

Abstract
Blood‐based protein biomarkers may be an attractive option for early detection of colorectal cancer (CRC). Here, we used a two‐stage design to measure 275 protein markers by proximity extension assay (PEA), first in plasma samples of a discovery set consisting of 98 newly diagnosed CRC cases and 100 age‐ and gender‐matched controls free of neoplasm at screening colonoscopy. An algorithm predicting the presence of early‐ or late‐stage CRC was derived by least absolute shrinkage and selection operator regression with .632+ bootstrap method, and the algorithms were then validated using PEA again in an independent validation set consisting of participants of screening colonoscopy with and without CRC (n = 56 and 102, respectively). Three different signatures for all‐, early‐, and late‐stage CRC consisting of 9, 12, and 11 protein markers were obtained in the discovery set with areas under the curves (AUCs) after .632 + bootstrap adjustment of 0.92, 0.91, and 0.96, respectively. External validation among participants of screening colonoscopy yielded AUCs of 0.76 [95% confidence interval (95% CI), 0.67–0.84], 0.75 (95% CI, 0.62–0.87), and 0.80 (95% CI, 0.68–0.89) for all‐, early‐, and late‐stage CRC, respectively. Although the identified protein markers are not competitive with the best available stool tests, these proteins may contribute to the development of powerful blood‐based tests for CRC early detection in the future.
Keywords: colorectal cancer, diagnostic biomarkers, early detection, proximity extension assay, screening, sensitivity and specificity
A signature consisting of 12 plasma protein biomarkers derived in a clinical setting and independently validated in a true screening setting (participants of screening colonoscopy) yielded area under the curves of 0.91 and 0.75 for distinguishing early‐stage colorectal cases from controls free of neoplasms. These 12 protein markers may contribute to the development of blood‐based tests for population‐based screening.

Abbreviations
- AA
advanced adenomas
- AREG
amphiregulin
- ASTER
Mit ASS Darmtumore früher erkennen
- AUC
area under the receiver operating curve
- AUCBS
.632+ bootstrap‐adjusted AUC
- BLITZ
Begleitende Evaluierung innovativer Testverfahren zur Darmkrebs‐Früherkennung
- CEA
carcinoembryonic antigen
- CRC
colorectal cancer
- CV
coefficient of variance
- GZMB
granzyme B
- iDa
Durch innovative Testverfahren Darmkrebs früher erkennen
- IL6
interleukin‐6
- ITGA11
integrin alpha 11
- ITGAV
integrin alpha V
- KRT19
keratin, type I cytoskeletal 19
- LASSO
least absolute shrinkage and selection operator
- MASP1
mannan‐binding lectin serine protease 1
- MCP1
monocyte chemotactic protein 1
- NPX
normalized protein expression
- NTproBNP
N‐terminal prohormone brain natriuretic peptide
- OPN
osteopontin
- PEA
proximity extension assay
- PON3
paraoxonase 3
- QCC
quality control criteria
- RARRES2
retinoic acid receptor responder protein 2
- S100A4
protein S100‐A4
- TR
transferrin receptor protein 1
- TRAIL
TNF‐related apoptosis‐inducing ligand
- TRAP
tartrate‐resistant acid phosphatase type 5
1. Introduction
With 1.85 million incident cases and ~ 880 000 deaths per year, colorectal cancer (CRC) is the third most common cancer and second leading cause of cancer mortality globally (Bray et al., 2018). Randomized trials and observational studies have established the potential of screening with stool‐based tests or endoscopy in reducing CRC incidence and mortality (Atkin et al., 2017; Brenner and Chen, 2018; Brenner et al., 2014; Shaukat et al., 2013; Zauber et al., 2012), and stool tests or endoscopy‐based CRC screening is offered in an increasing number of countries (Klabunde et al., 2015; Schreuders et al., 2015). However, the participation rates in screening programs are often low because of limitations such as invasiveness, costs and resources, inconvenience, and adherence (Bretthauer et al., 2016; Klabunde et al., 2015; Navarro et al., 2017; Pox et al., 2012; Schreuders et al., 2015; Segnan et al., 2007).
Minimally invasive blood‐based tests might improve the participation rates in population‐based screening programs, given their straightforward applicability in routine medical assessments. Extensive research has been conducted to identify blood‐based biomarkers for early detection of CRC. In particular, numerous studies searched for blood‐based protein biomarkers or biomarker signatures, and some of them reported seemingly good diagnostic performance (Bhardwaj et al., 2017a; Bhardwaj et al., 2017b). However, the majority of studies were conducted in clinical settings and lacked proper validation in true screening settings, which may have resulted in highly overoptimistic results of diagnostic performance. Furthermore, few studies reported on sensitivity by CRC stage.
Among the very few studies that performed both internal validation and external validation, two studies from our group in which 92 tumor‐associated proteins were screened simultaneously found promising results (Chen et al., 2017; Chen et al., 2015). However, compared with the current study both of the previous studies had smaller numbers of CRC cases in the screening cohort and no stage‐specific prediction algorithms were reported. Therefore, we prospectively evaluated a broader spectrum of 275 proteins with the objective to identify multimarker signature with optimal diagnostic performance for early detection of CRC in a discovery set. The estimates were independently validated in prospectively selected samples (from N = 9245) exclusively recruited in a true screening setting.
2. Materials and methods
2.1. Study design
The protein marker signature was developed in a two‐step approach, with construction of multimarker algorithms in a discovery set and evaluation and validation of findings in an independent validation set. The discovery set included CRC patients recruited in a clinical setting and compared them with participants found to be free of colorectal neoplasms at screening colonoscopy. The validation set was exclusively based on participants of screening colonoscopy in order to evaluate diagnostic performance in a true screening setting.
2.2. Study population: discovery set
The discovery set consisted of 98 CRC cases recruited prior to any therapeutic measures from the Durch innovative Testverfahren Darmkrebs früher erkennen (iDa) study in hospitals in southwestern Germany between 2013 and 2016. As controls, 100 participants of screening colonoscopy who were found to be free of neoplasms were selected using frequency matching by age and sex from the ‘Mit ASS Darmtumore früher erkennen’ (ASTER) study. The ASTER study, a multicenter prospective randomized controlled trial (EudraCT No. 2011‐005603‐32), recruited participants aged 40–80 with a planned screening (75%) or diagnostic (25%) colonoscopy from gastrointestinal practices in Germany from 2013 to 2016 to determine whether the application of a single 300 mg dose of aspirin before performing a fecal immunochemical tests (FIT) could improve FIT sensitivity for detecting advanced neoplasms (Tikk et al., 2018). For both iDa and ASTER, the use of samples for the evaluation of early detection markers for CRC has been approved by the ethics committees of the Medical Faculty Heidelberg (S‐489/2012 and AFmu‐271/2012, for iDa and ASTER, respectively) and from the responsible state medical boards. Both the studies were conducted adhering to the standards set by Declaration of Helsinki and were undertaken with the understanding and written consent of each subject. The complete Standards for the Reporting of Diagnostic Accuracy Studies (STARD) diagrams displaying the selection of study participants in iDa and ASTER studies are shown in Figs S1 and S2.
2.3. Study population: validation set
For the independent external validation of potentially promising signatures, blood samples were selected from participants of screening colonoscopy collected in the Begleitende Evaluierung innovativer Testverfahren zur Darmkrebs‐Früherkennung (BLITZ) study. Details of the BLITZ study design have been reported previously (Brenner et al., 2010b; Chen et al., 2017; Chen et al., 2015; Gies et al., 2018; Hundt et al., 2009). In brief, BLITZ is an ongoing screening study of participants of the German screening colonoscopy program that is offered to men and women aged 55 and older. Participants are recruited in 20 gastroenterology practices since end of the year 2005. By the end of June 2016, 9245 participants were recruited into the BLITZ study, and after the application of exclusion criteria (specified in Fig. 1), CRC and advanced adenomas (AA), the precursors of CRC had been detected in 56 and 623 participants, respectively. In the present study, the validation of protein signatures derived in the discovery set was carried out in blood samples from 56 participants with CRC and 102 controls who were free of colorectal neoplasm. Controls were frequency‐matched to the CRC cases by sex and age. In addition, we also selected 101 participants with AA (defined as adenoma with > 1 cm in diameter, tubulovillous or villous components, or high‐grade dysplasia (Brenner et al., 2010a)) who were likewise frequency‐matched to the CRC cases by sex and age. The BLITZ study and use of its samples for the evaluation of early detection markers for CRC have been approved by the ethics committees of the Medical Faculty Heidelberg (S‐178/2005), and of the physicians’ boards of Baden‐Wuerttemberg (M118‐05‐f), Rhineland‐Palatinate [837.047.06(5145)], Hessen (MC 254/2007), and Saarland (217/13). The BLITZ study adheres to the standards set by Declaration of Helsinki, and all the study participants have voluntarily given their written informed consent. The STARD diagram showing selection of study participants from the BLITZ study is presented in Fig. 1.
Figure 1.
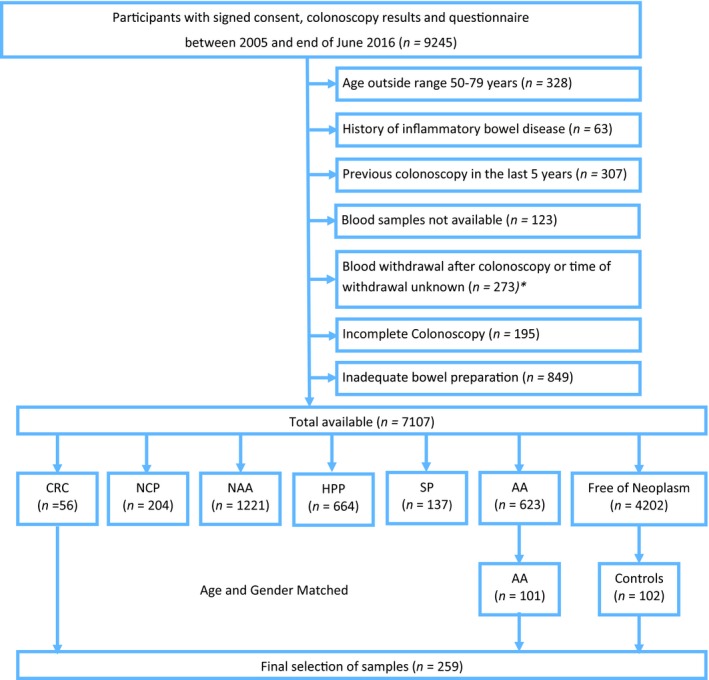
STARD flow diagram BLITZ study. HPP, hyperplastic polyps; NAA, nonadvanced adenoma; NCP, nonclassified polyp; SP, serrated poly. *The exclusion criteria for selection of CRC cases were not applicable after this point.
2.4. Sample collection and storage
The blood draw was performed at first diagnosis of CRC before any treatment for cancer in iDa and before colonoscopy in ASTER and BLITZ. After blood draw, EDTA plasma samples were transported to the laboratory while preserved in cold chain, followed by centrifugation at 2000–2500 g for 10 min, and were then stored at – 80 °C until the protein measurements. The laboratory staff was blind to any information regarding the study population.
2.5. Laboratory assay
Protein concentrations in plasma samples were measured utilizing the proximity extension assays (PEAs) offered by Olink. Olink’s multiplex panels allow simultaneous analysis of 92 biomarkers in 1 µl samples, and the full protocol of the PEA has been reported previously (Assarsson et al., 2014). Briefly, the 96 pairs of oligonucleotide‐labeled antibodies (92 biomarkers and four internal controls) are allowed to pairwise bind to target proteins and when in close proximity a PCR reporter sequence is formed due to DNA polymerization, which is quantified by real‐time PCR. For our study, we used the Olink multiplex panels ‘Oncology II’, ‘Immune response’, and ‘Cardiovascular III’ resulting in a total of 276 proteins analyzed. The full list of markers from each panel is provided in Table S1. Laboratory analyses were performed blinded with respect to disease status or findings at colonoscopy in the laboratory of the manufacturer of the panels. Each assay form these panels has been validated, and information on characteristics such as detection limits, dynamic range, repeatability, and reproducibility is available from the manufacturer’s website (https://www.olink.com/data-you-can-trust/validation/).
2.6. Statistical analysis
All linear protein values were log‐transformed to produce normalized protein expression (NPX). In the current study, the NPX values of each individual protein were compared between CRC and controls in the discovery set using Wilcoxon rank‐sum test with adjustment for multiple testing by the Benjamini and Hochberg method (Benjamini and Hochberg, 1995). For each individual protein biomarker, a logistic regression model was used to construct the prediction algorithm and .632+ bootstrap was applied to adjust for potential overestimation of diagnostic performance (Efron and Tibshirani, 1997). Areas under the receiver operating characteristic curves (AUCs) and their 95% confidence intervals (95% CI) and sensitivity (true positive rate) of each individual biomarker at cutoffs yielding 80% and 90% specificities (true‐negative rate) were calculated.
In order to derive multimarker algorithms for the prediction of the presence of CRC, least absolute shrinkage and selection operator (LASSO) logistic regression models were applied to markers that remained significant after multiple testing in the discovery set. The LASSO regression was adapted in order to obtain models with the best prediction accuracy and was combined with .632+ bootstrap (Efron and Tibshirani, 1997) to adjust for overfitting. Three prediction algorithms were derived, one each for all (I–IV)‐, early (I–II)‐, and late (III–IV)‐stage CRC vs controls, respectively. For evaluating the diagnostic performance of each algorithm, the sensitivity, specificity, and apparent AUC, that is, the AUC not adjusted for overfitting (AUC*) with 95% CI, as well as .632+ bootstrap‐adjusted AUC (AUCBS), were calculated. In order to externally validate the prediction algorithms in a real‐life screening setting, their performance was finally evaluated in the validation set that exclusively included participants of screening colonoscopy. The performance of CRC early‐stage algorithm was additionally evaluated for AA participants in the validation set.
In the validation set, diagnostic performance of the proteins and protein algorithms was evaluated separately for CRC and for AA, in each case in comparison with controls free of neoplasms. According to common practice in biomarker research, this comparison was done using controls that were matched to the CRC cases with respect to for age and sex distribution in the first place. However, the age and sex distribution of participants free of colorectal neoplasms actually differs from the corresponding distributions of CRC cases in a true screening setting. With the objective of providing valid estimates of the algorithms’ performance in a true screening setting, the observations from participants with AA and controls free of neoplasms were therefore weighted in the validation set of the current study in such a way that their distribution reflects the sex and age distribution of all participants with AA and controls free of neoplasms among the participants of screening colonoscopy, respectively (Brenner et al., 2013). All statistical analyses were performed with statistical software r language and environment (version 3.5.0, R core team) (R Core Team, 2016). For all tests, P‐values of 0.05 or less were considered to be statistically significant.
3. Results
3.1. Characteristics of the study populations
Figures S1 and S2 and Fig. 1 provide the STARD diagrams displaying the selection of study participants enrolled in iDa, ASTER, and BLITZ, respectively. The discovery set consisted of 98 and 100 clinically recruited CRC cases from iDa and controls from ASTER, respectively. The validation set included 56 and 101 participants of screening colonoscopy with CRC and AA, respectively, and 102 controls free of neoplasms from the BLITZ study. The characteristics of populations from both the discovery and the validation sets are shown in Table 1. The distribution of characteristics was largely similar across both sets with the median age being around 65 years and males representing ≥ 60% of population in both sets. Table 1 additionally includes the age and sex distribution of AA cases and controls in the entire screening population from which the matched AA cases and controls were drawn
Table 1.
Characteristics of the study population in both sets. Ca, cancer; N, number; SD, standard deviation.
| Group | Discovery set | Validation set | Participants of screening colonoscopy | ||||
|---|---|---|---|---|---|---|---|
| iDa (Clinical) CRC | ASTER (Mostly screening) controls | BLITZ matched set (Screening) | BLITZ (Screening) | ||||
| CRC | AA | Controls | AA | Controls | |||
| Total | N (%) | N (%) | N (%) | N (%) | N (%) | N (%) | N (%) |
| 98 | 100 | 56 | 101 | 102 | 623 | 4202 | |
| Age in years | |||||||
| 50–59 | 23 (23) | 29 (29) | 10 (18) | 22 (21) | 21 (21) | 237 (38) | 1916 (46) |
| 60–69 | 42 (43) | 45 (45) | 28 (50) | 49 (49) | 50 (49) | 247 (40) | 1614 (38) |
| 70–79 | 33 (34) | 26 (26) | 18 (32) | 30 (30) | 31 (30) | 139 (22) | 672 (16) |
| Mean | 64.6 | 64.0 | 66.0 | 65.5 | 65.4 | 63.3 | 61.9 |
| Median | 64.5 | 65.5 | 65.0 | 65.0 | 65.5 | 62.0 | 60.0 |
| SD | 6.9 | 7.5 | 5.8 | 6.6 | 6.9 | 5.9 | 6.5 |
| Gender distribution | |||||||
| Male | 60 (60) | 60 (60) | 36 (64) | 65 (64) | 66 (65) | 393 (63) | 1808 (43) |
| Female | 40 (40) | 40 (40) | 20 (36) | 36 (36) | 36 (35) | 230 (37) | 2394 (57) |
| Stage distribution | |||||||
| Stage I | 17 (17) | – | 17 (30) | – | – | – | – |
| Stage II | 32 (33) | – | 6 (11) | – | – | – | – |
| Stage III | 23 (23) | – | 26 (46) | – | – | – | – |
| Stage IV | 26 (27) | – | 7 (13) | – | – | – | – |
| Early stage (I/II) | 49 (50) | – | 23 (41) | – | – | – | – |
| Late stage (III/IV) | 49 (50) | – | 33 (59) | – | – | – | – |
| Cancer site | |||||||
| Proximal colon | 41 (42) | – | 9 (16) | – | – | – | – |
| Distal colon | 23 (23) | – | 24 (43) | – | – | – | – |
| Rectum | 33 (34) | – | 21 (38) | – | – | – | – |
| Unknown | 1 (1) | – | 2 (4) | – | – | – | – |
3.2. Assay performance
The quality control criteria (QCC) for both the datasets were considered good with 95–100% of the samples meeting QCC across the three Proseek panels and 82–100% of proteins being above the limit of detection. For the three multiplex panels, the average intra‐assay coefficient of variance (CV) was 5% and 7% and the average interassay CV was 19% and 15% for discovery and validation sets, respectively. Due to redundancy between panels and one analyte not meeting QCC, reportable results were obtained for 275 proteins. Amphiregulin (AREG) and interleukin‐6 (IL6) were the two proteins measured on multiple panels, and both showed good concordance (Pearson’s product–moment correlation, r = 0.99 and 0.98, respectively). The complete experimental workflow of the study has been described in Fig. S3.
3.3. Individual marker analysis for all‐stage CRC
For the discovery set, analysis was conducted with 97 CRC cases and 99 controls because two participants had to be excluded on account of several missing protein measurements. In the validation set, the analysis was based on 56 CRC, 101 AA, and 102 controls. Results of the univariate analysis comparing the expression difference in each marker in plasma between cases and controls in both sets are summarized in Table S2. When results for the 275 markers were adjusted for multiple testing, overall 83 markers showed statistically significant differences in expression levels in the discovery set (adjusted P‐values ≤ 0.05 in Table S2). After correction for overoptimism by .632+ bootstrap, AUCBS of these 83 markers ranged from 0.80 [0.73–0.89] to 0.58 [0.48–0.70] with sensitivities at cutoffs yielding 80% specificity ranging from 68% to 24%. However, in the validation set only nine of these 83 markers were successfully replicated with adjusted P‐values ≤ 0.05.
3.4. Multimarker predictor algorithms from the discovery set
The Lasso logistic regression model was applied on all 83 protein markers with adjusted P‐values ≤ 0.05 from the discovery set, in order to construct a multimarker prediction algorithm for comparing all‐, early‐, and late‐stage CRC to controls. Table 2 and Fig. 2 show that the best performances were obtained for 9‐, 12‐, and 11‐marker combinations. The 9‐marker algorithm for all stages selected the following proteins: AREG, carcinoembryonic antigen (CEA), granzyme B (GZMB), integrin alpha V (ITGAV), keratin, type I cytoskeletal 19 (KRT19), monocyte chemotactic protein 1 (MCP1), osteopontin (OPN), paraoxonase 3 (PON3), and transferrin receptor protein 1 (TR). The AUCBS was 0.92, and diagnostic sensitivities of 89% and 73% were obtained at cutoffs yielding 90% and 80% specificity, respectively.
Table 2.
Diagnostic performance of multimarker signatures for detecting CRC (all stages/early/late/AA) in discovery and validation sets. Se, sensitivity; Sp, specificity.
| Stage‐specific CRC vs controls free of neoplasms | Protein markers discovered in the algorithm/signatures | Discovery set |
Validation set (as in matched samples) |
Validation set (as in screening population) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUCBS | AUC* |
Se % at 80% Sp |
Se % at 90% Sp |
AUC (95% CI) |
Se % at 80% Sp |
Se % at 90% Sp |
AUC (95% CI) |
Se % at 80% Sp |
Se % at 90% Sp |
|||
| All‐stage CRC | AREG+ CEA+ GZMB+ ITGAV+ KRT19+ MCP1+ OPN+ PON3+ TR | 0.92 | 0.92 | 89 | 73 | 0.75 (0.67–0.82) | 50 | 41 | 0.76 (0.67–0.84) | 55 | 45 | |
| Early‐stage CRC | AREG+ CEA+ GZMB+ ITGAV+ KRT19+ MASP1+ MCP1+ PON3+ RARRES2+ S100A4+ TR+ TRAP | 0.91 | 0.91 | 82 | 74 | 0.73 (0.62–0.83) | 61 | 35 | 0.75 (0.62–0.87) | 61 | 35 | |
| Late‐stage CRC | AREG+ CEA+ IL6+ ITGA11+ ITGAV+ KRT19+ MCP1+ NTproBNP+ OPN+ TR+ TRAIL | 0.95 | 0.96 | 95 | 89 | 0.81 (0.72–0.89) | 71 | 51 | 0.80 (0.68–0.89) | 70 | 51 | |
| AA | AREG+ CEA+ GZMB+ ITGAV+ KRT19+ MASP1+ MCP1+ PON3+ RARRES2+ S100A4+ TR+ TRAP | – | – | – | – | 0.45 (0.37–0.53) | 13 | 5 | 0.58 (0.47–0.68) | 28 | 24 | |
Figure 2.
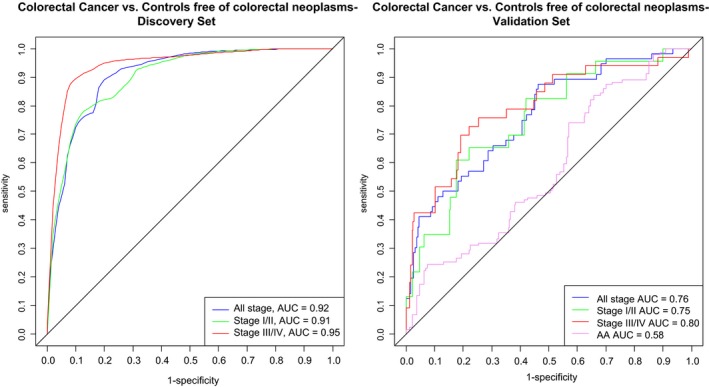
Comparison of the receiver operating characteristic curves for detecting (all/early/late) stage CRC and AA vs free of neoplasm controls for discovery and validation set with 9‐, 12‐, and 11‐marker signatures.
For early‐stage CRC, as shown in Table 2 the algorithm comprised 12 proteins, 8 of which were identical with algorithm from all stages, that is, AREG, CEA, GZMB, ITGAV, KRT19, MCP1, PON3, TR, and additionally mannan‐binding lectin serine protease 1 (MASP1), retinoic acid receptor responder protein 2 (RARRES2), protein S100‐A4 (S100A4), and tartrate‐resistant acid phosphatase type 5 (TRAP), respectively. The AUCBS for the 12‐marker algorithm for the comparison of early‐stage CRC cases vs controls was 0.91, and when defining cutoffs to 80% and 90% specificities, sensitivities of 82% and 74% were observed. For late stages, the AUCBS was 0.95 for an algorithm with 11 markers, namely AREG, CEA, ITGAV, KRT19, MCP1, OPN, TR, IL6, integrin alpha 11 (ITGA11), N‐terminal prohormone brain natriuretic peptide (NTproBNP), and TNF‐related apoptosis‐inducing ligand (TRAIL). The sensitivities for the 11‐marker algorithm for the comparison of late‐stage CRC cases vs controls were 95% and 89% at cutoffs yielding 80% and 90% specificity, respectively.
3.5. Diagnostic performance of multimarker signatures in the validation set
For external validation, the performance was evaluated in an independent population consisting of participants from a true screening study. The prediction algorithms derived from discovery set are presented in Table S3. Upon application of estimates from the predictor algorithms obtained from discovery set, the AUCs as shown in Table 2 and Fig. 2 for 9‐, 12‐, and 11‐marker signatures were 0.76 (95% CI, 0.67–0.84), 0.75 (95% CI, 0.62–0.87), and 0.80 (95% CI, 0.68–0.89). At cutoff yielding 80% specificity, sensitivities of 55%, 61%, and 70% and at defining cutoff at 90% specificity, sensitivities of 45%, 35%, and 51% were observed for comparing all‐, early‐, and late‐stage CRC cases vs controls, respectively. When the predictor algorithm for early stage was applied for comparing participants with AA to controls, an AUC of 0.58 (95% CI, 0.47–0.68) was observed.
3.6. Site‐specific diagnostic performance of markers in both sets
In order to observe the site‐specific diagnostic performances, further stratification was performed with respect to the location of cancer, that is, proximal colon, distal colon, or rectum, respectively. The results of the site‐specific analyses in the discovery and validation sets for individual markers and multimarker signature are presented in Tables S4 and S5, respectively. As shown in Table S4, when further stratification with respect to location was performed and all 275 markers were adjusted for multiple testing, overall 47, 30, and 25 protein markers showed statistically significant differences in expression levels for the proximal colon, distal colon, and rectum, respectively. In the validation set due to comparatively lower number of cases, only 1, 2, and 6 of these markers for the proximal colon, distal colon, and rectum, respectively, could be replicated. The site‐specific AUCs of the 9‐marker signature for the detection of proximal, distal, and rectal CRCs were 0.98, 0.95, and 0.95 in the discovery set and 0.70, 0.77, and 0.78 in the validation set, respectively (Table S5).
3.7. Stage‐specific diagnostic performance of identified markers in both sets
The individual marker performances of the 17 markers identified in all three prediction algorithms for early‐ and late‐stage CRC detection in discovery and validation sets are presented in Table 4. In the discovery set, diagnostic performance of 13 out of 17 markers was comparatively higher for late‐stage CRC detection and usually the performances were better in discovery than validation sets for both early‐stage CRC detection and late‐stage CRC detection. For early‐stage CRC, four markers that presented with AUC ≥ 0.7 in the discovery set were AREG, ITGA11, ITGAV, and PON3. However, in the validation set only AREG of these four markers presented with AUC < 0.7 and sensitivity of 55% at cutoff yielding 80% specificity. For late‐stage CRC, the four markers with AUC ≥ 0.7 in both the discovery and validation sets were AREG, CEA, KRT19, and TR. In validation set, the markers CEA (AUC = 0.76), KRT19 (AUC = 0.74), and TR (AUC = 0.74) best detected late‐stage CRC cases with sensitivities of 62%, 52%, and 50% at cutoff yielding 80% specificity.
Table 4.
Diagnostic performance of individual markers identified in signatures for detecting early‐ and late‐stage CRC in discovery and validation sets. Se, sensitivity; Sp, specificity.
| Marker | Early stages (Stages I and II) | Late stages (Stages III and IV | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Discovery set | Validation set | Discovery set | Validation set | |||||||||||||||||||||
| P‐val | P‐valadj | AUC* (95% CI) | AUCBS (95% CI) | Se% at 80% Sp | Se% at 90% Sp | P‐val | P‐valadj | AUC* (95% CI) | AUCBS (95% CI) | Se% at 80% Sp | Se% at 90% Sp | P‐val | P‐valadj | AUC* (95% CI) | AUCBS (95% CI) | Se% at 80% Sp | Se% at 90% Sp | P‐val | P‐valadj | AUC* (95% CI) | AUCBS (95% CI) | Se% at 80% Sp | Se% at 90% Sp | |
| AREG | <0.001 | <0.001 | 0.7 (0.6–0.79) | 0.68 (0.58–0.82) | 45 | 35 | <0.005 | <0.01 | 0.73 (0.6–0.85) | 0.7 (0.56–0.89) | 55 | 36 | <0.001 | <0.001 | 0.87 (0.81–0.93) | 0.86 (0.78–0.94) | 78 | 65 | <0.001 | <0.001 | 0.71 (0.61–0.81) | 0.69 (0.57–0.85) | 49 | 33 |
| CEA | <0.005 | <0.005 | 0.67 (0.58–0.77) | 0.64 (0.54–0.8) | 42 | 27 | <0.05 | 0.05 | 0.66 (0.51–0.81) | 0.64 (0.46–0.85) | 53 | 33 | <0.001 | <0.001 | 0.8 (0.71–0.88) | 0.79 (0.67–0.91) | 65 | 57 | <0.001 | <0.001 | 0.76 (0.65–0.87) | 0.74 (0.6–0.91) | 62 | 52 |
| GZMB | <0.05 | <0.05 | 0.63 (0.53–0.72) | 0.59 (0.49–0.76) | 28 | 12 | 0.06 | 0.11 | 0.63 (0.51–0.75) | 0.55 (0.4–0.79) | 25 | 12 | <0.005 | <0.005 | 0.66 (0.57–0.76) | 0.64 (0.53–0.79) | 33 | 12 | 0.65 | 0.65 | 0.53 (0.42–0.63) | 0.45 (0.33–0.6) | 14 | 7 |
| IL6 | <0.001 | <0.005 | 0.68 (0.59–0.77) | 0.65 (0.56–0.8) | 39 | 17 | <0.05 | 0.08 | 0.64 (0.52–0.76) | 0.54 (0.32–0.79) | 21 | 11 | <0.001 | <0.001 | 0.81 (0.74–0.89) | 0.8 (0.71–0.91) | 69 | 43 | 0.20 | 0.26 | 0.57 (0.47–0.68) | 0.5 (0.32–0.69) | 21 | 11 |
| ITGA11 | <0.001 | <0.001 | 0.73 (0.65–0.82) | 0.69 (0.58–0.84) | 47 | 33 | 1.00 | 1.00 | 0.5 (0.38–0.63) | 0.44 (0.29–0.6) | 13 | 5 | <0.001 | <0.001 | 0.86 (0.79–0.92) | 0.85 (0.75–0.93) | 69 | 59 | 0.07 | 0.13 | 0.6 (0.5–0.7) | 0.55 (0.44–0.72) | 20 | 9 |
| ITGAV | <0.001 | <0.001 | 0.7 (0.61–0.79) | 0.68 (0.56–0.81) | 45 | 33 | 0.66 | 0.75 | 0.53 (0.4–0.66) | 0.44 (0.28–0.64) | 15 | 5 | <0.001 | <0.001 | 0.86 (0.79–0.93) | 0.85 (0.76–0.94) | 75 | 65 | <0.005 | <0.005 | 0.69 (0.59–0.78) | 0.66 (0.56–0.81) | 37 | 22 |
| KRT19 | <0.001 | <0.005 | 0.68 (0.59–0.77) | 0.66 (0.55–0.8) | 42 | 25 | <0.05 | <0.05 | 0.68 (0.55–0.81) | 0.64 (0.51–0.85) | 44 | 30 | <0.001 | <0.001 | 0.84 (0.77–0.91) | 0.83 (0.74–0.93) | 67 | 60 | <0.001 | <0.001 | 0.74 (0.64–0.84) | 0.72 (0.59–0.86) | 52 | 39 |
| MASP1 | <0.005 | <0.005 | 0.66 (0.57–0.75) | 0.63 (0.53–0.77) | 26 | 16 | 0.50 | 0.69 | 0.54 (0.41–0.68) | 0.45 (0.26–0.65) | 16 | 6 | <0.01 | <0.01 | 0.64 (0.54–0.74) | 0.61 (0.51–0.78) | 38 | 25 | 0.06 | 0.11 | 0.61 (0.5–0.72) | 0.55 (0.39–0.75) | 28 | 15 |
| MCP1 | <0.001 | <0.005 | 0.69 (0.59–0.78) | 0.66 (0.56–0.81) | 42 | 24 | 0.66 | 0.75 | 0.53 (0.39–0.67) | 0.44 (0.27–0.6) | 14 | 8 | <0.01 | <0.01 | 0.64 (0.53–0.74) | 0.61 (0.5–0.79) | 40 | 28 | 0.18 | 0.26 | 0.58 (0.46–0.69) | 0.5 (0.3–0.69) | 22 | 12 |
| NTproBNP | 0.12 | 0.13 | 0.58 (0.47–0.68) | 0.52 (0.34–0.69) | 27 | 16 | 0.40 | 0.62 | 0.56 (0.43–0.68) | 0.45 (0.28–0.64) | 18 | 7 | <0.001 | <0.001 | 0.69 (0.6–0.79) | 0.68 (0.58–0.82) | 44 | 29 | 0.43 | 0.49 | 0.54 (0.42–0.67) | 0.49 (0.31–0.68) | 24 | 13 |
| OPN | <0.005 | <0.005 | 0.66 (0.56–0.75) | 0.62 (0.53–0.78) | 37 | 19 | <0.005 | <0.05 | 0.71 (0.6–0.82) | 0.68 (0.56–0.86) | 42 | 24 | <0.001 | <0.001 | 0.83 (0.76–0.9) | 0.82 (0.73–0.92) | 68 | 56 | 0.36 | 0.43 | 0.55 (0.44–0.67) | 0.49 (0.32–0.67) | 23 | 12 |
| PON3 | <0.001 | <0.001 | 0.73 (0.64–0.81) | 0.71 (0.6–0.84) | 45 | 35 | 0.26 | 0.44 | 0.58 (0.45–0.7) | 0.49 (0.3–0.71) | 16 | 9 | <0.001 | <0.001 | 0.77 (0.69–0.86) | 0.76 (0.66–0.89) | 62 | 47 | 0.06 | 0.11 | 0.61 (0.5–0.71) | 0.54 (0.34–0.73) | 23 | 9 |
| RARRES2 | <0.001 | <0.005 | 0.69 (0.6–0.78) | 0.67 (0.56–0.81) | 40 | 24 | <0.05 | <0.05 | 0.67 (0.57–0.78) | 0.62 (0.53–0.81) | 27 | 13 | <0.001 | <0.001 | 0.75 (0.66–0.84) | 0.54 (0.18–0.8) | 27 | 16 | <0.05 | 0.08 | 0.62 (0.51–0.73) | 0.54 (0.29–0.75) | 28 | 11 |
| S100A4 | <0.005 | <0.005 | 0.65 (0.56–0.74) | 0.63 (0.51–0.78) | 28 | 11 | 0.52 | 0.69 | 0.54 (0.42–0.66) | 0.47 (0.32–0.67) | 15 | 7 | <0.005 | <0.005 | 0.66 (0.56–0.75) | 0.63 (0.53–0.78) | 36 | 14 | 0.15 | 0.23 | 0.58 (0.47–0.69) | 0.51 (0.29–0.69) | 18 | 10 |
| TR | <0.05 | <0.05 | 0.62 (0.52–0.73) | 0.6 (0.49–0.76) | 38 | 29 | <0.001 | <0.01 | 0.74 (0.63–0.86) | 0.72 (0.58–0.9) | 48 | 35 | <0.001 | <0.001 | 0.76 (0.68–0.84) | 0.75 (0.66–0.87) | 53 | 43 | <0.001 | <0.001 | 0.74 (0.64–0.84) | 0.71 (0.61–0.87) | 50 | 28 |
| TRAP | <0.05 | <0.05 | 0.64 (0.53–0.74) | 0.61 (0.49–0.77) | 41 | 30 | 0.90 | 0.96 | 0.51 (0.39–0.63) | 0.44 (0.31–0.58) | 12 | 5 | 0.19 | 0.19 | 0.57 (0.46–0.67) | 0.49 (0.31–0.67) | 21 | 12 | 0.65 | 0.65 | 0.53 (0.42–0.64) | 0.44 (0.31–0.6) | 17 | 7 |
| TRAIL | 0.26 | 0.26 | 0.56 (0.45–0.66) | 0.51 (0.35–0.68) | 24 | 16 | <0.05 | 0.05 | 0.65 (0.54–0.77) | 0.59 (0.46–0.8) | 26 | 12 | <0.001 | <0.001 | 0.75 (0.67–0.83) | 0.74 (0.64–0.86) | 48 | 30 | <0.005 | <0.05 | 0.67 (0.56–0.77) | 0.63 (0.51–0.8) | 34 | 18 |
3.8. Function of the protein markers
There were in total 17 proteins found in three different prediction models, and as depicted in Table 3, these proteins have a wide variety of molecular functions with five, four, and three of them being cytokines, hydrolases, and receptors, respectively. As depicted in Fig. S4, when Ingenuity Pathway Analysis (Qiagen Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis, version 01‐10; Ingenuity Systems, Redwood City, CA, USA) was used to understand the interaction between these proteins in established canonical pathways, it was observed that though functionally different these proteins interact at cellular or extracellular level in several different pathways (Fig. S4). However, some proteins such as cytokines IL6 and MCP1 and receptors ITGAV and ITGA11 interact directly in more than one pathway. Additionally, when the role of these proteins in different organ toxicities was looked upon (Fig. S5), it was found that several proteins such as IL6, ITGAV, PON3, OPN, TR, and TRAIL are involved in toxicities such as liver necrosis/proliferation/fibrosis and renal necrosis.
Table 3.
Distribution of proteins from multimarker signatures across different panels and according to the stages involved and their functions. CVDIII, cardiovascular III panel; ID, identification; IR, immune response panel; ONCO II, oncology II panel.
| Name | Uniprot ID | Pea panel | Molecular function | Biological process | All stage | Early stage | Late stage |
|---|---|---|---|---|---|---|---|
| AREG | http://www.uniprot.org/uniprot/P15514 | ONCO II | Cytokine, growth factor | Cell–cell signaling, cell proliferation | x | x | x |
| CEA | http://www.uniprot.org/uniprot/P06731 | ONCO II | GPI anchor binding; glycoprotein | Homotypic cell–cell adhesion | x | x | x |
| GZMB | http://www.uniprot.org/uniprot/P10144 | ONCO II | Hydrolase, protease, serine protease | Apoptosis, cytolysis | x | x | |
| IL‐6 | http://www.uniprot.org/uniprot/P05231 | IR | Cytokine, growth factor | Acute‐phase response | x | ||
| Integrin alpha‐11 | http://www.uniprot.org/uniprot/Q9UKX5 | IR | Integrin, receptor | Cell adhesion | x | ||
| ITGAV | http://www.uniprot.org/uniprot/P06756 | ONCO II | Host cell receptor for virus entry, integrin, receptor | Calcium, metal binding | x | x | x |
| KRT19 | http://www.uniprot.org/uniprot/P08727 | IR | Protein complex binding | Host–virus interaction | x | x | x |
| MASP1 | http://www.uniprot.org/uniprot/P48740 | IR | Hydrolase, protease, serine protease | Complement activation lectin pathway, immunity, innate immunity | x | ||
| MCP1 | http://www.uniprot.org/uniprot/P13500 | CVD III | Cytokine/chemokine | Chemotaxis, inflammatory response | x | x | x |
| NTproBNP | http://www.uniprot.org/uniprot/NA | CVDIII | NA | NA | x | ||
| OPN | http://www.uniprot.org/uniprot/P10451 | CVD III | Cytokine | Biomineralization, cell adhesion | x | x | |
| PON3 | http://www.uniprot.org/uniprot/Q15166 | CVD III | Hydrolase | Calcium, metal binding | x | x | |
| RARRES2 | http://www.uniprot.org/uniprot/Q99969 | CVD III | Receptor binding | Chemotaxis, differentiation, inflammatory response | x | ||
| S100A4 | http://www.uniprot.org/uniprot/P26447 | ONCO II | Calcium‐dependent protein binding | Epithelial‐to‐mesenchymal transition | x | ||
| TR | http://www.uniprot.org/uniprot/P02786 | CVD III | Host cell receptor for virus entry, receptor | Endocytosis, host–virus interaction | x | x | x |
| TRAP | http://www.uniprot.org/uniprot/P13686 | CVD III | Hydrolase | Iron, metal binding | x | ||
| TRAIL | http://www.uniprot.org/uniprot/P50591 | ONCO II | Cytokine | Apoptosis | x |
4. Discussion
In this study, we performed PEA on two different sets of populations for the identification and validation of protein multimarker signatures for the detection of CRC and its precursors. The study was performed in a two‐stage design, and the independent external validation was performed on 56 CRC and 102 controls recruited among 9245 participants of a true screening study. We identified three different multimarker signatures for stage‐specific CRC. A total of 17 different proteins were identified in different prediction models. A 9‐marker panel with an AUCBS of 0.92 and an AUC 0.76 (95% CI, 0.67–0.84) in the discovery and the validation set, respectively, best detected CRC at all stage. For stage‐specific models, the best diagnostic performance was observed in forms of 12‐marker and 11‐marker signatures with AUCBS of 0.91 and 0.95 in the discovery set and AUC of 0.75 (95% CI, 0.62–0.87) and 0.80 (95% CI, 0.68–0.89) for detecting early‐ and late‐stage CRC in the validation set, respectively.
Previous research pertaining to blood‐based protein markers has been mostly conducted on samples from clinical settings (Bhardwaj et al., 2017a; Bhardwaj et al., 2017b). Only few studies went a step further and performed rigorous independent validation in true screening settings. The cases recruited in clinical settings are mostly symptomatic, have typically undergone several diagnostic procedures, and in several instances they may have undergone initial therapeutic intervention and lifestyle modification prior to sample collection. When such cases are compared to healthy controls, diagnostic performance indicators tend to be higher which might lead to spectrum bias (Kohn et al., 2013; Whiting et al., 2004). Likewise, the controls recruited in clinical settings often consist of patients suffering from some other systemic diseases. Such factors may influence the diagnostic performance indicators and may lead to false‐positive findings (Chen et al., 2016; Tao et al., 2011). Additionally, hospital controls usually have higher response rates than population controls, which might contribute to sampling bias. In our study, we therefore made major efforts to avoid such biases by externally validating the performance of the predictor algorithms in an independent set exclusively of participants recruited in a true screening setting, that is, before screening colonoscopy. The pre‐analytical processing of samples influences the measurements in protein biomarker research (Enroth et al., 2016; Lee et al., 2010; Shen et al., 2018). In the current study although participants in the discovery and validation sets were selected from three different studies, the collection, handling, processing, and storage of the samples across the three studies were performed with similar standardized operating procedure.
The PEAs utilize a pair of oligonucleotide‐labeled antibodies or probes for the detection of each protein. These probes have to be in close proximity, and only this dual recognition of the target protein leads to initiation of an amplified signal detection. PEA quantifies across five logs of abundance with good reproducibility (CV < 20%), and a very low volume of 1 µL is required per sample. The technical assay sensitivity for the PEAs is in the picogram·mL−1 range. The dual recognition, requirements of low sample volume, and good assay sensitivity make PEA a very target‐specific and efficient method. Nevertheless, the type and number of proteins included in each of these commercially offered PEA panels are predetermined and cannot be selected. Each panel allows simultaneous detection of only 92 proteins, so further developments are warranted for full proteome coverage. Additionally, even though the reproducibility of PEAs is comparatively better than other antibody‐based methods, replication of our findings using other quantification techniques should be aimed for in future studies.
In two former studies from our research group on protein marker signatures where external validation was conducted (Chen et al., 2015; Surinova et al., 2015), AUCs of 8‐marker and 6‐marker algorithms were 0.76 (Chen et al., 2015) and 0.84 (Surinova et al., 2015), respectively. However, unlike the current study, these studies included CRC cases recruited in clinical settings in the external validation. In another study from our group with a totally distinct discovery set but a partly overlapping external validation set (Chen et al., 2017) recruited in the BLITZ study, an AUC of 0.82 had been found for a five‐marker signature [AREG, growth differentiation factor 15, fas antigen ligand, fms‐related tyrosine kinase 3 ligand, and TP53 autoantibody (antiTP53)]. Even though the current study was based on a comparatively larger sample size and included approximately three times as many proteins (but not antiTP53) in three different panels, the AUC in the validation set was slightly lower. This was the case even if antiTP53 had been left out from our previously identified algorithm. Possibly, the combination of the previously detected and the newly detected proteins may allow for further improvement of diagnostic performance for early detection. Algorithms based on such combination would though have to be addressed in additional independent samples in order to provide unbiased estimates of diagnostic performance.
The diagnostic performance of our 12‐marker algorithm was comparable to that of FITs in the discovery set with 82% sensitivity at 80% specificity for early stage. Previous studies on blood‐based tests that are not exclusively based on proteins, such as COLODETECT (4 proteins + 3 phages) (Barderas et al., 2013), COLOX (gene expression of 29 genes) (Ciarloni et al., 2016), and CANCERSEEK (16 genes + eight proteins) (Cohen et al., 2018), recruited CRC cases in a complete or partial clinical setting and reported sensitivities of 89 %, 79.5%, and 64.9% at specificities of 90%, 90%, and 99.1%, respectively. However, the performance of these tests in screening settings remains unknown. When the performance of the blood‐based test COLOSENTRY (seven genes) was evaluated in screening setting, 61% sensitivity at 77% specificity for all stages was observed (Marshall et al., 2010; Yip et al., 2010). The FDA‐approved blood‐based test epi procolon 2.0 (Sept9 gene methylation) (Potter et al., 2014) showed 59% sensitivity for early‐stage CRC at 79% specificity which is very close to the results observed in the validation sample of our study (61% sensitivity at 80% specificity). Nevertheless, the combination of the markers from tests such as COLODETECT, COLOX, and CANCERSEEK with the protein markers identified in the current study might potentially boost the diagnostic potential of blood‐based markers for early detection of CRC.
The 17 proteins that turned out in different algorithms as demonstrated here and in previous research are involved in different biological processes and mechanisms and interact directly in several pathways. Some of the identified proteins AREG, CEA, ITGAV, KRT19, MCP1, and TR were present in all three prediction algorithms. However, biomarkers such as MASP1, RARRES2, S100A4 and IL6, ITGA11, and TRAIL were specific for early and late stages, respectively. As presented in Table 4, the individual marker performances of these 17 markers for early and late stages in discovery and validation sets were heterogeneous. However, all the markers typically showed higher performances in discovery than validation sets. This once again highlights the differences in efficacy of markers in the clinically recruited and screening samples and in addition displays the need for external validation of signatures in screening colonoscopy cohorts. As described before and shown in Table 4, the markers that showed AUC ≥ 0.70 in both discovery and validation sets for early‐stage (AREG) and late‐stage CRC (CEA, KRT19, and TR) can be considered as strong potential protein markers for inclusion into multimarker signatures. Our current analysis and the previous studies (Chen et al., 2017; Chen et al., 2015) confirm that AREG has excellent diagnostic potential for the detection of CRC. Even though CEA has undisputed value for monitoring tumor recurrence and metastasis, its potential for early detection of CRC remains questionable (Lech et al., 2013; Polat et al., 2014). KRT19 and TR, which are both involved in biological processes of host–virus interaction, have seldom been found associated with early detection of CRC.
Precursors of CRC, that is, AA, are at a risk of developing into invasive CRC over time. The detection of these precursors and their timely removal could contribute substantially to reduction in CRC risk. Detecting these noninvasive precursors with blood‐based tests is likely to be difficult. In the current study, the failure of early‐stage algorithm (that was evaluated for AA participants) is in line with results of the few studies validating diagnostic performance of blood‐based tests in true screening setting, such as the PRESEPT clinical trial on Sept9 gene methylation (Church et al., 2014; Potter et al., 2014). Possible combinations of protein biomarkers obtained with rigorous validation (as in the current study) with other types of genomic, epigenetic, or metabolomic biomarkers may bolster the diagnostic potential of blood‐based tests for early detection of CRC and its precursors.
A major strength of the current study is that we used a two‐stage design, with validation performed exclusively in a true screening setting. However, despite the overall very large size of the BLITZ study (N = 9245) the number of participants with CRC was still rather limited, a feature that is common for screening settings. Using cutting‐edge statistical machine learning algorithms, 275 markers were simultaneously evaluated for possible combinations and comparisons of the tested markers. In addition to an algorithm for overall prediction of CRC presence, signatures for stage‐specific detection were derived and internal as well as external validation was performed. For early stage in external independent validation, sensitivity of 61% was observed at 80% specificity, which is comparable to DNA‐Epi proColon 2.0, the only FDA‐approved blood‐based test for CRC detection. Major limitations include the limited sample size of CRC patients, especially for stage‐specific analyses, leading to rather wide CIs of the derived indicators of diagnostic performance. Given that no other cancers were included in our study, we could not evaluate whether and to what extent the identified markers are CRC‐specific. Therefore, further investigation with patients suffering from other cancers or systemic diseases would be essential. Further research including larger numbers of CRC cases should also address potential differences in detection of colon and rectum cancer. Furthermore, even though the reproducibility of PEAs is better compared with mass spectrometry‐based methods (Smith and Gerszten, 2017), replication of our findings using other quantification techniques should be aimed for. Additionally, further investigation should address to what extent the 17 identified proteins are secreted in vitro by tumor cells or immune cells.
5. Conclusion
We have identified several proteins that individually and in combination carry diagnostic potential for the detection of CRC. With 61% sensitivity at 80% specificity in a true screening setting, diagnostic performance of a 12‐marker algorithm was comparable to diagnostic performance of DNA‐Epi proColon 2.0, the only FDA‐approved blood‐based CRC screening test. Although not competitive with the best available stool‐based tests, the combination of identified protein markers with other informative blood‐based markers could contribute to the development of a promising blood‐based test for CRC screening.
Conflict of interest
The German Cancer Research Center has received industrial grants related to blood markers for early detection of CRC from Epigenomics, Applied Proteomics, and Roche Diagnostics.
Author contributions
HB conceived and supervised the studies. KT, KW, and HB coordinated the studies. MB planned and coordinated this project, selected and shipped the samples, conducted the statistical analyses, interpreted the results, and drafted the manuscript. MB, KW, KT, AB, PS‐K, and HB critically reviewed the manuscript, contributed to its revision, and approved the final version.
Supporting information
Fig. S1. STARD showing selection of study participants enrolled in the iDa Study.
Fig. S2. STARD showing selection of study participants enrolled in the ASTER Study during 2013–2016.
Fig. S3. Experimental workflow of the study.
Fig. S4. Interaction of identified markers from all predictor models in canonical pathways at subcellular level.
Fig. S5. Involvement of identified markers from all predictor models in different organ toxicities.
Table S1. List of 276 proteins measured in the three Olink Multiplex Panels.
Table S2. Diagnostic performance of all 275 proteins markers in discovery and validation set for all‐stage CRC detection.
Table S3. The algorithms identified from the discovery set for (all/early/late) stage CRC detection.
Table S4. Diagnostic performance of the individual markers for detection of CRC in discovery and validation sets stratified by cancer location.
Table S5. Diagnostic performance of the 9‐marker signature for detection of CRC in discovery and validation sets stratified by cancer location.
Acknowledgements
The authors acknowledge the cooperation of gastroenterology practices and clinics in patient recruitment and of Labor Limbach in sample collection. They also thank Katja Butterbach, Katarina Cuk, Ulrike Schlesselmann, Sabine Eichenherr, and Romana Kimmel for their excellent work in laboratory preparation of blood samples and Isabel Lerch, Susanne Köhler, Utz Benscheid, Jason Hochhaus, and Maria Kuschel for their contribution in data collection, monitoring, and documentation. The BLITZ study was partly funded by grants from the German Research Council (DFG, Grant No. BR1704/16‐1). The ASTER and iDa studies were funded by the German Federal Ministry of Education and Research through the German Cancer Consortium (DKTK).
References
- Assarsson E, Lundberg M, Holmquist G, Björkesten J, Bucht Thorsen S, Ekman D, Eriksson A, Rennel Dickens E, Ohlsson S, Edfeldt G et al (2014) Homogenous 96‐Plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 9, e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkin W, Wooldrage K, Parkin DM, Kralj‐Hans I, MacRae E, Shah U, Duffy S and Cross AJ (2017) Long term effects of once‐only flexible sigmoidoscopy screening after 17 years of follow‐up: the UK Flexible Sigmoidoscopy Screening randomised controlled trial. Lancet 389, 1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barderas R, Mendes M, Torres S, Bartolome RA, Lopez‐Lucendo M, Villar‐Vazquez R, Pelaez‐Garcia A, Fuente E, Bonilla F and Casal JI (2013) In‐depth characterization of the secretome of colorectal cancer metastatic cells identifies key proteins in cell adhesion, migration, and invasion. Mol Cell Proteomics 12, 1602–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y and Hochberg Y (1995) Controlling the false discovery rate ‐ a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57, 289–300. [Google Scholar]
- Bhardwaj M, Erben V, Schrotz‐King P and Brenner H (2017a) Cell line secretome and tumor tissue proteome markers for early detection of colorectal cancer: a systematic review . Cancers 9, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj M, Gies A, Werner S, Schrotz‐King P and Brenner H (2017b) Blood‐based protein signatures for early detection of colorectal cancer: a systematic review. Clin Transl Gastroenterol 8, e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA and Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424. [DOI] [PubMed] [Google Scholar]
- Brenner H, Altenhofen L and Tao S (2013) Matching of controls may lead to biased estimates of specificity in the evaluation of cancer screening tests. J Clin Epidemiol 66, 202–208. [DOI] [PubMed] [Google Scholar]
- Brenner H and Chen C (2018) The colorectal cancer epidemic: challenges and opportunities for primary, secondary and tertiary prevention. Br J Cancer 119, 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner H, Hoffmeister M, Arndt V, Stegmaier C, Altenhofen L and Haug U (2010a) Protection from right‐ and left‐sided colorectal neoplasms after colonoscopy: population‐based study. J Natl Cancer Inst 102, 89–95. [DOI] [PubMed] [Google Scholar]
- Brenner H, Stock C and Hoffmeister M (2014) Effect of screening sigmoidoscopy and screening colonoscopy on colorectal cancer incidence and mortality: systematic review and meta‐analysis of randomised controlled trials and observational studies. BMJ 348, g2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner H, Tao S and Haug U (2010b) Low‐dose aspirin use and performance of immunochemical fecal occult blood tests. JAMA 304, 2513–2520. [DOI] [PubMed] [Google Scholar]
- Bretthauer M, Kaminski MF, Løberg M, Zauber AG, Regula J, Kuipers EJ, Påhlman L, Hernán MA, McFadden E, Sunde A et al (2016) Population‐based colonoscopy screening for colorectal cancer: a European randomized trial. JAMA Intern Med 176, 894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Knebel P and Brenner H (2016) Empirical evaluation demonstrated importance of validating biomarkers for early detection of cancer in screening settings to limit the number of false‐positive findings. J Clin Epidemiol 75, 108–114. [DOI] [PubMed] [Google Scholar]
- Chen H, Qian J, Werner S, Cuk K, Knebel P and Brenner H (2017) Development and validation of a panel of five proteins as blood biomarkers for early detection of colorectal cancer. Clin Epidemiol 9, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Zucknick M, Werner S, Knebel P and Brenner H (2015) Head‐to‐head comparison and evaluation of 92 plasma protein biomarkers for early detection of colorectal cancer in a true screening setting. Clin Cancer Res 21, 3318–3326. [DOI] [PubMed] [Google Scholar]
- Church TR, Wandell M, Lofton‐Day C, Mongin SJ, Burger M, Payne SR, Castaños‐Vélez E, Blumenstein BA, Rösch T, Osborn N et al (2014) Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut 63, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarloni L, Ehrensberger SH, Imaizumi N, Monnier‐Benoit S, Nichita C, Myung SJ, Kim JS, Song SY, Kim TI, van der Weg B et al (2016) Development and clinical validation of a blood test based on 29‐gene expression for early detection of colorectal cancer. Clin Cancer Res 22, 4604–4611. [DOI] [PubMed] [Google Scholar]
- Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, Douville C, Javed AA, Wong F, Mattox A et al (2018) Detection and localization of surgically resectable cancers with a multi‐analyte blood test. Science 359, 926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B and Tibshirani R (1997) Improvements on cross‐validation: the 632+ bootstrap method. J Am Stat Assoc 92, 548–560. [Google Scholar]
- Enroth S, Hallmans G, Grankvist K and Gyllensten U (2016) Effects of long‐term storage time and original sampling month on biobank plasma protein concentrations. EBioMedicine 12, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gies A, Cuk K, Schrotz‐King P and Brenner H (2018) Direct comparison of diagnostic performance of 9 quantitative fecal immunochemical tests for colorectal cancer screening. Gastroenterology 154, 93–104. [DOI] [PubMed] [Google Scholar]
- Hundt S, Haug U and Brenner H (2009) Comparative evaluation of immunochemical fecal occult blood tests for colorectal adenoma detection. Ann Intern Med 150, 162–169. [DOI] [PubMed] [Google Scholar]
- Klabunde C, Blom J, Bulliard JL, Garcia M, Hagoel L, Mai V, Patnick J, Rozjabek H, Senore C and Tornberg S (2015) Participation rates for organized colorectal cancer screening programmes: an international comparison. J Med Screen 22, 119–126. [DOI] [PubMed] [Google Scholar]
- Kohn MA, Carpenter CR and Newman TB (2013) Understanding the direction of bias in studies of diagnostic test accuracy. Acad Emerg Med 20, 1194–1206. [DOI] [PubMed] [Google Scholar]
- Lech G, Slotwinski R and Krasnodebski IW (2013) The role of tumor markers and biomarkers in colorectal cancer. Neoplasma 61, 1–8. [PubMed] [Google Scholar]
- Lee DH, Kim JW, Jeon SY, Park BK and Han BG (2010) proteomic analysis of the effect of storage temperature on human serum. Ann Clin Lab Sci 40, 61–70. [PubMed] [Google Scholar]
- Marshall KW, Mohr S, Khettabi FE, Nossova N, Chao S, Bao W, Ma J, Li XJ and Liew CC (2010) A blood‐based biomarker panel for stratifying current risk for colorectal cancer. Int J Cancer 126, 1177–1186. [DOI] [PubMed] [Google Scholar]
- Navarro M, Nicolas A, Ferrandez A and Lanas A (2017) Colorectal cancer population screening programs worldwide in 2016: An update. World J Gastroenterol 23, 3632–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polat E, Duman U, Duman M, Atici AE, Reyhan E, Dalgic T, Bostanci EB and Yol S (2014) Diagnostic value of preoperative serum carcinoembryonic antigen and carbohydrate antigen 19–9 in colorectal cancer. Curr Oncol (Toronto, Ont.) 21, e1–e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter NT, Hurban P, White MN, Whitlock KD, Lofton‐Day CE, Tetzner R, Koenig T, Quigley NB and Weiss G (2014) Validation of a real‐time PCR‐based qualitative assay for the detection of methylated SEPT9 DNA in human plasma. Clin Chem 60, 1183–1191. [DOI] [PubMed] [Google Scholar]
- Pox CP, Altenhofen L, Brenner H, Theilmeier A, Von Stillfried D and Schmiegel W (2012) Efficacy of a nationwide screening colonoscopy program for colorectal cancer. Gastroenterology 142, 1460–1467.e1462. [DOI] [PubMed] [Google Scholar]
- R Core Team (2016) A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Schreuders EH, Ruco A, Rabeneck L, Schoen RE, Sung JJ, Young GP and Kuipers EJ (2015) Colorectal cancer screening: a global overview of existing programmes. Gut 64, 1637–1649. [DOI] [PubMed] [Google Scholar]
- Segnan N, Senore C, Andreoni B, Azzoni A, Bisanti L, Cardelli A, Castiglione G, Crosta C, Ederle A, Fantin A et al (2007) Comparing attendance and detection rate of colonoscopy with sigmoidoscopy and FIT for colorectal cancer screening. Gastroenterology 132, 2304–2312. [DOI] [PubMed] [Google Scholar]
- Shaukat A, Mongin SJ, Geisser MS, Lederle FA, Bond JH, Mandel JS and Church TR (2013) Long‐term mortality after screening for colorectal cancer. N Engl J Med 369, 1106–1114. [DOI] [PubMed] [Google Scholar]
- Shen Q, Bjorkesten J, Galli J, Ekman D, Broberg J, Nordberg N, Tillander A, Kamali‐Moghaddam M, Tybring G and Landegren U (2018) Strong impact on plasma protein profiles by precentrifugation delay but not by repeated freeze‐thaw cycles, as analyzed using multiplex proximity extension assays. Clin Chem Lab Med 56, 582–594. [DOI] [PubMed] [Google Scholar]
- Smith JG and Gerszten RE (2017) emerging affinity‐based proteomic technologies for large‐scale plasma profiling in cardiovascular disease. Circulation 135, 1651–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surinova S, Choi M, Tao S, Schueffler PJ, Chang C‐Y, Clough T, Vyslouzil K, Khoylou M, Srovnal J, Liu Y et al (2015) Prediction of colorectal cancer diagnosis based on circulating plasma proteins. EMBO Mol Med 7, 1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao S, Hundt S, Haug U and Brenner H (2011) Sensitivity estimates of blood‐based tests for colorectal cancer detection: impact of overrepresentation of advanced stage disease. Am J Gastroenterol 106, 242–253. [DOI] [PubMed] [Google Scholar]
- Tikk K, Czock D, Haefeli WE, Kopp‐Schneider A and Brenner H (2018) Clinical trial protocol of the ASTER trial: a double‐blind, randomized, placebo‐controlled phase III trial evaluating the use of acetylsalicylic acid (ASA) for enhanced early detection of colorectal neoplasms. BMC Cancer 18, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting P, Rutjes AW, Reitsma JB, Glas AS, Bossuyt PM and Kleijnen J (2004) Sources of variation and bias in studies of diagnostic accuracy: a systematic review. Ann Intern Med 140, 189–202. [DOI] [PubMed] [Google Scholar]
- Yip KT, Das PK, Suria D, Lim CR, Ng GH and Liew CC (2010) A case‐controlled validation study of a blood‐based seven‐gene biomarker panel for colorectal cancer in Malaysia. J Exp Clin Cancer Res 29, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zauber AG, Winawer SJ, O’Brien MJ, Lansdorp‐Vogelaar I, van Ballegooijen M, Hankey BF, Shi W, Bond JH, Schapiro M, Panish JF et al (2012) colonoscopic polypectomy and long‐term prevention of colorectal‐cancer deaths. N Engl J Med 366, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. STARD showing selection of study participants enrolled in the iDa Study.
Fig. S2. STARD showing selection of study participants enrolled in the ASTER Study during 2013–2016.
Fig. S3. Experimental workflow of the study.
Fig. S4. Interaction of identified markers from all predictor models in canonical pathways at subcellular level.
Fig. S5. Involvement of identified markers from all predictor models in different organ toxicities.
Table S1. List of 276 proteins measured in the three Olink Multiplex Panels.
Table S2. Diagnostic performance of all 275 proteins markers in discovery and validation set for all‐stage CRC detection.
Table S3. The algorithms identified from the discovery set for (all/early/late) stage CRC detection.
Table S4. Diagnostic performance of the individual markers for detection of CRC in discovery and validation sets stratified by cancer location.
Table S5. Diagnostic performance of the 9‐marker signature for detection of CRC in discovery and validation sets stratified by cancer location.