

SUMMARY
A subset of human neocortical neurons harbors complex karyotypes wherein megabase-scale copy-number variants (CNVs) alter allelic diversity. Divergent levels of neurons with complex karyotypes (CNV neurons) are reported in different individuals, yet genome-wide and familial studies implicitly assume a single brain genome when assessing the genetic risk architecture of neurological disease. We assembled a brain CNV atlas using a robust computational approach applied to a new dataset (>800 neurons from 5 neurotypical individuals) and to published data from 10 additional neurotypical individuals. The atlas reveals that the frequency of neocortical neurons with complex karyotypes varies widely among individuals, but this variability is not readily accounted for by tissue quality or CNV detection approach. Rather, the age of the individual is anti-correlated with CNV neuron frequency. Fewer CNV neurons are observed in aged individuals than in young individuals.
In Brief
Neurons with complex karyotypes harbor ≥1 large (>2 Mb) CNVs. Chronister et al. built a human brain CNV atlas using 1,285 single-cell libraries from 15 neurotypical individuals. The observed frequency of CNV neurons declines from >25% in young individuals to <10% in aged individuals.
Graphical Abstract
INTRODUCTION
Neocortical neurons are among the most diverse and longest-lived mammalian cells. The mammalian cerebral cortex is often put forward as a pinnacle of evolutionary complexity, and human-specific brain phenotypes are attributed to neocortical expansion during evolution (Geschwind and Rakic, 2013; Lui et al., 2011). Aberrant development and maturation of neocortical circuits are likewise associated with neuropsychiatric and neurodegenerative diseases (Del Pino et al., 2018; Morrison and Baxter, 2012; Südhof, 2017). Various approaches count ~15–20 billion neurons and as many as 35 billion glia in the human cerebral cortex (Pakkenberg et al., 2003; von Bartheld et al., 2016). Single-cell transcriptomic approaches are beginning to comprehensively catalog human neuronal diversity (Lake et al., 2016; Nowakowski et al., 2017) and identify new subtypes of human neurons (Boldog et al., 2018). After decades of debate, it is now clear that human neocortical neurons are not normally regenerated during the human lifespan (Bhardwaj et al., 2006; Rakic, 2006). With some exceptions (Spitzer, 2017), neuronal cell types are also generally thought to be stable throughout life, but neuronal genomes are surprisingly labile.
Every human neocortical neuron may contain private somatic variants. Single nucleotide variants (SNVs) are especially common, with hundreds per neuron reported (Bae et al., 2018; Lodato et al., 2015) and with frequencies of >3,000 SNVs per neuron observed in aged individuals (Lodato et al., 2018). Endogenous mobile elements such as long interspersed nuclear element 1 (LINE1) retrotransposons are also active during brain development (Coufal et al., 2009; Muotri et al., 2005). Reported frequencies of de novo mobile element events range from <1 to >7 per neuron (Baillie et al., 2011; Evrony et al., 2012; Upton et al., 2015). Mobile element activity has also been linked to the generation of copy-number variants (CNVs) (Erwin et al., 2016; Gilbert et al., 2002). Whole and subchromosomal CNVs bring about complex karyotypes through the duplication or deletion of several megabases (Mb) of genomic sequence in a subpopulation of neocortical neurons (Cai et al., 2014; Knouse et al., 2016; McConnell et al., 2013; Piotrowski et al., 2008). Gene density in the human genome averages >10 genes per Mb; thus, by contrast to other somatic variants, Mb-scale CNVs almost always affect multiple genes. A reanalysis of published data (Cai et al., 2014; Knouse et al., 2014; McConnell et al., 2013; van den Bos et al., 2016) herein found an average of 63 genes affected per neuronal CNV.
During the past decade, large CNVs have been recognized as major contributors to human genetic diversity (Conrad et al., 2010; Lupski, 2015; Redon et al., 2006). At the population level, SNVs are collectively more numerous than CNVs, but CNVs affect an order of magnitude more genome sequence (~10%), and some CNVs show evidence of positive selection during human evolution (Perry et al., 2007; Sudmant et al., 2015; Zarrei et al., 2015). In individuals, de novo CNVs represent rare variants with a strong contribution to the genetic risk of schizophrenia, autism, and other neurological disorders (Fromer et al., 2014; Iossifov et al., 2014; Marshall et al., 2017; Morrow, 2010; Sebat et al., 2007). Whereas the consequences of germline CNVs have been inferred from population-level studies, neuronal CNV studies to date have been underpowered to determine whether the genes affected by neuronal CNVs contribute to brain development, function, and disease.
We assembled a brain CNV atlas to evaluate how neuronal CNVs alter the genetic architecture of the neurotypical human cerebral cortex. A new dataset of 827 human cerebral cortical nuclei from 5 neurotypical individuals was combined with smaller published datasets (Cai et al., 2014; Knouse et al., 2014; McConnell et al., 2013; van den Bos et al., 2016) from 10 other neurotypical individuals. We developed an unbiased CNV detection approach based on population-level statistics and established a human neuronal CNV atlas with 507 CNVs. Initial analysis of the atlas identified substantial inter-individual variability in the frequency of neurons with complex karyotypes (CNV neurons), but also found support for the hypothesis (Weissman and Gage, 2016) that some long genomic loci shape the genetic architecture of neurotypical human brains.
RESULTS
Generation of Single-Cell Neuronal and Non-neuronal Genomic Data
Single neuronal and non-neuronal nuclei were isolated using fluorescence-activated nuclei sorting (FANS) of the prefrontal cortex from 5 non-diseased (neurotypical) male individuals aged 0.36, 26, 49, 86, and 95 years (Figure 1A; Table 1). Whole genome amplification (WGA) was performed using PicoPLEX (Rubicon Genomics), an approach that is similar to multiple annealing and loop-based amplification (MALBAC) (Zong et al., 2012), which produces Illumina-compatible libraries with 48 unique barcodes. Before library pooling and sequencing on Illumina platforms, we found that ~60% of WGA reactions produced a measurable product. Single-end or paired-end sequencing (50, 75, or 100 bp) of 48 pooled libraries on Illumina HiSeq Rapid platforms or of smaller pools (<17 libraries) on MiSeq platforms routinely produced >1 million reads per library after duplicate removal. Neither paired-end nor longer read sequencing altered the data quality. Reads were aligned to hg19 and read depth was calculated across 4,505 genomic bins, each containing ~500 kb of uniquely mappable sequence (mean bin size = 687 ± 1,072 kb). This approach (Figure 1B) generated >134.2 Gb of genomic sequence from 827 male single-cell genome libraries (162.3 ± 115.1 Mb/cell).
Figure 1. Optimization of Single-Cell CNV Detection.

(A and B) Representative NeuN+/− FANS (A) and summary of analysis pipeline (B).
(C) CNV profileoftest Neuron 1. Read depth-derived CN values of genomic bins are colored alternately (green, blue) by chromosome. Red line indicates DNAcopy segmentation.
(D) BIC scores for Neuron 1 across DNAcopy parameter space. Red diamond indicates lowest BIC score.
(E) Histogram of BIC scores for new dataset. Gaussian distributions (black and red) were used to establish BIC cutoff (−2.21).
(F) Segmentation output displays integer-like CN states (red, green, and blue) from which stringent (<1.14; >2.80; long dashes) and lenient (<1.34; >2.60; short dashes) CN state thresholds were established.
Table 1.
Summary of Brain CNV Atlas
| Age | Sex | Cell Type | Data Source | WGA | BIC Pass/Total Cells (%) | Cells with CNV(s) (%), Lenient | Cells with CNV(s) (%), Stringent | Total CNVs, Lenient | Total CNVs, Stringent |
|---|---|---|---|---|---|---|---|---|---|
| 0.36 | Male | Neuron | This paper | PicoPLEX | 46/138 (33.3) | 14 (30.4) | 7 (15.2) | 41 | 12 |
| 20 | Female | Neuron | McConnell et al., 2013 | GenomePlex | 50/50 (100) | 20 (40.0) | 12 (24.0) | 76 | 52 |
| 24 | Female | Neuron | McConnell et al., 2013 | GenomePlex | 18/19 (94.7) | 7 (38.9) | 3 (16.7) | 20 | 9 |
| 26* | Male | Neuron | This paper | PicoPLEX | 108/184 (58.7) | 25 (23.1) | 15 (13.9) | 76 | 33 |
| 26* | Male | Non-neuron | This paper | PicoPLEX | 43/63 (68.3) | 2 (4.7) | 0 (0) | 2 | 0 |
| 26* | Male | Neuron | McConnell et al., 2013 | GenomePlex | 41/41 (100) | 9 (22.0) | 6 (14.6) | 18 | 13 |
| 42 | Female | Neuron | Cai et al., 2014 | GenomePlex | 19/26 (73.1) | 7 (36.8) | 4 (21.1) | 57 | 27 |
| 48 | Female | Neuron | Knouse et al., 2014 | GenomePlex | 21/22 (95.5) | 0 (0) | 0 (0) | 0 | 0 |
| 49 | Male | Neuron | This paper | PicoPLEX | 99/101 (98.0) | 11 (11.1) | 8 (8.1) | 113 | 75 |
| 49 | Male | Non-neuron | This paper | PicoPLEX | 26/28 (92.9) | 2 (7.7) | 2 (7.7) | 4 | 2 |
| 52 | Male | Neuron | Knouse et al., 2014 | GenomePlex | 22/22 (100) | 4 (18.2) | 1 (4.5) | 4 | 1 |
| 68 | Male | Neuron | Knouse et al., 2014 | GenomePlex | 23/25 (92.0) | 3 (13.0) | 2 (8.7) | 4 | 2 |
| 69 | Male | Neuron | van den Bos et al., 2016 | Strand-seq | 78/81 (96.3) | 3 (3.8) | 2 (2.6) | 3 | 2 |
| 70 | Male | Neuron | Knouse et al., 2014 | GenomePlex | 20/20 (100) | 4 (20.0) | 4 (20.0) | 9 | 4 |
| 74 | Male | Neuron | van den Bos et al., 2016 | Strand-seq | 70/80 (87.5) | 6 (8.6) | 1 (1.4) | 6 | 1 |
| 81 | Female | Neuron | van den Bos et al., 2016 | Strand-seq | 43/72 (59.7) | 4 (9.3) | 2 (4.7) | 15 | 6 |
| 86 | Male | Neuron | This paper | PicoPLEX | 101/118 (85.6) | 4 (4.0) | 3 (3.0) | 20 | 15 |
| 86 | Male | Non-neuron | This paper | PicoPLEX | 46/55 (83.6) | 4 (8.7) | 2 (4.3) | 9 | 3 |
| 95 | Male | Neuron | This paper | PicoPLEX | 120/140 (85.7) | 8 (6.7) | 5 (4.2) | 45 | 28 |
Same individual.
Optimization of Read Depth-Based Single-Cell Genomic Segmentation
Neuronal CNV detection is inherently challenging because one cannot know the state of the genome before WGA, and neuronal CNVs are rarely clonal. For this reason, CNV calling approaches have been used conservatively, with bias toward avoiding type I errors at the risk of type II errors. We used a test dataset of 6 representative PicoPLEX WGA libraries (5 neurons and 1 trisomy 21 fibroblast) with varied subjective quality (Figures 1C and S1A–S1E) to optimize our approach.
Parameter space in DNAcopy (Olshen et al., 2004) is defined by 3 user-tunable parameters: significance threshold (alpha), minimum number of genomic bins required to call a copy number state change (min.width), and the number of SDs between the levels of copy-number states to maintain the copy-number state change (undo.SD). We assessed DNAcopy parameters using Bayesian information criterion (BIC) (Schwarz, 1978), a log-likelihood estimate of the performance of the segmentation algorithm, for dozens of parameter combinations. The lowest, or near-lowest, BIC scores for each test cell library were observed at alpha = 0.001, min.width = 5, and undo.SD = 0 (Figures 1D and S1G); these parameters identified monosomy X in all 6 male cells, and trisomy 21 in only the fibroblast. We also observed that minimum BIC scores were lower in WGA libraries with less overall bin-to-bin variation in read-depth (Figures 1C, S1A–S1E, and S1G), suggesting that low BIC scores represent an additional quality control filter.
We further assessed segmentation parameters with 2 in silico models (Figure S1F) built from the read-depth statistics (DNA copy segments, Gaussian noise, and autocorrelated noise [ϕ]) of each test cell. NULL model simulations contained no CNVs but did contain strong autocorrelated noise, a known source of DNAcopy false-positives (Muggeo and Adelfio, 2011). Alternative (ALT) model simulations harbored synthetic CNVs (i.e., DNAcopy calls) with residual autocorrelated noise. Segmentation outputs matched the ALT model, but not the NULL model, simulations well (Figures S1H and S1I). Concordance with ALT model cells (i.e., highest sensitivity and specificity) was also associated with the lowest BIC scores (Figure S1J).
Analysis of 827 PicoPLEX datasets identified additional population-based filters. First, we identified 101 genomic bins that routinely deviated from median read depth and confound segmentation (Figure S1K); these were excluded before the subsequent analysis. Second, we computed the 95th percentile of the low BIC score Gaussian distribution (−2.21) to establish an objective filter for the highest quality single-cell datasets (Figure 1E). Third, identified segments displayed 2 modes near integer-like copy number (CN) states of 2 (euploid, mean = 1.97) and 1 (deletion, mean = 1.12) and a heavy tail near the CN state of 3 (duplication, mean = 2.92) (Figure 1F). Lenient and stringent CN state thresholds were established, respectively, at a 2-tailed p value ≤ 0.05 (<1.34 for a deletion and >2.60 for a duplication) and ≤0.01 (<1.14 for a deletion and >2.80 for a duplication). The BIC threshold produced a final dataset of 589/827 (71.2%) neural nuclei, including 474/681 (69.6%) neuronal (NeuN+) nuclei and 115/146 (78.8%) non-neuronal (NeuN−) nuclei (Figures S1L and S1M). The stringent CN state threshold identified monosomy X in 52.8% of male neural nuclei, while the lenient threshold identified 99.5% of these true-positives (Figure S2A).
Additional NULL and ALT model simulations found that BIC cutoffs and CN state thresholds protect against false-positive CNVs brought about by simulated WGA-induced noise. We tested 6 neuronal WGA libraries in the BIC <−2.21 PicoPLEX dataset: 2 euploid, 2 with 1 CNV, 1 with 3 CNVs, and 1 with 6 CNVs. In the NULL model (Figure S2B), all of the simulated cells from neuron 10 were excluded by our BIC threshold as a consequence of strong autocorrelated noise (ϕ = 0.663; atlas mean ϕ = 0.1394). In the remaining 1,000 simulated cells, only 41 and 1 small (3.9 ± 0.7 Mb) segments, respectively, passed the lenient and stringent CN state thresholds. Thus, upper-bound estimates of false-positive CNV neuron detection rates are ~3% (lenient threshold) and <0.1% (stringent threshold). All ALT model simulations (Figure S2C) passed the BIC threshold, and only ~1% (14/1,200) of simulated cells contained DNAcopy segments that passed the lenient CN state thresholds without overlapping the synthetic CNV; these false-positive CNVs were also small (6.0 ± 2.7 Mb). As observed for monosomy X in the PicoPLEX dataset, improved detection of true-positives (synthetic CNVs) in ALT model cells was observed at lenient (95%) relative to stringent (81.8%) CN state thresholds.
Contribution of Mosaic CNVs to Neuronal Diversity
Large CNVs inevitably alter the CN state of a brain-expressed gene, as more of the genome is expressed in neurons than in other cell types (Uhlén et al., 2016). Our analysis (Table 1) identified 310 CNVs in 70 of 589 neural genomes (11.9%). CNVs ranged in size from 2.9 to 159.1 Mb (mean = 16.5 ± 20.1 Mb; Figure 2A). With stringent criteria that are prone to false-negatives, we still identify 168 CNVs (mean = 18.0 ± 22.3 Mb) in 42 neural nuclei (7.1%) (Figure S3A). CNVs were detected in each individual examined and, given their size and frequency (Figures 2B and S3D), represent a clear contribution to the genetic architecture of the brain (Figures 2C and S3G). We note that much smaller percentages of phenotypically distinct cells can bring about focal epilepsies (Marin-Valencia et al., 2014) and are essential for normal brain function (e.g., adult-born dentate granule neurons; Anacker and Hen, 2017; Christian et al., 2014).
Figure 2. Mosaic CNVs Contribute to Neuronal Diversity.

(A–C) Megabase-scale neuronal CNVs are observed across the human lifespan (ages 0.36–95 years) with varying size (A), number per cell (B), and percent genome coverage (C).
(D) Divergent CNV neuron (NeuN+), but similar non-neuronal (NeuN−), frequencies in 3 individuals.
(E–G) CNVs have an increased impact on genetic architecture in neuronal genomes compared to non-neuronal genomes, as measured by size (E), number per cell (F), and percent genome coverage (G).
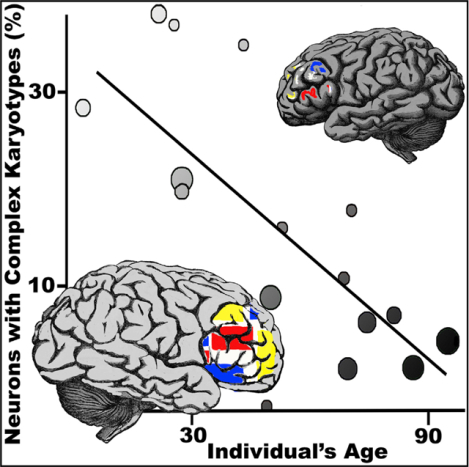
(H) Significant anti-correlation between age and CNV neuron frequency (linear fit, R2 = 0.9224, p = 0.0094). Lenient CN state thresholds throughout figure.
The 474 neuronal nuclei we analyzed represent the largest CNV dataset of neurotypical human brains generated by a single laboratory using a single WGA approach to date. We identified 62 CNV neurons (13.1%) with a mean CNV size of 16.8 Mb. We find that both neuronal and non-neuronal genomes harbor Mb-scale CNVs, which is consistent with previous reports (Cai et al., 2014; Knouse et al., 2016). The frequency (4%−23.1%) of complex karyotypes was more variable among neurons than among non-neuronal cells (4.7%−8.7%) from the same individuals (Figure 2D). CNV frequency in non-neuronal cells is similar to that reported in other somatic cells (e.g., keratinocytes). Relative to non-neuronal CNVs (Figures S3B, S3C, S3E, S3F, S3H, and S3I), neuronal CNVs are larger (Figure 2E), occur in greater numbers (Figure 2F), and affect more of the genome (1.3%−6% in neurons, 0.2%−0.8% in non-neurons; Figure 2G). Similar ratios of neuronal to non-neuronal CNVs were observed at stringent thresholds, but mean differences in non-euploid CN states were amplified (Figures S3K– S3M). A statistically significant linear decline in CNV neuron abundance was observed with age at both CN state thresholds (lenient: R2 = 0.9224, p = 0.0094, Figure 2H; stringent: R2 = 0.9434, p = 0.0058, Figure S3O).
Analysis and Integration of Publicly Available Data
Four previous studies used different WGA and CNV detection approaches on 458 additional single neurons from 6 male and 5 female neurotypical individuals of different ages (Cai et al., 2014; Knouse et al., 2014; McConnell et al., 2013; van den Bos et al., 2016). WGA was performed using GenomePlex (Cai et al., 2014; Knouse et al., 2014; McConnell et al., 2013), a degenerate oligonucleotide-primed (DOP)-PCR-based approach, or Strand-seq (van den Bos et al., 2016), a “pre-amplification free” approach. We harmonized these with our PicoPLEX dataset (e.g., alignment to hg19, exclusion of outlier bins in Figures S4A and S4B, BIC thresholds in Figures 3A, 3B, S4G, and S4H). GenomePlex and Strand-seq datasets displayed distinct outlier bin profiles. Analysis of RepeatMasker (University of California, Santa Cruz [UCSC]) genomic features in outlier bins found a depletion (log2 fold change <−0.25) of DNA, LINE, and long terminal repeat (LTR) features, and enrichment (log2 fold change >0.47) for short interspersed nuclear element (SINE) and simple repeat features (Wilcoxon, p < 2.2E−16) compared to remaining bins. BIC thresholds removed 11/225 GenomePlex cells (4.9%) and 41/233 Strand-seq cells (18.0%) from further analysis. The CN state thresholds reported above (Figure 1F) were inclusive of all of the WGA libraries passing BIC cutoffs. To further test the effectiveness of outlier bin removal, BIC cutoffs, and CNV thresholds, we re-analyzed split-amplification GenomePlex libraries from Knouse et al. (2016). One split-sample exceeded the BIC cutoff; the other with bad bins removed showed perfect concordance using our analysis pipeline (Figures S4C–S4F).
Figure 3. Brain CNV Atlas.

(A and B) BIC scores established from population distribution of GenomePlex (A, <−2.05) and Strand-seq (B, <− 1.93) WGA approaches.
(C–E) CNV attributes including size (C), number per cell (D), and percent genome coverage (E) were similar regardless of WGA approach.
(F) Significant atlas-wide anti-correlation between age and CNV neuron frequency (linear fit, R2 = 0.5521, p = 0.00097) Lenient CN state thresholds throughout figure.
The general characteristics of CNVs were similar regardless of WGA approach or sex (Figure 3C–3E). The mean length of single-cell CNVs was 14.8 Mb across all of the datasets, which is notably larger than most CNVs observed in bulk, germline human genomes (1–10 kb; MacDonald et al., 2014). As with PicoPLEX data, CNV size was variable among individuals (Figures S4J, S4K, S4M, S4N, S4P, and S4Q), but average CNV size was similar across WGA approaches (Figures 3C–3E). The average CNV size in GenomePlex libraries was 11.7 Mb, but it ranged from 4.1 to 15.9 Mb among individuals. The average CNV size in Strand-seq libraries was 13.2 Mb, but it ranged from 4.3 to 17.7 Mb among individuals. Similar but on average larger CNV sizes were observed at the stringent threshold (Figures S4I, S4L, and S4O). In contrast to a previous report (Knouse et al., 2016), no enrichment for LINE1 sequence was apparent in CNVs or CNV borders compared to chance, and equal rates of telomeric CNVs (~14%) were observed in young and aged neurons (see Method Details).
Anti-correlation between CNV neuron prevalence and individual age is roughly an order of magnitude more significant in the complete atlas (lenient: R2 = 0.5521, p = 0.00097, Figure 3F; stringent: R2 = 0.3941, p = 0.0092, Figure S4R). Despite narrower age ranges, CNV neuron prevalence was also anti-correlated with age in the published datasets (lenient: R2 = 0.5315, p = 0.011, Figure S4S; stringent: R2 = 0.3524, p = 0.054, Figure S4T). Potential confounding variables such as BIC scores or post-mortem interval showed no significant correlation with age (Figure S5).
Our observation of fewer CNV neurons in aged individuals contrasts with the concept of genosenium (Lodato et al., 2018), which states that the accumulation of somatic mutations over one’s lifetime is associated with aging-related cellular and molecular phenotypes. Thus, we also analyzed the number of CNVs per CNV neuron. Most CNV neurons (62.8%) contained only 1 or 2 CNVs, but the average number of CNVs per CNV neuron in the atlas was 3.9 (Figure 3D). Aged CNV neurons, although rare, had more irregular karyotypes. For example, the 20-year-old individual had the highest percentage of CNV neurons (40% CNV neurons, 3.8 CNVs/CNV neuron), but the 49-year-old (11.1% CNV neurons) had the most CNVs/CNV neurons (10.3). Other individuals, such as the 24-year-old and the 86-year-old, also support this trend; 38.9% of neurons in the 24-year-old had CNVs with an average of 2.8 CNVs/CNV neuron, whereas only 4% of neurons in the 86-year-old had CNVs, but these CNV neurons averaged 5.0 CNVs. The individual with the highest mean CNV size (37.2 Mb) was 86 years old (Figure S4J); this was due in large part to 1 neuron containing 2 trisomies and 1 monosomy (Figure S3N). Genosenium, if true, may operate on different somatic mutations in distinct ways.
Long Gene Mosaicism and Neuronal Diversity
Brain CNVs, like all non-V(D)J CNVs, occur because DNA repair is not perfect. Transcription and replication lead to DNA double-strand breaks (DSBs), which in turn sometimes lead to CNVs. Gene length increases susceptibility to transcriptional and replicative genomic stress. Genes encoded by >100 kb of genomic sequence (i.e., long genes) tend to be neuronally expressed genes with roles in neuronal connectivity and synaptic plasticity (Zylka et al., 2015). Long genes also overlap with DNA fragile sites, and replicative stress can lead to large CNVs that encompass these loci (Wilson et al., 2015). Likewise, transcriptional stress leads to DNA DSBs in neurons (Madabhushi et al., 2015) and has a predominant effect on long gene transcript abundance (King et al., 2013). Recent studies link these observations to DNA DSBs during mouse neurodevelopment (Wei et al., 2016) and motivate the hypothesis that somatic mutations affecting long genes mediate the functional consequences of brain somatic mosaicism (Weissman and Gage, 2016).
The assembled atlas identified 522 neural CNVs (Table 1). We performed overlap and random permutation analyses to determine whether subsets of 93 candidate long genes (Figure 4A) were associated with CNVs more frequently than expected by chance. Genomic regions (i.e., bins) that accumulated CNVs (i.e., duplications and deletions) at an increased population-wide frequency compared to the rest of the genome (i.e., hotspots; Figure 4B) were determined using different hotspot thresholds (see Method Details). Enrichment was assessed by calculating 184 raw p values (8 candidate gene lists, 23 hotspot thresholds) from 10,000 permutations per gene list per hotspot threshold. After correcting for multiple hypothesis testing (Benjamini-Hochberg false discovery rate [FDR], 5% FDR cutoff), dataset-wide significance was observed for the entire candidate gene list in some cases, and, notably, with putative hotspots that include 3 of the 4 common candidate genes: GPC6, NRXN3, and RBFOX1 (Figure 4C).
Figure 4. Long Gene Enrichment in Brain CNV Atlas.

(A) Venn diagram to define putative hotspots.
(B) Bin-level summary (lenient CN state threshold) of deletion (red) and duplication (green) occurrence in brain CNV atlas.
(C–E) Enrichment results for hotspots from all cells (C), individuals (D), and age groups (E). p values < 0.05 are red.
(F–H) REViGO plots of enriched Gene Ontology (GO) terms for all of the data analyzed. The relative size of each category reflects significance; the largest groups have the lowest p values. GO enrichment determined using PANTHER analysis of CNV-affected genes in all neural data (F), and neurons from age groups 68–74 (G) and 81–95 years old (H).
CNV neurons were rare in aged individuals, so we assessed whether the genetic architecture of CNV neurons may also change during the lifespan. We further tested each individual in the atlas for candidate gene enrichment using relevant thresholds for recurrent hotspots (see Method Details) at stringent and lenient criteria. Significant corrected p values were not observed in the youngest individuals, but they were observed in aged individuals (Figure 4D). When individuals were pooled based on age groups, significant p values were also observed only in the most aged group (Figure 4E).
CNV-affected loci may not be restricted to long genes. We assembled a comprehensive list of all of the genes affected in the brain CNV atlas and used PANTHER (Mi et al., 2013) to calculate enrichment statistics and plotting scripts from REViGO (Supek et al., 2011) to visualize these results. Gene Ontology categories associated with “sensory perception of smell” and “calcium-mediated signaling” were notably enriched in the atlas-wide gene set (Figure 4F). When assessed by age group, as in Figure 4E, these enrichments were only detected in the aged groups (Figures 4G and 4H).
DISCUSSION
Brain somatic mosaicism is a largely unexplored aspect of neuronal diversity (Harbom et al., 2018). In the human cerebral cortex, neuronal diversity is described in terms of electrophysio-logical properties (Contreras, 2004) and gene expression profiles (Lake et al., 2016) that are brought about by genetic programs (Lein et al., 2017). Current neurodevelopmental models implicitly assume that all somatic cells operate with identical genomes. In turn, population-based genetic studies of neurological disease typically sequence bulk blood DNA as a proxy for brain DNA. Somatic mutations affecting cell proliferation and survival pathways can alter neuronal diversity and lead to cortical overgrowth phenotypes ranging from hemimegalencephaly to focal dysplasia (Jamuar et al., 2014; Lee et al., 2012; Mirzaa et al., 2016; Poduri et al., 2012). Elevated levels of somatic mutations have also been associated with Rett syndrome (Muotri et al., 2010), neurodegenerative disease (Bushman et al., 2015; Iourov et al., 2009; Lee et al., 2018; Lodato et al., 2018; McConnell et al., 2004), schizophrenia (Bundo et al., 2014), and altered behavior (Bedrosian et al., 2018). The Brain Somatic Mosaicism Network is an ongoing multi-site effort that aims to define how brain somatic mutations affect the genetic architecture of psychiatric disease (McConnell et al., 2017). However, the consequences of somatic mutations in neurotypical human brains remain a central unaddressed question. We report a neurotypical human brain somatic CNV atlas to begin to assess how mosaic somatic mutations affect neuronal diversity.
We assembled a brain CNV atlas from 1,285 single brain nucleus libraries, the accumulated work of our laboratory and 3 other laboratories. Development of a single, robust computational pipeline that both protected against false-positive CNV calls and minimized false-negative calls was essential. First, we identified and excluded a small group (101/4,505) of genomic bins that were consistently non-euploid (i.e., outliers) across our 5 analyzed individuals. Second, we applied an objective BIC cutoff to exclude WGA libraries that would be the most prone to aberant segmentation calls. Third, we evaluated published datasets from 4 other sources that used different WGA methods. Population-level statistics were generally similar, but each WGA approach identified unique outlier bins and slightly different BIC cutoffs. Fourth, CN state distributions centered near CN = 1, 2, and 3 were apparent across all of the WGA approaches, so we used 2-tailed p values (lenient = 0.05, stringent = 0.01) from pan-method DNAcopy segmentation calls to define 2 sets of CN state thresholds. Fifth, we showed that these filters (outlier bin removal, BIC cutoffs, and CN state thresholds) effectively eliminate false-positive CNV calls (15/2,400; <1%). Lenient CN state thresholds are mildly (<3%) more prone to short (~<6 Mb) false-positive calls, but far better at identifying true-positives (>95%). Our central findings are unchanged when only the largest (>6 Mb) CNVs are considered. The brain CNV atlas comprises 879 neuronal and 115 non-neuronal genomic libraries; Mb-scale CNVs were identified in 129 neurons and 8 non-neurons.
The most salient feature of the brain CNV atlas is an anti-correlation between the age of an individual and the percentage of CNV neurons in the frontal cortex of that individual. This finding was significant in our new PicoPLEX dataset and in the published GenomePlex and Strand-seq datasets, and is highly significant using all of the available data (p = 0.00097). By contrast, the initial assessment of CNV location finds evidence for the enrichment of a subset of long genes and neurally associated Gene Ontology categories only in aged brains. Given the enrichment of these CNVs in aged, not young, neurons, CNVs affecting some genomic loci may be more compatible with neural survival than others. We found similar rates of CNV non-neurons at different ages; however, it will be interesting to determine whether other long-lived cells (e.g., cardiomyocytes) show a similar change in mosaic composition during aging.
We provide evidence that a functional consequence of CNV neurons may be selective vulnerability to aging-related cell death. Age-related cognitive decline is associated with notable decreases in cerebral cortical thickness, myelination, and synapse number accompanied by ex vacuo enlargement in ventricular volume (Morrison and Baxter, 2012). Although neuronal cell death is generally considered to be minimal in the healthy mature brain, rates of ~10% cerebral cortical neuron loss during adulthood are consistent with stereological counts in neurotypical individuals (Pakkenberg et al., 2003). The decline in CNV neuron prevalence that we observe between individuals <30 years old and individuals >70 years old is also strikingly consistent with selective CNV neuron loss during a person’s adult lifetime. We conclude that the most parsimonious interpretation of these data is that many, but not all, CNV neurons are selectively vulnerable to aging-associated atrophy. This cross-sectional finding highlights the unmet need for an increased longitudinal understanding of human neuronal genome dynamics during an individual’s health span. Human pluripotent stem cell-based models (Brennand et al., 2015) represent a straightforward means to this end.
STAR* METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mike McConnell (mikemc@virginia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
Brain tissue was taken from five human individuals. Descriptive details of these individuals, such as age, sex, and number and type of cells, are available in Table 1. Four samples (Lieber) were collected under the IRB approved Protocol Title: Collection of Postmortem Human Brain, Blood and Scalp Samples for Neuropsychiatric Research’, registered to Thomas Hyde, MD, PhD, BA, Located on the Third Floor, 855 N. Wolfe Street, Baltimore, MD 21205, United States. Executed board action date: 06/19/2017. STUDY NUM: 1126332; ONLINE TRACKING: INVEST NUM: 165719.WO NUM: 1-1010280-1. STUDY APPROVAL EXPIRES: 07/18/2019 and is renewed annually. Informed consent was obtained from all subjects. The Western Institutional Review Board® (1019 39th Avenue SE. Suite 120 | Puyallup, WA 98374–2115; Office: (360) 252–2500 | Toll Free: (800) 562–4789; http://www.wirb.com/Pages/default.aspx) certifies the protocols for collecting and transferring clinical samples in the context of this study are in full compliance with good clinical practices as defined under the U.S. Food and Drug Administration regulations, and the International Conference on Harmonisation guidelines. These statements are true and correct as reflected in the records of the Western Institutional Review Board (WIRB), OHRP/FDA parent organization number IORG 0000432, IRB registration number IRB00000533. The fifth was obtained from the National Institute for Child Health and Human Development (NIH) Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD, contract HHSN2752009000011C, ref. no. N01-HD-9–011.
METHOD DETAILS
Single nuclei isolation
All procedures were done on ice. Solutions were made using nuclease-free water and supplemented with freshly prepared 50 X proteinase inhibitor just before use. Nuclei isolation media, optiprep nuclei diluent, nuclei storage buffer and blocking buffer (prior to adding tween) were 0.22 μm filtered.
Buffers used:
Nuclei Isolation Media (NIM): 25 mM KCl, 5 mM MgCl2, 10 mM Tris-Cl (pH 8.8), 250 mM sucrose, 1mM dithiothreitol (DTT).
OptiPrep Density Gradient Medium (ODGM): 60% Iodixanol solution.
OptiPrep Nuclei Diluent (OND): 150 mM KCl, 30 mM MgCl2, 60 mM Tris-Cl (pH 8.8), 250 mM sucrose.
Blocking Buffer (BB): 1x PBS, 1.0% Bovine Serum Albumin (BSA), and 0.1% Tween 20.
Nuclei Storage Buffer (NSB): 5 mM MgCl2, 50 mM Tris-Cl (pH 8.8), 166 mM sucrose, 1 mM dithiothreitol (DTT).
50 X EDTA-free Protease Inhibitor Cocktail in nuclease-free water.
10% Triton x-100 in nuclease-free water.
Post-mortem cortex was stored at −80°C until fragmented while frozen on dry ice in a pre-chilled mortar and pestle. Fragments (~100 mg) were completely solubilized in 1 mL of nuclei isolation media (NIM) by gently triturating using a 1000 μL pipette tip. We transferred slurry to a 5 mL round bottom, polypropylene tube and homogenized using a Polytron PT 1300 D tissue disruptor for 2 minutes. The sample appeared opaque and homogeneous. We added 1 μl of 10% Triton X-100 (final conc. 0.01%) and gently mixed by rotation before transferring to a dounce with large and small pestle clearance (0.0030–0.0050 in. and 0.0005–0.0025 in., respectively). After 15 stokes with each clearance we microscopically verified cell disruption with 0.4% trypan blue solution. We centrifuged samples at 1,000 × g for 8 minutes at 4°C and re-suspended the pellet in 1 mL of 6:5:1 NIM:ODGM:OND (25% iodixanol). We used a 1 mL syringe (without needle) to apply the suspension onto 1 mL of 29:31 ODGM:OND solution (29% iodixanol) in a 5 mL thin-wall polyallomer ultracentrifuge tube (13 × 51 mm). Samples were centrifuged at 10,300 × g for 20 minutes at 4°C in a Beckman L8-M ultracentrifuge using a SW55 Ti rotor. The supernatant containing cell debris was removed, leaving ~50 μL in the bottom of the tube. We confirmed the presence of nuclei by microscopy using 0.4% trypan blue and used immediately or stored up to 1 week in NSB at 4°C without reduction in whole genome amplification efficiency.
Single nuclei genome sequencing
We labeled nuclei derived from neurons by incubating with mouse monoclonal anti-human NeuN IgG clone A60 (Alexa Fluor 555 conjugate) diluted 1:250 in blocking buffer overnight at 4°C. We verified that NeuN+ nuclei also contained dsDNA by co-staining with either SYTO 13 green fluorescent nucleic acid stain at 500 nM or standard DAPI. We isolated individual NeuN+, DNA+ nuclei by flow sorting into 8-well thin-wall PCR tube strips. At certain points in the execution of these procedures we also isolated individual nuclei from the flow-sorted NeuN+, DNA+ nuclei pool using the CellRaft system (Cell Microsystems) to verify integrity of the nuclei and quality of the WGA on a small scale. There are videos and extensive literature on the use of the CellRaft at https://www.cellmicrosystems.com and Wierman et al. (2017). These quality control steps ensured the nuclei were intact and contained genomic material suitable for WGA. The PicoPLEX Whole Genome Amplification Kit was applied to single nuclei according to manufacturer’s instructions (Rubicon Genomics, Ann Arbor, MI). The PicoPLEX reaction enzymatically copies elements across the entire genome, fragments DNA products, and bar-codes the fragments with unique Illumina i5 and i7 index sequences. We confirmed reactions produced high molecular weight DNA by 1 X TBE, 1% agarose gel electrophoresis containing 1 mg/ml ethidium bromide. Productive reactions were purified using QIAquick PCR purification columns. We quantified yields using the high-sensitivity (HS) DNA Qubit 3.0 assay and combined equimolar portions of each purified PicoPLEX product into pools of 48 nuclei with compatible index combinations for multiplex sequencing. We electrophoresed pooled libraries into 0.75 mm thick 1 X TBE, 7.5% polyacrylamide (37.5:1 acrylamide:bisacrylamide) gels at 35 mA for 20 minutes and incubated with SYBR Gold DNA stain diluted 1:10,000 in 1 X TBE for 5 minutes before excising sections containing 450 bp to 800 bp DNA fragments on a UV transilluminator (long wave). We electro-eluted DNA from polyacrylamide sections into 1 X TAE, 1% low-melt agarose gel containing 1 μg/ml ethidium bromide at 100 V for 15 minutes and isolated chunks containing DNA on a UV transilluminator (long wave). DNA was purified from agarose using QG buffer according to the QIAquick PCR column protocol (QIAGEN). We quantified size-selected pooled library DNA by HS DNA Qubit 3.0 assay and diluted to 6 nM prior to sequencing on the Illumina (San Diego, CA) platform. We sequenced a total of 829 brain nuclei and used these for BIC and copy number state population statistics, including threshold determination. Two single nuclei were flagged for ambiguous provenance. The CNV atlas includes 827 male brain nuclei that were amplified by the DNA WGA PicoPLEX kit. These single nucleus genome sequencing procedures were previously described in McConnell et al. (2013) and in greater detail in Wierman et al. (2017).
Analysis of single cell sequencing data
Sequence reads from Illumina were trimmed of PicoPLEX primers using the fastx_trimmer command (hannonlab.cshl.edu/fastx_toolkit/). Reads were then aligned to the human genome (version hg19) with BWA-aln V0.7.12 using default options (Li and Durbin, 2009) and converted to BAM format using Samtools V1.1 (Li et al., 2009). Duplicates were removed using MarkDuplicates (Picard tools V1.105, broadinstitute.github.io/picard). Using a 40-mer mappability track (UCSC, wgEncodeCrgMapabilityAlign40-mer.bigWig) to determine uniquely mappable bases, we divided the genome into 4,505 dynamically sized genomic bins, each containing 500kb of mappable sequence. The mean bin size was 687 kb. Read counts for each bin were determined by Bedtools V2.17.0 coverageBed (Quinlan and Hall, 2010). To avoid read count bias arising from GC content, bins were grouped into 16 roughly equal size groups according to GC percentage and each read count within a GC group was divided by the median read count of the group and multiplied by two.
Following analysis of several hundred single cell datasets, we observed that certain genomic bins were consistently above or below the euploid state, most likely due to biases arising from alignment or artifacts generated during WGA. To avoid biases in segmentation introduced by these outlier bins, namely false positive and false negative CNVs, we used Tukey’s Outlier Method on the median log-copy number values of all 4,505 bins in a sex and WGA-specific fashion, resulting in between 101 and 153 bins to be excluded from segmentation, depending on the sex of the individual and WGA used. In addition to the bins excluded by the outlier detection method, two Y chromosome bins were manually excluded from female Strand-seq dataset. Single cell datasets were segmented using DNAcopy (Seshan and Olshen, 2018), an R package (www.R-project.org) that implements circular binary segmentation (CBS) to detect copy number “changepoints” in genomic data. DNAcopy was run on the normalized bin data using parameters alpha = 0.001, undo.SD = 0, and min.width = 5.
BIC scoring and filtering
To determine which samples were of sufficient quality to merit further analysis, we implemented Bayesian Information Criterion (BIC) (Schwarz, 1978)
as a scoring metric where σ2 is the variance of the data points about their respective segment means, kp is equal to 1 + 2*(number of changepoints), and n is the number of bins (4,505) assuming a piecewise-constant, Gaussian error segmentation model (see Muggeo and Adelfio (2011) for details of segmentation modeling and parameterization). The equation above shows the usual BIC score divided by the number of bins, which is fixed across samples. BIC penalizes a segmentation that under-fits the data (i.e., allows high variance of data within segments) or over-fits the data (i.e., creates too many segments); thus, a cell with properly fitted data and relatively low bin value variance will receive a low BIC score.
To define threshold BIC scores for inclusion in further analysis, a histogram of BIC scores was generated for each WGA method. Using the R package mixtools (Benaglia et al., 2009), we fit two Gaussian distributions to the PicoPLEX and Strand-seq histograms, and one Gaussian distribution to the GenomePlex histogram due to its displaying a single mode. Using the Gaussian distribution with the lower mean (or, in the case of GenomePlex, the lone distribution), we set the threshold BIC score for inclusion to correspond to p = 0.05 on the upper tail. For PicoPLEX, cells scoring below −2.21 were selected for further analysis (Figure 1E); for GenomePlex, the threshold was −2.05 (Figure S4G); for Strand-seq, the cutoff was −1.93 (Figure S4H).
Defining CNVs
Because DNAcopy does not assign integer copy number values to the segments it outputs, it was necessary to define threshold copy number values for a segment to be considered a CNV. We set CNV thresholds by evaluating autosomal segments of sizes ranging from 5, our minimum number of bins required to call a CNV, and 45 bins, a length smaller than the shortest autosome, resulting in a distribution of copy numbers excluding the high number of whole chromosomes at or near copy number two. The segments were plotted by copy number value in a histogram, and we fit a three-Gaussian mixture model using mixtools (Benaglia et al., 2009) and plotted the resulting mixed Gaussian model of three distributions centered at the local peaks near copy number 1, 2, and 3. Using the central Gaussian, centered near 2, we calculated two sets of thresholds: the stringent thresholds, 2.80 and 1.14, the result of a cumulative two-tailed probability of 0.01; and the lenient thresholds, 2.60 and 1.34, determined by a two-tailed cumulative probability of 0.05. Throughout our CNV analyses, we used both thresholds in order to give a wider range of possible results and avoid any influence of false positive or false negative CNVs. Additionally, CNVs were required to be at least 5 bins in length, and chromosomes X and Y were not examined for CNVs.
Test data simulation
DNAcopy is prone to calling CNV events using correlated noise as input (Muggeo and Adelfio, 2011), so we sought to determine the degree to which our single cell data contains correlated noise. We selected six single cells to simulate test data. In our “NULL model,” we assumed that the data for our six cells were described by correlated noise and Gaussian noise about the euploid copy number state, and contained no real CNV events. In the “alternative model,” we assumed that the six cells were described by real CNV events identified by DNAcopy and residual correlated and Gaussian noise. We simulated 200 test data cells for each of the cells under each of the models and found that the CNVs produced by the NULL model rarely matched those of the original cell in size or divergence from 2 (Figure S1H), leading us to conclude that the ALT model was a more accurate representation of our data (Figure S1I). To determine the best DNAcopy segmentation parameters, we used simulation data of the “alternative model” to explore values of alpha ranging from 0.05 to 10−5, undo.SD ranging from 0 to 5, and min.width ranging from 2 to 5, and calculated a BIC score for each. We also tested the performance of each segmentation using a receiver operating characteristic (ROC) curve to determine the parameters at which sensitivity was maximized and false positive CNVs were minimized (Figure S1J). We concluded that the best parameters for segmentation were alpha = 0.001, undo.SD = 0, and min.width = 5.
We later simulated data based on 6 additional cells to assess the effect of our BIC and CNV thresholds on false positives and false negatives using ALT and NULL model simulations as described above (Figure S2B–C).
RepeatMasker analysis of outlier bins
To see if outlier bins showed any differences in genomic features compared to normal bins, we selected 100 outlier bins and 100 normal bins of similar size and calculated base overlap with RepeatMasker (UCSC) feature types, such as LINE, SINE, simple repeats, long terminal repeats, etc. Following normalization of overlapping bases by bin size, we performed a Wilcoxon rank-sum test for each RepeatMasker feature type to test whether there was a difference in the fraction of overlapping bases between the bin groups.
RepeatMasker enrichment analysis of CNVs
To check for overlap of genomic features in the CNVs that we detected, we selected a subsample of CNVs corresponding to the 5th, 25th, 50th, 75th, and 95th percentiles of CNV size and used Bedtools shuffle (Quinlan and Hall, 2010) to generate 1000 randomized genomic locations for each CNV. We then computed the percent overlap of each simulated CNV with RepeatMasker (UCSC) feature types. From this, we computed the mean and standard deviation percent overlap of the randomized CNVs which we used to calculate a z-score associated with the actual overlap as a function of CNV size. We also checked for border region enrichment by calculating the percent overlap of RepeatMasker feature types with the first and last megabase of each simulated CNV and deriving the associated z-score of the actual CNV border overlap as a function of CNV size.
Gene set enrichment
To explore the biological significance of large scale CNVs in the brain, we sought to follow up on the work of two papers that identified lists of long genes that may be predisposed to DNA breaks in mice (King et al., 2013; Wei et al., 2016). Building upon this idea, we also examined a third list containing the 50 longest human genes obtained using biomaRt (Smedley et al., 2015). These gene lists shared genes in common with one another. There were ultimately seven gene lists drawn from one, two, or three of the original lists, as well as an eighth gene list containing all 93 genes gathered from all three sources. To test for enrichment, we collected hotspot coordinates corresponding to a minimum number of CNV events, which varied depending on the subset of data being tested and the CNV threshold used. For each data subset, we set hotspot cutoffs for the required number of CNVs such that no cutoff should lead to greater than 20% coverage of the human genome. The resulting cutoffs for each subset and threshold are listed in the table below:
| Individual(s) | Threshold | CNVs | Dels. | Dups. |
|---|---|---|---|---|
| All | Lenient | 0, 5–8, 6–8, 7–8, 8 | 0, 4–6, 5–6, 6 | 2–5, 3–5, 4–5, 5 |
| 0.36–26 | Lenient | 3–5, 4–5, 5 | 2–5, 3–5, 4–5, 5 | 2–4, 3–4, 4 |
| 42–52 | Lenient | 3–4, 4 | 3–4, 4 | 1 |
| 68–74 | Lenient | 1–2, 2 | 1–2, 2 | 1–2, 2 |
| 81–95 | Lenient | 2–4, 3–4, 4 | 2–3, 3 | 1–2, 2 |
| 0.36 | Lenient | 1–3, 2–3, 3 | 1–2, 2 | 1–2, 2 |
| 20 | Lenient | 2–4, 3–4, 4 | 2 | 1–2, 2 |
| 24 | Lenient | 1 | 1 | 1 |
| 26 NeuN+ | Lenient | 2–5, 3–5, 4–5, 5 | 1–3, 2–3, 3 | 1–4, 2–4, 3–4, 4 |
| 26 NeuN- | Lenient | NA | NA | 1 |
| 42 | Lenient | 2 | 2 | 1 |
| 48 | Lenient | NA | NA | NA |
| 49 NeuN+ | Lenient | 2–3, 3 | 3 | 1 |
| 49 NeuN- | Lenient | 1 | 1 | 1 |
| 52 | Lenient | 1–2, 2 | 1–2, 2 | 1 |
| 68 | Lenient | 1 | 1 | 1 |
| 69 | Lenient | 1 | 1 | 1 |
| 70 | Lenient | 1 | ||
| 74 | Lenient | 1–2, 2 | 1–2, 2 | 1 |
| 81 | Lenient | 1 | 1 | 1 |
| 86 NeuN+ | Lenient | 2 | 1 | 1 |
| 86 NeuN- | Lenient | 1 | 1 | 1 |
| 95 | Lenient | 1–3, 2–3, 3 | 1–2, 2 | 1 |
| All | Stringent | 0, 4–6, 5–6, 6 | 3–5, 4–5, 5 | 2–4, 3–4, 4 |
| 0.36–26 | Stringent | 2–5, 3–5, 4–5, 5 | 2 | 1–3, 2–3, 3 |
| 42–52 | Stringent | NA | 2–3, 3 | NA |
| 68–74 | Stringent | 1–2, 2 | 1 | 1–2, 2 |
| 81–95 | Stringent | 2–4, 3–4, 4 | 2–3, 3 | 1 |
| 0.36 | Stringent | 1–3, 2–3, 3 | 1 | 1–2, 2 |
| 20 | Stringent | 2–4, 3–4, 4 | 1–2, 2 | 1–2, 2 |
| 24 | Stringent | NA | 1 | NA |
| 26 NeuN+ | Stringent | 1–3, 2–3, 3 | 1–2, 2 | 1–2, 2 |
| 26 NeuN- | Stringent | NA | NA | NA |
| 42 | Stringent | NA | 1 | NA |
| 48 | Stringent | NA | NA | NA |
| 49 NeuN+ | Stringent | NA | 2 | NA |
| 49 NeuN- | Stringent | NA | 1 | NA |
| 52 | Stringent | NA | 1 | NA |
| 68 | Stringent | 1 | 1 | 1 |
| 69 | Stringent | 1 | 1 | 1 |
| 70 | Stringent | NA | 1 | NA |
| 74 | Stringent | NA | NA | 1 |
| 81 | Stringent | 1 | 1 | 1 |
| 86 NeuN+ | Stringent | NA | 1 | 1 |
| 86 NeuN- | Stringent | NA | 1 | NA |
| 95 | Stringent | 1–3, 2–3, 3 | 1–2, 2 | 1 |
For each set of hotspots, we randomly shuffled the hotspot loci within the genome 10,000 times to generate a null model of CNV coverage. We then determined the number of genes of interest found in the actual hotspots and the range of genes of interest found in the null model to generate enrichment p values, which were then FDR-corrected using the Benjamini-Hochberg method. Owing to the considerable inter-dependence among hotspot sets being tested, these p values were FDR-corrected within stratified sub-groups; for example, p values for hotspots derived from individuals were corrected separately from p values for hotspots derived from age groups. Likewise, p values for hotspots defined as regions of 3–5 CNVs were corrected separately from regions defined as regions of 2–5, 4–5, or 5 CNVs.
Gene Ontology (GO) term analysis
We compiled a list of genes with genomic coordinates overlapping each CNV and separated these lists by individual and, where applicable, cell type. These lists of CNV-affected genes were submitted to PANTHER (Mi et al., 2013) (pantherdb.org) to determine if any GO terms were enriched. The resulting GO terms and corresponding p values were then submitted to REViGO (Supek et al., 2011) (revigo.irb.hr) to aid visualization via downloadable plotting scripts.
QUANTIFICATION AND STATISTICAL ANALYSIS
Most statistical analyses were performed using R (version 3.4.1, www.R-project.org), utilizing base and downloaded packages. BIC score cutoffs and CNV thresholds were defined as explained in Method Details. Statistical tests and outlier detection are explained in figure legends and in Results text. Stratification of p value correction in gene set enrichment is described above in Method Details. Statistical significance was defined as p < 0.05.
DATA AND SOFTWARE AVAILABILITY
Single cell sequencing data for PicoPLEX neurons and non-neurons is available through Synapse (https://www.synapse.org/#!Synapse:syn16803262) and the NIH data archive (collections 2963 and 2458). The R script used for simulating data using ALT and NULL models is available at https://github.com/mcconnell-lab/scripts/blob/master/ALT_NULL_dataset_simulation.R.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse monoclonal anti-human NeuN IgG Alexa Fluor 555 Conjugate clone A60 | EMD via Millipore | Cat. # MAB377A5 |
| Biological Samples | ||
| Human pre-frontal cortex, Frozen 0.36 year-old | Lieber Institute for Brain Development | Anonymized Index #1845 |
| Human pre-frontal cortex, Frozen 49 year-old | Lieber Institute for Brain Development | Anonymized Index #5154 |
| Human pre-frontal cortex, Frozen 86 year-old | Lieber Institute for Brain Development | Anonymized Index #5401 |
| Human pre-frontal cortex, Frozen 95 year-old | Lieber Institute for Brain Development | Anonymized Index #5570 |
| Human pre-frontal cortex, Frozen 26 year-old | University of Maryland Brain and Tissue Bank | Anonymized Index #1583 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| OptiPrep iodyxonol solution | Sigma-Aldrich | D1556–250ML |
| Acrylamide solution 37.5:1 (Acrylamide:Bis-acrylamide) | Bio-Rad | Cat. #1610158 |
| TEMED, 50 mL | Bio-Rad | Cat. #1610801 |
| Ammonium Persulfate (APS) | Bio-Rad | Cat. #1610700 |
| GelPilot DNA Loading Dye, 5x | QIAgen | Cat. # 239901 |
| 10 X Tris-Boric Acid-EDTA buffer | ThermoFisher | Cat. # B52 |
| 10 X Tris-Acetate-EDTA buffer | ThermoFisher | Cat. # B49 |
| Magnesium Chloride anhydrous, ≥ 98% | Sigma-Aldrich | Cat. # M8266 |
| Potassium chloride BioXtra, ≥ 99.0% | Sigma-Aldrich | Cat. # P9333 |
| Sodium chloride BioXtra, ≥ 99.5% | Sigma-Aldrich | Cat. # S7653 |
| Sodium hydroxide | Sigma-Aldrich | Cat. # S8045 |
| Polyoxyethylenesorbitan monolaurate CAS Number: 9005-64-5 | Sigma-Aldrich | Cat. #P1379 |
| Tris(hydroxymethyl)aminomethane hydrochloride reagent grade, ≥ 99.0% | Sigma-Aldrich | Cat. # T3253 |
| Sucrose for molecular biology, ≥ 99.5% (GC) | Sigma-Aldrich | Cat. # S0389 |
| DL-Dithiothreitol BioUltra, for molecular biology, ≥ 99.5% (RT) (Sigma) | Sigma-Aldrich | Cat. #43815 |
| Trypan Blue Solution of 0.4% | ThermoFisher | Cat. #15250061 |
| Bovine Serum Albumin, lyophilized powder, suitable for (for molecular biology) | Sigma-Aldrich | Cat. # B6917 |
| Hydrochloric acid ACS reagent, 37% | Sigma-Aldrich | Cat. # 320331 |
| Glacial acetic acid | Sigma-Aldrich | Cat. # A6283 |
| Ethylenediaminetetraacetic acid | Sigma-Aldrich | Cat. #EDS-100G |
| Ultra-Pure Low Melting Point agarose | ThermoFisher | Cat. #16520050 |
| EDTA-free Protease Inhibitor Cocktail; Tablets | Roche via Sigma-Aldrich | Cat. #11873580001 |
| SYTO 13 green fluorescent nucleic acid stain | ThermoFisher | Cat. # S7575 |
| SYBR Gold nucleic acid gel stain | ThermoFisher | Cat. #S11494 |
| Ethidium Bromide Solution (10 mg/mL) | ThermoFisher | Cat. #17898 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | ThermoFisher | Cat. #D1306 |
| UltraPure DNase/RNase-Free Distilled Water | ThermoFisher | Cat. #10977015 |
| Phosphate Buffered Saline, PBS (10X), pH 7.4 | ThermoFisher | Cat. #70011044 |
| IsoFlow Sheath Fluid | Beckman Coulter | Cat. # 8546859 |
| Critical Commercial Assays | ||
| Rubicon PicoPLEX WGA Kit | via Agilent | Cat. #5190–9533 |
| QIAquick PCR purification columns Kit | QIAgen | Cat. #28106 |
| MiSeq Reagent Kit v2 (50 cycle) | Illumina | MS-102–2001 |
| HiSeq PE Rapid Cluster Kit v2 | Illumina | PE-402–4002 |
| Qubit dsDNA HS Assay Kit | ThermoFisher | Cat. # Q32851 |
| Critical Commercial Assays | ||
| Rubicon PicoPLEX WGA Kit | via Agilent | Cat. #5190–9533 |
| QIAquick PCR purification columns Kit | QIAgen | Cat. #28106 |
| MiSeq Reagent Kit v2 (50 cycle) | Illumina | MS-102–2001 |
| HiSeq PE Rapid Cluster Kit v2 | Illumina | PE-402–4002 |
| Qubit dsDNA HS Assay Kit | ThermoFisher | Cat. # Q32851 |
| Deposited Data | ||
| Single cell sequencing data | This study (NDA Study ID 636) | https://dx.doi.org/10.15154/1503237 |
| Software and Algorithms | ||
| Bedtools version 2.17.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/index.html |
| biomaRt (R package) version 2.36.1 | Smedley et al., 2015 | https://bioconductor.org/packages/release/bioc/html/biomaRt.html |
| BWA version 0.7.12 | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| DNAcopy (R package) version 1.50.1 | Seshan and Olshen, 2018 | https://bioconductor.org/packages/release/bioc/html/DNAcopy.html |
| FASTX Toolkit version 0.0.13 | http://hannonlab.cshl.edu/ | http://hannonlab.cshl.edu/fastx_toolkit/download.html |
| mixtools (R package) version 1.1.0 | Benaglia et al., 2009 | https://cran.r-project.org/web/packages/mixtools/index.html |
| PANTHER | Mietal., 2013 | http://www.pantherdb.org/ |
| Picard Tools version 1.105 | https://broadinstitute.github.io/picard/ | https://github.com/broadinstitute/picard/releases |
| Python version 2.6.6 | https://www.python.org/ | https://www.python.org/downloads/ |
| R version 3.4.1 | https://www.r-project.org/ | https://cran.r-project.org/src/base/R-3/ |
| REViGO | Supek et al., 2011 | http://revigo.irb.hr/ |
| Samtools version 1.1 | Li et al., 2009 | http://www.htslib.org/download/ |
| Simulation script | This study | https://github.com/mcconnell-lab/scripts/blob/master/ALT_NULL_dataset_simulation.R |
| Other | ||
| 0.2 mL TempAssure PCR 8-tube strips | USA Scientific | Cat. #1402–2700 |
| Gloves large nitrile | Denville Scientific | Cat. #G4163 |
| Dounce Tissue grinder Pestle A clearance 0.0030–0.0050 in. Pestle B clearance 0.0005–0.0025 in. | Kimble via Sigma-Aldrich | Cat. # D8938 |
| SW 55 Ti Swinging-Bucket Rotor | Beckman-Coulter | Cat. #342196 |
| 13 × 51 mm, 5 mLThinwall polyallomer tubes | Beckman-Coulter | Cat. #326819 |
| Qubit Reader 3.0 | ThermoFisher | Cat. #Q33216 |
| Polytron PT 1300 D Manual Disperser | Kinematica via Fisher Scientific | Cat. #08-451-71 |
| 100 bp DNA ladder | New England Biolabs | Cat. # N3231 |
| Corning 0.22 μn bottle top filter | Sigma-Aldrich | Cat. # CLS430769 |
| CellRaft System Kit for Inverted Microscopes | Cell Microsystems | P/N: CRK |
| Syringe PP/PE without needle | Sigma-Aldrich | Cat. # Z230723 |
Highlights.
A robust computational approach was developed and applied to single-cell datasets
In cross-sectional data, the frequency of CNV neurons declines continuously with age
CNV neurons may be selectively vulnerable to aging-related atrophy
CNVs in aged neurons frequently overlap long neuronal genes
ACKNOWLEDGMENTS
We thank J. Lannigan and M. Solga (University of Virginia [UVA] fluorescence-activated cell sorting [FACS] core), Y. Bao (UVA genome analysis and technology core), and A. Koeppel (UVA bioinformatics core) for their contributed expertise. We thank P. Lansdorp (University of British Columbia) for prompt sharing of unpublished metadata, and F.H. Gage (Salk Institute) and J.V. Moran (University of Michigan) for critical feedback. Human tissue was obtained from the National Institute for Child Health and Human Development (NIH) Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore (contract HHSN2752009000011C, ref. no. N01-HD-9–011). Data are documented through the Brain Somatic Mosaicism Network Knowledge Portal (synapse.org/BSMN) and made available at the National Institute of Mental Health (NIMH) Data Archive. NIH funding to M.J.M. (U01 MH106882), to D.R.W. (U01 MH106893), and to M.J.M. and S.B. (U01 MH106882–03S1) supported this work. I.E.B. received support from the McDonnell Foundation, and W.D.C. received support from NIH T32 GM008136–30.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.107.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anacker C, and Hen R (2017). Adult hippocampal neurogenesis and cognitive flexibility - linking memory and mood. Nat. Rev. Neurosci 18, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae T, Tomasini L, Mariani J, Zhou B, Roychowdhury T, Franjic D, Pletikos M, Pattni R, Chen BJ, Venturini E, et al. (2018). Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science 359, 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M, et al. (2011). Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedrosian TA, Quayle C, Novaresi N, and Gage FH (2018). Early life experience drives structural variation of neural genomes in mice. Science 359, 1395–1399. [DOI] [PubMed] [Google Scholar]
- Benaglia T, Chauveau D, Hunter DR, and Young DS (2009). mixtools: an R Package for Analyzing Mixture Models. J. Stat. Softw 32, 29. [Google Scholar]
- Bhardwaj RD, Curtis MA, Spalding KL, Buchholz BA, Fink D, Björk-Eriksson T, Nordborg C, Gage FH, Druid H, Eriksson PS, and Frisén J (2006). Neocortical neurogenesis in humans is restricted to development. Proc. Natl. Acad. Sci. USA 103, 12564–12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldog E, Bakken TE, Hodge RD, Novotny M, Aevermann BD, Baka J, Bordé S, Close JL, Diez-Fuertes F, Ding SL, et al. (2018). Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat. Neurosci 21, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Marchetto MC, Benvenisty N, Brüstle O, Ebert A, Izpisua Belmonte JC, Kaykas A, Lancaster MA, Livesey FJ, McConnell MJ, et al. (2015). Creating Patient-Specific Neural Cells for the In Vitro Study of Brain Disorders. Stem Cell Reports 5, 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundo M, Toyoshima M, Okada Y, Akamatsu W, Ueda J, Nemoto-Miyauchi T, Sunaga F, Toritsuka M, Ikawa D, Kakita A, et al. (2014). Increased I1 retrotransposition in the neuronal genome in schizophrenia. Neuron 81, 306–313. [DOI] [PubMed] [Google Scholar]
- Bushman DM, Kaeser GE, Siddoway B, Westra JW, Rivera RR, Rehen SK, Yung YC, and Chun J (2015). Genomic mosaicismwith increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. eLife 4 10.7554/eLife.05116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Evrony GD, Lehmann HS, Elhosary PC, Mehta BK, Poduri A, and Walsh CA (2014). Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep. 8, 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian KM, Song H, and Ming GL (2014). Functions and dysfunctions of adult hippocampal neurogenesis. Annu. Rev. Neurosci 37, 243–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. ; Wellcome Trust Case Control Consortium (2010). Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D (2004). Electrophysiological classes of neocortical neurons. Neural Netw. 17, 633–646. [DOI] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O’Shea KS, Moran JV, and Gage FH (2009). L1 retrotransposition in human neural progenitor cells. Nature 460, 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Pino I, Rico B, and Marín O (2018). Neural circuit dysfunction in mouse models of neurodevelopmental disorders. Curr. Opin. Neurobiol 48, 174–182. [DOI] [PubMed] [Google Scholar]
- Erwin JA, Paquola AC, Singer T, Gallina I, Novotny M, Quayle C, Bedrosian TA, Alves FI, Butcher CR, Herdy JR, et al. (2016). L1-associated genomic regions are deleted in somatic cells of the healthy human brain. Nature Neurosci. 19, 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrony GD, Cai X, Lee E, Hills LB, Elhosary PC, Lehmann HS, Parker JJ, Atabay KD, Gilmore EC, Poduri A, et al. (2012). Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, and Rakic P (2013). Cortical evolution:judgethe brain by its cover. Neuron 80, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N, Lutz-Prigge S, and Moran JV (2002). Genomic deletions created upon LINE-1 retrotransposition. Cell 110, 315–325. [DOI] [PubMed] [Google Scholar]
- Harbom LJ, Michel N, and McConnell MJ (2018). Single-cell analysis of diversity in human stem cell-derived neurons. Cell Tissue Res. 371, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iourov IY, Vorsanova SG, Liehr T, Kolotii AD, and Yurov YB (2009). Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. Hum. Mol. Genet 18, 2656–2669. [DOI] [PubMed] [Google Scholar]
- Jamuar SS, Lam AT, Kircher M, D’Gama AM, Wang J, Barry BJ, Zhang X, Hill RS, Partlow JN, Rozzo A, et al. (2014). Somatic mutations in cerebral cortical malformations. N. Engl. J. Med 371, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, et al. (2013). Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, and Amon A (2014). Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. USA 111, 13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouse KA, Wu J, and Amon A (2016). Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res. 26, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB,Ai R, Kaeser GE, Salathia NS,Yung YC, Liu R,Wildberg A, Gao D, Fung HL, Chen S, et al. (2016). Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, et al. (2012). De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet 44, 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Siddoway B, Kaeser GE, Segota I, Rivera R, Romanow WJ, Liu CS, Park C, Kennedy G, Long T, and Chun J (2018). Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 563, 639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Belgard TG, Hawrylycz M, and Molnár Z (2017). Transcriptomic Perspectives on Neocortical Structure, Development, Evolution, and Disease. Annu. Rev. Neurosci 40, 629–652. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato MA,Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, Lee S, Chittenden TW, D’Gama AM, Cai X, et al. (2015). Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava C, et al. (2018). Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JH, Hansen DV, and Kriegstein AR (2011). Development and evolution of the human neocortex. Cell 146, 18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR (2015). Structural variation mutagenesis of the human genome: impact on disease and evolution. Environ. Mol. Mutagen 56, 419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, and Scherer SW (2014). The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 42, D986–D992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, et al. (2015). Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 161, 1592–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Valencia I, Guerrini R, and Gleeson JG (2014). Pathogenetic mechanisms of focal cortical dysplasia. Epilepsia 55, 970–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, Antaki D, Shetty A, Holmans PA, Pinto D, et al. ; Psychosis Endophenotypes International Consortium; CNV and Schizophrenia Working Groups of the Psychiatric Genomics Consortium (2017). Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet 49, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Kaushal D, Yang AH, Kingsbury MA, Rehen SK, Treuner K, Helton R, Annas EG, Chun J, and Barlow C (2004). Failed clearance of aneuploid embryonic neural progenitor cells leads to excess aneuploidy in the Atm-deficient but not the Trp53-deficient adult cerebral cortex. J. Neurosci 24, 8090–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM, and Gage FH (2013). Mosaic copy number variation in human neurons. Science 342, 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Moran JV, Abyzov A, Akbarian S, Bae T, Cortes-Ciriano I, Erwin JA, Fasching L, Flasch DA, Freed D, et al. ; Brain Somatic Mosaicism Network (2017). Intersection of diverse neuronal genomes and neuropsychiatric disease: the Brain Somatic Mosaicism Network. Science 356, eaal1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, and Thomas PD (2013). Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc 8, 1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa GM, Campbell CD, Solovieff N, Goold C, Jansen LA, Menon S, Timms AE, Conti V, Biag JD, Adams C, et al. (2016). Association of MTOR Mutations With Developmental Brain Disorders, Including Megalencephaly, Focal Cortical Dysplasia, and Pigmentary Mosaicism. JAMA Neurol. 73, 836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, and Baxter MG (2012). The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat. Rev. Neurosci 13, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM (2010). Genomic copy number variation in disorders of cognitive development. J. Am. Acad. Child Adolesc. Psychiatry 49, 1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muggeo VM, and Adelfio G (2011). Efficient change point detection for genomic sequences of continuous measurements. Bioinformatics 27, 161–166. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, and Gage FH (2005). Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435, 903–910. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Marchetto MC, Coufal NG, Oefner R, Yeo G, Nakashima K, and Gage FH (2010). L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, and Wigler M (2004). Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572. [DOI] [PubMed] [Google Scholar]
- Pakkenberg B, Pelvig D, Marner L, Bundgaard MJ, Gundersen HJ, Nyengaard JR, and Regeur L (2003). Aging and the human neocortex. Exp. Gerontol 38, 95–99. [DOI] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, and Šmuc T (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Hallström BM, Lindskog C, Mardinoglu A, Pontén F, and Nielsen J (2016). Transcriptomics resources of human tissues and organs. Mol. Syst. Biol 72, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton KR, Gerhardt DJ, Jesuadian JS, Richardson SR, Sánchez-Luque FJ, Bodea GO, Ewing AD, Salvador-Palomeque C, van der Knaap MS, Brennan PM, et al. (2015). Ubiquitous L1 mosaicism in hippocampal neurons. Cell 161, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bos H, Spierings DC, Taudt AS, Bakker B, Porubský D, Falconer E, Novoa C, Halsema N, Kazemier HG, Hoekstra-Wakker K, et al. (2016). Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer’s disease neurons. Genome Biol. 17, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS, Bahney J, and Herculano-Houzel S (2016). The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J. Comp. Neurol 524, 3865–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei PC, Chang AN, Kao J, Du Z, Meyers RM, Alt FW, and Schwer B (2016). Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 164, 644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman IL, and Gage FH (2016). A Mechanism for Somatic Brain Mosaicism. Cell 164, 593–595. [DOI] [PubMed] [Google Scholar]
- Wierman MB, Burbulis IE, Chronister WD, Bekiranov S, and McConnell MJ (2017). Single-Cell CNV Detection in Human Neuronal Nuclei In Genomic Mosaicism in Neurons and Other Cell Types, Frade JM and Gage FH, eds. (Springer; New York: ), pp. 109–131. [Google Scholar]
- Wilson TE, Arlt MF, Park SH, Rajendran S, Paulsen M, Ljungman M, and Glover TW (2015). Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 25, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrei M, MacDonald JR, Merico D, and Scherer SW (2015). A copy number variation map of the human genome. Nat. Rev. Genet 16, 172–183. [DOI] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman AR, and Xie XS (2012). Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338, 1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylka MJ, Simon JM, and Philpot BD (2015). Gene length matters in neurons. Neuron 86, 353–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, Redon R, Werner J, Villanea FA, Mountain JL, Misra R, et al. (2007). Diet and the evolution of human amylase gene copy number variation. Nat. Genet 39, 1256–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski A, Bruder CE, Andersson R, Diaz de Ståhl T, Menzel U, Sandgren J, Poplawski A, von Tell D, Crasto C, Bogdan A, et al. (2008). Somatic mosaicism for copy number variation in differentiated human tissues. Hum. Mutat 29, 1118–1124. [DOI] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, et al. (2012). Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P (2006). Neuroscience. No more cortical neurons for you. Science 313, 928–929. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. (2006). Global variation in copy number in the human genome. Nature 444, 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz G (1978). Estimating the Dimension of a Model. Ann. Stat 6, 461–464. [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshan VE, and Olshen A (2018). DNAcopy: DNA copy number data analysis R package version 1.56.0 (Bioconductor; ). [Google Scholar]
- Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, Arnaiz O, Awedh MH, Baldock R, Barbiera G, et al. (2015). The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 43 (W1), W589–W598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer NC (2017). Neurotransmitter Switching in the Developing and Adult Brain. Annu. Rev. Neurosci 40, 1–19. [DOI] [PubMed] [Google Scholar]
- Südhof TC (2017). Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 171, 745–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Mallick S, Nelson BJ, Hormozdiari F, Krumm N, Huddleston J, Coe BP, Baker C, Nordenfelt S, Bamshad M, et al. (2015). Global diversity, population stratification, and selection of human copy-number variation. Science 349, aab3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single cell sequencing data for PicoPLEX neurons and non-neurons is available through Synapse (https://www.synapse.org/#!Synapse:syn16803262) and the NIH data archive (collections 2963 and 2458). The R script used for simulating data using ALT and NULL models is available at https://github.com/mcconnell-lab/scripts/blob/master/ALT_NULL_dataset_simulation.R.