

Abstract
Background
The swimming crab, Portunus trituberculatus, is an important commercial species in China and is widely distributed in the coastal waters of Asia-Pacific countries. Despite increasing interest in swimming crab research, a high-quality chromosome-level genome is still lacking.
Findings
Here, we assembled the first chromosome-level reference genome of P. trituberculatus by combining the short reads, Nanopore long reads, and Hi-C data. The genome assembly size was 1.00 Gb with a contig N50 length of 4.12 Mb. In addition, BUSCO assessment indicated that 94.7% of core eukaryotic genes were present in the genome assembly. Approximately 54.52% of the genome was identified as repetitive sequences, with a total of 16,796 annotated protein-coding genes. In addition, we anchored contigs into chromosomes and identified 50 chromosomes with an N50 length of 21.80 Mb by Hi-C technology.
Conclusions
We anticipate that this chromosome-level assembly of the P. trituberculatus genome will not only promote study of basic development and evolution but also provide important resources for swimming crab reproduction.
Keywords: Portunus trituberculatus, genome assembly, crab, chromosome, evolution
Introduction
The swimming crab, Portunus trituberculatus (NCBI:txid210409, marinespecies.org:taxname:1061762), belonging to Brachyura, Portunidae, Portunus, is named for its shuttle-shaped head breastplate and 3 verrucous bumps on the back of the stomach and heart regions [1, 2]. The chelipeds of swimming crabs are well developed for feeding and attacking, with the first 3 pairs and last pair used for crawling and swimming, respectively [3, 4]. Male and female crabs are distinguished by the shape of their abdomen, with the male having a triangular abdomen and the female having an almost circular one [5]. Owing to their lack of drilling ability, swimming crabs often live in soft mud or sand [6] or in seagrass near the shore, and also show a certain level of phototaxis, spending time on the sea floor during the day and foraging at night [5]. Swimming crabs are also omnivorous, feeding on shellfish, small fish, shrimp, algae, and decomposing animal and plant carcasses [7].
The swimming crab is widely distributed in the coastal waters of Korea, Japan, China, and Southeast Asia and is one of the most valuable marine crustaceans in Asia [8]. It is widely found in Chinese coastal waters of the Bohai Sea, Yellow Sea, East China Sea, and South China Sea and is an important commercially cultured species [9]. Swimming crabs are considered highly nutritious, especially in regard to crab cream, and are very popular in China [10, 11]. As a result, the crab has been heavily overfished, resulting in substantial declines in its natural population [12] and initiation of artificial breeding [13, 14]. With continued research on the crab, its morphological and physiological characteristics have become clear, but the genetic changes are poorly understood. At present, several genomic studies of swimming crab have been carried out [15–18], but the high-quality chromosome-level genome is still lacking.
In the present study, we constructed a chromosome-level genome assembly of P. trituberculatus by combining short reads, Nanopore long reads, and Hi-C sequencing data. This chromosome-level genome will not only promote study on development and evolution but also provide important resources for reproductive studies of P. trituberculatus and other crab species.
Methods
Sampling, library construction, and sequencing
A male swimming crab was collected in Bohai Bay, Hebei Province, China, for sequencing (Fig. 1). To obtain sufficient high-quality DNA for the Oxford Nanopore (Oxford, UK) and BGISEQ-500 platforms (BGI, Qingdao, China), the swimming crab was rinsed 5 times with clean water and dissected immediately. Fresh muscle tissue was collected and snap-frozen in liquid nitrogen. The samples were then used to extract DNA with a Qiagen Blood & Cell Culture DNA Mini Kit and prepared for Nanopore, BGISEQ-500, and Hi-C sequencing. Using the same individual, muscle RNA was also extracted using TRIzol (Invitrogen) according to the manufacturer's instructions. To obtain an overview of the transcriptome, polyadenylated RNA was chosen by oligo (dT) purification and reverse-transcribed to complementary DNA and sequenced using the BGISEQ-500 platform.
Figure 1:

Swimming crab, Portunus trituberculatus. The adult male swimming crab collected from Bohai Bay, Hebei Province.
Extracted DNA was sequenced using both the BGISEQ and Oxford Nanopore platforms. The short reads generated from the BGISEQ platform were used for estimation of genome size and error correction of the assembled genome, and the Nanopore long reads were used for genome assembly. To this end, 1 library with insertion lengths of ∼300 bp was sequenced on the BGISEQ-500 platform, and another library with an average length of 20 kb was constructed using the Oxford Nanopore platform according to the manufacturers’ protocols.
Data filtering
Three different sources of reads were used to achieve the high-quality genome assembly, i.e., Nanopore long reads, short reads, and Hi-C reads. Thus, we used different methods for filtering. For the Nanopore long reads, any reads <1 kb or with a mean quality value of <7 were removed. For the short reads, any read with >10% unknown bases (usually designated “N”) or with >50% low-quality bases was removed, and its paired-end read was also removed. All adaptor sequences and duplicated reads produced by PCR were removed. The low-quality Hi-C reads were filtered using HiC-Pro v2.10.0 [19] with default parameters.
Genome characteristic estimation
All filtered BGISEQ short reads were used for estimation of genome size and other characteristics. In addition, 17-mer was chosen for k-mer analysis and the 17-mer depth frequency distribution was calculated using the k-mer method. Genome size was estimated as follows: genome size = TKN17-mer/PKFD17-mer, where TKN17-mer is the total k-mer number and PKFD17-mer is the peak k-mer frequency depth of 17-mer. The estimated genome size was used to determine subsequent genome assembly results.
Genome assembly
To improve the quality of the genome and reduce the error ratio, self-error correction of all Nanopore long reads was performed using NextDenovo software [20]. The error-corrected Nanopore long reads were then used to assemble the raw genome via contig construction with WTDBG software (WTDBG, RRID:SCR_017225) [21] and the following parameters: -p 0 -k 15 -AS 2 -E 1 -s 0.05 -L 5000. The assembled genomic sequences were further polished by Racon v1.2.1 [22] with 4 iterations using all the error-corrected Nanopore long reads with default parameters. After this, all filtered BGISEQ short reads were polished by Pilon v1.21 (Pilon, RRID:SCR_014731) [23] at the single-base level with default parameters. After completion of the error correction steps, the Hi-C data were used to obtain a chromosome-level genome assembly. All Hi-C sequencing data were first filtered by HiC-Pro v2.10.0 [19] with default parameters and then mapped to the polished swimming crab genome to improve the connection integrity of the contigs. Finally, 3D de novo assembly software (v180419) [24] with default parameters was used to determine contig location and direction.
Genome assembly evaluation
Three different strategies were used to evaluate the completeness and accuracy of the assembled genome. First, the quality of the assembled genome and gene completeness were assessed using BUSCO (BUSCO, RRID:SCR_015008) [25] with the core gene sets of the eukaryote and metazoan databases, respectively. Second, all filtered short reads generated by BGISEQ were mapped to the assembled genome using BWA-MEM v0.7.12 [26] to detect genome integrity with default parameters. Third, transcripts were mapped to the assembled genome using BLAT software (BLAT, RRID:SCR_011919) [27] with e-value <10−5.
Repetitive element annotation
Tandem repeats and transposable elements (TEs) were also annotated in the chromosome-level genome. Tandem repeats were annotated using Tandem Repeat Finder v4.04 [28] with default parameters. The TEs were annotated at the protein level using RepeatProteinMask (RM-BLASTX) to search the protein database and at the DNA level using RepeatMasker (open-4.0.7) (RepeatMasker, RRID:SCR_012954) [29] to search the de novo libraries and repbase. The de novo repeat libraries were constructed using RepeatModeler (RepeatModeler, RRID:SCR_015027) [30], with consensus sequences used for de novo library construction, and all software using the default parameters.
Gene structure prediction and function annotation
After repetitive element annotation, the repeat-masked genome was used for gene set annotation with 3 different methods, i.e., de novo prediction, RNA-seq–based annotation, and homology-based annotation. We first assembled the RNA-seq reads into transcripts using Bridger r2014–12-01 (Bridger, RRID:SCR_017039) [31]. The assembled genome and transcripts were then used for Augustus training to obtain an accurate Augustus annotation species model. Augustus v2.5.5 (Augustus, RRID:SCR_008417) [32] was used for de novo prediction of coding genes with the previous training results. Second, proteins of Bicyclus anynana (GCF_900239965.1) [33], Bombus terrestris (GCF_000214255.1) [34], Drosophila melanogaster (GCA_000001215.4) [35], Mus musculus (GCF_000001635.26) [36], Stegodyphus mimosarum (GCA_000611955.2), Penaeus vannamei (GCA_003789085.1), Mesobuthus martensii [37], Eriocheir japonica sinensis (i.e., Eriocheir sinensis) (GigaDB: 100186) [38–43], and Tachypleus tridentatus (GCA_004102145.1) [44] were downloaded from the NCBI, GigaDB, or their own databases. The longest transcript of each gene was selected for further annotation and phylogenetic analysis. All filtered genes were searched with an e-value cutoff of 1e−5, with the blast results then formatted and prepared for Genewise [45] prediction of the gene structure of the swimming crab genome. Third, for the RNA-seq–based method, all assembled transcripts were aligned against the genome using BLAT [27] (identity > 90% and coverage > 90%), with PASA used to filter overlaps to link the spliced alignments. Finally, EvidenceModeler (EVM; EVidenceModeler, RRID:SCR_014659) v1.1.1 was used to integrate the above data into an EVM-derived gene set [46].
Five different public protein databases were used for gene functional annotation of the swimming crab, with InterProScan v4.8 (InterProScan, RRID:SCR_005829) [47] used to screen proteins against the 5 databases (Pfam, release 27.0; PRINTS, release 42.0; PROSITE, release 20.97; ProDom, 2006.1; and SMART, release 6.2) to determine the number of InterPro- and GO-predicted protein-coding genes. In addition, the KEGG, UniProt/SwissProt, and UniProt/TrEMBL databases were also used for functional annotation with BLAST v2.3.0 [48]. Blastp (BLASTP, RRID:SCR_001010) was used in this step, and the e-value was set as 10−5 and other parameters were set as defaults.
Identification of orthologous genes
The annotated genes in the swimming crab and 6 other species, including Aedes aegypti (GCF_002204515.2), B. anynana, D. melanogaster, S. mimosarum, P. vannamei, and E. j. sinensis, were used for orthologous gene identification with OrthoMCL v2.0.9 [49] with default parameters. The identified genes were then used to run reciprocal alignment and pairwise relationship analysis. The reciprocal best similarity pairs in different species were considered as putative orthologous genes, and reciprocal better similarity pairs in 1 species were considered as paralogous genes. The 1:1:1:1:1:1:1 single-copy genes in the 7 species were also identified for further phylogenetic and divergence time estimation analysis.
Phylogenetic analysis and divergence time estimation
Using the single-copy genes of the 7 species (P. trituberculatus, A. aegypti, B. anynana, D. melanogaster, S. mimosarum, P. vannamei, and E. j. sinensis), we connected the genes in each species into 1 super-gene for phylogenetic tree building. Maximum likelihood–based phylogenetic analysis was conducted using RAxML v8.2.10 (RAxML, RRID:SCR_006086) [50] with default parameters. The MCMCTREE program in the PAML package v4.8 [51] was then used to calculate divergence time, with all fossil records downloaded from the TIMETREE website [52] for calibration.
Relative evolution rate
The relative evolution rate of species was analyzed with LINTRE software (version 1) [53] using the "tpcv" model and S. mimosarum as an outgroup. Using the default parameters of LINTRE, we then evaluated the relative evolution rate between the swimming crab and other related species.
Gene family expansion and contraction
Using the divergence time results calculated by MCMCTREE and the gene pairwise relationships calculated by OrthoMCL [49], we determined gene family expansion and contraction for each node using CAFÉ v3.1 (CAFÉ, RRID:SCR_005983) [54]. The expansion and contraction genes of the swimming crab were extracted for GO/KEGG enrichment analysis [55, 56].
Results
Chromosome-level genome assembly
To obtain a high-quality chromosome-level swimming crab genome, we extracted high-quality DNA from the muscle tissue and constructed libraries for genome sequencing. To estimate the genome characteristics of the swimming crab, we generated 205.40 Gb of BGISEQ data (Table S1), with 17-mer analysis indicating a genome size of ∼918.52 Mb and a heterozygosity rate of ∼0.9% (Fig. S1). In total, we generated 54.97 Gb (54.75-fold coverage) of Nanopore long-read data with N50 >20 kb (Table S2). The Nanopore long reads were assembled into contigs using WTDBG software [21] (genome size: 1.00 Gb; N50: 4.12 Mb) (Table 1). To further improve genome accuracy, we aligned all corrected Nanopore long reads to the assembled genome and conducted error correction using Racon [22] with 4 iterations. The genome was subsequently corrected using all filtered BGISEQ clean reads via Pilon [23] with 2 iterations. We then constructed the chromosome-level genome with 95.95 Gb of Hi-C sequencing data (Table S3) by 3D de novo assembly [24]. Finally, we obtained 50 chromosomes and a mounting rate (total length of the contigs that anchored to chromosomes divided by the total length of all assembled contigs) of 97.80% (Fig. 2; Table S4), which is the first chromosome-level crab genome with N50 of 21.79 Mb (Table 1). The high mounting rate suggested successful assembly of the swimming crab genome at the chromosome level. We also compared our assembled genome to the published swimming crab genome; the assembly quality of our genome is better than the previous one (Table S5). Because the previous study has the genomic markers, we also mapped all the markers to our genome, and we found that 99.40% (10,897 of 10,963) markers can be mapped to our genome. Among these mapped genome markers, 98.83% (10,769 of 10,897) are exactly mapped to our assembled 50 chromosomes (Table S6). All these results show that we obtained a high-quality and quite complete chromosome-level genome.
Table 1:
Assembly of swimming crab genome
| Term | Contig phase | Hi-C phase | ||
|---|---|---|---|---|
| Size (bp) | Number | Size (bp) | Number | |
| N90 | 439,683 | 334 | 11,273,125 | 41 |
| N80 | 1,225,551 | 203 | 14,151,211 | 33 |
| N70 | 2,035,154 | 141 | 16,942,622 | 27 |
| N60 | 2,950,146 | 100 | 19,786,189 | 21 |
| N50 | 4,121,416 | 71 | 21,793,880 | 17 |
| Maximum length | 17,984,318 | - | 42,710,960 | - |
| Total length | 1,004,084,521 | - | 1 005,046,021 | - |
| No. ≥100 bp | - | 2,446 | - | 523 |
| No. ≥10 kb | - | 1,756 | - | 314 |
Note: Contig phase represents results assembled by WTDBG software, and Hi-C phase represents scaffold statistics of genome after chromosome assembly.
Figure 2:

Genome characteristics of swimming crab. From outer circle to inner circle: gene distribution, tandem repeats (TRP), long tandem repeats (LTR), long interspersed nuclear elements (LINE) and short interspersed nuclear elements (SINE), the DNA elements, and the GC content of the genome.
Genome quality evaluation
We next assessed the completeness of the swimming crab genome by BUSCO [25] and identified 94.7% Eukaryota and 92.9% Metazoa conserved core genes in the genome (Table 2). We checked the mapping rates of the BGISEQ short reads to our genome and found that 95.85% of reads were properly pair-mapped to the genome (Table S7). We then de novo assembled the transcripts using the RNA-seq data (Table S8) with Bridger software [31] and an N50 length of 2,124 bp (Table S9). After transcript mapping, we found that 97.80% of the transcripts could be mapped to the swimming crab genome (Table S10). We also analyzed the genome quality of previously published high-quality genomes from closely related species and determined that the quality of the assembled chromosome-level swimming crab genome was markedly higher or comparable to that of other species (Table S11). In summary, these results indicated that we acquired a high-quality swimming crab genome. To investigate genome characteristics, such as GC content, we analyzed the GC distribution in the genome with a slide-window method. The peak value of GC content was ∼41%, which agrees with the average GC content in the swimming crab genome. We also found that the GC content in the swimming crab was closer to that of mouse than of shrimp (Fig. S2).
Table 2:
Quality evaluation of assembled swimming crab genome by BUSCO
| Library | Eukaryota | Metazoa |
|---|---|---|
| Complete BUSCO (C) | 287 | 909 |
| Complete and single-copy BUSCO (S) | 283 | 903 |
| Complete and duplicated BUSCO (D) | 4 | 6 |
| Fragmented BUSCO (F) | 2 | 19 |
| Missing BUSCO (M) | 14 | 50 |
| Total BUSCO groups searched | 303 | 978 |
| Percentage of complete BUSCO (%) | 94.7 | 92.9 |
Genome annotation
The repetitive sequences of the swimming crab genome were identified through 4 different methods, resulting in 547.39 Mb of repeated sequences and accounting for 54.52% of the assembled genome (Table S12). Among the repeated sequences, 19.28% (∼193.56 Mb) were tandem repeats and 52.29% (∼525.49 Mb) were TEs (Table S12; Table 3). The TEs could be further divided into 4 main types, including 0.014% (∼142.88 kb) of short interspersed nuclear elements (SINEs), 15.23% (∼153.03 Mb) of long interspersed nuclear elements (LINEs), 14.90% (∼149.71 Mb) of DNA elements, and 4.50% (∼45.19 Mb) of long terminal repeats (LTRs) (Table 3).
Table 3:
Statistics on transposable elements in swimming crab genome
| Type | Repbase TEs | TE proteins | De novo | Combined TEs | ||||
|---|---|---|---|---|---|---|---|---|
| Length (bp) | % in genome | Length (bp) | % in genome | Length (bp) | % in genome | Length (bp) | % in genome | |
| DNA | 131,799,733 | 13.11 | 2,434,533 | 0.24 | 19,288,080 | 1.92 | 149,711,951 | 14.90 |
| LINE | 16,171,649 | 1.61 | 75,759,827 | 7.54 | 131,530,457 | 13.09 | 153,027,744 | 15.23 |
| SINE | 142,878 | 0.01 | 0 | 0 | 0 | 0 | 142,878 | 0.014 |
| LTR | 26,546,055 | 2.64 | 10,195,324 | 1.01 | 18,421,957 | 1.83 | 45,189,365 | 4.50 |
| Other | 89,969,319 | 8.95 | 0 | 0 | 211,157,523 | 21.01 | 230,116,216 | 22.90 |
| Unknown | 34,752 | 0.0035 | 0 | 0 | 90,989,908 | 9.05 | 91,007,921 | 9.06 |
| Total | 213,558,503 | 21.25 | 88,375,336 | 8.79 | 464,908,824 | 46.26 | 525,492,271 | 52.29 |
After masking the repeated sequences, we annotated the protein-coding genes using de novo prediction, homology-based prediction, and transcript-based prediction. We merged the results and obtained 16,791 protein-coding genes. We checked the quality of the annotated genes by comparing with several closely related species. Results showed that the messenger RNA, CDS, exon, and intron length distributions of the swimming crab were similar to those of the closely related species, suggesting that the swimming crab annotation results were dependable (Fig. 3).
Figure 3:

Annotation quality comparison of protein-coding genes. We compared the messenger RNA (mRNA) length, CDS length, exon length, and intron length among 5 species: P. trituberculatus, A. aegypti, S. mimosarum, D. melanogaster, and P. vannamei.
We also performed functional annotation of the 16,791 genes with InterPro, GO, KEGG, SwissProt, and TrEMBL. The highest annotation rate (74.77%) was found for SwissProt, in which 12,558 genes were annotated. In total, 16,053 genes (∼95.58%) were annotated, indicating that most genes could be found in the public protein databases (Table 4). Thus, taken together, we acquired a high-quality protein-coding gene set for the swimming crab.
Table 4:
Functional annotation of predicted protein-coding genes
| Term | Gene number | Percentage (%) |
|---|---|---|
| GO | 8,712 | 51.87 |
| InterPro | 11,691 | 69.61 |
| KEGG | 10,880 | 64.78 |
| SwissProt | 12,558 | 74.77 |
| TrEMBL | 12,256 | 72.97 |
| Annotated | 16,053 | 95.58 |
| Unannotated | 743 | 4.42 |
| Total | 16,796 | 100 |
Orthologous identification and gene family analysis
For comparative genomics analysis of the swimming crab, we analyzed the orthologous gene relationships among several species, including A. aegypti, B. anynana, D. melanogaster, S. mimosarum, P. vannamei, and E. j. sinensis using OrthoMCL. In total, 15,503 gene families were clustered in the 7 species and 1,018 one-to-one single-copy genes were identified (Fig. 4A). Because the swimming crab has several unique characteristics, we performed gene family analysis and found 8,832 gene families shared among the 7 species, with 328 gene families unique to the swimming crab (Fig. 4B). We then performed functional analysis and identified 34 enriched KEGG terms (Table S13), suggesting that these unique gene families play important roles in the swimming crab.
Figure 4:
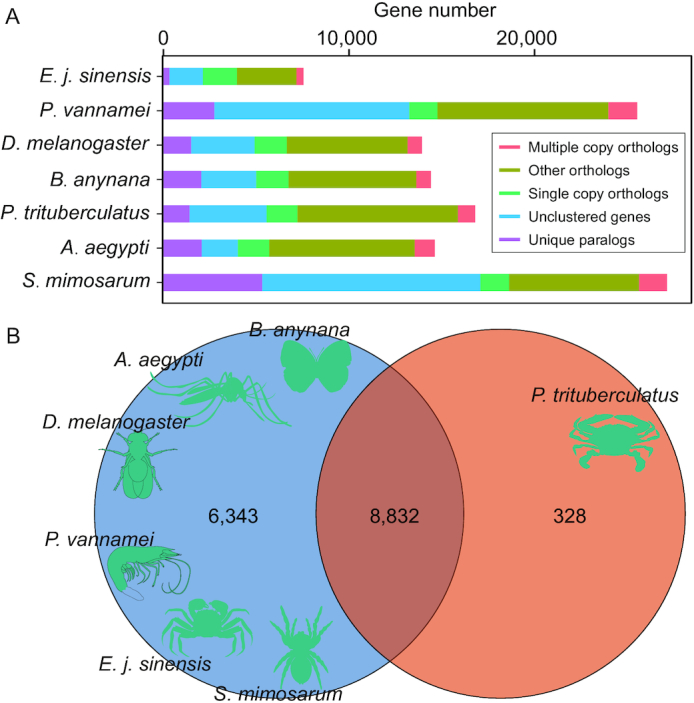
Gene family analysis of swimming crab. A. Orthologous genes among species. The multiple-copy orthologs are orthologs that have multiple copies in 1 species, the single-copy orthologs are orthologs that have only 1 copy in 1 species, the other orthologs are the rest of the orthologs, the unclustered genes are genes that have no homology with others, and the unique paralogs are genes that only exist in 1 specific species. B. Unique and common gene families among these species, including B. anynana, A. aegypti, D. melanogaster, P. vannamei, E. j. sinensis, S. mimosarum, and P. trituberculatus.
Phylogenetic relationships and divergence time
Although the phylogenetic relationships of the swimming crab and closely related species have been analyzed in previous studies, most used few nuclear and mitochondrial genes. To determine the evolutionary relationship of the swimming crab, we analyzed all single-copy genes using RAxML software [50], with the spider used as the outgroup species. Results showed that the swimming crab has a close relationship with the Chinese mitten crab and shrimp (Fig. 5A). The 7 species of pancrustaceans—P. trituberculatus, A. aegypti, B. anynana, D. melanogaster, S. mimosarum, P. vannamei, and E. j. sinensis—formed 2 clades: i.e., Hexapoda and Crustacea. The Hexapoda group consisted of all lepidopteran and dipterous insects, whereas the second clade comprised all other crustaceans, with P. trituberculatus and E. j. sinensis forming a Pleocyemata clade, followed by Dendrobranchiata shrimp (P. vannamei). In addition, Hexapoda and Crustacea were both found to be monophyletic (Fig. 5A). To determine divergence time, we employed MCMCTREE analysis in the PAML package [51] and found that the Chinese mitten crab and swimming crab diverged ∼183.5 million years ago, and diverged from shrimp ∼428.5 million years ago (Fig. 5A).
Figure 5:

Phylogenetic relationships, divergence time, and evolution rate analysis. A. Phylogenetic relationship and divergence time of species. Red dot represents fossil record used here, and numbers in parentheses indicate 95% confidence interval. B. Relative evolution rate of species.
Relative evolution rate
Species in different environments can experience different survival pressures. As such, we conducted relative evolution rate analysis in LINTRE (version 1) [53], with spider as the outgroup species and swimming crab as the reference species. Results showed that the shrimp had the slowest evolution rate among the 7 species, whereas the fruit fly and butterfly exhibited relatively fast evolution rates (Fig. 5B; Table S14). Interestingly, the slowest evolution rates were found among the Malacostraca (Fig. 5B; Table S14), suggesting that the specific environments or habitats caused their different evolution rates.
Gene family expansion and contraction
We performed gene family expansion and contraction analysis of the 7 species using CAFÉ v4.0 and identified 148 and 25 expanded and contracted gene families (P < 0.05) in the swimming crab, respectively. We then performed KEGG functional enrichment analysis of the expanded gene families and found that the HIF-1 signaling pathway (Q-value = 0.000109025), focal adhesion (Q-value = 0.000135977), Hippo signaling pathway (Q-value = 0.000184649), and insulin signaling pathway (Q-value = 0.000357592) were enriched (Table S15). These biological processes are related to early development, hypoxia adaptation, and other key processes, which may help us better understand the evolution of the swimming crab.
Conclusions
Based on BGISEQ, Nanopore, and Hi-C sequencing data, we assembled a chromosome-level high-quality genome of the swimming crab. Evaluation results indicated that the genome quality of swimming crab was comparable to that of most high-quality model species. We also successfully obtained 16,791 high-quality protein-coding genes by integrating 3 different methods. The genome and annotation data will help researchers better understand the evolution of crabs and improve their economic value. The phylogenetic results indicated that the swimming crab is closely related to the Chinese mitten crab, from which it diverged ∼183.5 million years ago. The unique and/or expanded gene family analysis provides clues to swimming crab development and environmental adaptation.
Availability of Supporting Data and Materials
The raw sequencing data were deposited in the NCBI database under accession number PRJNA555262. The genome assembly and annotation results are available via the GigaScience repository GigaDB [57].
Additional Files
Table S1: Statistics on genome sequencing data from BGISEQ platform.
Table S2: Statistics on sequencing reads from Oxford Nanopore platform.
Table S3: Statistics on Hi-C sequencing data.
Table S4: Statistics on assembled chromosome-level genome by 3D de novo assembly software.
Table S5: The quality comparison of these 2 genomes.
Table S6: The mapping results of genomic markers to the assembled genome.
Table S7: Statistics on mapping ratio of the BGISEQ short reads to swimming crab genome.
Table S8: Statistics on RNA-seq data.
Table S9: Statistics on assembled transcripts by Bridger software.
Table S10: Statistics on transcript mapping ratio of swimming crab genome.
Table S11: Genome quality comparison of swimming crab with other species.
Table S12: Statistics on annotated repetitive sequences using different software.
Table S13: KEGG enrichment analysis of unique gene families in swimming crab relative to 6 other species.
Table S14: Two-cluster analysis of swimming crab and other species.
Table S15: KEGG enrichment analysis of expanded gene families in swimming crab.
Figure S1: 17-mer analysis of swimming crab genome.
Figure S2: GC distribution in species.
Hugues Roest Crollius -- 10/4/2019 Reviewed
Joseph F. Ryan -- 10/4/2019 Reviewed
Abbreviations
BLAST: Basic Local Alignment Search Tool; BLAT: BLAST-Like Alignment Tool; bp: base pairs; BUSCO: Benchmarking Universal Single-Copy Orthologs; BWA: Burrows-Wheeler Aligner; CDS: coding DNA sequence; Gb: gigabase pairs; GC: guanine-cytosine; GO: Gene Ontology; Hi-C: High-throughput chromosome conformation capture; kb: kilobase pairs; KEGG: Kyoto Encyclopedia of Genes and Genomes; Mb: megabase pairs; LINE: long interspersed nuclear element; LTR: long terminal repeat; NCBI: National Center for Biotechnology Information; PASA: Program to Assemble Spliced Alignments; RAxML: Randomized Axelerated Maximum Likelihood; RNA-seq: RNA sequencing; SINE: short interspersed nuclear element; TE: transposable element.
Competing Interests
The authors declare that they have no competing interests.
Funding
This study was supported by the National Natural Science Foundation of China (31672267, 31640074), Jiangsu Agriculture Science and Technology Innovation Fund (CX(18)3027, CX(18)2027), Natural Science Foundation of Jiangsu Province (BK20171276, BK20160444), “Qing Lan Project” of Daizhen Zhang and China Postdoctoral Science Foundation (2018M642105).
Authors' Contributions
Y.R., Q.L., Y. Li, and X.L. conceived the project. B.T., D.Z., S.S., H.Z., Y. Liu, and S.J. collected and dissected the samples. H.L., Zhongkai Wang, K.W., Y.S., Q.Q., C.L., and Y. Li estimated genome size. F.X., Y.C., W.J., and H.J. assembled the genome. B.G., Zhengfei Wang, Z.S., and B.T. performed genome assembly, genome annotation, and evolution analysis. Y.L., B.T., Q.Q., and W.W. wrote the manuscript. Y.R. and W.W. revised the manuscript.
References
- 1. Spiridonov VA, Neretina TV, Schepetov D. Morphological characterization and molecular phylogeny of Portunoidea Rafinesque, 1815 (Crustacea Brachyura): Implications for understanding evolution of swimming capacity and revision of the family-level classification. Zool Anz. 2014;253(5):404–29. [Google Scholar]
- 2. Dai A, Yang S, Song Y. Marine Crabs in China Sea. Beijing: Marine Publishing Co; 1986. [Google Scholar]
- 3. Okamoto K. Malformed regeneration of partly cut swimming leg as a marker for swimming crab Portunus trituberculatus. Fish Sci. 2010;72(5):1121–3. [Google Scholar]
- 4. Hazlett BA. Interspecific fighting in three species of brachyuran crabs from Hawaii. Crustaceana. 1971;20(3):308–14. [Google Scholar]
- 5. Xue J, Du N, Wei L, et al.. A review of studies on Portunus trituberculatus in China. Donghai Marine Science, 1997;4:60–65. [Google Scholar]
- 6. Sakai T. Crabs of Japan and the Adjacent Seas. Tokyo: Kodansha; 1976. [Google Scholar]
- 7. Wu RSS, Shin PKS. Food segregation in three species of portunid crabs. Hydrobiologia. 1997;362(1–3):107–13. [Google Scholar]
- 8. Marine Species Identification Portal: Portunus trituberculatus. http://species-identification.org/species.php?species_group=crabs_of_japan&menuentry=soorten&id=1106&tab=beschrijving. Accessed December 27, 2019. [Google Scholar]
- 9. Qi JB, Gu XL, Ma LB, et al.. The research progress on food organism culture and technology utilization in crab seed production in ponds in China. Agric Sci. 2013;4(10):563–9. [Google Scholar]
- 10. Su XR, Li TW, Ding MJ, et al.. Evalution on nutritive value of Portunus trituberculatus. Chin J Oceanol Limnol. 1997;15(2):168–72. [Google Scholar]
- 11. Wang Z, Sun L, Guan W, et al.. De novo transcriptome sequencing and analysis of male and female swimming crab (Portunus trituberculatus) reproductive systems during mating embrace (stage II). BMC Genetics. 2018;19(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dan S, Oshiro M, Ashidate M, et al.. Starvation of Artemia in larval rearing water affects post-larval survival and morphology of the swimming crab, Portunus trituberculatus (Brachyura, Portunidae). Aquaculture. 2016:S0044848615300442. [Google Scholar]
- 13. Hamasaki K, Obata Y, Dan S, et al.. A review of seed production and stock enhancement for commercially important portunid crabs in Japan. Aquac Int. 2011;19(2):217–35. [Google Scholar]
- 14. Liu S, Sun J, Hurtado LA. Genetic differentiation of Portunus trituberculatus, the world\"s largest crab fishery, among its three main fishing areas. Fish Res. 2013;148(148):38–46. [Google Scholar]
- 15. Lv J, Ping L, Yu W, et al.. Transcriptome analysis of Portunus trituberculatus in response to salinity stress provides insights into the molecular basis of osmoregulation. PLoS One. 2013;8(12):e82155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang Y, Wang J, Han T, et al.. Ovarian transcriptome analysis of Portunus trituberculatus provides insights into genes expressed during phase III and IV development. PLoS One. 2015;10(10):e0138862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meng X, Liu P, Jia F, et al.. Correction: De novo transcriptome analysis of Portunus trituberculatus ovary and testis by RNA-Seq: identification of genes involved in gonadal development. PLoS One. 2015;10(7):e0133659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lv J, Gao B, Liu P, et al.. Linkage mapping aided by de novo genome and transcriptome assembly in Portunus trituberculatus: applications in growth-related QTL and gene identification. Sci Rep. 2017;7(1):7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Servant N, Varoquaux N, Lajoie B, et al.. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16(1):259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. NextDenovo. https://github.com/Nextomics/NextDenovo/ Nextomics; 2019. Accessed December 27, 2019. [Google Scholar]
- 21. Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2019, doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Talay AC, Altilar DT. RACON: a routing protocol for mobile cognitive radio networks. In: CoRoNet '09: Proceedings of the 2009 ACM workshop on Cognitive Radio Networks, Beijing, China. New York: ACM; 2009. [Google Scholar]
- 23. Walker BJ, Abeel T, Shea T, et al.. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dudchenko O, Batra SS, Omer AD, et al.. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356(6333):92–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simao FA, Waterhouse RM, Ioannidis P, et al.. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2. [DOI] [PubMed] [Google Scholar]
- 26. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kent WJ. BLAT - The BLAST-like alignment tool. Genome Res. 2002;12(4):656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bedell JA, Korf I, Gish W. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics. 2000;16(11):1040. [DOI] [PubMed] [Google Scholar]
- 30. RepeatModeler. http://www.repeatmasker.org/RepeatModeler. Accessed December 27, 2019. [Google Scholar]
- 31. Chang Z, Li G, Liu J, et al.. Bridger: a new framework for de novo transcriptome assembly using RNA-seq data. Genome Biol. 2015;16(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(suppl_2):215–25. [DOI] [PubMed] [Google Scholar]
- 33. Nowell RW, Elsworth B, Oostra V, et al.. A high-coverage draft genome of the mycalesine butterfly Bicyclus anynana. Gigascience. 2017;6(7), doi: 10.1093/gigascience/gix035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sadd BM, Barribeau SM, Bloch G, et al.. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 2015;16(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adams MD, Celniker SE, Holt RA, et al.. The genome sequence of Drosophila melanogaster. Science. 2000;287(5461):2185. [DOI] [PubMed] [Google Scholar]
- 36. Waterston RH, Lindblad-Toh K, Birney E, et al.. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–62. [DOI] [PubMed] [Google Scholar]
- 37. Ma J, Shi YB. The Mesobuthus martensii genome reveals the molecular diversity of scorpion toxins. Cell Biosci. 2014;4(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Linsheng S, Chao B, Yongju L, et al.. Draft genome of the Chinese mitten crab,Eriocheir sinensis. Gigascience. 2016;5, doi: 10.1186/s13742-016-0112-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu RY. Introduction to the classification of recent crustacean. In: Transactions of the Chinese Custacean Society No. 4. Beijing: Chinese Crustacean Society, Science Press; 2003:76–86. [Google Scholar]
- 40. Tang B, Zhou K, Song D, et al.. Molecular systematics of the Asian mitten crabs, genus Eriocheir (Crustacea: Brachyura). Mol Phylogenet Evol. 2003;29(2):309–16. [DOI] [PubMed] [Google Scholar]
- 41. Song DX, Yang SL. The Crustacea Fauna of Hebei. Shijiazhuang: Hebei Science and Technology Publishing House; 2009:1–772. [Google Scholar]
- 42. Tang BP, Xin ZZ, Liu Y, et al.. The complete mitochondrial genome of Sesarmops sinensis reveals gene rearrangements and phylogenetic relationships in Brachyura. PLoS One. 2017;12(6):e0179800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xuan F, Wu X, Liu N, et al.. Reproductive potential of individual male Chinese mitten crabs Eriocheir japonica sinensis in a local pond‐reared broodstock: Implications for parent crab selection and sex ratio optimization. Aquac Res. 2018;49:3498–507. [Google Scholar]
- 44. Gong L, Fan G, Ren Y, et al.. Chromosomal level reference genome of Tachypleus tridentatus provides insights into evolution and adaptation of horseshoe crabs. Mol Ecol Resour. 2019;19(3):744–56. [DOI] [PubMed] [Google Scholar]
- 45. Birney E, Durbin R. Using genewise in the Drosophila annotation experiment. Genome Res. 2000;10(4):547–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haas BJ, Salzberg SL, Zhu W, et al.. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9(1):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17(9):847–8. [DOI] [PubMed] [Google Scholar]
- 48. Altschul SF. Basic Local Alignment Search Tool (BLAST). J Mol Biol. 2012;215(3):403–10. [DOI] [PubMed] [Google Scholar]
- 49. Li L. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appli Biosci. 1997;13(5):555–6. [DOI] [PubMed] [Google Scholar]
- 52. TIMETREE. http://www.timetree.org. Accessed December 27, 2019. [Google Scholar]
- 53. Takezaki N, Rzhetsky A, Nei M. Phylogenetic test of the molecular clock and linearized trees. Mol Biol Evol. 1995;12(5):823–33. [DOI] [PubMed] [Google Scholar]
- 54. De Bie T, Cristianini N, Demuth JP, et al.. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22(10):1269–71. [DOI] [PubMed] [Google Scholar]
- 55. Beissbarth T, Speed TP. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20(9):1464–5. [DOI] [PubMed] [Google Scholar]
- 56. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ren Y, Tang B, Li H, et al.. Supporting data for “Chromosome-level genome assembly reveals adaptive evolution of the swimming crab (Portunus trituberculatus).”. GigaScience Database. 2019. 10.5524/100678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Hugues Roest Crollius -- 10/4/2019 Reviewed
Joseph F. Ryan -- 10/4/2019 Reviewed