramer SL, Saha A, Liu J, et al. Systemic depletion of L‐cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017;23:120‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials