

Abstract
Ambient ozone (O3) exposure has serious consequences on respiratory health, including airway inflammation and injury. Decades of research have yielded thorough descriptions of these outcomes; however, less is known about the molecular processes that drive them. The aim of this study was to further describe the cellular and molecular responses to O3 exposure in murine airways, with a particular focus on transcriptional responses in 2 critical pulmonary tissue compartments: conducting airways (CA) and airway macrophages (AM). After exposing adult, female C57BL/6J mice to filtered air, 1 or 2 ppm O3, we assessed hallmark responses including airway inflammation (cell counts and cytokine secretion) and injury (epithelial permeability), followed by gene expression profiling of CA and AM by RNA-seq. As expected, we observed concentration-dependent increases in airway inflammation and injury. Conducting airways and AM both exhibited changes in gene expression to both 1 and 2 ppm O3 that were largely compartment-specific. In CA, genes associated with epithelial barrier function, detoxification processes, and cellular proliferation were altered, while O3 affected genes involved in innate immune signaling, cytokine production, and extracellular matrix remodeling in AM. Further, CA and AM also exhibited notable differences in concentration–response expression patterns for large numbers of genes. Overall, our study has described transcriptional responses to acute O3 exposure, revealing both shared and unique gene expression patterns across multiple concentrations of O3 and in 2 important O3-responsive tissues. These profiles provide broad mechanistic insight into pulmonary O3 toxicity, and reveal a variety of targets for focused follow-up studies.
Keywords: ozone, air pollution, transcriptomics, lung, inflammation, injury, mouse
Ozone (O3) is a common urban air pollutant generated by photochemical reactions of primary pollutants including volatile organic compounds and nitrogen oxides. Exposure to O3 is associated with a variety of adverse health outcomes, including increased cardiorespiratory morbidity and mortality (Day et al., 2017; Ito et al., 2005; Mirowsky et al., 2017). Upon inhalation, O3 causes pulmonary inflammation (Aris et al., 1993; Devlin et al., 1996), decreased lung function (Kim et al., 2011; Schelegle et al., 2009), and impaired epithelial barrier integrity (Devlin et al., 1997; Kehrl et al., 1987). Together, these effects contribute to exacerbation, and potentially onset of chronic respiratory diseases including asthma (Akinbami 2010; Anenberg 2018; Gent et al., 2003; Greer et al., 1993; McConnell 2002; McDonnell et al., 1999; Nishimura 2016; Tetreault et al., 2016; Thurston et al., 1997) and chronic obstructive pulmonary disease (Medina-Ramon et al., 2006; Strosnider et al., 2019; Wang 2019). Regulatory measures have led to considerable improvements in air quality in recent decades and, consequently, decreases in O3-related adverse health outcomes (Cromar et al., 2019; Garcia et al., 2019); however, O3 exposure remains a persistent problem (Cromar et al., 2019) and concentrations are expected to rise with climate change-associated warming temperatures (Bernstein and Rice 2013; Pfister et al., 2014).
Inhaled O3 readily reacts with cellular membranes and components of the lung lining fluid to generate bioactive mediators that induce oxidative stress, tissue injury, and innate immune signaling (Mudway and Kelly 2000; Pryor et al., 1995). Airway epithelial cells and resident airway macrophages (AM) are the first 2 pulmonary cell types that encounter O3 and its reaction products, and previous work has established their critical roles in initiating and resolving O3-induced airway inflammation (Bauer et al., 2015; Mathews et al., 2015; Sunil et al., 2015, 2012). Further, O3 exposure causes damage to the airway epithelium and impairs macrophage phagocytic and efferocytic function, which can cause prolonged injury and inflammation (Becker et al., 1991; Devlin et al., 1994; Gilmour et al., 1991). Though previous studies have extensively described these processes (reviewed in Bromberg 2016), the exact molecular mechanisms that drive them have not been completely elucidated.
Using transcriptomic approaches is a powerful method to thoroughly probe responses to a given stimulus (Sweeney et al., 2017). In the case of examining toxicant-induced responses, genomic profiling studies are useful for identifying markers of exposure and early effect and comprehensively describing a toxicant’s effects at the transcriptional level. Previous studies that investigated transcriptional responses in whole lung tissue and in inflammatory cells recruited to the lungs following O3 exposure have broadened our appreciation of O3-response pathways and mechanisms of toxicity, including the involvement of heat-shock proteins (Hspa1), extracellular matrix remodeling enzymes (Mmp2, Mmp9), and various proinflammatory signaling pathways (Tnfr family members, Rela), amongst others (Backus et al., 2010; Bauer et al., 2011; Ciencewicki et al., 2016; Gabehart et al., 2014; Gohil et al., 2003; Kooter et al., 2007; Leroy et al., 2015; Nadadur et al., 2005; Verhein et al., 2015; Ward et al., 2015). Because whole lung tissue is composed of a complex mixture of many (perhaps > 40) cell types (Franks et al., 2008), bulk transcriptomics may reflect only the most marked alterations in gene expression; more subtle effects, including those that are tissue-specific, may be obscured. Therefore, approaches that focus on specific target tissues and/or cell types are required to resolve heterogeneity in gene expression responses across individual compartments and facilitate their clearer interpretation. To this end, we designed a study to examine transcriptional responses in the conducting airways (CA) and AM after exposure to multiple concentrations of O3.
We exposed adult, female C57BL/6J mice to filtered air (FA), 1 or 2 ppm O3 for 3 h and evaluated hallmark pulmonary inflammation and injury responses 21 h later. In tandem, we performed gene expression profiling of the CA and AM. We found that the expression of a large number of genes was altered in a concentration- and tissue-dependent manner, including many genes not reported previously as O3-responsive.
MATERIALS AND METHODS
Animals
Adult (8 weeks of age) female C57BL6/J mice were purchased from the Jackson Laboratory (Bar Harbor, Maine) and acclimated at the University of North Carolina at Chapel Hill for 3 weeks. Because sex is known to influence responses to O3 exposure (Birukova et al., 2019; Cabello et al., 2015; Cho et al., 2019; Fuentes et al., 2019), we chose to use a single sex to maximize our ability to define a homogeneous transcriptional response. Previous research has established that female mice have more severe inflammatory and injury-associated responses to acute O3 exposure (Cabello et al., 2015; Fuentes et al., 2019; Mishra et al., 2016), hence we chose to use female mice to increase the biological signal. All animals were housed in groups of 3 or more in polycarbonate cages on ALPHA-Dri bedding (Shepard), under normal 12-h light/dark cycles with ad libitum food (Envigo 2929) and water. The Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina at Chapel Hill approved all experiments.
Ozone Exposure
Mice were exposed to FA, 1 or 2 ppm ozone (O3), concentrations that produce inflammation and/or injury. By comparison, these concentrations are approximately 10- to 30-fold higher than the 8-h National Ambient Air Quality Standard (NAAQS) for O3 (0.07 ppm). Previous studies have established that these concentrations of O3 are roughly 4–5 times higher than the concentration required to induce comparable inflammation in exercising human subjects (ie, 1 ppm O3 for rodents is roughly equivalent to 0.2 ppm O3 for humans, and 2 ppm O3 is roughly equivalent to 0.4 ppm O3) (Hatch et al., 2014; Hatch et al., 2013, 1994).
Mice were exposed for 3 h in individual wire-mesh chambers without access to food or water, as described previously (Smith et al., 2019). Exposures at each concentration were performed on separate days to ensure that the exposure time (9 am–12 pm) was kept consistent for each exposure group. A set of mice was exposed to FA each day, and data from each was combined to form a single control group. At the end of the 3-h exposure period, mice were returned to their normal housing.
Phenotyping
Lung phenotyping
Nine to 11 mice per treatment group were used for lung phenotyping and gene expression analyses. Twenty-one hours after exposure, mice were anesthetized (2 g/kg urethane) and sacrificed by exsanguination via the abdominal aorta/inferior vena cava. Bronchoalveolar lavage (BAL) was performed by cannulating the trachea and instilling phosphate-buffered saline supplemented with cOmplete protease inhibitor cocktail (Roche) (1 x 0.5 ml, 1 x 1 ml). The right upper and middle lobes were snap-frozen in liquid nitrogen and stored at −80°C. The remaining lobes and a section of the trachea were transferred to RNAlater solution (Sigma Aldrich) and stored at 4°C until microdissection and RNA extraction. The recovered BAL fluid was centrifuged at 400 × g for 10 min. The supernatant from the first fraction was saved and stored at −80°C as 2 aliquots for protein and cytokine analysis. Pellets from both fractions were pooled, washed once in red blood cell lysis buffer, and centrifuged again as 400 × g for 10 min. Pellets were resuspended in 500 µl of HBSS, a 20 µl cell suspension was added to 20 µl of Trypan blue solution, and total numbers of viable cells were determined by counting on a hemacytometer. A 100 µl aliquot of the cell suspension was used to prepare cytospin slides. The remaining cell suspension was used for isolating AM.
Airway Macrophage Isolation
The remaining BAL cell suspension was plated in FBS-containing RPMI-1640 and rested in a cell culture incubator for 4 h. Non-adherent cells were decanted, while an enriched fraction of adherent AM was frozen in RLT lysis buffer (Qiagen) and stored at −80°C until RNA extraction. We have chosen to broadly define these cells as “airway” macrophages as they represent cells from both the large airways and the alveoli, and in O3-exposed mice, this mixture may contain both resident macrophages and inflammatory monocytes.
Protein and Cytokine Analysis
Total protein was measured using the Qubit Total Protein Quantification kit and Qubit 2.0 fluorometer (Thermo Scientific). Cytokines were measured using a Milliplex immunoassay kit (Millipore) on a Bio-Rad Bio-Plex 200 multiplex suspension array system.
Histology
Mice exposed exclusively for histological analyses (n = 5 per treatment group) were not subjected to BAL to avoid eliminating infiltrating immune cells and disrupting the natural architecture of the lungs. Mice were sacrificed as described in the previous section. Lung tissue was inflated in situ with 10% neutral-buffered formalin (NBF) through the tracheal cannula at 25 cm of static fluid pressure. The lungs were removed and immersed in 10% NBF for 24 h overnight, followed by washing and dehydration in 70% ethanol. After fixation, lungs were embedded in paraffin and sectioned along the main axial airway, as described previously (Farraj et al., 2003). Paraffin sections were stained with hematoxylin and eosin and were immunostained with antibodies specific for FOXJ1 (Abcam, cat# ab235445, clone EPR21874, 1:1000 dilution), CCSP (Abcam, cat# ab40873, 1:2000 dilution), or proliferating cell nuclear antigen (PCNA) (Agilent/Dako, cat# M0879, clone PC10, 1:180 dilution).
Statistical Analysis
For analysis presented in Figures 1 and 2, all raw data were subjected to Box-Cox power transformations to reduce heteroscedasticity and to conform to a normal distribution. Subsequently, we performed ANOVA and pairwise t-tests in R (version 3.5.3), and test results were considered significant if the resulting p-value was < 0.05.
Figure 1.

O3 exposure induces inflammation and injury in C57BL/6J mice. Eleven-week-old female C57BL/6J mice were exposed to filtered air (FA), 1 or 2 ppm O3 for 3 h, sacrificed 21 h subsequently, and cell types, cell numbers, and total protein concentration were measured in bronchoalveolar lavage fluid. (A) Total cell number (106), (B) neutrophil number (105), and (C) % neutrophils were measured by differential cell counting. (D) Total protein in BAL was measured using a fluorometric Qubit assay. Results are displayed as box-and-whisker plots depicting the distribution of the data as the minimum, first quartile, median, third quartile, and maximum with all points overlaid (n = 11 for 1 and 2 ppm O3 exposure, n = 9 for FA exposure); a: p < 0.05 compared to FA group, b: p < 0.05 compared to 1 ppm group.
Figure 2.

Ozone induced changes in cytokine and chemokine concentrations in BAL. A multiplex cytokine detection assay was used to measure the concentration of (A) CCL11 (eotaxin), (B) G-CSF, (C) IL-6, (D), IL-10, (E) CXCL10 (IP-10), (F) CXCL1 (KC), and (G) CXCL5 (LIX) in BAL fluid. Results are displayed as box-and-whisker plots depicting the distribution of the data as the minimum, first quartile, median, third quartile, and maximum with all points overlaid (n = 10 for 1 and 2 ppm O3 exposure, n = 9 for FA exposure, and points below limited of detection were excluded from analysis); a: p < 0.05 compared to FA group, b: p < 0.05 compared to 1 ppm group.
RNA Isolation and Sequencing
Airway Macrophage Gene Expression Analysis
Airway macrophages enriched from the BAL cell suspension were lysed directly with 350 µl of RLT buffer (Qiagen). Lysed AM were combined to create 4 pools per treatment group which were frozen for later RNA isolation. Total RNA was isolated using the Qiagen RNeasy Micro Kit. Libraries were prepared using the Takara Bio Low Input SMARTer Stranded RNA-Seq Library Kit. Libraries were pooled and sequenced on one lane of an Illumina HiSeq 4000 to generate single-end, 50-bp reads.
Conducting Airway Gene Expression Analysis
Four lung samples were selected from each treatment group, representative of the 4 pools of AMs. Conducting airways were isolated from lungs preserved in RNAlater using a previously published method (Baker et al., 2003). Briefly, the left lung lobe was immersed in RNAlater for 1 week to rubberize the tissue. Then, under a dissecting stereomicroscope, the parenchyma was manually separated from the CA. Total RNA was extracted using the Qiagen RNeasy Mini Kit. Libraries were prepared from poly A-enriched RNA using the Kapa Stranded RNA-Seq Library Kit. Libraries were pooled and sequenced on one lane of an Illumina HiSeq 4000 to generate single-end, 50-bp reads.
qRT-PCR Gene Expression Validation
In an independent experiment, we exposed mice to FA or 2 ppm O3 and extracted total RNA as described above from n = 6 CA and n = 4 AM individual samples per treatment (rather than pools). cDNA was reversed-transcribed using the High-Capacity RNA-to-cDNA kit using 100 ng RNA. All reactions were performed in triplicate with 1 ng cDNA input using the Taqman Universal PCR Master Mix (Applied Biosystems) and plates were read using a Bio-Rad CFX384 Touch Thermocycler. In CA, we quantified abundance of Scgb1a1 (Mm00442046_m1), Saa3 (Mm00441203_m1), Eif3f (Mm00517953_m1), and Mt2 (Mm04207591_g1), and in AM, Scgb1a1, Ccl3 (Mm99999057_m1), Marco (Mm00440265_m1), and Tns1 (Mm00452886_m1). The expression levels of all genes (except Mt2) were quantified relative to the abundance of Actb (Mm01205647_g1). Sdhc (Mm00481172_m1) was used as a normalization gene for Mt2 in CA.
Gene Expression Analysis
Given the differences in the library preparation methods, analysis of CA and AM samples were performed separately rather than jointly, and only post hoc comparisons between the 2 compartments were made.
Sequence Alignment and Transcript Quantification
After sequencing, reads were de-multiplexed and deposited as fastq files. Reads were aligned to the C57BL/6J [mm9, GENCODE release M18] reference genome using STAR v.2.6.0a (Dobin et al., 2013). Transcripts were quantified with Salmon v0.9.1 using default parameters (Patro et al., 2017).
Differential Expression Analysis
tximport was used to import and summarize Salmon quantification files. Differentially expressed genes (DEGs) were identified using the standard differential expression analysis using DESeq2 (Love et al., 2014), extracting pairwise comparisons between each treatment group within a tissue compartment. A gene was considered differentially expressed if the absolute log2 fold-change was greater than 1 and the Benjamini–Hochberg adjusted p-value (false discovery rate (FDR)) was less than 0.05.
Literature Search and Meta-Analysis
PubMed and Google Scholar were searched for studies that evaluated gene expression responses to acute O3 exposure. We excluded studies that used lower (<0.5 ppm) or higher (>2 ppm) doses of O3 than our study, chronic exposure, or involved species other than mice. Some studies included in this analysis included genetically engineered mice. For these studies, only results from wild-type animals were used. For studies that published expression of microarray probes that had not been assigned final gene annotations (ie, were published with accession numbers only), the genes were systematically assigned symbol names using the DAVID Gene ID Conversion Tool or manually assigned symbol names using Ensembl. From the current study, we used genes that were differentially expressed in the 1 ppm versus FA contrast. A hypergeometric test (using a total background gene set size of 20 000) was performed to determine the significance of overlap between our results and a given study’s results, and we used all reported significant DEGs from the study, regardless of the magnitude of fold-change or the directionality of effect. The input lists of genes and all comparisons with p-values are included in the Supplementary material. We also evaluated how consistently genes were differentially expressed across all 3 published studies and the current study using the “vote counting” method of meta-analysis (Ramasamy et al., 2008) in which we tallied the number of studies in which a gene was considered differentially expressed.
Pathway Analysis
We used Gene Set Variation Analysis (GSVA) (Hänzelmann et al., 2013) to identify pathways that were differentially expressed in CA and AM due to O3 exposure. The variance-stabilized transformation of the gene expression count matrix from DESeq2 was used as input, and gene set libraries were downloaded from the Enrichr website (date: January 22, 2019, URLs in Supplementary Table 1) (Kuleshov et al., 2016). Resulting pathway enrichment scores were tested for differential enrichment using limma (Ritchie et al., 2015). Given the exploratory nature of GSVA, we used a less stringent FDR of 0.1 for these analyses.
Ordered Factor Model for Categorizing Gene Expression
We used a Bayesian model selection approach to explore patterns in gene expression responses across the FA, 1 and 2 ppm groups. This approach utilizes the order of the treatment groups, defining incremental effects for the differences between FA→1 ppm and 1→2 ppm. We fit a collection of models that constrain these incremental effects to be positive, negative, or unchanged, and the appropriate model is inferred probabilistically for each gene. Genes that share the same model with high probability display similar response patterns across the treatment groups.The models of expression for each gene are of the form
where is the expression of a particular gene for sample i (normalized and covariate-adjusted using DESeq2), is an indicator denoting membership in the 1 or 2 ppm groups, and is an indicator denoting membership in the 2 ppm group. The incremental effects, and are constrained to be positive via a truncated normal prior distribution:
Each model is then specified by , where is equal to one of . specifies the direction of each effect (−1, negative; 1, positive) or removes it from the model (0, unchanged). There are 9 possible settings of , each one specifying a pattern of response (Supplementary Table 2). This model is similar to a previously described isotonic model (Neelon and Dunson 2004), but allows non-isotonic effects and treats as an ordered factor instead of a continuous variable. The model parameters and are given a proper conjugate prior distribution:
This ensures that the prior distribution is uninformative with respect to the location and scale of the gene expression data.
The hyperparameter controls the prior standard deviation of the incremental treatment effects relative to error. We used an empirical Bayes approach to set this hyperparameter, evaluating each model over a grid of values (1 to 2 by 0.1) and selecting the value of that maximized the total likelihood for all genes, integrated over all models, within each data set (AM or CA). This approach resulted in setting for the AM data set and for the CA data set.
In Bayesian model selection, we are interested in the posterior of :
which involves calculating the marginal likelihood, conditional on each , and also specifying a prior distribution, , over the possible settings of . We specify a prior distribution over that assumes 50% prior probability of no change in gene expression across the treatment groups , with the remaining 50% prior probability distributed uniformly over the other non-null models.
We implemented this model in R using the RStan package and obtained marginal likelihoods using the bridgesampling package (Carpenter et al., 2017).
RESULTS
Ozone (O3) Induces Inflammation, Injury and Altered Cytokine Production at 2 Concentrations
We exposed 11-week-old, female C57BL/6J mice to FA, 1 or 2 ppm O3 for 3 h. Twenty-one hours after exposure, the total number of cells and neutrophils (as well as the percentage of neutrophils) in BAL were significantly increased in mice exposed to either 1 or 2 ppm O3 compared to mice exposed to FA (Figure 1A–C). Additionally, total cells and total number of neutrophils in BAL were significantly increased in mice exposed to 2 ppm O3 compared to those exposed to 1 ppm O3 (Figure 1A and B), providing evidence of a roughly linear concentration–response relationship for these endpoints. In contrast to inflammation, lung injury (as measured by total protein in BAL fluid) was only apparent after exposure to 2 ppm O3, indicating a threshold type effect.
To further characterize airway inflammatory responses, we measured the concentrations of a panel of cytokines. We observed concentration-dependent increases in canonical O3-responsive cytokines (Che et al., 2016; Johnston et al., 2007; Lu et al., 2006; Mathews et al., 2017) including eotaxin (CCL11), G-CSF, IL-6, IP-10 (CXCL10), and KC (CXCL1) (Figure 2A–C, E and F). Contrary to expectation, we did not detect an increase in IL-10 or LIX (CXCL5), 2 cytokines that have been previously implicated in O3 responses (Backus et al., 2010; Kasahara et al., 2012; Kierstein et al., 2006). IL-1β, IL-12p70, MCP-1 (CCL2), MIP-1α (CCL3), MIP-1β (CCL4), MIP-2 (CXCL2), and TNF-α were also included in our multiplex panel but were not detected above the minimum threshold (data not shown).
Ozone Induces Epithelial Injury and Morphological Changes to the Airways
We performed histopathological analysis to assess airway morphological changes induced by O3 exposure (Figure 3). At both concentrations of O3, we observed denudation of the epithelium and exfoliation of ciliated cells, both of which were more severe in mice exposed to 2 ppm O3 (Figure 3A–C). Using immunohistochemical approaches, we found a marked loss of FOXJ1 protein, a marker of airway ciliated cells, in the axial airways of mice exposed to 1 and 2 ppm O3 (Figure 3D–F). In contrast, while there was an appreciable change in the pattern of the secretory cell marker CCSP, its expression remained high in airway epithelial cells. This may be attributable to new, proliferating cells (Figure 3G–I), which is supported by the concentration-dependent increase in PCNA staining observed in the epithelium (Figure 3J–K).
Figure 3.

O3 exposure causes epithelial damage in the upper airways. Light photomicrographs of the respiratory epithelium lining the axial airway of the left lung lobe from mice exposed to 0 (A,D,G,J), 1 (B,E,H,K) or 2 ppm (C,F,I,L) ozone. Tissues were histochemically stained with hematoxylin and eosin (A, B, C) or immunohistochemically stained for (D, E, F) FOXJ1, (G, H, I) club cell secretory protein (CCSP), or (J, K, L) proliferating cell nuclear antigen (PCNA). Abbreviations: a, alveolar parenchyma; arrow *, exfoliating epithelium; arrow C (red chromogen), CCSP; arrow F (brown chromogen), FOXJ1; arrow P (brown chromogen), PCNA; e, respiratory epithelium; sm, airway smooth muscle.
Marked Transcriptional Alterations in Both Conducting Airways and Airway Macrophages Following O3 Exposure
We sought to examine transcriptional activity in CA tissue and AM 21 h after FA, 1 ppm or 2 ppm O3 exposure. Here, we defined CA as the large airway tree beginning at the trachea and ending at the terminal bronchioles, which captures 4 generations of branching airways (Baker et al., 2003) and is highly susceptible to O3 toxicity (Plopper et al., 1994). We use the term AM to describe the enriched population of adherent cells we collected from BAL. In O3-exposed mice, this likely includes both resident macrophages (Tighe et al., 2011) (including alveolar macrophages (Birukova et al., 2019; Sunil et al., 2012)) and recruited monocyte-derived macrophages (Francis et al., 2017).
RNA-seq was performed on 12 CA samples and 12 pooled AM samples (n = 4/treatment group), and yielded an average of 27 million reads per sample, after preprocessing. Of these, an average of 81% of reads in CA and 75% of reads in AM were uniquely mapped, without any evidence of differential alignment across treatment groups. Additional mapping statistics are provided in Supplementary Table 3. Principal components analysis (PCA) of normalized gene expression profiles revealed a clear separation between the 3 treatment groups in both CA and AM samples (Figures 4A and 5A). Interestingly, the PCA for CA mirrors the percent neutrophils concentration–response pattern. More specifically, samples from 1 and 2 ppm-exposed mice cluster nearer to each other than to the samples from FA-exposed mice, and this threshold-like exposure–response pattern can largely be explained by PC1. The PCA for AM samples is more complex, with treatment effects reflected in both PC1 and PC2.
Figure 4.

O3 exposure induces altered gene expression in conducting airways (CA). Gene expression of CA tissue from mice exposed to FA, 1 or 2 ppm O3. (A) Principal components analysis shows separation of the 3 treatment groups. Differential expression analysis revealed (B) 903 DEGs in 1 ppm versus FA, (C) 2148 DEGs in 2 ppm versus FA, and (D) 188 DEGs in 2 ppm versus FA (n = 4 per treatment group, fold-change cutoff = 2, Benjamini–Hochberg adjusted p-value < 0.05).
Figure 5.

Airway macrophage (AM) gene expression is altered by O3 exposure. Gene expression in pooled AM from mice exposed to FA, 1 or 2 ppm O3. (A) Principal components analysis displays separation of AMs across all 3 treatment groups. Differential expression analysis revealed (B) 693 DEGs in 1 ppm versus FA, (C) 971 DEGs in 2 ppm versus FA, and (D) 467 DEGs in 2 versus 1 ppm (n = 4 pools per treatment group, fold-change cutoff = 2, Benjamini–Hochberg adjusted p-value < 0.05).
We identified DEGs by performing all pairwise comparisons between treatment groups within a tissue compartment. A summary of results from this analysis is included in Table 1, and all significantly DEGs (defined as FDR < 0.05 and absolute log2 fold-change > 1) for each treatment comparison and compartment are provided in the Supplementary material. In CA samples, we identified 903 DEGs between 1 ppm and FA, 2148 DEGs between 2 ppm and FA, and 188 DEGs between 2 ppm and 1 ppm (Figure 4B–D). Generally, fold-changes were greater in magnitude within the 2 ppm versus FA comparison than within the 1 ppm versus FA comparison. In AM samples, we identified 694 DEGs between 1 ppm and FA, 972 DEGs between 2 ppm and FA, and 467 DEGs between 2 ppm and 1 ppm (Figure 5B–D). Similar to CA samples, the range of fold-changes was wider in the 2 ppm versus FA comparison than within the 1 ppm versus FA comparison in AM samples.
Table 1.
The Number of Differentially Expressed Genes, by Tissue Compartment and Treatment Comparison
| Tissue | Comparison | FDR < 0.05 | FDR < 0.1 |
|---|---|---|---|
| Conducting airways | 1 ppm vs FA | 903 | 1048 |
| 2 ppm vs FA | 2148 | 2382 | |
| 2 vs 1 ppm | 188 | 255 | |
| Airway macrophages | 1 ppm vs FA | 693 | 859 |
| 2 ppm vs FA | 971 | 1216 | |
| 2 vs 1 ppm | 467 | 585 |
All genes had an absolute fold-change cutoff of 2 (ie, absolute log2FC > 1).
Many of the top genes with altered expression in the CA samples (Table 2) are those commonly associated with barrier function and epithelial organization (Gjb3-5, Cldn2/4, Adam12, Tgm1) (Esseltine and Laird 2016; Frank 2012), detoxification (Mt1/2, Ugt1a6a/b, Gstm1/2/6), and DNA replication (Mcm5-7, Cdt1, Orc1) (Riera et al., 2017). Notably, within CA samples, we observed an increase in expression of genes associated with proliferative responses (Pcna, Cdh3) concordant with our histopathological analysis. Furthermore, we noted a concentration-dependent decrease in expression of ciliated and club cell markers (Foxj1, Cyp2f2) Supplementary Figure 1, corresponding with the loss of ciliated cells observed in the airways (Figure 3). While we observed no loss in protein expression of CCSP in our histopathological analysis, we did see a significant decrease in expression of Scgb1a1 (the gene encoding CCSP) as well as Scgb3a2 (Supplementary Figure 1).
Table 2.
Top 50 Most Differentially Expressed Genes (25 Downregulated, 25 Upregulated) For Each Treatment Comparison Within Conducting Airways
| 1 ppm vs FA |
2 ppm vs FA |
2 vs 1 ppm |
||||||
|---|---|---|---|---|---|---|---|---|
| Symbol | FC | FDR | Symbol | FC | FDR | Symbol | FC | FDR |
| Cyp1a1 | −26.99 | 2.60E−04 | D7Ertd443e | −32.67 | 2.83E−05 | Trpm1 | −15.30 | 2.55E−04 |
| Ucp1 | −24.07 | 1.93E−02 | Cdh26 | −27.47 | 3.30E−06 | Dthd1 | −9.98 | 5.92E−04 |
| D7Ertd443e | −17.28 | 1.16E−03 | Gys2 | −22.66 | 1.12E−05 | Gpr37 | −4.43 | 7.11E−04 |
| H3f3aos | −16.51 | 1.72E−02 | Igkv3-2 | −22.62 | 6.05E−06 | Tsnaxip1 | −4.15 | 4.83E−06 |
| Igkv3-2 | −10.76 | 1.26E−03 | Dthd1 | −21.05 | 5.68E−05 | Nusap1 | −3.86 | 2.76E−05 |
| Gys2 | −8.74 | 5.39E−03 | Anks1b | −18.81 | 5.16E−03 | Atp6v1b1 | −3.83 | 2.27E−05 |
| Oxtr | −8.04 | 3.27E−04 | Cnpy1 | −17.54 | 5.78E−04 | Erich5 | −3.78 | 3.34E−04 |
| Hectd2os | −6.42 | 6.54E−04 | Lyzl4 | −17.10 | 3.93E−08 | Ccna1 | −3.78 | 7.43E−07 |
| Snx31 | −6.00 | 3.18E−02 | Gfi1b | −16.14 | 5.21E−03 | Cryaa | −3.75 | 2.00E−05 |
| Glb1l3 | −5.95 | 3.11E−05 | Ampd1 | −14.13 | 9.10E−11 | Mcidas | −3.74 | 8.11E−04 |
| Islr2 | −5.43 | 5.04E−03 | Gpr37 | −13.46 | 2.85E−08 | Shisa8 | −3.70 | 5.25E−04 |
| Nat8l | −5.37 | 4.21E−02 | Calml3 | −13.42 | 3.31E−06 | Uox | −3.69 | 5.39E−04 |
| Gabrb1 | −5.30 | 1.40E−02 | S100a7a | −12.22 | 6.37E−03 | Gtsf1l | −3.65 | 4.84E−04 |
| Col26a1 | −5.17 | 8.84E−03 | Pih1h3b | −11.96 | 2.94E−16 | Ube2c | −3.64 | 5.68E−04 |
| Thrsp | −5.13 | 4.10E−09 | Iqch | −10.95 | 1.13E−07 | Krt1 | −3.58 | 2.97E−05 |
| Ampd1 | −5.13 | 6.44E−05 | Atp6v1b1 | −9.72 | 4.48E−11 | Rsg1 | −3.53 | 2.10E−05 |
| Lyzl4 | −5.11 | 2.30E−03 | Gtsf1l | −9.68 | 1.51E−08 | Plk5 | −3.52 | 3.09E−04 |
| Itgad | −5.06 | 4.43E−02 | Tmprss6 | −9.62 | 7.48E−04 | Ttc39d | −3.47 | 8.21E−04 |
| Col6a5 | −5.03 | 1.53E−02 | Cyp1a1 | −8.82 | 1.52E−02 | Wfdc13 | −3.27 | 7.61E−05 |
| Otop1 | −4.90 | 2.68E−04 | Cacna1i | −8.60 | 2.50E−02 | Kif2c | −3.12 | 4.01E−04 |
| Sema5b | −4.78 | 4.34E−02 | Cryaa | −8.30 | 2.20E−10 | Prss35 | −3.03 | 4.49E−04 |
| Marc1 | −4.76 | 3.66E−04 | Unc79 | −8.28 | 4.90E−04 | Calcoco2 | −3.02 | 6.49E−05 |
| Dyx1c1 | −4.76 | 1.10E−02 | Slc38a11 | −8.05 | 2.20E−07 | Ccdc33 | −3.00 | 2.93E−05 |
| Kcnj13 | −4.55 | 7.58E−03 | Cyp4a12b | −8.03 | 1.44E−04 | Cyp4f15 | −3.00 | 1.19E−05 |
| Pih1h3b | −4.46 | 4.00E−07 | Btg4 | −7.85 | 1.32E−07 | Prr11 | −2.91 | 5.44E−04 |
| Rad54b | 9.41 | 1.85E−09 | Hells | 14.66 | 3.16E−05 | Lipt2 | 1.59 | 3.91E−02 |
| Serpina3m | 10.01 | 6.36E−25 | Serpina3m | 14.86 | 1.00E−34 | Gadd45g | 1.59 | 1.43E−02 |
| Grpr | 10.58 | 1.26E−02 | Orc1 | 15.17 | 7.55E−03 | Cd200 | 1.61 | 7.48E−03 |
| Hells | 10.59 | 8.78E−04 | Gcnt3 | 15.59 | 4.55E−10 | Rev1 | 1.62 | 1.67E−02 |
| Upk3bl | 10.68 | 1.30E−10 | Mt2 | 15.67 | 1.68E−30 | Plk3 | 1.65 | 4.12E−02 |
| Pbp2 | 10.74 | 2.26E−06 | Gjb4 | 15.74 | 1.06E−13 | Pmvk | 1.66 | 1.43E−03 |
| Gsta1 | 10.89 | 5.67E−14 | Dio3 | 16.60 | 5.77E−07 | Mettl22 | 1.66 | 5.08E−03 |
| Il24 | 10.91 | 1.06E−03 | Ptx3 | 17.02 | 2.64E−09 | Plaur | 1.70 | 4.78E−02 |
| Mymk | 11.43 | 1.14E−08 | Pbp2 | 17.36 | 7.97E−10 | Sphk1 | 1.81 | 7.68E−03 |
| Saa1 | 11.85 | 1.16E−04 | Prss22 | 17.43 | 9.19E−13 | Prmt8 | 1.82 | 2.47E−03 |
| Krt36 | 11.94 | 7.06E−04 | Upk3bl | 17.89 | 1.43E−16 | Klhdc1 | 1.82 | 3.32E−02 |
| Prss22 | 12.01 | 3.39E−09 | Gsta1 | 18.52 | 1.66E−21 | Rgcc | 1.87 | 3.60E−02 |
| Krt13 | 12.67 | 1.05E−04 | Krt13 | 18.80 | 9.22E−07 | Fgfbp3 | 1.88 | 2.23E−02 |
| Sectm1b | 12.78 | 9.19E−05 | Saa1 | 19.22 | 3.29E−07 | Psca | 2.18 | 2.82E−02 |
| Sgo1 | 13.75 | 2.40E−06 | Grpr | 19.45 | 2.89E−04 | Mt1 | 2.21 | 5.20E−04 |
| Ankrd2 | 14.79 | 9.79E−05 | Sprr2a2 | 19.93 | 6.94E−12 | Fmo4 | 2.21 | 4.33E−02 |
| Fgf23 | 14.95 | 3.72E−02 | Mymk | 23.67 | 1.75E−15 | S100a9 | 2.25 | 4.96E−02 |
| Psca | 16.40 | 1.35E−15 | Il24 | 24.73 | 6.45E−07 | Slc30a2 | 2.30 | 1.29E−02 |
| Orc1 | 18.13 | 8.45E−03 | Saa3 | 27.34 | 7.66E−12 | Dancr | 2.56 | 4.55E−03 |
| Pdyn | 20.39 | 1.22E−04 | Cwh43 | 33.09 | 3.65E−06 | Hsd17b14 | 2.61 | 1.63E−02 |
| Saa3 | 25.39 | 9.99E−11 | Psca | 35.69 | 1.38E−26 | Mt2 | 2.84 | 2.43E−03 |
| Kif14 | 26.41 | 4.53E−04 | Serpina3k | 44.37 | 2.65E−05 | Cxcl2 | 3.87 | 4.33E−02 |
| Sprr2a2 | 31.38 | 5.63E−15 | Ankrd2 | 44.39 | 7.37E−10 | Glb1l3 | 4.56 | 5.74E−03 |
| Krt6a | 33.61 | 1.36E−02 | Fgf23 | 48.25 | 3.41E−04 | Ptx3 | 4.86 | 1.29E−02 |
| Serpina3k | 36.67 | 2.42E−04 | Krt6a | 341.68 | 8.98E−07 | Gpr21 | 22.07 | 4.85E−03 |
Within AM samples, genes involved in canonical immunological functions (Ccl17, Slpi, and Ccl22) and extracellular matrix organization and remodeling (Krt7, Krt8, Krt18) were highly differentially expressed (Table 3). Additionally, we observed differential expression of a number of genes that have been previously implicated in AM-mediated O3 responses including macrophage polarization genes (M1: Cd80, Tnf, Ccl5, Tlr2; M2: Arg1, Retnla, Ccl24, Cxcl10), proteases (Mmp8, Mmp9, Mmp12, Ctsd, Ctse, Ctsf), and surfactant proteins (Sftpa1, Sftpc, Sftpd) (Supplementary Figure 2) (Groves et al., 2012; Laskin et al., 2019; Laskin et al., 2011).
Table 3.
Top 50 Most Differentially Expressed Genes (25 Downregulated, 25 Upregulated) For Each Treatment Comparison Within Airway Macrophages
| 1 ppm vs FA |
2 ppm vs FA |
2 vs 1 ppm |
||||||
|---|---|---|---|---|---|---|---|---|
| Symbol | FC | FDR | Symbol | FC | FDR | Symbol | FC | FDR |
| Sptbn4 | −72.13 | 3.27E−02 | Cyp4f37 | −60.73 | 2.54E−05 | Ndnf | −40.77 | 1.71E−03 |
| Cyp4f37 | −38.76 | 3.60E−05 | Ndnf | −15.77 | 2.01E−02 | Uchl1 | −31.40 | 7.11E−03 |
| Zfp990 | −23.31 | 2.85E−02 | Hamp | −14.46 | 6.55E−06 | Hamp | −30.05 | 1.07E−08 |
| Abcb1a | −22.80 | 3.41E−03 | Dnah2 | −9.84 | 6.33E−03 | Kcnh3 | −27.74 | 1.79E−02 |
| Dnah2 | −15.87 | 1.17E−03 | Tcim | −9.14 | 3.15E−18 | Col5a3 | −11.58 | 1.74E−02 |
| Mamdc4 | −10.74 | 3.96E−02 | Fam19a3 | −8.90 | 1.55E−02 | Ocstamp | −9.53 | 1.45E−03 |
| Rasd1 | −9.29 | 1.73E−04 | Has1 | −7.69 | 6.30E−03 | Cd99l2 | −8.78 | 6.24E−07 |
| Slc2a5 | −8.62 | 5.75E−03 | Dact2 | −7.45 | 1.09E−07 | Tmem184a | −8.13 | 3.70E−03 |
| Hes1 | −7.23 | 9.43E−17 | Ocstamp | −7.43 | 3.06E−03 | Il7 | −8.12 | 3.49E−04 |
| Tpsab1 | −6.81 | 1.47E−02 | Klrb1c | −6.73 | 2.39E−04 | Ppfia3 | −7.97 | 1.23E−08 |
| Phlda2 | −6.15 | 1.52E−05 | Rnu3b1 | −6.55 | 1.03E−02 | Itgb4 | −7.89 | 8.47E−03 |
| Hist4h4 | −6.03 | 1.43E−04 | Ntn4 | −5.79 | 3.42E−03 | Pard3b | −7.77 | 4.08E−02 |
| Hist1h4m | −5.84 | 3.09E−08 | Mroh2a | −5.78 | 6.32E−04 | Tcim | −6.93 | 4.63E−13 |
| Prss36 | −5.60 | 2.46E−02 | Cd83 | −5.37 | 4.91E−21 | Perm1 | −6.63 | 7.95E−04 |
| Hfm1 | −5.37 | 1.77E−02 | Adgrl3 | −5.25 | 1.35E−07 | Vasn | −6.62 | 6.92E−08 |
| Zfp14 | −5.27 | 2.50E−03 | Pdgfb | −4.89 | 1.88E−06 | Has1 | −6.50 | 2.28E−02 |
| Adora3 | −5.16 | 1.05E−02 | Fam19a5 | −4.84 | 1.67E−03 | Lad1 | −6.34 | 2.78E−03 |
| Ifnb1 | −4.87 | 3.37E−05 | Fzd8 | −4.75 | 7.45E−22 | Nfkbid | −6.32 | 6.37E−05 |
| Fbxo10 | −4.81 | 2.71E−02 | Fhl1 | −4.70 | 3.34E−02 | Pdgfb | −6.13 | 1.04E−07 |
| Bcl9l | −4.78 | 4.44E−03 | Tppp3 | −4.51 | 1.36E−16 | Osbpl3 | −5.78 | 2.17E−04 |
| Sbk1 | −4.69 | 1.48E−09 | Bicd1 | −4.49 | 5.22E−03 | Hrc | −5.77 | 3.22E−02 |
| Lims2 | −4.62 | 7.60E−03 | Spag11b | −4.37 | 3.28E−16 | Cnn3 | −5.59 | 1.77E−03 |
| Tbc1d16 | −4.43 | 3.62E−05 | Vasn | −4.34 | 2.46E−05 | Zbtb18 | −5.39 | 1.07E−03 |
| Mpv17l | −4.40 | 4.80E−02 | Lims2 | −4.24 | 9.40E−03 | H2−M2 | −5.04 | 2.19E−05 |
| Ahdc1 | −4.26 | 2.78E−04 | Tmem53 | −4.13 | 1.13E−02 | Lamc2 | −4.75 | 4.22E−03 |
| Prok2 | 21.19 | 2.81E−09 | Mt3 | 36.57 | 7.94E−06 | Fbxo10 | 6.28 | 9.47E−03 |
| Pkd1l2 | 21.45 | 2.59E−06 | Cd177 | 37.67 | 1.97E−06 | Klf8 | 6.35 | 4.50E−03 |
| Ston2 | 21.51 | 3.39E−07 | Col1a1 | 43.44 | 6.81E−07 | Runx3 | 6.51 | 3.14E−03 |
| Ifit1bl2 | 21.96 | 1.28E−09 | Pkd1l2 | 47.62 | 2.71E−10 | Rrad | 6.52 | 4.00E−24 |
| Mt3 | 21.99 | 3.20E−04 | Egln3 | 53.26 | 1.27E−10 | Vaultrc5 | 6.54 | 5.14E−07 |
| Ms4a4a | 22.77 | 3.17E−04 | Ppbp | 53.65 | 1.40E−10 | Rpp25 | 6.72 | 4.74E−03 |
| Ppbp | 25.29 | 9.02E−07 | Cxcr3 | 54.83 | 2.62E−05 | Zscan20 | 6.74 | 8.68E−06 |
| Uchl1 | 27.61 | 1.04E−02 | Esrp2 | 57.33 | 1.68E−02 | Rmrp | 6.83 | 2.87E−03 |
| Ednrb | 36.40 | 7.60E−04 | Cmklr1 | 60.80 | 3.20E−04 | Sel1l3 | 6.87 | 4.71E−03 |
| F13a1 | 36.51 | 1.53E−10 | Ifit1bl2 | 61.05 | 1.21E−17 | Cxcr3 | 6.98 | 2.61E−03 |
| Col1a1 | 38.39 | 3.32E−06 | Msantd3 | 69.52 | 4.24E−04 | Zfp882 | 7.39 | 2.07E−02 |
| Kcnh3 | 41.38 | 8.48E−03 | Angptl2 | 71.23 | 3.18E−13 | Saa3 | 8.47 | 3.30E−18 |
| Galnt9 | 42.85 | 1.05E−06 | Ccl24 | 74.00 | 1.39E−16 | Angptl2 | 9.15 | 2.69E−06 |
| Fgf13 | 50.21 | 9.20E−03 | Olfm4 | 74.52 | 2.11E−07 | Phlda2 | 9.32 | 5.59E−08 |
| Ly6c2 | 51.94 | 1.49E−04 | Ms4a4a | 79.84 | 3.24E−08 | Aqp3 | 10.49 | 4.80E−12 |
| Cnn3 | 56.47 | 4.40E−13 | Gdf3 | 84.01 | 3.52E−05 | Abcb1a | 14.13 | 2.09E−02 |
| Retnla | 56.77 | 1.82E−15 | Carmil1 | 84.15 | 2.66E−03 | Rasd1 | 15.94 | 1.33E−06 |
| Itgb6 | 58.48 | 2.37E−02 | Ngf | 91.61 | 4.00E−06 | Cfap157 | 16.03 | 6.51E−05 |
| Esrp2 | 60.29 | 2.09E−02 | F13a1 | 92.18 | 3.73E−17 | Pianp | 20.95 | 2.38E−02 |
| Olfm4 | 61.74 | 1.59E−06 | Retnla | 93.49 | 5.75E−20 | Tpsab1 | 21.05 | 1.77E−05 |
| Mrgpra2a | 73.32 | 1.69E−05 | Mrgpra2a | 100.82 | 1.43E−06 | Zfp990 | 23.15 | 3.32E−02 |
| Carmil1 | 90.34 | 3.36E−03 | Galnt9 | 120.43 | 3.73E−11 | Notch4 | 27.16 | 4.79E−03 |
| Gucy2c | 150.07 | 2.18E−08 | Ednrb | 122.85 | 7.85E−07 | Kcnj15 | 32.65 | 2.04E−02 |
| Wnt7a | 152.95 | 2.05E−02 | Ly6c2 | 137.25 | 3.94E−07 | Kcng2 | 33.55 | 1.30E−02 |
| Ngf | 180.67 | 1.05E−07 | Gucy2c | 485.08 | 3.91E−13 | Tiam1 | 36.90 | 4.96E−02 |
To validate our findings, we performed qRT-PCR on CA and AM from an independent experiment of mice exposed to FA or 2 ppm O3. RNA was isolated from AM for individual mice rather than in pooled AM samples; likewise, CA samples from individual mice were used. For each tissue type, we reproduced the differential expression patterns previously identified in the RNA-seq analysis for 4 out of 4 genes (Scgb1a1, Saa3, Eif3f, and Mt2 in CA, and Scgb1a1, Ccl3, Marco, Tns1 in AM, Supplementary Table 4).
Conducting Airways and Airway Macrophages Display Distinct Transcriptional Responses to O3 Exposure
Previous studies have described aspects of the cellular responses to O3 exposure in airway epithelial cells and AM, primarily through the use of in vitro cultures (Bauer et al., 2015; Gilmour et al., 1991; Jakab et al., 1995; Leikauf et al., 1995); however, no studies to-date have provided direct comparison of transcriptional responses in CA and AM. Hence, we compared lists of DEGs across tissue compartments to identify unique and shared properties of their transcriptional responses (Figure 6, Supplementary Table 5). We identified 121 genes that were DE in both CA and AM comparing 1 ppm versus FA (Figure 6A). For the 1 ppm versus FA contrast, only 13% of CA DEGs were shared with AM DEGs. Likewise, for the 2 ppm versus FA contrast, 8% of CA DEGs were shared with AM DEGs. Genes that were differentially expressed in both CA and AM after 1 ppm O3 included those involved in antioxidant responses (Gsta1), cell cycle progression and DNA replication (Cdk1, Mcm2, Mcm6, Mcm8, Mcm10), and acute phase proteins (Clu, Lcn2, Saa3). After 2 ppm O3 exposure, DEGs for both CA and AM included those involved in immune signaling (Ccl17, Slpi, Cx3cl1) in addition to some of the cell cycle-associated and acute phase genes (Mcm2, Mcm10, Lcn2, Saa3) that were differentially expressed after 1 ppm O3 exposure. Lastly, only 1 gene (Hsph1), a heat-shock protein, was differentially expressed in both CA and AM when comparing 2 ppm versus 1 ppm O3, albeit discordantly (upregulated in CA, downregulated in AM).
Figure 6.
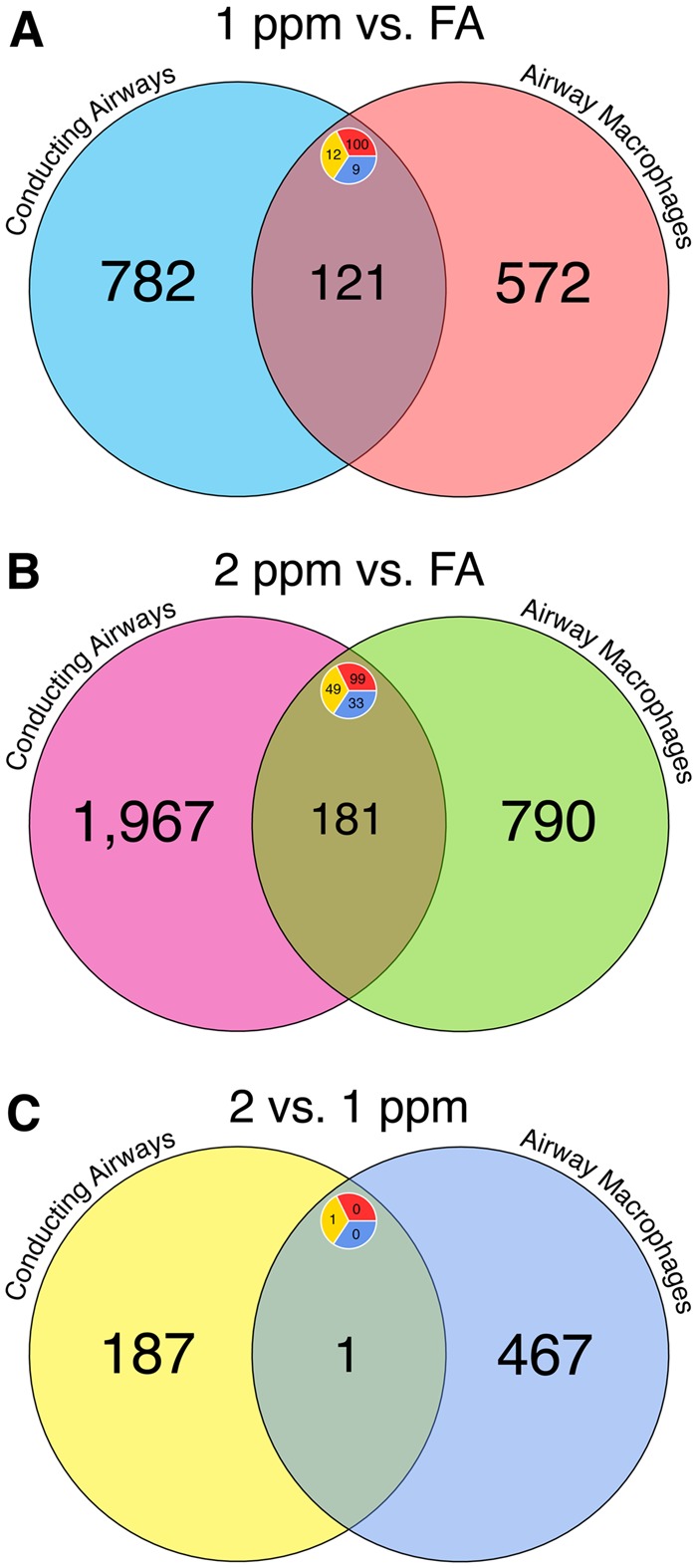
Comparison of differential gene expression results shows shared and unique responses to O3 across tissue compartments. Venn diagrams comparing lists of differentially expressed (DE) genes in conducting airway tissue and airway macrophages from (A) the 1 ppm versus FA comparison, (B) the 2 ppm versus FA comparison, and (C) the 2 versus 1 ppm comparison. Labels display the number of genes within a set. Inset circles display the number of upregulated (red top right), down regulated (blue bottom right), or genes with contrasting expression patterns (yellow left) within an intersecting set.
Comparison of Gene Expression in Conducting Airways and Airway Macrophages to Previous Studies Identifies Common O3-Responsive Genes
In order to identify broadly generalizable O3-responsive genes, we compared our lists of DEGs to those published in 3 previous studies of whole lung tissue (Gabehart et al., 2014; Gohil et al., 2003; Kooter et al., 2007) (Supplementary Tables 6 and 7). First, we found a significant degree of overlap in our lists of DEGs (CA and AM) with the DEGs from each individual study, with a notably stronger enrichment for CA DEGs (3- to 13-fold enrichment) compared to AM DEGs (2.5- to 5-fold) (Supplementary Table 8). Then, using the simple meta-analysis method of vote counting, we categorized genes as consistent if they were differentially expressed across the majority of studies, ie, 3 out 4 studies (including our own) (see Supplementary Table 9). Using our list of CA DEGs, 5 genes were consistent, namely Cdk1, Lcn2, Mt1, Saa3, and Serpina3n; 4 genes were consistent using our AM DEGs: Cdk1, Lcn2, S100a9, and Saa3.
Pathway Level Effects of O3 Exposure
In order to assess pathway level changes due to O3 in CA or AM, we performed GSVA, which has been shown to be more powerful at detecting subtle changes in pathway level expression compared to previous methods (Hänzelmann et al., 2013). This is because in GSVA, for each sample, the expression scores of a predefined set of genes (ie, pathways) are summed and then differential expression analysis is performed on these aggregate scores. Thus, modest differences in the expression of genes that may not have reached statistical significance (for each gene individually) can still cumulatively result in differential expression of a gene set. The results of these analyses are summarized in Supplementary Tables 10–12 (all significantly enriched gene sets are reported in Supplementary material).
We noted a clear decrease in expression of gene sets related to cilia function in CA, in line with the epithelial injury and ciliated cell loss in our histological analysis. In contrast, there were multiple gene sets related to DNA replication and DNA damage repair that were increased in response to 1 ppm (vs FA), and pathways involved in proteasome function and activity were upregulated in response to 2 ppm O3.
Within AM, many pathways involved in oxidative stress and detoxification responses including metallothionein and alcohol dehydrogenase activity were increased due to 1 and 2 ppm O3. Fc-gamma-regulated phagocytosis, positive regulation of vesicle fusion and other endocytic pathways were downregulated after 1 and 2 ppm O3, in alignment with previous reports that O3 disrupts AM phagocytosis (Gilmour et al., 1991; Jakab et al., 1995). Antigen processing and presentation by MHC-I and other related pathways were also decreased after 2 ppm O3. Pathways that were upregulated in AM after 2 versus 1 ppm include erythrocyte O2/CO2 exchange and arachidonic acid metabolism, while cellular communication pathways including secretion of various soluble mediators (eg, growth factors, cytokines, and hormones) were downregulated.
Relationships Between Gene Expression and O3 Concentration
Non-monotonic dose–response relationships have been documented in the literature for a number of environmental toxicants (Calabrese and Baldwin 2001; Vandenberg et al., 2012). As such, we were interested in determining whether non-monotonic O3 concentration–response relationships with gene expression (ie, biomarkers of effect) were present in CA or AM. To facilitate this analysis, we developed a Bayesian model selection approach that used all the gene expression data (excluding unexpressed transcripts) from mice exposed to FA, 1 or 2 ppm O3. For each gene, we fit a collection of models (9 total, including a null model) that constrained each incremental effect (FA → 1 ppm and 1 → 2 ppm) to be positive, negative, or unchanged, and generated the marginal likelihood for each model. The collection of models, depicted in Figure 7A, includes: monotonic, in which genes exhibited directionally consistent changes in expression across both FA → 1 ppm and 1 → 2 ppm increments; 2 threshold models, in which genes exhibited either a change from FA → 1 ppm but no further change from 1 → 2 ppm (threshold 1) or unchanged expression from FA → 1 ppm but changed from 1 → 2 ppm (threshold 2); and finally, genes that exhibited a change from FA → 1 ppm and a change in the opposite direction for 1 → 2 ppm. These 4 trends are further subdivided into 8 models based on the directionality (positive versus negative) of the incremental effects (Figure 7A).
Figure 7.

Concentration-response patterns of gene expression in conducting airways and airway macrophages. A subset of genes in both conducting airways and airway macrophages were probabilistically assigned into 1 of 8 distinct expression patterns. Schematic graphs of each category are displayed in panel A, and their order and the color assigned to each pattern is consistent throughout the figure. The frequency of genes assigned into each trend category with a marginal likelihood greater than 0.5 within (B) conducting airways and (C) airway macrophages. Heatmaps depict median-centered expression of categorized gene from (D) conducting airways and (E) airway macrophages, arranged by expression pattern. Boxplots of genes exemplifying each pattern are arranged to the right of each heatmap.
Overall, the number of genes that were classified into 1 of the 8 (non-null) models was similar between CA and AM (3587 and 3040, respectively; Figure 7B and C, Supplementary Table 13). For both compartments, this represented ∼13.7% of expressed genes (total transcripts expressed in one or more samples: 26 204 in CA and 22 216 in AM). Within both CA and AM, most genes were categorized as either positive or negative monotonic; however, in AM there was also an abundance of genes classified into non-monotonic models, particularly the “peak” and “trough” models. Given that the majority of genes in both tissue compartments were classified into the monotonic models (positive or negative), one expectation would be that that all of genes in these 2 model categories would have been declared differentially expressed in the prior analyses (Figures 4 and 5); however, this was not the case, particularly for the 2 versus 1 ppm contrast. This is attributable to differences between these 2 analyses. In our differential expression analysis, we applied stringent requirements for the fold-change and adjusted p-values for multiple tests, thereby excluding genes whose expression patterns conformed to 1 of the 8 models but that were not statistically significant for any or all pairwise contrasts between O3 concentrations. The Bayesian model selection approach does not evaluate pairwise differences between O3 concentrations in the same way, instead considering all possible changes between adjacent concentrations simultaneously. Further, because the purpose of the Bayesian approach was to categorize individual genes rather than report differential expression over many genes, we specified a prior distribution that was less stringent than in the differential expression analysis.
After assigning genes to the 8 models outlined above, we tested for enrichment of functional annotations in genes from each expression model (Supplementary Table 14). In CA, genes that fit the negative monotonic model were associated with cilia development and function, consistent with the loss of airway epithelial cells noted by our histological analyses. Additionally, canonical immune signaling pathways including those mediated by TNF-α, NF-κB, IL-1, and IL-12 were enriched in the positive monotonic model. Genes classified into the positive threshold 1 model (genes that are upregulated similarly after both 1 and 2 ppm O3 exposure), were associated with a variety of pathways including DNA damage repair and replication. For AM genes classified into the negative monotonic model, many pathways associated with stress responses and neutrophil activation/degranulation were enriched. NF-κB signaling, TNF-α biosynthesis, and cholesterol biosynthetic and metabolic pathways were enriched among genes fitting the positive monotonic model. Finally, the glutathione metabolic pathway was enriched among genes fitting the trough model (ie, downregulated after only 1 ppm O3 exposure), while mitosis and DNA replication-associated gene sets were enriched for genes fitting the peak model.
DISCUSSION
In the present work, we describe O3-concentration-dependent increases in inflammation and tissue injury which have been thoroughly documented by other researchers. Because mechanistic information about these responses is more limited, the primary goal of this study was to characterize the transcriptional response of 2 target tissues, the CA and AM. Overall, CA tissue had a more dynamic transcriptional response to O3 exposure than AM as judged by the number of DEGs when comparing 1 ppm or 2 ppm versus FA exposure, though AM had more DEGs when comparing 2 versus 1 ppm exposure. These observed differences may be attributable to the heterogeneous cellular composition of CA compared to AM.
Previous studies of airway epithelial responses to O3 exposure have identified the importance of epithelial-derived immune signaling and oxidative stress responses (Devlin et al., 1994; Wang et al., 2006). Our results confirm that genes critical for these processes (eg, Krt6a, Orc1, Gsta1, Mt2) are upregulated in CA after O3 exposure, along with enrichment of pathways associated with tissue regeneration and cellular proliferation. Together, this supports the findings of our histological analyses: namely, the airways were denuded, which increased in severity in a concentration-dependent manner.
Within CA, many genes were differentially expressed that have not been previously associated with O3 responses, including Oxtr and Ptx3. Oxtr (oxytocin receptor) was significantly downregulated (8-fold) after both 1 and 2 ppm O3 exposure versus FA. Downregulation of Oxtr has been associated with behavioral changes after diesel exhaust exposure in mice (Win-Shwe et al., 2014), and may bind to RAGE (receptor for advanced glycation endproducts) (Yamamoto et al., 2019), a receptor involved in recognizing damage-associated molecular patterns (DAMPs) that has been implicated in inflammatory responses to acute lung injury (Blondonnet et al., 2017; Griffiths and McAuley 2008). Ptx3, the gene encoding pentraxin 3, was upregulated ∼17-fold after 2 ppm O3 exposure. This gene belongs to a family of widely evolutionarily conserved proteins called pentraxins, among which are the acute phase proteins C-reactive protein (CRP) and its murine cognate serum amyloid component P (SAP) (Doni et al., 2019). Ptx3 is less well characterized but is induced by pro-inflammatory cytokines such as TNF, along with regulating chronic inflammation and responses to sterile tissue damage (Kunes et al., 2012). Though Ptx3 is known to be expressed by innate immune cells, including macrophages (Imamura et al., 2007), Ptx3 was selectively expressed by CA in our study suggesting that it may have specific roles in epithelial responses to O3 exposure.
Airway macrophages are known to have dual pathogenic and protective roles after O3 exposure, including secretion of both pro- and anti-inflammatory mediators (Pendino et al., 1995; Sunil et al., 2012) and scavenging of cellular debris (Arredouani et al., 2007). O3 exposure is also associated with AM dysfunction, including decreased phagocytic and efferocytic function (Gilmour et al., 1991; Jakab et al., 1995), and increased susceptibility to respiratory infections (Mikerov et al., 2008). Thus, identifying molecular mechanisms that drive these functional outcomes is critical. Many of the most significantly upregulated DEGs were involved in immune signaling including genes encoding chemokines- and cytokines (eg, Ccl24, Cxcl3, Ccl17) and canonical markers of macrophage polarization (eg, Retnla, Arg1, Cd80), consistent with the findings of previous studies (Laskin et al., 2019; Laskin et al., 2011; Sunil et al., 2012).
As in CA, we identified a number of novel DEGs in AM after O3 exposure including Hamp and Hes1. Hamp (hepcidin) is a hormone primarily produced in the liver (though also produced by lung macrophages (Nguyen et al., 2006)) and operates in a positive feedback loop with circulating iron to regulate serum iron homeostasis. Interestingly, Hamp is known to be upregulated in the context of infection and inflammation but was highly downregulated (∼15-fold) after 2 ppm O3 exposure in our study. Hes1 (hairy and enhancer of split 1) was significantly downregulated in AM after 1 ppm O3 exposure (7-fold), but not after 2 ppm O3 exposure. Hes1 encodes a suppressor of transcriptional elongation of macrophage-derived chemokine genes, including CXCL1 (Shang et al., 2016), a well-established neutrophil chemokine.
On the whole, CA and AM exhibited unique transcriptional responses to O3. However, we also identified genes that were concordantly upregulated or downregulated in both CA and AM samples. Genes that were upregulated in both compartments included cyclin family members and kinetochore components (Cdc45, Cdk18, Cenpk) which are involved in DNA replication, proteases and anti-proteases (Timp1, Ctse, Prss27) which might regulate tissue regeneration and repair, and Spp1 (osteopontin), a gene that has been previously studied in the context of O3 responses (Barreno et al., 2013; Leroy et al., 2015). A smaller proportion of genes were downregulated in both compartments and spanned a wide range of categories including kinases (Nek3), actin- and microtubule-associated proteins (Espn, Tppp3, Dnah2), and chromatin-associated enzymes (Usp11, Hr).
The vote counting meta-analysis we conducted led to the identification of genes that are consistently altered in response to O3 exposure across different studies and tissue types in mice, namely Cdk1, Lcn2, and Saa3. The fact that the expression of these genes was reproducibly altered despite there being design differences between studies demonstrates that some genes can be broadly classified as “O3-responsive” genes. We also found that the overlap of CA DEGs with whole lung tissue DEGs was greater than that observed for AM DEGs, which was not unexpected given that the proportion of CA-derived cells in whole lung is greater than the proportion of AMs.
We also examined differences in concentration-response gene expression patterns in CA and AM using a Bayesian approach. Overall, a similar number of genes from CA and AM could be categorized into 1 of 8 different concentration response models with high certainty, but the relative proportions of each genes fitting each model differed considerably by compartment. While the majority of genes were categorized as positive or negative monotonic in both CA and AM, many genes in AM displayed non-monotonic expression patterns. In combination with global expression response revealed by principal components analysis, these results point to more complex exposure–gene expression response relationships in AM relative to CA.
While the methods we used enabled us to describe the transcriptional responses of 2 important lung compartments to O3, both tissue compartments are heterogeneous, and the bulk RNA-seq analysis approach we used may have masked differential contributions of particular cell types. Future studies stand to improve upon our description of gene expression changes by resolving this heterogeneity through the use of flow sorting or single-cell RNA-seq. This would be particularly useful for assigning AM origin and phenotype, particularly after O3 exposure when inflammatory monocytes have been recruited from the periphery (Francis et al., 2017). Similarly, these approaches could be applied for the many cell types that comprise the airways and resident and/or recruited immune cell populations that are tightly associated with or interdigitated within the epithelium. This approach would also address the challenge of interpreting whether gene expression in CA of O3-exposed mice is primarily due to altered transcriptional activity or a result of changes in cell composition. We also opted to use only female mice, and sex is known to have an impact on gene expression responses in many contexts, including after O3 exposure (Cabello et al., 2015; Fuentes et al., 2019; Fuentes et al., 2018; Mishra et al., 2016). Thus, additional studies will be required to determine the impact of sex on transcriptional responses in the airway epithelium and macrophages.
In summary, we have identified genes that change in the airway after O3 exposure in parallel to concentration-dependent increases in airway neutrophilia, pro-inflammatory cytokine secretion, and epithelial tissue injury. Additionally, we demonstrated that the CA and AM have both common and distinct transcriptional signatures following O3 exposure. The altered genes and pathways presented in our study increase our understanding of the molecular mechanisms that underlie respiratory toxicity to O3, and provide candidate genes and pathways for focused future study.
Data and Code Availability
All raw data and code used for analysis are deposited on Dryad (provisional doi: https://datadryad.org/review?doi=doi:10.5061/dryad.31nb080). Additionally, Supplementary material including tables, data, and figures are available at the same link. All .fastq files are deposited in the Sequence Read Archive (BioProject accession: PRJNA552482).
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to acknowledge the assistance of Kathryn McFadden (laboratory animal support), Michala Patterson (conducting airway microdissection and RNA isolation), Carlton Anderson (UNC Advanced Analytics Core, Luminex processing), the UNC High-Throughput Sequencing Facility (library preparation and RNA-seq), Amy Porter (Michigan State University Investigative Histopathology Laboratory, histology and immunohistochemistry), and Courtney Nesline and the UNC Division of Comparative Medicine.
Funding
Research reported in this publication was supported by the National Institute of Environmental Health Sciences of the National Institute of Health under awards ES024965, ES024965-S1, and P30ES010126 (University of North Carolina Center for Environmental Health and Susceptibility). Additional funds were provided by the Albert C. and Lois E. Dehn Endowment at Michigan State University for Veterinary Medicine (Pathobiology and Diagnostic Investigation).
References
- Akinbami L. J., Lynch C. D., Parker J. D., Woodruff T. J. (2010). The association between childhood asthma prevalence and monitored air pollutants in metropolitan areas, united states, 2001-2004. Environ. Res. 110, 294–301. [DOI] [PubMed] [Google Scholar]
- Anenberg S. C., Henze Daven K., Tinney V., Kinney P. L., Raich W., Fann N., Malley C. S., Roman H., Lamsal L., Duncan B., et al. (2018). Estimates of the global burden of ambient pm2.5, ozone, and no2 on asthma incidence and emergency room visits. Environ. Health Perspect. 126, 107004.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aris R. M., Christian D., Hearne P. Q., Kerr K., Finkbeiner W. E., Balmes J. R. (1993). Ozone-induced airway inflammation in human subjects as determined by airway lavage and biopsy. Am. Rev. Respir. Dis. 148, 1363–1372. [DOI] [PubMed] [Google Scholar]
- Arredouani M. S., Franco F., Imrich A., Fedulov A., Lu X., Perkins D., Soininen R., Tryggvason K., Shapiro S. D., Kobzik L. (2007). Scavenger receptors SR-AI/II and MARCO limit pulmonary dendritic cell migration and allergic airway inflammation. J. Immunol. 178, 5912–5920. [DOI] [PubMed] [Google Scholar]
- Backus G. S., Howden R., Fostel J., Bauer A. K., Cho H. Y., Marzec J., Peden D. B., Kleeberger S. R. (2010). Protective role of interleukin-10 in ozone-induced pulmonary inflammation. Environ. Health Perspect. 118, 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker G. L., Shultz M. A., Fanucchi M. V., Morin D. M., Buckpitt A. R., Plopper C. G. (2003). Assessing gene expression in lung subcompartments utilizing in situ RNA preservation. Toxicol. Sci. 77, 135–141. [DOI] [PubMed] [Google Scholar]
- Barreno R. X., Richards J. B., Schneider D. J., Cromar K. R., Nadas A. J., Hernandez C. B., Hallberg L. M., Price R. E., Hashmi S. S., Blackburn M. R., et al. (2013). Endogenous osteopontin promotes ozone-induced neutrophil recruitment to the lungs and airway hyperresponsiveness to methacholine. Am. J. Physiol. Lung Cell. Mol. Physiol. 305, L118–L129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A. K., Rondini E. A., Hummel K. A., Degraff L. M., Walker C., Jedlicka A. E., Kleeberger S. R. (2011). Identification of candidate genes downstream of tlr4 signaling after ozone exposure in mice: A role for heat-shock protein 70. Environ. Health Perspect. 119, 1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer R. N., Muller L., Brighton L. E., Duncan K. E., Jaspers I. (2015). Interaction with epithelial cells modifies airway macrophage response to ozone. Am. J. Respir. Cell Mol. Biol. 52, 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S., Madden M. C., Newman S. L., Devlin R. B., Koren H. S. (1991). Modulation of human alveolar macrophage properties by ozone exposure in vitro. Toxicol. Appl. Pharmacol. 110, 403–415. [DOI] [PubMed] [Google Scholar]
- Bernstein A. S., Rice M. B. (2013). Lungs in a warming world: Climate change and respiratory health. Chest 143, 1455–1459. [DOI] [PubMed] [Google Scholar]
- Birukova A., Cyphert-Daly J., Cumming R. I., Yu Y. R., Gowdy K. M., Que L. G., Tighe R. M. (2019). Sex modifies acute ozone-mediated airway physiologic responses. Toxicol. Sci. 169, 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondonnet R., Audard J., Belville C., Clairefond G., Lutz J., Bouvier D., Roszyk L., Gross C., Lavergne M., Fournet M., et al. (2017). Rage inhibition reduces acute lung injury in mice. Sci. Rep. 7, 7208.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg P. A. (2016). Mechanisms of the acute effects of inhaled ozone in humans. Biochim. Biophys. Acta 1860, 2771–2781. [DOI] [PubMed] [Google Scholar]
- Cabello N., Mishra V., Sinha U., DiAngelo S. L., Chroneos Z. C., Ekpa N. A., Cooper T. K., Caruso C. R., Silveyra P. (2015). Sex differences in the expression of lung inflammatory mediators in response to ozone. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L1150–L1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese E. J., Baldwin L. A. (2001). Hormesis: A generalizable and unifying hypothesis. Crit. Rev. Toxicol. 31, 353–424. [DOI] [PubMed] [Google Scholar]
- Carpenter B., Gelman A., Hoffman M. D., Lee D., Goodrich B., Betancourt M., Brubaker M., Guo J., Li P., Riddell A. (2017). Stan: A probabilistic programming language. J. Stat. Soft. 76, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che L., Jin Y., Zhang C., Lai T., Zhou T., Xia L., Tian B., Zhao Y., Liu J., Wu Y., et al. (2016). Ozone-induced IL-17A and neutrophilic airway inflammation is orchestrated by the caspase-1-IL-1 cascade. Sci. Rep. 6, 18680.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y., Abu-Ali G., Tashiro H., Brown T. A., Osgood R. S., Kasahara D. I., Huttenhower C., Shore S. A. (2019). Sex differences in pulmonary responses to ozone in mice. Role of the microbiome. Am. J. Respir. Cell. Mol. Biol. 60, 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciencewicki J. M., Verhein K. C., Gerrish K., McCaw Z. R., Li J., Bushel P. R., Kleeberger S. R. (2016). Effects of mannose-binding lectin on pulmonary gene expression and innate immune inflammatory response to ozone. Am. J. Physiol. Lung Cell. Mol. Physiol. 311, L280–L291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromar K. R., Gladson L. A., Ewart G. (2019). Trends in excess morbidity and mortality associated with air pollution above American thoracic society-recommended standards, 2008-2017. Ann. Am. Thorac. Soc. 16, 836–845. [DOI] [PubMed] [Google Scholar]
- Day D. B., Xiang J., Mo J., Li F., Chung M., Gong J., Weschler C. J., Ohman-Strickland P. A., Sundell J., Weng W., et al. (2017). Association of ozone exposure with cardiorespiratory pathophysiologic mechanisms in healthy adults. JAMA Intern. Med. 177, 1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin R. B., Folinsbee L. J., Biscardi F., Hatch G., Becker S., Madden M. C., Robbins M., Koren H. S. (1997). Inflammation and cell damage induced by repeated exposure of humans to ozone. Inhal. Toxicol. 9, 211–235. [Google Scholar]
- Devlin R. B., McKinnon K. P., Noah T., Becker S., Koren H. S. (1994). Ozone-induced release of cytokines and fibronectin by alveolar macrophages and airway epithelial cells. Am. J. Physiol. 266, L612–L619. [DOI] [PubMed] [Google Scholar]
- Devlin R. B., McDonnell W. F., Becker S., Madden M. C., McGee M. P., Perez R., Hatch G., House D. E., Koren H. S. (1996). Time-dependent changes of inflammatory mediators in the lungs of humans exposed to 0.4 ppm ozone for 2 hr: A comparison of mediators found in bronchoalveolar lavage fluid 1 and 18 hr after exposure. Toxicol. Appl. Pharmacol. 138, 176–185. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R. (2013). Star: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doni A., Stravalaci M., Inforzato A., Magrini E., Mantovani A., Garlanda C., Bottazzi B. (2019). The long pentraxin ptx as a link between innate immunity, tissue remodeling, and cancer. Front. Immunol. 10, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esseltine J. L., Laird D. W. (2016). Next-generation connexin and pannexin cell biology. Trends Cell. Biol. 26, 944–955. [DOI] [PubMed] [Google Scholar]
- Farraj, A. K., Harkema, J. R., Jan, T. R., Kaminski, N. E. (2003). Immune responses in the lung and local lymph node of A/J mice to intranasal sensitization and challenge with adjuvant-free ovalbumin. Toxicol Pathol 31,432–447. [DOI] [PubMed] [Google Scholar]
- Francis M., Groves A. M., Sun R., Cervelli J. A., Choi H., Laskin J. D., Laskin D. L. (2017). Editor's highlight: CCR2 regulates inflammatory cell accumulation in the lung and tissue injury following ozone exposure. Toxicol. Sci. 155, 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J. A. (2012). Claudins and alveolar epithelial barrier function in the lung. Ann. N. Y. Acad. Sci. 1257, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks T. J., Colby T. V., Travis W. D., Tuder R. M., Reynolds H. Y., Brody A. R., Cardoso W. V., Crystal R. G., Drake C. J., Engelhardt J., et al. (2008). Resident cellular components of the human lung: Current knowledge and goals for research on cell phenotyping and function. Proc. Am. Thorac. Soc. 5, 763–766. [DOI] [PubMed] [Google Scholar]
- Fuentes N., Cabello N., Nicoleau M., Chroneos Z. C., Silveyra P. (2019). Modulation of the lung inflammatory response to ozone by the estrous cycle. Physiol. Rep. 7, e14026.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes N., Roy A., Mishra V., Cabello N., Silveyra P. (2018). Sex-specific microRNA expression networks in an acute mouse model of ozone-induced lung inflammation. Biol. Sex Differ. 9, 18.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabehart K., Correll K. A., Yang J., Collins M. L., Loader J. E., Leach S., White C. W., Dakhama A. (2014). Transcriptome profiling of the newborn mouse lung response to acute ozone exposure. Toxicol. Sci. 138, 175–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia E., Berhane K. T., Islam T., McConnell R., Urman R., Chen Z., Gilliland F. D. (2019). Association of changes in air quality with incident asthma in children in California, 1993-2014. JAMA 321, 1906–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gent J. F., Triche E. W., Holford T. R., Belanger K., Bracken M. B., Beckett W. S., Leaderer B. P. (2003). Association of low-level ozone and fine particles with respiratory symptoms in children with asthma. JAMA 290, 1859–1867. [DOI] [PubMed] [Google Scholar]
- Gilmour M. I., Hmieleski R. R., Stafford E. A., Jakab G. J. (1991). Suppression and recovery of the alveolar macrophage phagocytic system during continuous exposure to 0.5 ppm ozone. Exp. Lung Res. 17, 547–558. [DOI] [PubMed] [Google Scholar]
- Gohil K., Cross C. E., Last J. A. (2003). Ozone-induced disruptions of lung transcriptomes. Biochem. Biophys. Res. Commun. 305, 719–728. [DOI] [PubMed] [Google Scholar]
- Greer J. R., Abbey D. E., Burchette R. J. (1993). Asthma related to occupational and ambient air pollutants in nonsmokers. J. Occup. Med. 35, 909–915. [DOI] [PubMed] [Google Scholar]
- Griffiths M. J., McAuley D. F. (2008). Rage: A biomarker for acute lung injury. Thorax 63, 1034–1036. [DOI] [PubMed] [Google Scholar]
- Groves A. M., Gow A. J., Massa C. B., Laskin J. D., Laskin D. L. (2012). Prolonged injury and altered lung function after ozone inhalation in mice with chronic lung inflammation. Am. J. Respir. Cell Mol. Biol. 47, 776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S., Castelo R., Guinney J. (2013). GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch G. E., Duncan K. E., Diaz-Sanchez D., Schmitt M. T., Ghio A. J., Carraway M. S., McKee J., Dailey L. A., Berntsen J., Devlin R. B. (2014). Progress in assessing air pollutant risks from in vitro exposures: Matching ozone dose and effect in human airway cells. Toxicol. Sci. 141, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch G. E., McKee J., Brown J., McDonnell W., Seal E., Soukup J., Slade R., Crissman K., Devlin R. (2013). Biomarkers of dose and effect of inhaled ozone in resting versus exercising human subjects: Comparison with resting rats. Biomark. Insights 8, 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch G. E., Slade R., Harris L. P., McDonnell W. F., Devlin R. B., Koren H. S., Costa D. L., McKee J. (1994). Ozone dose and effect in humans and rats. A comparison using oxygen-18 labeling and bronchoalveolar lavage. Am. J. Respir. Crit. Care Med. 150, 676–683. [DOI] [PubMed] [Google Scholar]
- Imamura M., Kawasaki T., Savchenko A. S., Ohashi R., Jiang S., Miyamoto K., Ito Y., Iwanari H., Sagara M., Tanaka T., et al. (2007). Lipopolysaccharide induced expression of pentraxin 3 in human neutrophils and monocyte-derived macrophages. Cell. Immunol. 248, 86–94. [DOI] [PubMed] [Google Scholar]
- Ito K., De Leon S. F., Lippmann M. (2005). Associations between ozone and daily mortality: Analysis and meta-analysis. Epidemiology 16, 446–457. [DOI] [PubMed] [Google Scholar]
- Jakab G. J., Spannhake E. W., Canning B. J., Kleeberger S. R., Gilmour M. I. (1995). The effects of ozone on immune function. Environ. Health Perspect. 103(Suppl. 2), 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston R. A., Mizgerd J. P., Flynt L., Quinton L. J., Williams E. S., Shore S. A. (2007). Type I interleukin-1 receptor is required for pulmonary responses to subacute ozone exposure in mice. Am. J. Respir. Cell Mol. Biol. 37, 477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara D. I., Kim H. Y., Williams A. S., Verbout N. G., Tran J., Si H., Wurmbrand A. P., Jastrab J., Hug C., Umetsu D. T., et al. (2012). Pulmonary inflammation induced by subacute ozone is augmented in adiponectin-deficient mice: Role of IL-17A. J. Immunol. 188, 4558–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrl H. R., Vincent L. M., Kowalsky R. J., Horstman D. H., O'Neil J. J., McCartney W. H., Bromberg P. A. (1987). Ozone exposure increases respiratory epithelial permeability in humans. Am. Rev. Respir. Dis. 135, 1124–1128. [DOI] [PubMed] [Google Scholar]
- Kierstein S., Poulain F. R., Cao Y., Grous M., Mathias R., Kierstein G., Beers M. F., Salmon M., Panettieri R. A., Haczku A. (2006). Susceptibility to ozone-induced airway inflammation is associated with decreased levels of surfactant protein D. Resp. Res. 7, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C. S., Alexis N. E., Rappold A. G., Kehrl H., Hazucha M. J., Lay J. C., Schmitt M. T., Case M., Devlin R. B., Peden D. B., et al. (2011). Lung function and inflammatory responses in healthy young adults exposed to 0.06 ppm ozone for 6.6 hours. Am. J. Respir. Crit. Care Med. 183, 1215–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooter I. M., Pennings J. L. A., Fokkens P. H. B., Leseman D. L. A. C., Boere A. J. F., Gerlofs-Nijland M. E., Cassee F. R., Schalk J. A. C., Orzechowski T. J. H., Schaap M. M., et al. (2007). Ozone induces clear cellular and molecular responses in the mouse lung independently of the transcription-coupled repair status. J. Appl. Physiol. 102, 1185–1192. [DOI] [PubMed] [Google Scholar]
- Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., et al. (2016). Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunes P., Holubcova Z., Kolackova M., Krejsek J. (2012). Pentraxin 3(PTX 3): An endogenous modulator of the inflammatory response. Mediators Inflamm. 2012, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin D. L., Malaviya R., Laskin J. D. (2019). Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicol. Sci. 168, 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin D. L., Sunil V. R., Gardner C. R., Laskin J. D. (2011). Macrophages and tissue injury: Agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 51, 267–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikauf G. D., Simpson L. G., Santrock J., Zhao Q., Abbinante-Nissen J., Zhou S., Driscoll K. E. (1995). Airway epithelial cell responses to ozone injury. Environ. Health Perspect. 103(Suppl. 2), 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy P., Tham A., Wong H., Tenney R., Chen C., Stiner R., Balmes J. R., Paquet A. C., Arjomandi M. (2015). Inflammatory and repair pathways induced in human bronchoalveolar lavage cells with ozone inhalation. PLoS One 10, e0127283.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F. L., Johnston R. A., Flynt L., Theman T. A., Terry R. D., Schwartzman I. N., Lee A., Shore S. A. (2006). Increased pulmonary responses to acute ozone exposure in obese db/db mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L856–L865. [DOI] [PubMed] [Google Scholar]
- Mathews J. A., Krishnamoorthy N., Kasahara D. I., Cho Y., Wurmbrand A. P., Ribeiro L., Smith D., Umetsu D., Levy B. D., Shore S. A. (2017). Il-33 drives augmented responses to ozone in obese mice. Environ. Health Perspect. 125, 246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews J. A., Kasahara D. I., Ribeiro L., Wurmbrand A. P., Ninin F. M. C., Shore S. A. (2015). Γδ T cells are required for M2 macrophage polarization and resolution of ozone-induced pulmonary inflammation in mice. PLoS One 10, e0131236.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell R., Berhane K., Gilliland F., London S. J., Islam T., Gauderman W. J., Avol E., Margolis H. G., Peters J. M. (2002). Asthma in exercising children exposed to ozone: A cohort study. Lancet 359, 386–391. [DOI] [PubMed] [Google Scholar]
- McDonnell W. F., Abbey D. E., Nishino N., Lebowitz M. D. (1999). Long-term ambient ozone concentration and the incidence of asthma in nonsmoking adults: The AHSMOG study. Environ. Res. 80, 110–121. [DOI] [PubMed] [Google Scholar]
- Medina-Ramon M., Zanobetti A., Schwartz J. (2006). The effect of ozone and PM10 on hospital admissions for pneumonia and chronic obstructive pulmonary disease: A national multicity study. Am. J. Epidemiol. 163, 579–588. [DOI] [PubMed] [Google Scholar]
- Mikerov A. N., Gan X., Umstead T. M., Miller L., Chinchilli V. M., Phelps D. S., Floros J. (2008). Sex differences in the impact of ozone on survival and alveolar macrophage function of mice after Klebsiella pneumoniae infection. Respir. Res. 9, 24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirowsky J. E., Carraway M. S., Dhingra R., Tong H., Neas L., Diaz-Sanchez D., Cascio W., Case M., Crooks J., Hauser E. R., et al. (2017). Ozone exposure is associated with acute changes in inflammation, fibrinolysis, and endothelial cell function in coronary artery disease patients. Environ. Health 16, 126.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra V., DiAngelo S. L., Silveyra P. (2016). Sex-specific IL-6-associated signaling activation in ozone-induced lung inflammation. Biol. Sex Differ. 7, 16.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudway I. S., Kelly F. J. (2000). Ozone and the lung: A sensitive issue. Mol. Aspects Med. 21, 1–48. [DOI] [PubMed] [Google Scholar]
- Nadadur S. S., Costa D. L., Slade R., Silbjoris R., Hatch G. E. (2005). Acute ozone-induced differential gene expression profiles in rat lung. Environ. Health Perspect. 113, 1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelon B., Dunson D. B. (2004). Bayesian isotonic regression and trend analysis. Biometrics 60, 398–406. [DOI] [PubMed] [Google Scholar]
- Nguyen N. B., Callaghan K. D., Ghio A. J., Haile D. J., Yang F. (2006). Hepcidin expression and iron transport in alveolar macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L417–L425. [DOI] [PubMed] [Google Scholar]
- Nishimura K. K., Iwanaga K., Oh S. S., Pino-Yanes M., Eng C., Keswani A., Roth L. A., Nguyen E. A., Thyne S. M., Farber H. J., et al. (2016). Early-life ozone exposure associated with asthma without sensitization in Latino children. J. Allergy Clin. Immunol. 138, 1703–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C. (2017). Salmon: Fast and bias-aware quantification of transcript expression using dual-phase inference. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendino K. J., Meidhof T. M., Heck D. E., Laskin J. D., Laskin D. L. (1995). Inhibition of macrophages with gadolinium chloride abrogates ozone-induced pulmonary injury and inflammatory mediator production. Am. J. Respir. Cell Mol. Biol. 13, 125–132. [DOI] [PubMed] [Google Scholar]
- Pfister G. G., Walters S., Lamarque J.-F., Fast J., Barth M. C., Wong J., Done J., Holland G., Bruyère C. L. (2014). Projections of future summertime ozone over the U.S. J. Geophys. Res. Atmos. 119, 2013JD020932. [Google Scholar]
- Plopper C. G., Chu F. P., Haselton C. J., Peake J., Wu J., Pinkerton K. E. (1994). Dose-dependent tolerance to ozone. I. Tracheobronchial epithelial reorganization in rats after 20 months’ exposure. Am. J. Pathol. 144, 404–420. [PMC free article] [PubMed] [Google Scholar]
- Pryor W. A., Squadrito G. L., Friedman M. (1995). The cascade mechanism to explain ozone toxicity: The role of lipid oxidation products. Free Radic. Biol. Med. 19, 935–941. [DOI] [PubMed] [Google Scholar]
- Ramasamy A., Mondry A., Holmes C. C., Altman D. G. (2008). Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 5, e184.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera A., Barbon M., Noguchi Y., Reuter L. M., Schneider S., Speck C. (2017). From structure to mechanism-understanding intiation of DNA replication. Genes Dev. 31, 1073–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelegle E. S., Morales C. A., Walby W. F., Marion S., Allen R. P. (2009). 6.6-hour inhalation of ozone concentrations from 60 to 87 parts per billion in healthy humans. Am. J. Respir. Crit. Care Med. 180, 265–272. [DOI] [PubMed] [Google Scholar]
- Shang Y., Coppo M., He T., Ning F., Yu L., Kang L., Zhang B., Ju C., Qiao Y., Zhao B., et al. (2016). The transcriptional repressor Hes1 attenuates inflammation via regulating transcriptional elongation. Nat. Immunol. 17, 930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. J., Walsh L., Higuchi M., Kelada S. (2019). Development of a large-scale computer-controlled ozone inhalation exposure system for rodents. Inhal. Toxicol. 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strosnider H. M., Chang H. H., Darrow L. A., Liu Y., Vaidyanathan A., Strickland M. J. (2019). Age-specific associations of ozone and fine particulate matter with respiratory emergency department visits in the United States. Am. J. Respir. Crit. Care Med. 199, 882–890. [DOI] [PubMed] [Google Scholar]
- Sunil V. R., Francis M., Vayas K. N., Cervelli J. A., Choi H., Laskin J. D., Laskin D. L. (2015). Regulation of ozone-induced lung inflammation and injury by the β-galactoside-binding lectin galectin-3. Toxicol. Appl. Pharmacol. 284, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil V. R., Patel-Vayas K., Shen J., Laskin J. D., Laskin D. L. (2012). Classical and alternative macrophage activation in the lung following ozone-induced oxidative stress. Toxicol. Appl. Pharmacol. 263, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney T. E., Lofgren S., Khatri P., Rogers A. J. (2017). Gene expression analysis to assess the relevance of rodent models to human lung injury. Am. J. Respir. Cell Mol. Biol. 57, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetreault L. F., Doucet M., Gamache P., Fournier M., Brand A., Kosatsky T., Smargiassi A. (2016). Childhood exposure to ambient air pollutants and the onset of asthma: An administrative cohort study in Quebec. Environ. Health Perspect. 124, 1276–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston G. D., Lippmann M., Scott M. B., Fine J. M. (1997). Summertime haze air pollution and children with asthma. Am. J. Respir. Crit. Care Med. 155, 654–660. [DOI] [PubMed] [Google Scholar]
- Tighe R. M., Li Z., Potts E. N., Frush S., Liu N., Gunn M. D., Foster W. M., Noble P. W., Hollingsworth J. W. (2011). Ozone inhalation promotes CX3CR1-dependent maturation of resident lung macrophages which limit oxidative stress and inflammation. J. Immunol. 187, 4800–4808. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Vandenberg L. N., Colborn T., Hayes T. B., Heindel J. J., Jacobs D. R., Lee D.-H., Shioda T., Soto A. M., vom Saal F. S., Welshons W. V., et al. (2012). Hormones and endocrine-disrupting chemicals: Low-dose effects and nonmonotonic dose responses. Endocr. Rev. 33, 378–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhein K. C., McCaw Z., Gladwell W., Trivedi S., Bushel P. R., Kleeberger S. R. (2015). Novel roles for notch3 and notch4 receptors in gene expression and susceptibility to ozone-induced lung inflammation in mice. Environ. Health Perspect. 123, 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Wang S., Manzer R., McConville G., Mason R. J. (2006). Ozone induces oxidative stress in rat alveolar type II and type I-like cells. Free Radic. Biol. Med. 40, 1914–1928. [DOI] [PubMed] [Google Scholar]
- Wang M., Aaron C. P., Madrigano J., Hoffman E. A., Angelini E., Yang J., Laine A., Vetterli T. M., Kinney P. L., Sampson P. D., et al. (2019). Association between long-term exposure to ambient air pollution and change in quantitatively assessed emphysema and lung function. JAMA 322, 546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward W. O., Ledbetter A. D., Schladweiler M. C., Kodavanti U. P. (2015). Lung transcriptional profiling: Insights into the mechanisms of ozone-induced pulmonary injury in Wistar Kyoto rats. Inhal. Toxicol. 27, 80–92. [DOI] [PubMed] [Google Scholar]
- Win-Shwe T.-T., Fujitani Y., Kyi-Tha-Thu C., Furuyama A., Michikawa T., Tsukahara S., Nitta H., Hirano S. (2014). Effects of diesel engine exhaust origin secondary organic aerosols on novel object recognition ability and maternal behavior in BALB/c mice. Int. J. Environ. Res. Public Health 11, 11286–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y., Liang M., Munesue S., Deguchi K., Harashima A., Furuhara K., Yuhi T., Zhong J., Akther S., Goto H., et al. (2019). Vascular rage transports oxytocin into the brain to elicit its maternal bonding behaviour in mice. Commun. Biol. 2, 76.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data and code used for analysis are deposited on Dryad (provisional doi: https://datadryad.org/review?doi=doi:10.5061/dryad.31nb080). Additionally, Supplementary material including tables, data, and figures are available at the same link. All .fastq files are deposited in the Sequence Read Archive (BioProject accession: PRJNA552482).