

Abstract
Two of the most common and well-characterized epigenetic changes, DNA methylation and histone modifications, occur in leukemia. Decitabine (5-aza-2’-deoxycytidine, DAC), as a hypomethylating agent (HMA), and chidamide (CS055), as a histone deacetylase inhibitor (HDACi), each demonstrate effects against leukemia. However, whether the combination of low-dose DAC with chidamide constitutes an effective epigenetic regimen for the treatment of myeloid leukemia is currently unknown. In this study, the combination of DAC at low doses and chidamide showed enhanced inhibition of myeloid leukemia cell (K562, THP-1) growth. As a novel HDACi, chidamide increased the level of ace-H3K18 expression. Combined use of low-dose DAC and chidamide arrested the cell cycle at the G0/G1 phase by upregulating p21 expression, and the combination also suppressed PI3K/AKT/mTOR signaling pathway. Furthermore, chidamide enhanced the apoptotic effect of DAC by downregulating expression of Bcl-2 and pro-caspase-3 and upregulating that of Bax, cleaved PARP-1, and caspase-9. Moreover, the mitochondrial transmembrane potential was significantly decreased in DAC-, chidamide-, or combination-treated leukemia cells. These results suggest that targeting the leukemia epigenome through the combination of low-dose DAC and chidamide is a promising approach.
Keywords: Decitabine, chidamide, myeloid leukemia cells, apoptosis
Introduction
Myeloid leukemia (ML) is a malignant disease caused by both genetic and epigenetic changes [1]. Aberrant DNA methylation and histone modifications, which are prevalent in cancers including leukemia, are the two most well-known and substantially characterized epigenetic changes [2,3]. It has been reported that aberrant DNA promoter methylation of tumor-suppressor genes (TSGs) and histone acetylation can regulate pathways involved in regulating the cell cycle and apoptosis in leukemia [4-6], and the reversible nature of these modifications renders the leukemia epigenome a potential target of epigenetic therapies, such as treatment with hypomethylating agents (HMAs) and histone deacetylase inhibitors (HDACis) [7,8].
Decitabine (5-aza-2’-deoxycytidine, DAC) is one of the most widely used DNA methyltransferase inhibitors (DNMTis) and can reactivate epigenetically silenced TSGs through demethylation [9]. Several clinical trials have reported that low-dose decitabine showed promising activity in myeloid leukemia patients [10,11]. Indeed, decitabine was approved in the European Union in 2012 for the treatment of acute myeloid leukemia (AML) patients ≥ 65 years old. Nevertheless, decitabine cannot reactivate all TSGs because some are silenced by other epigenetic mechanisms, e.g., chromatin compaction and histone acetylation [12]. This less-than-ideal situation calls for the development of novel treatment approaches, especially in combination epigenetic therapy [13]. For example, Shi et al. have shown that low-dose decitabine can enhance chidamide-induced apoptosis in acute lymphoblastic leukemia (ALL) [14]. Chidamide is a novel HDACi drug developed wholly in China; in 2015, oral administration of the drug for treating recurrent or refractory peripheral T-cell lymphoma (PTCL) was approved [15]. Recently, chidamide has been reported to inhibit the viability of AML cells [16] and to target AML stem and progenitor cells [17]; hence, it may be effective to combine decitabine with chidamide to treat leukemia cells. In this study, we sought to determine the antileukemic effects of low-dose decitabine combined with chidamide on myeloid leukemia cells by detecting cell proliferation, cell cycle distribution and apoptosis to provide a promising regimen for clinical application.
Materials and methods
Reagents
Chidamide was provided by professor Kai Sun and dissolved in dimethyl sulfoxide (DMSO) (Sigma, USA) at a 25 mg/mL concentration for stock solution. Decitabine was provided by Qilu Pharmaceutical Co., Ltd. (Shandong, China) and dissolved in DMSO at 8 mmol/L for a stock solution. All stock solutions were stored at -20°C and diluted with growth media to working concentrations for experiments.
Cell lines and cell culture
Myeloid leukemia cell lines K562 and THP-1 were purchased from the China Center for Type Culture Collection (Wuhan, China). The cells were cultured in Roswell Park Memorial Institute 1640 medium (RPMI 1640, HyClone, USA) containing 10% fetal bovine serum (FBS, SeraPro, Germany), 100 U/mL penicillin (Wuhan Servicebio Technology Co., Ltd., China) and 100 μg/mL streptomycin (Servicebio, China) at 37°C in a humidified atmosphere with 5% CO2.
Immunocytochemistry staining analysis
Cells were washed and centrifuged at 1800 rpm at 4°C for 5 min to remove the culture medium, fixed with 4% paraformaldehyde for 20 min, washed, centrifuged and mixed with PBS. The cell suspension was coated onto a slide overnight at room temperature until the PBS was completely evaporated, after which 50-100 μL stationary solution was added. Twenty minutes later, the cells were washed, and membrane breaking working fluid was added for 20 min, followed by 3% BSA for 30 min at room temperature for blocking. The primary antibody was diluted as indicated in PBS and added to the slide, followed by overnight incubation at 4°C. After washing, the secondary antibody was incubated with the slide at room temperature for 50 min. The slides were then washed, dried slightly and stained with DAPI dye solution at room temperature avoiding light for 10 min. The slides were sealed with anti-fluorescence quenching sealing tablets and observed under a fluorescence microscope to collect images.
Cell viability assay
The cell counting kit-8 (CCK-8, Dojindo, Japan) was used to measure the effects of chidamide or decitabine alone or in combination on cell viability. Cells were seeded into a 96-well plate at a density of 3-5 × 104 cells/mL with 100 μL of complete medium per well. After exposure to chidamide or decitabine at different concentrations or a combination of the two for the indicated time, 10 μL of CCK-8 reagent was added to each well and incubated for 2 h. Absorbance detection was performed at 450 nm using a microplate reader (Rayto, USA). All experiments were repeated three times. Based on the results, the cell inhibition rate (%) was calculated as follows: [1-(OD450test group - OD450blank)/(OD450control group - OD450blank)] × 100%. All experiments were repeated three times.
Cell cycle analysis
After 72 h of treatment, cells were collected and washed with phosphate-buffered saline (PBS, Servicebio, China) and fixed overnight in 75% precooled ethanol at 4°C. The cells were washed again and suspended in 100 μL of PBS. RNA was removed by adding 2 μL 1.0 mg/mL RNaseA (Solarbio, China) for 40 min at 37°C. The cells were harvested and stained with 100 μL 100 μg/mL propidium iodide (PI, Servicebio, China) for 20 min of incubation in the dark. The DNA content was assessed by flow cytometry (Beckman, USA) at an excitation wavelength of 488 nm and an emission wavelength of 585 ± 21 nm. ModfitLT software version 2.0 (Verity Software House, Inc., Topsham, ME, USA) was used for the analysis of DNA distributions. All experiments were repeated three times.
Apoptosis assay
After 72 h of treatment, cells were collected at 1500 rpm for 5 min, washed with PBS and resuspended in 200 μL of binding buffer. The cells were incubated with 5 μL of Annexin V-FITC and 5 μL of PI for 15 min in the dark. Apoptosis was detected by flow cytometry at an excitation wavelength of 488 nm and emission wavelengths of 525 ± 20 nm and 585 ± 20 nm. FlowJo software was used to analyze the number and percentage of apoptotic cells. All experiments were repeated three times.
Total RNA isolation and real-time quantitative PCR
Total cellular RNA was isolated using RNA extraction solution (Wuhan Goodbio Technology Co., Ltd.) after 72 h of treatment. RNA dissolved in RNase-free water was quantified using an ultra-micro spectrophotometer (NanoDrop 2000, Thermo, USA) and reverse transcribed into cDNA using RevertAid First Strand cDNA Synthesis Kit (Thermo). Real-time quantitative PCR (RT-qPCR) was performed using the cDNA as the template and FastStart Universal SYBR Green Master (Rox) (Roche, Switzerland) according to the manufacturer’s instructions. Gene-specific primers were synthesized by Goodbio (Wuhan, China) (Table 1). The RT-qPCR conditions were as follows: one cycle at 95°C for 10 min and 40 cycles at 95°C for 15 sec and 60°C for 60 sec; melting curve analysis was from 60°C to 95°C at a 0.3°C increase per 15 sec. The results were analyzed using the 2-ΔΔCt method and ΔΔCt = Cttarget gene of sample - Ctβ-actin of sample - (Cttarget gene of control - Ctβ-actin of control). All experiments were repeated twice.
Table 1.
Gene-specific primers designed for the designated human genes and used to determine expression levels by RT-qPCR
| Gene name | Gene ID | Sequence (5’→3’) | Product length (bp) |
|---|---|---|---|
| β-Actin | NM_001101 | CACCCAGCACAATGAAGATCAAGAT | 317 |
| CCAGTTTTTAAATCCTGAGTCAAGC | |||
| Bax | NM_004324.3 | TTTTGCTTCAGGGTTTCATCCA | 215 |
| TGCCACTCGGAAAAAGACCTC | |||
| Bcl-2 | NM_000633.2 | TCGCCCTGTGGATGACTGA | 130 |
| GACAGCCAGGAGAAATCAAACAG |
Western blotting analysis
After 72 h of treatment, cells were lysed using RIPA buffer (Servicebio, China). The supernatant was collected via centrifugation at 12,000 rpm at 4°C for 10 min, and the total protein in the sample was quantified using a BCA kit (Servicebio, China). Equal amounts of proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto PVDF membranes (Millipore, USA). The membranes were blocked with 5% skim milk and incubated overnight at 4°C with primary antibodies against mouse anti-β-actin (Servicebio, GB12001, 1:3000 dilution), mouse anti-GAPDH (Servicebio, GB12002, 1:1000 dilution), rabbit anti-Ki67 (Servicebio, GB11030, 1:1000 dilution), rabbit anti-Histone H3 (acetyl K18) (Abcam, ab40888, 1:1000 dilution), rabbit anti-p21 (Proteintech, Wuhan, China, 10355-1-AP, 1:1000 dilution), mouse anti-PI3K (Servicebio, GB13161-1, 1:1000 dilution), rabbit anti-p-PI3K (Bioss, BS-5570R, 1:1000 dilution), rabbit anti-AKT (Proteintech, 10176-2-AP 1:1000 dilution), rabbit anti-p-AKT (Ser473, Affinity, AF0908, 1:1000 dilution), rabbit anti-p-AKT (Thr308, Affinity, AF0832, 1:1000 dilution), rabbit anti-mTOR (CST, 2983, 1:1000 dilution), rabbit anti-p-mTOR (Ser2481, Bioss, BS-3495R, 1:1000 dilution), rabbit anti-cyclin D1 (Abcam, ab134175, 1:1000 dilution), rabbit anti-pro-caspase-3 (CST, 9665, 1:1000 dilution), rabbit anti-PARP-1 (Bioworld, Nanjing, China, BS70047, 1:1000 dilution), and rabbit anti-caspase-9 (Servicebio, GB11053-1, 1:1000 dilution). The membranes were washed with Tris-buffered saline plus Tween 20 (TBST) and incubated at room temperature for 30 min with secondary antibodies of horseradish peroxidase (HRP)-conjugated goat anti-mouse and anti-rabbit IgG (Servicebio, GB23301 and GB23303, respectively, 1:3000 dilution). Bands were visualized using ECL and exposed to X-ray film in the dark.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was measured using a JC-1 kit (Beyotime, Shanghai, China). After 72 h of incubation, cells were collected at 1000 rpm for 5 min, washed with PBS and counted using a blood cell counter. The JC-1 probe was diluted to a working concentration of 10 μg/mL and incubated with the cells at a density of 0.5-1 × 106/mL for 20 min. The cells were washed again to remove the probe, and flow cytometry at an excitation wavelength of 488 nm and emission wavelengths of 525 ± 20 nm and 585 ± 20 nm was performed. The results were analyzed using FlowJo software version 10.0 (Tree Star, Inc., Ashland, OR, USA). The experiments were repeated twice.
Statistical analysis
All data in this study are presented as the means ± standard deviation (SD) of three independent experiments. Statistical analysis was performed using IBM SPSS Statistics version 19.0 software (SPSS Inc.). Significant differences in the mitochondrial membrane potential assay were analyzed by one-way analysis of variance (ANOVA) followed by the LSD test. The results of the cell cycle analysis, apoptosis assay and RT-qPCR were analyzed by Student’s t-test. A P value < 0.05 was considered statistically significant.
Results
Chidamide and decitabine inhibited the proliferation of myeloid leukemia cells
K562 cells were treated with chidamide alone at concentrations of 0.5, 1, 2, 4, 8 μg/mL and decitabine alone at concentrations of 0.5, 1, 2, 4, 8 μmol/L for 24, 48, 72, and 96 h; DMSO was used as the negative control. The results showed that chidamide or decitabine alone inhibited K562 cell viability in a dose- and time-dependent manner (Figure 1A and 1B). Furthermore, K562 cells were treated with chidamide at 8 μg/mL in combination with decitabine at 8 μmol/L for 24, 48, 72, 96 h, with DMSO as the negative control. The results indicated that the inhibitory effect of chidamide on K562 cell viability was higher than that of decitabine and that the inhibitory effect of the combination of chidamide with decitabine was obviously higher than that of chidamide alone or decitabine alone (Figure 1C). The IC50 for chidamide was 2.56 μg/mL and for decitabine was 5.60 μmol/L after 72 h of treatment. For THP-1 cells, the concentration gradient of chidamide was the same as that used in K562 cells, whereas the concentration gradient of decitabine was increased to 0.5, 1, 5, 10, 20 μmol/L to observe whether the inhibitory effect of decitabine on leukemia cells was enhanced by chidamide. According to the results, chidamide or decitabine alone inhibited THP-1 cell viability in a dose- and time-dependent manner (Figure 1D and 1E), and the inhibitory effect of the combination was markedly higher than that of chidamide alone and slightly higher than that of decitabine alone (Figure 1F). Moreover, the results of immunofluorescence analysis via Ki67 staining after 72 h of treatment showed that cell proliferation was inhibited by chidamide, decitabine alone or the combination, with the combination causing the greatest effect (Figure 1G), which was consistent with the results of western blotting analysis of Ki67 expression (Figure 1H). Altogether, the results demonstrated that when used alone, the DNMTi decitabine and the HDACi chidamide had an inhibitory effect in the proliferation of myeloid leukemia cells and that the combination of the two enhanced this effect.
Figure 1.

Chidamide in combination with decitabine inhibited the proliferation of myeloid leukemia cells. A-C. Cell viability was detected by CCK-8 assay in K562 cells treated with chidamide (0, 0.5, 1, 2, 4, or 8 μg/mL) or decitabine (0, 0.5, 1, 2, 4, or 8 μmol/L) alone for 24, 48, 72 and 96 h or treated with 8 μg/mL chidamide combined with 8 μmol/L decitabine for 24, 48, 72, 96 and 120 h. D-F. Cell viability was detected by the CCK-8 assay in THP-1 cells treated with chidamide (0, 0.5, 1, 2, 4, or 8 μg/mL) or decitabine (0, 0.5, 1, 5, 10, or 20 μmol/L) alone for 24, 48, 72 and 96 h, or with 0.5 μg/mL chidamide combined with 20 μmol/L decitabine for 24, 48, 72, and 96 h. Cell inhibition rate (%) was calculated as: [1-(OD450test group - OD450blank)/(OD450control group - OD450blank)] × 100%. G. Immunofluorescence staining analysis was performed by detecting Ki67 after 72 h of treatment. H. Western blotting analysis of Ki67 expression.
Effects of chidamide and decitabine on histone H3 acetylation
Histone H3 acetylation levels were detected via western blotting to explore the effects of chidamide in combination with decitabine on leukemia cells. As shown in Figure 2, the acetylation levels of histone H3 in both K562 and THP-1 cells were strongly increased only in the chidamide-treated group (chidamide alone or chidamide in combination with decitabine). Conversely, the acetylation levels of histone H3 in K562 and THP-1 cells were not changed compared with the control group. These results indicate that chidamide, and not decitabine, had pronounced effects on histone H3 acetylation levels in leukemia cells.
Figure 2.
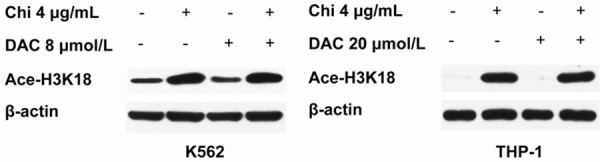
The effects of chidamide in combination with decitabine on histone H3 acetylation in myeloid leukemia cell lines. K562 cells were treated with 4 μg/mL chidamide or 8 μmol/L decitabine alone or 4 μg/mL chidamide in combination with 8 μmol/L decitabine for 72 h; THP-1 cells were treated with 4 μg/mL chidamide or 20 μmol/L decitabine alone or 4 μg/mL chidamide in combination with 20 μmol/L decitabine for 72 h. The expression level of acetylated histone H3 (K18) was analyzed by western blotting.
The combination of chidamide and decitabine arrested the cell cycle at the G0/G1 phase
The cell cycle distribution in myeloid leukemia cells was also investigated by flow cytometry. K562 cells were treated with 4 μg/mL chidamide alone, 8 μmol/L decitabine alone, and the combination for 72 h; THP-1 cells were treated with 4 μg/mL chidamide alone, 20 μmol/L decitabine alone, and the combination for 72 h. Compared with the control or decitabine-treated group, the percentage of cells in G0/G1 phase was significantly increased by the combined use of chidamide and decitabine (44.86 ± 0.43%/46.08 ± 1.57% vs. 84.37 ± 0.94% in K562 cells, 60.07 ± 2.15%/50.70 ± 1.51% vs. 76.90 ± 0.58% in THP-1 cells, Figure 3A-C). In addition, the combined use of the two significantly increased the percentage of THP-1 cells at G2/M phase compared with the chidamide-treated group (11.01 ± 0.73% vs. 4.89 ± 1.95%, Figure 3C). Western blotting showed that expression of p21 was upregulated in both chidamide-treated cells and cells treated with the combination (Figure 3D), which may help to explain the mechanism for cell cycle arrest at G0/G1.
Figure 3.

Chidamide in combination with decitabine arrested the cell cycle at G0/G1 phase in myeloid leukemia cells. A-C. The combination of 4 μg/mL chidamide and 8 μmol/L decitabine (in K562 cells) or 20 μmol/L decitabine (in THP-1 cells) induced cell cycle arrest at G0/G1 phase after 72 h of treatment. D. Expression of the cell cycle protein p21 was increased at 72 h compared with the control group based on western blotting analysis in both K562 and THP-1 cells. “*” indicates a significant difference compared with the control group (*P<0.05, **P<0.01, ***P<0.001), “#” indicates a significant difference compared with the decitabine-treated group (###P<0.001), and “&” indicates a significant difference compared with the chidamide-treated group (&P<0.05).
The combination of chidamide and decitabine inhibited the PI3K/AKT/mTOR signaling pathway
To further explore the inhibitory mechanism of cell proliferation, we examined the levels of non- and phosphorylated forms of PI3K, AKT and mTOR in the PI3K/AKT/mTOR signaling pathway. After 72 h of combined treatment with low-dose decitabine and chidamide, levels of phosphorylated PI3K, AKT and mTOR as well as the downstream target protein cyclin D1 were decreased compared with that of the negative control in both K562 (Figure 4A) and THP-1 cells (Figure 4B). Therefore, we speculate that the combined treatment inhibited cell proliferation by downregulating the phosphorylation of PI3K/AKT/mTOR pathway components PI3K, AKT and mTOR.
Figure 4.
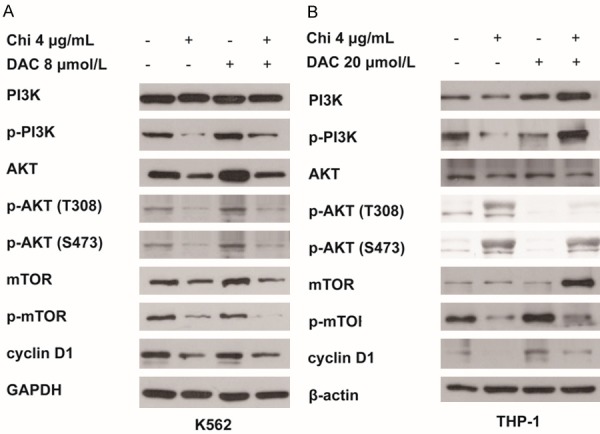
Chidamide in combination with low-dose decitabine inhibited the PI3K/AKT/mTOR signaling pathway. After 72 h of treatment, expression of PI3K, p-PI3K, AKT, p-AKT, mTOR, p-mTOR and cyclin D1 in both K562 (A) and THP-1 cells (B) was determined by western blotting.
Combination of chidamide and decitabine induced apoptosis in myeloid leukemia cells
To further investigate the mechanism by which proliferation was inhibited and the cell cycle was arrested in myeloid leukemia cells, apoptosis assays were performed using flow cytometry. The results showed that the apoptosis rate was significantly increased when cells were treated with the combination of chidamide and decitabine compared with DMSO-treated cells (Figure 5A-C). Compared with decitabine alone-treated cells, the percentage of apoptotic cells was markedly increased by the combined use of chidamide and decitabine, which indicated that chidamide enhanc-ed the apoptotic effect of decitabine on both K562 and THP-1 cells (Figure 5A-C). Furthermore, based on RT-qPCR, relative gene expression of Bax was notably increased and that of Bcl2 reduced when K562 and THP-1 cells were exposed to these two compounds (Figure 5B and 5C). As shown in Figure 5D, western blotting analysis also revealed that expression of apoptotic markers pro-caspase-3 and PARP-1 was downregulated but that of cleaved PARP-1 and caspase-9 was upregulated.
Figure 5.

Chidamide enhanced the effect of decitabine on apoptosis in myeloid leukemia cells. A-C. The combination of 4 μg/mL chidamide and 8 μmol/L decitabine (in K562 cells) or 20 μmol/L decitabine (in THP-1 cells) induced apoptosis after 72 h of treatment. Relative gene expression levels of the apoptotic gene Bax and the antiapoptotic gene Bcl-2 were analyzed by RT-qPCR. D. Expression of cell apoptosis-related proteins pro-caspase-3, PARP-1, cleaved PARP-1 and caspase-9 was analyzed after 72 h of treatment by western blotting. “*” indicates a significant difference compared with the control group (*P<0.05, **P<0.01, ***P<0.001), “#” indicates a significant difference compared with the decitabine-treated group (###P<0.001), and “&” indicates a significant difference compared with the chidamide-treated group (&P<0.05).
Effects of chidamide and decitabine on the mitochondrial membrane potential of myeloid leukemia cells
Decreased mitochondrial membrane potential (MMP) is an early manifestation of apoptosis, after which cells begin an irreversible apoptotic process. To assess the mitochondrial membrane potential, K562 and THP-1 cells were treated with chidamide or decitabine alone or both for 72 h, and the JC-1 kit was employed to measure MMP. The results demonstrated that compared with the control group, MMP was significantly decreased in all treated cells; however, no significant difference was observed between the cells in the combination group and those treated with chidamide or decitabine alone (Figure 6).
Figure 6.

Chidamide in combination with decitabine downregulated the mitochondrial membrane potential of myeloid leukemia cells. A, B. Chidamide (4 μg/mL) or decitabine (8 μmol/L in K562 cells or 20 μmol/L in THP-1 cells) alone or in combination downregulated the mitochondrial membrane potential after 72 h of treatment. “*” indicates a significant difference compared with the control group (***P<0.001).
Discussion
Epigenetic alterations in DNA methylation [18] and histone posttranslational modifications, such as histone methylation and acetylation [19], occur in the cells of leukemia patients and are promising targets for treatment [20]. Although HMAs or HDACis have been used alone as epigenetic therapies in clinical practice, the effects have not been satisfactory [14,21]. Combination trials of HMAs and HDACis have been performed by independent groups, and significant antileukemia effects were observed [22,23]. In this study, we investigated the effects of a combination of decitabine and chidamide on myeloid leukemia cells. As mechanisms responsible for the effects on leukemia of decitabine and chidamide when used alone differ, the combined use of the two may be an effective therapy for AML.
In the current study, CCK-8 assay results showed that low-dose decitabine and chidamide inhibited K562 and THP-1 cell proliferation in a time- and dose-dependent manner (Figure 1). Additionally, the combination of the two exhibited enhanced inhibitory effects on cell proliferation compared with that of each drug alone. Consistent with the results of previous studies [20], as an HDAC inhibitor, chidamide significantly increased the acetylation level of histone H3 with or without the addition of decitabine (Figure 2), though the mechanism of the synergistic effect on cell proliferation needs to be clarified.
It has been reported that decitabine induces cell cycle arrest at G0/G1 and G2/M phases in solid tumor and hematologic malignancies [24-26] and that chidamide induces cell cycle arrest at G0/G1 phase as well as ROS-dependent apoptosis in leukemia cells [27,28]. In the present study, the results of the cell cycle distribution assay demonstrated that the combination of low-dose decitabine with chidamide also resulted in G0/G1 cell cycle arrest (Figure 3). Expression of p21, a negative regulator of the cell cycle, was increased in response to the addition of chidamide, with or without decitabine. The PI3K/AKT/mTOR signaling pathway is reported to be activated in many human cancers, including leukemia. It is known to play key roles in numerous cellular functions to control cell growth, metabolism and survival in cancer development, and its inhibition is regarded as an important therapeutic approach [29,30]. Chidamide reportedly inhibits the PI3K/AKT signaling pathway, resulting in cell cycle arrest at G1 phase and apoptosis promotion in colon cancer cells [31]. It was also reported that decitabine in combination with the mTOR inhibitor displays synergistic activity in medullary thyroid carcinoma cell lines to overcome drug resistance [32]. In this study, low-dose decitabine had a synergistic effect with chidamide on leukemia cell proliferation and cell cycle arrest at G0/G1 phase (Figure 4), providing a promising approach to treat leukemia patients in clinical practice.
The results of apoptosis assays indicated that chidamide enhanced the apoptotic effect of decitabine in myeloid leukemia cells (Figure 5). The expression levels of several apoptotic regulators were detected by RT-qPCR, showing that Bcl-2, a negative regulator of ell apoptosis, was downregulated and that Bax, a positive regulator of apoptosis, was upregulated in combination-treated cells. Furthermore, according to western blotting assays, expression of pro-caspase-3 was decreased, that of caspase-9 was increased, and that of cleaved PARP-1, which functions in single-stranded DNA repair, was increased in chidamide-treated cells, with or without the addition of decitabine. These results suggest that the combination of decitabine and chidamide may activate caspase-9 and caspase-3 via the mitochondria-dependent signaling pathway. Potential changes in the mitochondrial membrane during apoptosis [33,34] were also detected after 72 h of treatment. Indeed, MMP was significantly decreased in cells treated with decitabine or chidamide alone or in combination (Figure 6), damaging the mitochondrial membrane. As MMP was decreased, the apoptotic cascade was activated. The level of reactive oxygen species (ROS) production was also detected, though a significant difference was only observed in THP-1 cells treated with chidamide compared with the control (Supplementary Figure 1). ROS production is a dynamic biological process, and a cellular increase in ROS production can induce apoptosis. Consistently, HDACi has been reported to induce the production of cellular ROS [27,35]. The results indicate that chidamide may play an important role in acute myeloid leukemia apoptosis, yet this needs to be observed in more AML cell lines.
In summary, the combination of low-dose decitabine and chidamide was shown to enhance cell growth inhibition and apoptosis and to induce cell cycle arrest in myeloid leukemia cells. The results of this study provide a promising chemotherapy strategy that combines epigenetic agents at low doses for leukemia in clinical treatment. The in vivo efficacy of such a treatment regimen will be the focus of future studies.
Additional files: All original western images were showed in Supplementary Figure 2.
Acknowledgements
We thank Qilu Pharmaceutical Co., Ltd. for providing the decitabine used in the study. This work was partially supported by grants from the National Natural Science Foundation of China (No. 81971508, No. 81471589, No. 81273259) and Promotion Projects in Henan Province (132102310120) and also supported by both doctoral and postdoctoral research start-up funds of Henan Provincial People’s Hospital.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Gutierrez SE, Romero-Oliva FA. Epigenetic changes: a common theme in acute myelogenous leukemogenesis. J Hematol Oncol. 2013;6:57–70. doi: 10.1186/1756-8722-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yen CY, Huang HW, Shu CW, Hou MF, Yuan SS, Wang HR, Chang YT, Farooqi AA, Tang JY, Chang HW. DNA methylation, histone acetylation and methylation of epigenetic modifications as a therapeutic approach for cancers. Cancer Lett. 2016;373:185–192. doi: 10.1016/j.canlet.2016.01.036. [DOI] [PubMed] [Google Scholar]
- 3.Tsai CT, So CW. Epigenetic therapies by targeting aberrant histone methylome in AML: molecular mechanisms, current preclinical and clinical development. Oncogene. 2017;36:1753–1759. doi: 10.1038/onc.2016.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodoor K, Haddad Y, Alkhateeb A, Al-Abbadi A, Dowairi M, Magableh A, Bsoul N, Ghabkari A. DNA hypermethylation of cell cycle (p15 and p16) and apoptotic (p14, p53, DAPK and TMS1) genes in peripheral blood of leukemia patients. Asian Pac J Cancer Prev. 2014;15:75–84. doi: 10.7314/apjcp.2014.15.1.75. [DOI] [PubMed] [Google Scholar]
- 5.Koprinarova M, Schnekenburger M, Diederich M. Role of histone acetylation in cell cycle regulation. Curr Top Med Chem. 2016;16:732–744. doi: 10.2174/1568026615666150825140822. [DOI] [PubMed] [Google Scholar]
- 6.Valiulienė G, Stirblytė I, Jasnauskaitė M, Borutinskaitė V, Navakauskienė R. Anti-leukemic effects of HDACi Belinostat and HMTi 3-Deazaneplanocin A on human acute promyelocytic leukemia cells. Eur J Pharmacol. 2017;799:143–153. doi: 10.1016/j.ejphar.2017.02.014. [DOI] [PubMed] [Google Scholar]
- 7.Jain N, Rossi A, Garcia-Manero G. Epigenetic therapy of leukemia: an update. Int J Biochem Cell Biol. 2009;41:72–80. doi: 10.1016/j.biocel.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kikuchi J, Furukawa Y. DNA methyltransferase inhibitors * histone deacetylase inhibitors. Nihon Rinsho. 2014;72:1136–1142. [PubMed] [Google Scholar]
- 9.Hackanson B, Daskalakis M. Decitabine. Recent Results Cancer Res. 2014;201:269–297. doi: 10.1007/978-3-642-54490-3_18. [DOI] [PubMed] [Google Scholar]
- 10.Cashen AF, Schiller GJ, O’Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J. Clin. Oncol. 2010;28:556–561. doi: 10.1200/JCO.2009.23.9178. [DOI] [PubMed] [Google Scholar]
- 11.Phillips CL, Davies SM, McMasters R, Absalon M, O’Brien M, Mo J, Broun R, Moscow JA, Smolarek T, Garzon R, Blum W, Schwind S, Marcucci G, Perentesis JP. Low dose decitabine in very high risk relapsed or refractory acute myeloid leukaemia in children and young adults. Br J Haematol. 2013;161:406–410. doi: 10.1111/bjh.12268. [DOI] [PubMed] [Google Scholar]
- 12.Momparler RL, Côté S, Momparler LF, Idaghdour Y. Epigenetic therapy of acute myeloid leukemia using 5-aza-2’-deoxycytidine (decitabine) in combination with inhibitors of histone methylation and deacetylation. Clin Epigenetics. 2014;6:19–30. doi: 10.1186/1868-7083-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grishina O, Schmoor C, Döhner K, Hackanson B, Lubrich B, May AM, Cieslik C, Müller MJ, Lübbert M. Decider: prospective randomized multicenter phase ii trial of low-dose decitabine (DAC) administered alone or in combination with the histone deacetylase inhibitor valproic acid (VPA) and all-trans retinoic acid (ATRA) in patients >60 years with acute myeloid leukemia who are ineligible for induction chemotherapy. BMC Cancer. 2015;15:430–437. doi: 10.1186/s12885-015-1432-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi P, Zhang L, Chen K, Jiang Z, Deng M, Zha J, Guo X, Li P, Xu B. Low-dose decitabine enhances chidamide-induced apoptosis in adult acute lymphoblast leukemia, especially for p16-deleted patients through DNA damage. Pharmacogenomics. 2017;18:1259–1270. doi: 10.2217/pgs-2017-0061. [DOI] [PubMed] [Google Scholar]
- 15.Lu X, Ning Z, Li Z, Cao H, Wang X. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis Res. 2016;5:185–191. doi: 10.5582/irdr.2016.01024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao S, Guo J, Zhao Y, Fei C, Zheng Q, Li X, Chang C. Chidamide, a novel histone deacetylase inhibitor, inhibits the viability of MDS and AML cells by suppressing JAK2/STAT3 signaling. Am J Transl Res. 2016;8:3169–3178. [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Chen K, Zhou Y, Xiao Y, Deng M, Jiang Z, Ye W, Wang X, Wei X, Li J, Liang J, Zheng Z, Yao Y, Wang W, Li P, Xu B. A new strategy to target acute myeloid leukemia stem and progenitor cells using chidamide, a histone deacetylase inhibitor. Curr Cancer Drug Targets. 2015;15:493–503. doi: 10.2174/156800961506150805153230. [DOI] [PubMed] [Google Scholar]
- 18.Rahmani M, Talebi M, Hagh MF, Feizi AAH, Solali S. Aberrant DNA methylation of key genes and acute lymphoblastic leukemia. Biomed Pharmacother. 2018;97:1493–1500. doi: 10.1016/j.biopha.2017.11.033. [DOI] [PubMed] [Google Scholar]
- 19.Neff T, Armstrong SA. Chromatin maps, histone modifications and leukemia. Leukemia. 2009;23:1243–1251. doi: 10.1038/leu.2009.40. [DOI] [PubMed] [Google Scholar]
- 20.Mao J, Li S, Zhao H, Zhu Y, Hong M, Zhu H, Qian S, Li J. Effects of chidamide and its combination with decitabine on proliferation and apoptosis of leukemia cell lines. Am J Transl Res. 2018;10:2567–2578. [PMC free article] [PubMed] [Google Scholar]
- 21.DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, Frankfurt O, Konopleva M, Wei AH, Kantarjian HM, Xu T, Hong WJ, Chyla B, Potluri J, Pollyea DA, Letai A. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17. doi: 10.1182/blood-2018-08-868752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brodská B, Holoubek A. Generation of reactive oxygen species during apoptosis induced by DNA-damaging agents and/or histone deacetylase inhibitors. Oxid Med Cell Longev. 2011;2011:253529. doi: 10.1155/2011/253529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao Y, Zhou J, Wang L, Gao X, Ning Q, Jiang M, Wang J, Wang L, Yu L. Increased PRAME-specific CTL killing of acute myeloid leukemia cells by either a novel histone deacetylase inhibitor chidamide alone or combined treatment with decitabine. PLoS One. 2013;8:e70522. doi: 10.1371/journal.pone.0070522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lavelle D, DeSimone J, Hankewych M, Kousnetzova T, Chen YH. Decitabine induces cell cycle arrest at the G1 phase via p21WAF1 and the G2/M phase via the p38 MAP kinase pathway. Leuk Res. 2003;27:999–1007. doi: 10.1016/s0145-2126(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 25.Shin DY, Sung Kang H, Kim GY, Kim WJ, Yoo YH, Choi YH. Decitabine, a DNA methyltransferases inhibitor, induces cell cycle arrest at G2/M phase through p53-independent pathway in human cancer cells. Biomed Pharmacother. 2013;67:305–311. doi: 10.1016/j.biopha.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Shang D, Han T, Xu X, Liu Y. Decitabine induces G2/M cell cycle arrest by suppressing p38/NF-κB signaling in human renal clear cell carcinoma. Int J Clin Exp Pathol. 2015;8:11140–11148. [PMC free article] [PubMed] [Google Scholar]
- 27.Gong K, Xie J, Yi H, Li W. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem J. 2012;443:735–746. doi: 10.1042/BJ20111685. [DOI] [PubMed] [Google Scholar]
- 28.Liu Z, Chen J, Wang H, Ding K, Li Y, de Silva A, Sehgal V, Burbano JL, Sundararaj R, Gamage J, Audu V, Fu R. Chidamide shows synergistic cytotoxicity with cytarabine via inducing G0/G1 arrest and apoptosis in myelodysplastic syndromes. Am J Transl Res. 2017;9:5631–5642. [PMC free article] [PubMed] [Google Scholar]
- 29.Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs. 2005;16:797–803. doi: 10.1097/01.cad.0000173476.67239.3b. [DOI] [PubMed] [Google Scholar]
- 30.Bertacchini J, Heidari N, Mediani L, Capitani S, Shahjahani M, Ahmadzadeh A, Saki N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci. 2015;72:2337–2347. doi: 10.1007/s00018-015-1867-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L, Chen B, Qin S, Li S, He X, Qiu S, Zhao W, Zhao H. A novel histone deacetylase inhibitor Chidamide induces apoptosis of human colon cancer cells. Biochem Biophys Res Commun. 2010;392:190–195. doi: 10.1016/j.bbrc.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 32.Vitale G, Dicitore A, Pepe D, Gentilini D, Grassi ES, Borghi MO, Gelmini G, Cantone MC, Gaudenzi G, Misso G, Di Blasio AM, Hofland LJ, Caraglia M, Persani L. Synergistic activity of everolimus and 5-aza-2’-deoxycytidine in medullary thyroid carcinoma cell lines. Mol Oncol. 2017;11:1007–1022. doi: 10.1002/1878-0261.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scarlett JL, Sheard PW, Hughes G, Ledgerwood EC, Ku HH, Murphy MP. Changes in mitochondrial membrane potential during staurosporine-induced apoptosis in Jurkat cells. FEBS Lett. 2000;475:267–272. doi: 10.1016/s0014-5793(00)01681-1. [DOI] [PubMed] [Google Scholar]
- 34.Yun X, Rao W, Xiao C, Huang Q. Apoptosis of leukemia K562 and Molt-4 cells induced by emamectin benzoate involving mitochondrial membrane potential loss and intracellular Ca2+ modulation. Environ Toxicol Pharmacol. 2017;52:280–287. doi: 10.1016/j.etap.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 35.Wu YF, Ou CC, Chien PJ, Chang HY, Ko JL, Wang BY. Chidamide-induced ROS accumulation and miR-129-3p-dependent cell cycle arrest in non-small lung cancer cells. Phytomedicine. 2018;56:94–102. doi: 10.1016/j.phymed.2018.09.218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.