

Abstract
It is now becoming clear that human metabolism is extremely plastic and varies substantially between healthy individuals. Understanding the biochemistry that underlies this physiology will enable personalized clinical interventions related to metabolism. Mitochondrial quality control and the detailed mechanisms of mitochondrial energy generation are central to understanding susceptibility to pathologies associated with aging including cancer, cardiac and neurodegenerative disease. A precision medicine approach is also needed to evaluate the impact of exercise or caloric restriction on health. In this review, we discuss how technical advances in assessing mitochondrial genetics, cellular bioenergetics and metabolomics offer new insights into developing metabolism-based clinical tests and metabolotherapies. We discuss informatics approaches, which can define the bioenergetic-metabolite-interactome and how this can help define healthy energetics. We propose that a personalized medicine approach that integrates metabolism and bioenergetics with physiologic parameters is central for understanding the pathophysiology of diseases with a metabolic etiology. New approaches that measure energetics and metabolomics from cells isolated from human blood or tissues can be of diagnostic and prognostic value to precision medicine. This is particularly significant with the development of new metabolotherapies, such as mitochondrial transplantation, which could help treat complex metabolic diseases.
Keywords: translational research, mitochondria, metabolomics, bioenergetic-metabolite interactome, personalized medicine
INTRODUCTION
Worldwide demographic changes and general improvements in healthcare have resulted in a major shift in causes of human morbidity and mortality, from acute illnesses to chronic, degenerative diseases. A quarter of all Americans and two-thirds of older adults suffer from such conditions, which include diabetes, cardiovascular disease, and neurological disorders. Although treatment of these diseases and the aging population account for >70% of the healthcare budget in the developed world, there appears to be no long-term plan for how to deal with the current and impending healthcare burden. Personalized medicine offers a unique solution. Tailoring therapies to each individual patient should increase the efficacy and efficiency of medical treatment and alleviate strain on the healthcare system. This approach, however, has mainly been limited to the use of genetic information to characterize or diagnose a patient or population, which is not sensitive to environmental systems or ‘upstream’ pathophysiologic changes. Because metabolism represents the convergence of all signals—consisting of genomic, proteomic, environmental, and metabolic effectors—assessing the metabolotype of patients would provide the most complete and useful information possible to scientists and clinicians. Not only could such an approach be useful for diagnosing or predicting disease, but it could also be used to examine toxicity due to environmental exposures, assess the relative efficacy (or toxicity) of therapies and relate metabolism and energetics to healthy aging.
The etiology underlying individual susceptibility to diseases with a metabolic basis involves a complex interaction among genetic, lifestyle and environmental factors. Importantly, “normal” genetic polymorphisms (i.e. not pathogenic mutations), that under conditions of environmental stress and aging are linked with increased inflammation and metabolic dysfunction, will affect bioenergetics. It follows that we need to establish clear definitions of the bioenergetic phenotypes that discriminate healthy and diseased human populations. This is a challenging problem because, unlike laboratory animal models or cell monocultures, there is an intrinsic variability in metabolism governed by genetic and environmental factors in the absence of any underlying pathology. Understanding what contributes to metabolic variability or plasticity could lead to a mechanistic approach to assess risk for the development of chronic pathologies such as diabetes, cardiovascular disease and neurodegenerative disease. Underlying the maintenance of this metabolic plasticity are a variety of quality control mechanisms which both sense and dynamically regulate bioenergetic function and correct defective processes.
These critical factors include changes in cellular metabolism that are responsive to changes in the extra- and intra-cellular environment (e.g. caloric intake and physical activity). “Normal” metabolic responses can be genetically influenced by both the mitochondrial and nuclear genomes and thus may be linked not only to racial differences in disease susceptibility, but to differences in oxidant and/or inflammatory response between individuals (Dunham-Snary et al., 2015). Consequently, a broader understanding of metabolic plasticity and its control will be important therapeutically. It has been widely assumed that bioenergetic health assessments have to be localized to specific organs or tissues (e.g., skeletal muscle). However, the clinical utility of these tests, particularly in preventative medicine, is severely limited because of significant risk for the patient and the high cost. An alternative approach is to use tests of bioenergetic function in circulating cells (e.g. peripheral blood leukocytes, platelets) as a surrogate for metabolic diseases having a systemic impact. We discuss progress in this area in the context of the role of metabolic measures in precision medicine.
In this review, we also develop the concept that both environment and genetics influence cellular bioenergetics, mitochondrial function, and inflammatory processes and are dependent on the individual. We outline how emerging technologies make quantification of metabolic health feasible by establishing new clinical parameters, which characterize mitochondrial genetics, bioenergetics and metabolomics in human subjects, thereby introducing the concept of the bioenergetic-metabolite-interactome. Metabolotherapies are of particular interest because they represent an unusual pharmacological paradigm in which a class of agent can be applied with equal efficacy to pathologies that share common metabolic mechanisms but are otherwise different. We will discuss the emerging notion that mitochondria can be transferred between cells through a controlled process and the implications for mitochondrial transplantation. These concepts and some key examples will be discussed. In the first section, we outline the current understanding of mitochondrial genetics and some of the implications for translational research.
Mitochondrial Genetics in Metabolic Plasticity
In humans, the mitochondrial DNA (mtDNA) is a double-stranded, 16.5 kb circular molecule that encodes 37 genes, 13 of which are subunits for the electron transport chain and ATP synthase, in addition to 22 tRNAs and 2 rRNAs that are required as translational machinery (the mtDNA has a different genetic code). Each mitochondrion contains ~4–10 copies of mtDNA, and each cell can contain a few to thousands of mitochondria; consequently a single cell can possess a few hundred to thousands of mtDNA copies (Krzywanski et al., 2011). Pathogenic mtDNA mutations associated with disease were reported almost 3 decades ago, and from a genetic perspective, confirmed the importance of the mtDNA in human disease (Cortopassi et al., 1990; Wallace et al., 1988). Diseases caused by pathogenic mtDNA mutations commonly present as complex syndromes with diverse clinical presentations including variability in the age of onset, symptoms, and severity (Krzywanski et al., 2011). Patients with more severe presentations of mitochondrial disease commonly display symptoms associated with defects in their muscle and central nervous tissues, such as weakness, loss of vision, cognitive delay, dementia and seizures (Bray et al., 2017). The contributing factors to this variability are due to: i) the type of mtDNA mutation, ii) age of the individual, iii) gender, iv) pre-existent risk factors, v) “heteroplasmy” and different energetic requirements of the affected tissues. Pathogenic missense mutations in the mtDNA tend to be associated with less severe disease phenotypes relative to those involving tRNAs, rRNAs, or deletions (Wallace, 2015). This is because a single mutation within a coding gene impacts a single polypeptide, whereas mutations involving the translational machinery (tRNAs and rRNAs) or mtDNA deletions can impact multiple genes. Mitochondrial function naturally declines with age, and hence disease development and severity usually advance with age (Trounce et al., 1989; Wallace et al., 1995). In general, it has also been noted that males have greater sensitivity to the clinical effects of pathogenic mtDNA mutations; possibly due to the hormonal differences between males and females that have been noted to preserve aspects of mitochondrial function (reviewed in (Demarest et al., 2015)). However, additional mechanisms are likely since sex differences can be modeled in the absence of circulating hormones (Li et al., 2005). Due to its electrochemical polarity, high concentration of metalloproteins and high membrane content, many environmental toxicants also accumulate in the mitochondrion, and/or directly inhibit mitochondrial metabolism (e.g., arsenic, cadmium, cyanide, azide, rotenone, etc.); therefore, by inhibiting organelle function, the impact of pathogenic mutations is increased (Fetterman et al., 2017; Prezant et al., 1993). Similarly, increased mitochondrial dysfunction and damage have been linked to many disease risk factors (e.g., hypercholesterolemia, diabetes, smoking). Finally, when more than one type of mtDNA sequence occurs within a cell or tissue, this heteroplasmy adds an additional complexity to the response to xenobiotics (Sharpley et al., 2012). For example, if one of these mtDNAs contains a pathogenic mutation, the ratio of pathogenic to “normal” influences the capacity of the mitochondrion to meet cellular demands which are dictated by the minimal requirements of that tissue (Krzywanski et al., 2011). Collectively, these factors influence mitochondrial function, disease susceptibility and the penetrance of a clinical phenotype.
The majority of studies examining the role of the mtDNA in disease have focused upon pathogenic mtDNA mutations. As summarized above, these findings have provided insights about how pathogenic mtDNA mutations play an important role in cellular homeostasis and their relevance to disease. However, the role of “normal” or non-pathogenic mtDNA sequence variation and its role on metabolic plasticity has been a more difficult concept to embrace from a molecular medicine perspective. This was a concept originally proposed by Wallace and colleagues based upon evolutionary genetic considerations and the biological fact that the eukaryotic cell is the consequence of an estimated 1.5 billion years of co-evolution between the mitochondrion and nucleus (Ballinger, 2013; Wallace, 2005). These “mito-Mendelian” genetic interactions enabled genetic adaptation to change in environment by modulating the utilization of electron flow molecular and thermal energy (ATP and heat), and signaling (oxidants and metabolites) important for survival (Figure 1). This paradigm for explaining the genetic – physiologic basis in medicine remains controversial despite a growing number of studies showing the impact of different mtDNA backgrounds on disease, using both in vitro and in vivo approaches (Dunham-Snary et al., 2018; Fetterman et al., 2013; Ishikawa et al., 2008). Early studies in the cancer literature were among the earliest reports showing the influence of the mtDNA on tumor growth and metastasis using in vitro approaches (Ishikawa et al., 2008; Petros et al., 2005). Additional studies have shown mitochondrial – nuclear incompatibility (Roubertoux et al., 2003), and adaptation (Scott et al., 2011). These findings are consistent with the hypothesis that genes associated with metabolism (both mitochondrial and nuclear encoded) are important for adaptation.
Figure 1: Mitochondrial – Nuclear genetic interactions.

(A) Endosymbiotic origins of the eukaryote. Aerobic α-proteobacteria entered into an endosymbiotic relationship with a “primitive” host cell ~1.5 billion years ago, creating a “proto-eukaryote”. As the endosymbiont and host genomes co-evolved, the former lost or transferred the majority of its genetic material to the “nucleus”, while retaining key catalytic subunits required to electron transport, forming the contemporary eukaryote, whereby mitochondrial and nuclear genomes co-evolve in response to environmental challenges. (B) Example of how this co-evolution manifests in metabolic changes – under low (blue) versus high (orange) conditions of environmental stress (e.g. nutrient availability), co-evolution of both genomes associate with differences in mitochondrial metabolism (low or high economy). Economy is linked to dedicated electron (e-) flow for ATP production, in that low economies require greater dedicated e- flow for ATP production to generate equivalent amounts of molecular energy (ATP), compared to those with high economy. Similarly, under conditions of positive energy balance, high and low economy is associated with increased and decreased oxidant production, respectively. Economy is also linked with energy loss (heat). Collectively, these differences may impact inflammatory response in that a pro-oxidative mitochondrion is more prone to generate oxidants and incur damage, releasing components (e.g. damage associated molecular patterns - DAMPs) known to trigger host immune response. (C) Mitochondrial – nuclear genetic interaction and disease susceptibility. The figure illustrates three different mitochondrial genetic combinations with the same nuclear genetic background (Nuclear Genotype A). Each different mtDNA mutation (indicated by red, white, and green stars) associated with the same nuclear background exhibits different degrees of biologic fitness and metabolism (e.g. dedicated e- flow). These differences will influence cellular response to a stress factor and the penetrance of the mito-nuclear genetic based response, and thus disease risk (indicated by black arrows).
These interactions can be demonstrated experimentally in the Mitochondrial – Nuclear eXchanged (MNX) models. These systems directly generate reciprocal combinations of different mtDNA-nuclear DNA combinations and have shown that mtDNA background significantly influences disease development in mice having isogenic nuclear backgrounds in vivo (Dunham-Snary et al., 2018; Fetterman et al., 2013). They also provide evidence that mtDNA – nDNA combination can be important for overall mitochondrial “economy” and thus, impacts organelle bioenergetic efficiency and oxidant generation (Fetterman et al., 2013). It has recently been shown that whole body metabolism, body composition and nuclear gene expression are influenced by mtDNA background between isogenic (nuclear) animals (Dunham-Snary et al., 2018). The observation that nuclear transcriptome can be influenced by mtDNA background provides a complementary explanation for the “common genes, common disease” hypothesis employed to explain individual variation in common disease susceptibility (Hirschhorn et al., 2005). As illustrated in Figure 1, changes in mitochondrial function consequent on mitochondrial–nuclear genetic co-evolution impact metabolic homeostasis, and thus, can have a myriad of downstream consequences including redox signaling, innate immunity, and gene expression that collectively can modulate common disease susceptibility. The significance of observing mtDNA–linked differential gene expression in the nuclear genome could be altered “penetrance” of the nuclear genotype (Dunham-Snary et al., 2018). These concepts are consistent with the studies showing that different mtDNA–nDNA combinations in MNX models can have altered oxidative stress levels, resistance to surgically induced heart failure, different serum lipid levels, adiposity, and differential tumor latency and metastasis relative to nuclear isogenic controls having different mtDNA backgrounds (Betancourt et al., 2014; Feeley et al., 2015; Fetterman et al., 2013). One of the interesting features found in these studies is that certain mtDNA backgrounds (e.g. the C57/BL6) appear to have a “hyper-responder” effect to pro-inflammatory stimuli (Dunham-Snary et al., 2018).
Mitochondrial Genetics and Innate Immunity
Associations with mtDNA sequence have also been linked to sepsis susceptibility and other inflammatory disorders [reviewed in (West et al., 2017)]. This suggests a more pivotal role for mtDNA sequences in controlling metabolic plasticity (and thus, mitochondrial health). The mitochondrion has characteristics reminiscent of its bacterial ancestry. For example, similar to prokaryotes, initiation of translation of mtDNA encoded proteins requires N-formylated methionine; under conditions of stress, N-formyl peptides can be released from the organelle and bind specific receptors that trigger an inflammatory response (Spencer et al., 2004). Similarly, the mitochondrial inner membrane contains a unique phospholipid, cardiolipin, which is also found in prokaryotic membranes, but is limited to the mitochondrion in eukaryotes. While primarily considered a structural component in the inner membrane, cardiolipin can also drive the innate immune response upon its release or externalization from the organelle (Dudek, 2017; Ren et al., 2014; Shen et al., 2015). It is also clear that outside the mitochondrion, whether intra- or inter-cellular, the mtDNA serves as an endogenous ligand that activates innate immune response pathways (Zhang et al., 2010). A number of studies have now shown that mtDNA outside the mitochondrion in the cytoplasm, extracellular space or circulation, interacts with multiple receptors to activate pro-inflammatory and type I interferon responses (Lood et al., 2016; Rongvaux et al., 2014; West et al., 2015; White et al., 2014). While beyond the scope of this review, many reviews are available that address the role of these mitochondria-derived agonists, collectively considered damage associated molecular patterns (DAMPs), in innate immunity (Banoth et al., 2018; West et al., 2017). While it has become evident that the mitochondrion plays an important role in the regulation of host immune response (reviewed in (Banoth et al., 2018; Picca et al., 2017; West et al., 2017)), the basis for these differences is not yet clear; one possibility is that because different mtDNA–nDNA combinations can modulate the metabolic characteristics of the organelle, they indirectly influence of immune response.
Mito-Nuclear Epigenetics
As outlined above, mitochondrial function is dependent upon the coordinated expression of gene products from both the mtDNA and nDNA, which occurs in a bidirectional manner, and is influenced by changes in environmental stimuli and cellular metabolism. Collectively, this “Mitonuclear” communication can regulate gene expression via changes in the epigenome. While reviewed in greater detail elsewhere (Matilainen et al., 2017), mitochondrial metabolism can both regulate, and be regulated by, the epigenome. For example, the degree of histone acetylation influences nuclear gene expression through the regulation of chromatin structure (Allis et al., 2016; Zentner et al., 2013). Because histone acetylation utilizes acetyl-CoA, mitochondrial metabolism (which regulates intracellular ATP, and thus acetyl-CoA, and the levels of metabolites required for histone acetyltransferases and deacetylases) has the capacity to regulate acetyl-CoA levels. Similarly, mitochondrial function can also regulate histone/DNA methylation by controlling the levels of S-adenosyl methionine (SAM; generation of SAM is dependent upon both the folate cycle and ATP levels, which are controlled by mitochondrial metabolism) which is required for both histone and DNA methylation. Finally, a variant of DNA demethyltransferase 1 (DNMT1), and enzymes important for demethylation have been found in the mitochondrion—consistent with reports that the mtDNA has distinct methylation patterns (no or very low levels of CpG methylation) from the nuclear DNA (Bellizzi et al., 2013; Hong et al., 2013; Shock et al., 2011). The degree of mtDNA methylation, and the physiologic significance of mtDNA methylation, still needs to be resolved. Interestingly, it has been suggested that epigenetic regulation of the mtDNA is a factor in aging, and its methylation status may be a biomarker for disease onset and/or progression (Iacobazzi et al., 2013). Because mitochondrial function has been shown to decline with age, and differences in metabolism and gene expression linked with mtDNA–nDNA background combination, it is reasonable to consider they are integrated in a manner that can also impact the epigenome (Dunham-Snary et al., 2018).
Integration of Metabolism with Redox Biology
The understanding of metabolism has progressed from the early studies of Lavoisier, Liebig and Voit, which focused on primary roles of oxygen (Lusk), to the expanded roles of metabolism in the current era. The late 1930s through the early 1960s witnessed a dramatic escalation in metabolic research, primarily due to discovery of metabolic pathways. The identification of the citric acid cycle (Krebs et al.), the establishment of reproducible methods for studying mitochondrial metabolism (Chance et al.; Chance et al.; Lardy et al.; Schneider), and the introduction of the chemiosmotic hypothesis for oxidative phosphorylation (Mitchell) paved the way for experimental interrogation of metabolism in biological systems. More recently, technological advances in high throughput bioenergetics measurements, metabolomics, and stable isotope tracing have expanded our awareness of the mechanisms by which metabolism modulates tissue health and remodeling. These new methodologies also paved the way for the paradigm that loss of metabolic network fidelity, or plasticity, is a cause of chronic disease and emphasizes the need to understand metabolic networks in healthy subjects.
These new methodologies also paved the way for the paradigm that loss of metabolic network fidelity, or plasticity, is a cause of chronic disease and emphasizes the need to understand metabolic networks in healthy subjects. This paradigm is based on the idea that, in health, the fluxes of intermediary metabolic pathways are appropriately balanced, and that prior to and during disease, these pathways become dysregulated, which results in progressive cell and tissue dysfunction. The concept has both predictive and explanatory value, and the implications of sustained or progressive defects in metabolic pathways are considerable. Similarly, (Lopez-Otin et al.), have recently proposed that aging favors “an imbalance in [the] metabolic landscape that self-amplifies and eventually becomes clinically manifest.” This is interesting because many of the chronic diseases that plague our society (e.g., diabetes, cardiovascular disease, neurodegeneration) are, at a fundamental level, diseases of accelerated aging. Defects in the metabolic system or exposure to a toxicological challenge from the environment could dysregulate metabolism and the networks associated with them; it might even lead to the structurally different metabolites being made, a phenomenon that has been termed metabolic error (Nadtochiy et al.; Rzem et al., 2015).
Metabolic plasticity or fidelity could be changed or compromised in several ways. Because metabolism and its regulatory networks are needed to not only provide useable energy, i.e., ATP, but to also build or repair cells and tissues, both catabolic and anabolic metabolic pathways should be synchronized and programmed to fulfill specific cellular tasks. For example, for proliferation, nucleotides must be synthesized to meet the demands for DNA synthesis and repair, and amino acids are required to make proteins and enzymes. Phospholipids must be made to renovate membranes and provide the building blocks for new ones. Systemically, the circulating substrates for intracellular metabolism (i.e. dietary sugars, fats, and proteins) are also regulated to optimize cell growth and resistance to stress (Fulghum et al., 2018; Gibb et al., 2018). Thus, the system is finely tuned to consume and produce the appropriate metabolites, in the optimal amounts, at defined times, and in the appropriate relationships with other metabolites. Environmental or genetic factors that dysregulate the bioenergetic-metabolite interactome could decrease the capacity of the metabolic network to adapt, thereby causing progressive loss of cell and tissue function.
Beyond energy provision and building block synthesis, metabolism is also critical for regulating the redox state of the cell. Several metabolic pathways regulate the NAD+/NADH and NADP+/NADPH redox couples in the cell. Whereas catabolism couples substrate oxidation (e.g., via glycolysis and the Krebs cycle) to NADH generation, which is used to support Complex I-driven oxidative phosphorylation, anabolism requires NADP(H) for the de novo biosynthesis of nucleic acids, fatty acid acyl chains, cholesterol and steroid hormones (Fessel et al., 2018; Nakamura et al., 2012). NADPH is also used to power redox defenses and maintain protein thiol redox status (Jones et al., 2015). In mammals, NAD+ is synthesized through three pathways: the de novo synthesis pathway, the Preiss-Handler pathway, and the salvage pathway, utilizing four precursor molecules: tryptophan, nicotinic acid, nicotinamide, and nicotinamide riboside. NADP+ is biosynthesized via NAD kinase, and is also generated via metabolic sources (Xiao et al., 2018). The largest source of cytosolic NADPH in proliferating cells appears to be the oxidative pentose phosphate pathway, where glucose 6-phosphate dehydrogenase catalyzes the conversion of glucose 6-phosphate to 6-phosphogluconate, which is then further metabolized to ribose 5-phosphate by 6-phosphogluconate dehydrogenase (Lunt et al., 2011). These reactions couple the oxidation of glucose 6-phosphate and 6-phosphogluconate to the reduction of NADP+ (to NADPH). Interestingly, serine-driven one-carbon metabolism also contributes significantly to NADPH levels: methylene tetrahydrofolate oxidation to 10-formyl tetrahydrofolate is coupled to the reduction of NADP+ to NADPH (Fan et al., 2014). Other enzymes that contribute to NAD(P)/NAD(P)H balance in cells include anaplerotic enzymes (e.g., malic enzyme, glutamate dehydrogenase), the nicotinamide nucleotide transhydrogenase, and several shuttle systems (e.g., the malate-aspartate, glycerol 3-phosphate, isocitrate-α-ketoglurarate shuttles) (Xiao et al., 2018).
Changes in metabolic flux configurations must occur for a cell to respond to stress, to grow, or to proliferate. Although the catabolic processes that support energy production are important for maintaining cell viability under conditions of stress (Dranka et al., 2010; Hill et al., 2009; Sansbury et al., 2011), the changes in anabolic pathways that must occur for a cell to grow, proliferate, or secrete extracellular matrices are less well understood. Studies in cancer cells show that although ATP is required for normal homeostasis, there is little need for additional ATP to fuel for proliferation. Rather, proliferating cells exhibit increases in anabolic metabolism (Vander Heiden et al., 2009). The synthesis of nucleotides, sugar donors (UDP-GlcNAc), amino acids, and phospholipids integrate tightly with the catabolic pathways that provide ATP to fuel cellular work. Moreover, phosphoenolpyruvate carboxykinase isoforms can drive carbon from the Krebs cycle back into the glycolytic pathway, where the carbon can be allocated for biosynthetic reactions (Montal et al., 2015; Vincent et al., 2015). Such metabolic reprogramming is consistent with the need for building blocks for cell membranes, proteins, and nucleic acids in daughter cells (Figure 2). In most proliferating cells, glucose and glutamine contribute most to the core metabolic functions that support proliferation; however, recent studies indicate that other substrates, including branched chain amino acids (BCAAs), lactate, serine, and glycine, also provide material resources required for replication (DeBerardinis et al., 2016; White, 2013). Further studies are required to elucidate how changes in metabolism regulate not only cell proliferation, but also tissue growth and adaptation.
Figure 2: Simplified model of the metabolic changes occurring in non-proliferative and proliferative cells.

To support proliferation, cells take up more glucose and amino acids such as glutamine, which provide the carbon and nitrogen sources for catabolic processes and biosynthetic reactions. The pentose phosphate pathway (blue) supports nucleotide biosynthesis and regenerates NADPH, which provides reducing power to biosynthetic and redox pathways. The hexosamine biosynthetic pathway (green) produces UDP-GlcNAc, which is important for the glycosylation reactions that regulate protein folding and function. While the glycerol backbone of phospholipids is synthesized via the glycolytic precursor dihydroxacetone phosphate (DHAP), the fatty acyl chains can be synthesized via carbon engendered in the Krebs cycle (brown). The serine biosynthesis pathway is also important not only for serine and glycine synthesis, but for the formation of methyl donors such as methylene tetrahydrofolate and for NADPH generation (yellow).
Metabolite signaling is an additional way that metabolism contributes to cell proliferation and tissue growth and adaptation. Metabolites contribute to both extracellular and intracellular signaling processes. For example, intracellular metabolites such as glucose 6-phosphate, 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR), and AMP regulate the activities of mammalian target of rapamycin (mTOR) and AMP-activated kinase (AMPK) (Hurlimann et al., 2011; Kundu et al., 2015; Roberts et al., 2014; Sen et al., 2013; Sullivan et al., 1994). Similarly, circulating metabolites can function as robust signaling molecules. Fatty acids, such as palmitoleate (C16:1n7) liberated during exercise, promote cardiac growth potentially by activating G-protein-coupled receptors (GPCRs), Akt, or nuclear receptors (Foryst-Ludwig et al., 2015). As has recently been recognized, gut microbiota-derived metabolites, bile acids, and numerous metabolites of intermediary metabolism (e.g., succinate, lactate, kynurenic acid) as extracellular signaling molecules through binding to cognate GPCRs (Husted et al., 2017). This realization has led to a new, burgeoning field that is likely to provide many exciting insights into the regulation of metabolism.
Platelets and Peripheral Blood Cells as Metabolic Biomarkers
The concept of measuring “bioenergetic health” as a diagnostic and/or prognostic clinical tool has gained significant traction over recent years (Cardenes et al., 2014; Chacko et al., 2014; Kramer et al., 2014; Tyrrell et al., 2016; Zharikov et al., 2013). However, a major barrier to understanding the role of mitochondria in disease pathogenesis is the complexity of assessing mitochondrial function in humans. Traditionally, muscle biopsies, skin grafts to culture fibroblasts, and 31P-NMR have been utilized to measure mitochondrial function. However, these methods are either highly invasive or cost and expertise prohibitive, making them impractical for clinical throughput and precision medicine. Thus, it has become necessary to explore and develop alternative tools to assess bioenergetics in human cohorts. The measurement of bioenergetics/mitochondrial function in circulating blood cells has emerged as a viable option in this regard and represents a minimally invasive method that could be utilized for translational research, particularly in large clinical trials with multiple time points, as well as ultimately developed for clinical use in personalized medicine.
Circulating blood cells and platelets are an attractive source of human mitochondria due to the relative ease of acquiring them via a simple blood draw. While red blood cells lose their mitochondria during maturation (Moras et al., 2017), platelets and leukocytes are abundant within the blood and contain fully functional mitochondria and glycolytic machinery (Chacko et al., 2013; Kramer et al., 2014). Studies as early as the 1960s optimized mitochondrial isolation from circulating cells primarily as a technique to study oxidative phosphorylation in a human tissue (Fukami et al., 1973; Holmsen et al., 1969). Subsequent studies focused on determining whether mitochondrial functional changes could be measured in leukocytes and platelets isolated from patients with diagnosed diseases (Montagna et al., 1988; Parker et al., 1989; Shi et al., 2008). These studies demonstrated the ability to measure specific parameters of mitochondrial dysfunction in blood cells from patients with various diagnosed pathologies. For example, in peripheral blood mononuclear cells (PBMC), ATP synthase expression and activity was found to be decreased in individuals hospitalized for sepsis (Japiassu et al., 2011), while a separate study demonstrated increased mitochondrial mass and hyperpolarization in PBMC from subjects with Type 2 Diabetes (Kizhakekuttu et al., 2010). Studies of platelets from patients with neurological diseases have shown changes in specific mitochondrial electron transport chain complex activities in syndromes ranging from acute migraine headaches (Montagna et al., 1988) to chronic neurodegenerative conditions including Parkinson’s Disease (Parker et al., 1989), Alzheimer’s Disease (Shi et al., 2008; Wilkins et al., 2017), and Huntington’s Disease (Ehinger et al., 2016; Silva et al., 2013). Cumulatively, these early studies set the stage for the utilization of blood cells as a biomarker of mitochondrial function. However, broader clinical and translational application of this concept was stymied by the lack of high throughput, unified, and reproducible methodology for the measurement of multiple bioenergetic parameters in small volumes of blood.
With the advent of platforms such as the XF96 extracellular flux analyzer, dynamic measures of cellular bioenergetics and mitochondrial function can be determined by the sequential addition of defined metabolic inhibitors (Dranka et al., 2011). Using well-validated and defined methods, it is possible to measure integrated bioenergetic metabolism in intact cells/platelet in a sensitive and reproducible manner (Chacko et al., 2013; Ravi et al., 2015). In this methodology, a “bioenergetic profile” can be generated through application of the mitochondrial stress test (MST). In this assay the response of cellular oxygen consumption to the sequential addition of mitochondrial inhibitors is used as shown in Figure 3A (Cardenes et al., 2014; Chacko et al., 2014; Chacko et al., 2013; Hill et al., 2012). These methods have now been extended to allow the measurement of both cellular bioenergetics, the corresponding mitochondrial complex activities and metabolites in the same cell or platelet preparations (Chacko et al., 2019). The schematic for the mitochondrial stress test is shown in Figure 3A.
Figure 3: Translational Bioenergetics and Metabolomics in Human Platelets.

A: The key features of the MST are shown derived from the sequential addition of oligomycin (O-a complex V inhibitor, FCCP (F-an uncoupler) and Antimycin (A-an inhibitor of complex III. The integration of these parameters into the bioenergetic health index (BHI) is also shown (Chacko et al., 2014). B: The relationship between ATP linked and Basal OCR in platelets isolated from a cohort of healthy volunteers where each (ο) represents the values for a single individual. The regression line is shown with the corresponding R2 value and representative individuals indicated by the arrows. C: metabolomics analysis of platelet samples integrated with energetic parameters using the xMWAS analysis to define the bioenergetic-metabolite interactome (Chacko et al., 2019). The bioenergetic parameters are represented by the labeled rectangles and the metabolic features by the circles. Each edge (colored line) represents a positive (blue) or negative (red) association and the communities C1-C4 are represented by different colors. D: The regression analysis for platelets isolated from 85 healthy individuals of the indicated parameters from the MST. ATP linked respiration is highly correlated with basal respiration whereas the correlation with proton leaks is much weaker (adapted from (Chacko et al., 2019)
From the MST assay, several parameters can be obtained which give insight into mitochondrial function. These are listed below with some of the possible interpretations.
-
➢
Basal OCR (oxygen consumption rate): This serves as an individual baseline for each subject and varies considerably between individuals as is shown in Figure 3B and as discussed below.
-
➢
ATP-linked OCR: This value is proportional to the basal activity that is linked to ATP production and the level of proton leak (after injecting oligomycin in the medium). This value can decrease if complex V is inhibited but a lower level can also be related to a lower cellular ATP demand.
-
➢
Proton-leak OCR: This parameter reflects any mitochondrial OCR that is not oligomycin-sensitive and may reflect movement of any cations or substrates across the mitochondrial inner membrane including protons. An increase in proton leak is consistent with the mitochondria becoming less efficient. This may be associated with pathological conditions but, as will be discussed later, may also be a control mechanism to allow the TCA cycle flux to be differentially regulated from oxidative phosphorylation.
-
➢
Maximal OCR: The maximal oxygen respiration (after injecting FCCP in the medium). A decrease in this parameter is consistent with a deficit in mitochondrial biogenesis, damage to mtDNA or the respiration machinery, or limitations in substrate availability or transport.
-
➢
Reserve capacity: (maximal minus basal respiration). This determines the threshold at which bioenergetic dysfunction occurs. When reserve capacity is zero or negative, the cell cannot satisfy the bioenergetic demand through mitochondrial respiration and the bioenergetic threshold is exceeded (Hill et al., 2012). Reserve capacity (sometimes-called spare capacity) is also used to counteract oxidative stress (Chacko et al., 2016; Dranka et al., 2011; Ravi et al., 2016).
Non-mitochondrial OCR: This represents reactive oxygen species (ROS) generation or other oxygen-consuming processes, including pro-inflammatory enzymes such as the cyclooxygenases and lipoxygenases. For example, in the platelets isolated from sickle cell patients, this was ascribed to xanthine oxidase.
-
➢
Respiratory chain complex activities: These values are obtained from permeabilized platelets/cells and allow discrete measurement of Complexes I-V and coupling efficiency.
As shown in Figure 3 the parameters from the mitochondrial stress test (MST) can be integrated as a bioenergetic health index (BHI) and can be related to the ability to combat stress and fatigue (Chacko et al., 2014; Chacko et al., 2016; Tyrrell et al., 2015).
These relationships between the bioenergetic parameters from the MST suggest that the basal metabolome should vary in a systematic manner between individuals as their bioenergetic program, determined by epigenetics, environment and lifestyle factors, changes. For example, the data in Figure 3B suggest that since the amount of oxygen needed to generate the same amount of ATP varies between individuals, then this should be reflected in changes in the steady state levels of metabolites in glycolysis and the TCA cycle. Using this untargeted metabolomics approach over 2700 molecular features were identified. To determine the associations between the basal metabolome and 6 bioenergetic parameters, we used xMWAS analysis (Chacko et al., 2019; Hu et al., 2019). This statistical technique has been successfully used to identify relationships between parameters, including mitochondrial function, in complex data sets (Hu et al., 2019). The results of this analysis are shown in Figure 3C and revealed four distinct communities defining the bioenergetic-metabolite interactome. Remarkably, these communities segregated the bioenergetic parameters on the basis of the metabolome with the same relationships established from multivariate analysis of the MST parameters alone (Chacko et al., 2019) (eg Figure 3D). The implications of these finding are that the basal metabolome is predictive of the response to mitochondrial stressors. Another interesting feature of this data was that environmental toxins and natural products in isolated platelets were among the metabolites most associated with changes in bioenergetics. These findings indicate that environment has a large effect on bioenergetics and likely platelet function. In support of this concept, a recent study has shown that pesticides in the platelets isolated from human subjects are associated with decreased mitochondrial function (Bronstein et al., 2015). It is now becoming evident that the modulation of bioenergetics in human populations is dependent on factors from the diet, environment and the microbiome.
Measures of Bioenergetic Health and Precision Medicine
From a translational perspective, it is important to define “normal bioenergetic health” and how this changes in disease in human subjects. This requires a detailed understanding of how bioenergetic programs vary between healthy individuals on a shifting background of genetic programs, environment and lifestyle. Can these variations be understood and exploited in clinical research? In addition, the question of whether circulating cells provide relevant information about other organs needs to be addressed and is being investigated in a number of independent laboratories. To test this, samples need to be taken from tissues such as skeletal muscle and compared with bioenergetics in platelets or PBMC. This is now being undertaken in well-controlled studies with non-human primate populations which have shown that the maximal respiratory capacity of monocytes and platelets is well correlated with parameters of mitochondrial function in permeabilized skeletal or cardiac muscle from the same subject (Tyrrell et al., 2016). Similar primate studies measuring brain frontal cortex mitochondrial function showed a significant correlation between brain mitochondrial respiratory capacity and monocyte maximal respiratory capacity (Tyrrell et al., 2017). It is important to note that this correlation between circulating cells and other tissues is not limited to highly metabolic organs such as muscle and brain. In a study comparing respiration of platelets versus lung airway epithelium in healthy humans and those with asthma, platelet maximal respiratory capacity correlated significantly with airway epithelial maximal capacity in the same individual (Winnica et al., 2019). Further, airway epithelial cells from asthmatic individuals showed increased basal and maximal respiration compared with healthy subjects, an effect that was recapitulated in platelets from the same cohort (Winnica et al., 2019).
Utilization of bioenergetic profiling methods by several independent groups has shown significant alterations in the bioenergetic profile of platelet, monocyte, or mixed population PBMC in a number of human disease cohorts including sickle cell disease (Cardenes et al., 2014), pulmonary arterial hypertension (PAH) (Nguyen et al., 2017), Type 2 diabetes (Avila et al., 2012; Czajka et al.), asthma (Winnica et al., 2019; Xu et al., 2015), porphyria(Chacko et al., 2019), post cardiac surgery patients (Datta et al., 2015), as well as in aging populations (Tyrrell et al., 2015) (See Figure 4 for a summary of the bioenergetic changes in these human populations). Further, in many of these diseases and in natural populations, parameters of blood cell bioenergetics correlate well with clinical or physical parameters. For example, in a population of older adults, PBMC maximal respiratory capacity correlated with gait speed, a physical parameter that predicts morbidity and mortality in this population (Tyrrell et al., 2015). Similarly, in a population of patients with PAH, platelet maximal respiratory capacity was significantly increased in PAH patients compared with healthy controls, and this bioenergetic parameter was positively associated with mean pulmonary artery pressure and right ventricular stroke work index (Nguyen et al., 2017), hemodynamic parameters used for PAH diagnosis and prognosis, respectively (Figure 4).
Figure 4: Blood based bioenergetics in human populations:
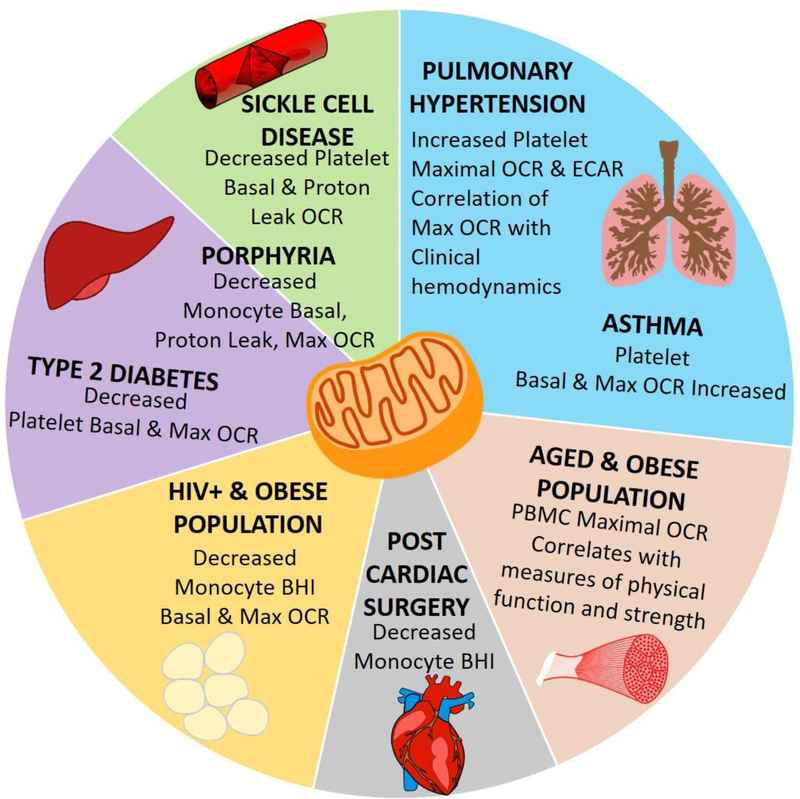
Summary of studies in which bioenergetic changes were measured in monocytes, PBMCs, or platelets isolated from human subjects with diagnosed pulmonary hypertension (Nguyen et al., 2017), asthma (Winnica et al., 2019; Xu et al., 2015), type 2 diabetes (Avila et al., 2012; Czajka et al.), porphyria (Chacko et al., 2019), sickle cell disease (Cardenes et al., 2014), or after cardiac surgery (Kramer et al., 2015). Bioenergetics were also studied in an HIV positive population with differing body mass (Willig et al., 2017) and in older obese adults (Tyrrell et al., 2015).
The studies highlighted above clearly demonstrate that circulating cell respirometry can report bioenergetic function in other tissues. This seemingly contradicts the existing paradigm in the field, which suggests that mitochondria are specialized and variable in function depending on their cellular localization (Fernandez-Vizarra et al., 2011). However, although strong correlations exist between circulating cells and cells from solid tissues, the absolute rate of respiration differs between cell types. For example, in the study of primates, brain mitochondrial maximal capacity ranged from 50–400 pmol O2/min/250,000 cells, while monocyte respiratory capacity was lower, ranging from ~25–150 pmol O2/min/250,000 cells (Tyrrell et al., 2017). Additionally, while specific bioenergetic parameters (such as maximal respiratory capacity) correlate well between cell types, other parameters (such as basal respiration rate) may have a weaker correlation (Tyrrell et al., 2016; Tyrrell et al., 2017). These differences could reconcile the divergent ideas of mitochondria being highly specialized to the tissues in which they are contained, versus a commonality in function between mitochondria in circulating cells and solid tissues.
The factors that account for similarity between bioenergetics in blood cells and other tissues remain unclear, but likely include influence from genetic as well as biochemical, paracrine, and environmental factors. Pathogenic processes specific to different diseases may also regulate both tissue and circulating cell bioenergetic function. For example, in patients with sickle cell disease, circulating free hemoglobin due to erythrocyte hemolysis causes a significant alteration in the platelet bioenergetic profile (Cardenes et al., 2014; Chacko et al., 2019). Similarly, in asthmatic individuals, a global increase in arginine bioavailability drives increased basal and maximal respiration in both airway epithelial cells and platelets (Winnica et al., 2019). Effective utilization of blood-based bioenergetics as a translational or precision medicine tool requires the ability to determine variability in bioenergetics among individuals and more importantly, assess the sensitivity of bioenergetics to physiologic factors and pathologic stressors, as well as determine the downstream metabolic response. In this regard, we have recently taken an approach to integrate bioenergetics with measurements of oxidative phosphorylation and untargeted metabolomics in platelets from healthy individuals (Chacko et al., 2019). This study confirms that a large degree of variability exists in bioenergetics between different healthy individuals. However, key correlations were identified between energetic parameters in platelets from the same individuals. For example, strong correlations were reported between basal respiration and ATP-linked and maximal respiratory capacity (Figure 3B). Notably, many of these relationships (i.e. the correlation between basal respiration and maximal capacity) were disrupted in platelets from a cohort of sickle cell disease patients, indicative of pathogenic alterations in platelet metabolism. Further study in healthy controls showed that the activities of mitochondrial complexes I-IV did not correlate with measures of bioenergetics. This was shown to likely be due to the fact that mitochondrial capacity is well in excess of what is required to sustain metabolism in the platelet and is consistent with the concept that factors beyond mitochondrial enzyme activity regulate the cellular bioenergetic response (Chacko et al., 2019).
Systems level metabolic measurements for precision medicine
The strategies mentioned above use parameters such as oxygen consumption to assess bioenergetic phenotypes and stress responses. Although integrated readouts such as OCR enable the widespread use of technologies for categorizing individual ‘metabotypes’, they have limited resolution for understanding metabolic dysfunction outside the mitochondrion. This is important since the field of metabolomics focuses on understanding metabolic changes at a systems level. Unlike respirometry or linear pathway flux measurements, metabolomics is capable of identifying diverse metabolic biomarkers of disease, revealing differences in anabolic pathway activity, and distinguishing sites of metabolic dysfunction. However, the translational applications of metabolomics to identify and understand the roles of metabolic differences have challenges at both the technical and informatics level.
The conceptual advances provided by metabolomics have stimulated new interest in metabolism, which was considered by many a mature field. For example, the “comprehensive” metabolic charts of the 1960s, which began as renderings of approximately 20 metabolic pathways (Zamboni et al., 2015), have been revised and expanded: maps consisting of a few hundred metabolites now contain thousands (Ogata et al., 1999; Thiele et al., 2013), and it is now estimated that the human metabolome comprises nearly 20,000 metabolites (Wishart et al., 2013). Progress in metabolite detection, pathway tracing, and bioinformatics analysis has stimulated enthusiasm for discovering new metabolites, identifying novel biochemical pathways, and understanding how metabolic changes can be used in precision medicine.
The precedent for using metabolites as a means of diagnosing or treating disease grew from glucose test strips for diabetes in the 1950s (Clarke et al., 2012), to phenylalanine measurements to screen for phenylketonuria in the 1960s (Guthrie et al., 1963), to using metabolomics to identify biomarkers and mechanisms of cardiovascular disease, cancer, and diabetes. This approach has continued with the recent identification of trimethylamine N-oxide (TMAO) as a diet and microbiome-engendered ‘atherotoxin’ that contributes to atherosclerosis (Wang et al., 2011); the identification of ‘oncometabolites’ that initiate, sustain, or propagate tumor growth and metastasis (Ward et al., 2010); and the recognition of metabolite profiles that predict and may contribute to diabetes (Abu Bakar et al., 2015). Each of these findings was possible in part through metabolomics. For example, untargeted and targeted metabolomics strategies were used to identify higher plasma levels of TMAO in atherosclerotic rats (Wang et al., 2011). Subsequent studies showed that microbial breakdown of products found in dietary meat leads to formation of trimethylamine (TMA), which is then converted to TMAO in the liver (Gregory et al., 2015; Koeth et al., 2013; Wang et al., 2014). High circulating levels of TMAO were found to be associated with adverse myocardial events in humans (Wang et al., 2014) and to be produced by particular strains of gut organisms (Gregory et al., 2015). Similarly, oncometabolites such as sarcosine (Sreekumar et al., 2009) and 2-hydroxglutarate (2-HG) (Ward et al., 2010) have been found via metabolomics strategies to be increased in the context of cancer. The causal links of these and other oncometabolites in cancer initiation and progression, or their use as cancer biomarkers, is an area of intense inquiry (Collins et al., 2017; Sciacovelli et al., 2016; Wishart, 2016).
Metabolomics has also revealed new elements of biology relevant to cardiac disease and diabetes. For example, targeted and untargeted metabolomics analyses revealed that 2-HG is not only a cancer-associated metabolite but is also elevated in the hearts of mice subjected to transverse aortic constriction (Sansbury et al., 2014) and to ischemic preconditioning (Nadtochiy et al., 2015). This may be important to cardiac biology because 2-HG inhibits several metabolic enzymes (Wishart, 2016), including α-ketoglutarate dehydrogenase, leading to loss of myocardial contractile function (Karlstaedt et al., 2016). Metabolomics has also identified metabolic footprints of heart failure (Lai et al., 2014; Sansbury et al., 2014; Sun et al., 2016), which developed further into a mechanistic understanding of how changes in glucose metabolism and BCAAs regulate cardiac hypertrophic remodeling (Gibb et al., 2017; Shao et al., 2018). In the context of metabolic disease, metabolomics has unveiled unique metabolic signatures of obesity and diabetes. Branched chain amino acids as well as aromatic amino acids (phenylalanine, tyrosine) and atypical amino acids (aminoadipic acid) have been found to be predictive and to play causal roles in the development of diabetes (Petersen et al., 2018; Wang et al., 2013; Wurtz et al., 2013).
While metabolomic measurements provide relative abundances of numerous metabolites, static snapshots of the metabolome are difficult to translate into an understanding of pathway flux. In some cases, metabolomics data can be used in concert with modeling strategies to develop an understanding of pathway activity [e.g., (Cortassa et al., 2015)]; however, tracer strategies empower metabolomics with the capacity to resolve differences in metabolic pathway flux and to assess the contribution of different nutrient sources to catabolic and anabolic pathways. Provision of 13C-labeled glucose (or other labeled nutrients) to cells, tissues, or organisms leads to time-dependent incorporation of stable label into metabolic intermediates or biosynthetic end products. The metabolite labeling patterns reflect shifts in the mass of metabolites caused by incorporation of the isotope label. Assessed from such data are changes in the labeling pattern, temporal differences in isotope enrichment, or differences in the contribution of nutrients to a metabolite pool, which provide knowledge of the state of metabolism in cells or tissues (Buescher et al., 2015; Jang et al., 2018).
Stable isotope metabolomics can be used to identify loci of metabolic dysfunction and to develop a deeper understanding of metabolism. For example, in experiments using 13C6-glucose, the abundance and patterns of 13C metabolite labeling helped to identify the PFK step of glycolysis as a locus of metabolic dysfunction in diabetic cardiac mesenchymal cells (Salabei et al., 2016) and to show that PFK is a major regulator of ancillary biosynthetic pathway activity in cardiomyocytes (Gibb et al., 2017). Moreover, the use of multiple tracers in metabolomics approaches has the capacity to reveal new facets of metabolism [e.g., NADPH metabolism (Fan et al., 2014), anaplerotic metabolism (Le et al., 2012; Sellers et al., 2015)]. Deep network tracing, which introduces stable tracers via a stress-free, ad libitum diet, can reveal changes in numerous anabolic and catabolic pathways simultaneously in vivo (Sun et al., 2017). This approach may be especially useful for understanding how pathways with lower flux rates, e.g., some biosynthetic pathways of glucose metabolism, change with physiological and pathological stress.
Transfer of mitochondria between cells and the coming era of mitochondrial transplantation
While it is well accepted that mitochondria regulate cellular homeostasis at many levels within cells through retrograde signaling, accumulating studies suggest that these organelles may also be central to signaling between cells. This can be achieved by the cellular release of mitochondrial components, such as mtDNA and cardiolipin, which have been shown to act as DAMPs that stimulate the inflammatory response (Dela Cruz et al., 2018; Hauser et al., 2018; Nakayama et al., 2018; Wilkins et al., 2017), or even by the release of intact mitochondria from cells, through exosomes or other mechanisms, to serve a similar function (Boudreau et al., 2014; Hough et al., 2018). Numerous studies now demonstrate that mitochondria can be transferred from one cell to another (Herst et al., 2018; Murray et al., 2019; Russo et al., 2018; Torralba et al., 2016), demonstrating mitochondrially driven cell to cell signaling and raising the possibility for the potential replacement of dysfunctional mitochondria with healthy organelles; mitochondrial transplantation!
Indeed, early reports of mitochondrial transfer involved transfer of healthy mitochondria from stem cells to cells with damaged or dysfunctional mitochondria (Alvarez-Dolado et al., 2003; Oh et al., 2003; Spees et al., 2006). For example, Spees et al demonstrated that lung epithelial cells that were treated with ethidium bromide to deplete mtDNA regained bioenergetic function after being incubated with human mesenchymal stem cells, and this was due to transfer of mitochondria (Spees et al., 2006). Since then a number of studies demonstrate the transfer of mitochondria from one mammalian cell to another both in vitro and in vivo (Alvarez-Dolado et al., 2003; Lou et al., 2012; McCully et al., 2009; Moschoi et al., 2016; Oh et al., 2003; Spees et al., 2006; Tan et al., 2015; Torralba et al., 2016). These studies demonstrate the potential for mitochondrial transfer for purposes such as the rescue of dysfunctional cells, mtDNA transfer, and immune activation. Notably, mitochondrial transfer between cell types has been shown to serve both physiological and pathological functions. For example, in a murine model of stroke, transfer of mitochondria from astrocytes to damaged neurons was shown to improve neuronal survival, suggesting this transfer as an adaptive, protective mechanism after stress (Hayakawa et al., 2016). However, multiple studies demonstrate that cancer cells with chemotherapy-induced mitochondrial damage can gain bioenergetic function through the transfer of mitochondria from host cells (Moschoi et al., 2016; Tan et al., 2015), demonstrating a potentially pathogenic role for mitochondrial transfer.
A number of mechanism by which mitochondria are transferred between cells have been demonstrated (Figure 5). Tunneling nanotubes are ultrafine structures that form between two cells to facilitate the transfer of organelles. A number of stressors, including hydrogen peroxide (Wang et al., 2011), serum starvation (Lou et al., 2012), and ethidium bromide (Cho et al., 2012), have been shown to induce tunneling nanotube formation and mitochondrial transfer. Extracellular vesicles are small vesicles up to 100nm in size that are secreted a majority of cell types. These vesicles, which encompass exosomes and apoptotic bodies, can contain a variety of cargo including mitochondria and facilitate transport between two cell types (Hayakawa et al., 2016; Hough et al., 2018; Phinney et al., 2015). For example, it was recently demonstrated that exosomes mediate the transfer of mitochondria from airway myeloid-derived regulatory cells to T-cells in the lung. Interestingly, in this study, the number of exosomes containing functional mitochondria was increased in the bronchial alveolar lavage fluid from asthmatics compared to healthy controls, suggesting this type of exosomal transfer mediates the asthmatic inflammatory response (Hough et al., 2018). Finally, while it occurs less frequently, cell fusion is an additional mechanism of mitochondrial transfer. This mechanism involves the fusion of two cells such that the two nuclei remain intact but organelles and cytoplasmic contents are shared. A number of studies have reported cell fusion occurring in vitro between stem cells and a variety of differentiated cell types (Alvarez-Dolado et al., 2003; Oh et al., 2003). However, the evidence for this mechanism in vivo is limited. Mitochondrial transfer between cells appears to be a normal biological process which can mediate adaption/repair and also contribute to pathophysiology.
Figure 5: Mechanisms of mitochondrial transfer between cells.

Mitochondria can be transferred by (A) tunneling nanotubes, (B) extracellular vesicles, or (C) cell fusion.
Reports of physiological transfer of mitochondria between cells has sparked interest in the concept of mitochondrial transplantation as a therapeutic strategy to repair damaged mitochondria. Several groups have performed animal studies to test the ability and efficacy of transferring mitochondria in in vivo models. For example, Hayakawa and colleagues stimulated astrocytes to generate extracellular particles containing mitochondria and injected these into the infarcted area of mice subjected to focal cerebral ischemia. Incorporation of the fluorescently labeled astrocyte mitochondria into the neurons was observed 24 hours after injection (Hayakawa et al., 2016). In a murine model of Parkinson’s disease, Shi and colleagues demonstrated that isolated mitochondria injected via tail vein were incorporated into neurons as well as other organs (Shi et al., 2017). Others have taken the approach of utilizing autologous isolated mitochondria in models of myocardial ischemia/reperfusion. In a rabbit model of myocardial infarction, mitochondria isolated from the rabbit’s pectoralis major muscle was injected into the ischemic myocardium. Mitochondrial injection decreased infarct size and improved cardiomyocytes viability compared to control animals in this model (Cowan et al., 2016; Masuzawa et al., 2013; McCully et al., 2009). More recently mitochondrial transfer has been tested in a small pilot human trial of pediatric patients requiring extracorporeal membrane oxygenation (ECMO) for ischemia-associated myocardial dysfunction after surgical procedures. Mitochondria were isolated from autologous non-ischemic skeletal muscle and injected into the myocardium. Of the five patients receiving mitochondrial transplant, all had improvement in myocardial systolic function and all but one patient was weaned off ECMO at 24 hours after transplant (Emani et al., 2017).
Despite the apparent beneficial effects of mitochondrial transplant outlined in the studies above, several scientific concerns have raised skepticism about the efficacy and utility of this approach (Bertero et al., 2018). Specific concerns include the fact that it is unclear how isolated mitochondria, which undergo permeability transition at high concentrations of calcium, could survive in the high calcium concentrations present in the blood and extracellularly. Current studies have demonstrated that mitochondria are functional prior to injection (Emani et al., 2018; Emani et al., 2017), but viability studies have not been performed in vivo after transplant. Moreover, if these mitochondria do remain functional until transport to the cardiomyocytes or other target cell, it is unknown how and whether they are incorporated into the cell. Early studies suggest that less than 10% of transplanted mitochondria enter the cardiomyocyte (Cowan et al., 2016; Masuzawa et al., 2013). Skepticism remains around the fact that incorporation of such a low number of mitochondria into a cell that contains millions of mitochondria could significantly alter the functionality of the cell. However, this position does not take into account the potential of a signaling gain of function from the transplanted organelle or mitochondrial fragment (Hough et al., 2018). Beyond these concerns, study design of the clinical trial of only five subjects raises skepticism. In this study, one of the five subjects was not weaned off ECMO after mitochondrial transplantation. While this was reported as a 20% mortality rate (Emani et al., 2017), the small sample size raises concerns as to whether this is really a significant improvement over the ~40% mortality rate in much larger cohorts of similar subjects undergoing ECMO. Taken together, these concerns suggest that while there may exist potential for the therapeutic utilization of mitochondrial transplant, far more research is required to test and understand the molecular mechanisms of mitochondrial viability, transfer, incorporation and function after transplant. Further, well designed controlled clinical trials will then no doubt build on more rigorous pre-clinical studies to thoroughly test these protocols.
Metabolotherapies and Mitochondria as a Therapeutic Target
Mitochondrial therapeutics now encompass some interesting new pharmacological agents which are entering the clinical arena and also the repurposing of drugs which work through a metabolic mechanism (Logan et al., 2017; Manczak et al., 2010; Szeto et al., 2014). One interesting approach is to enhance the mitochondrial quality control process. This involves mitochondrial fission and fusion, the mitochondrial proteases and the process of mitochondrial autophagy (mitophagy), which are regulated by diverse signals such as nutrient availability, reactive species, and mitochondrial inhibition (Giordano et al., 2014; Lee et al., 2012; Redmann et al., 2014). Targeting the mitochondrial quality control machinery by genetic and pharmacological means is of tremendous interest, particularly in the field of neurodegenerative diseases (Brown et al., 2017; Lee, 2016; Mitchell et al., 2013; Palikaras et al., 2017) (Figure 6).
Figure 6: Metabolotherapies and Mitochondria as a Therapeutic Target.

Efforts have been made to target mitochondria and mitochondrial quality control for therapeutic application to various diseases. Shown highlighted those that inhibit fission, activate mitochondrial proteases, enhancing mitophagy, enhancing general autophagy, enhancing mitochondrial biogenesis and inhibition of the overproduction of the toxic TCA cycle metabolite succinate.
Proteins involved in mitochondrial dynamics are involved in mitochondrial quality control. A quinazolinone derivative identified from a chemical library screen to influence mitochondrial morphology, mdivi-1, has been shown to be protective against ischemia-reperfusion injuries, traumatic brain injury and Parkinson’s disease in preclinical models (Grohm et al., 2012; Ong et al., 2010; Rappold et al., 2014; Wu et al., 2016), although follow-up studies questioned the impacts of mdivi-1 on fission and instead demonstrated its inhibition on ETC complex I dependent oxygen consumption in neurons and COS-7 cells (Bordt et al., 2017). A peptide inhibitor of fission protein Drp1, P110, has been shown to inhibit neurotoxin or pathogenic protein-induced fission and promote cell survival in SH-SY5Y cells, G93A SOD1 motor neuron-like cell line and transgenic mice, and Huntington’s disease patient iPS cell-derived neurons (Guo et al., 2013; Joshi et al., 2018; Joshi et al., 2018; Qi et al., 2013). P110 enhanced State 3, State 4 and CCCP-mediated uncoupling state oxygen consumption rate in isolated cardiac mitochondria of post-myocardial infarction animals (Disatnik et al., 2013). In human iPSC-derived cardiomyocytes expressing expanded glutamine repeats, p110 has been shown to improve basal, maximal, ATP linked and reserve capacity (Joshi et al., 2019). A new peptide inhibitor DA1 has also been developed to decrease Drp1 interaction with ATAD3A, and suppress mitochondrial fragmentation and bioenergetic deficits as assessed by the mitochondrial stress test in HD mouse and patient-derived cells (Zhao et al., 2019). P110 as well as Mdivi-1 (another Drp1 inhibitor) abolished circadian dependent oscillation of total cellular ATP in human skin fibroblasts and in mouse hippocampi in constant darkness (Schmitt et al., 2018).
Mitochondrial proteases are important for mitochondrial quality control (Quiros et al., 2015). LonP, ATP-dependent Clp protease, human presequence protease (hPreP), and high temperature requirement A (HTRA) degrade misfolded, oxidized or damaged proteins. One example of targeting some of these mitochondrial proteases is the development of hPreP agonists (Vangavaragu et al., 2014). Increased neuronal PreP activity has been shown to decrease Aβ accumulation, attenuate neuroinflammation and improves cognition in AD mouse models and increases mitochondrial complex IV activity in hippocampal neurons (Fang et al., 2015). Thus, hPreP agonists may have the potential to be neuroprotective in certain neurodegenerative diseases.
Mitophagy uses both the core autophagy machinery and the mitochondrial specific target and adaptor proteins. Of those important in mitophagy, Parkin, PINK1 and DJ-1 have been shown to be genetic factors for familial Parkinson’s disease (Obeso et al., 2017). PINK1 has been shown to be stabilized in the mitochondria in response to decreased mitochondrial membrane potential (Jin et al., 2012). The majority of PINK1 mutant proteins associated with Parkinson’s disease exhibit loss of kinase activity (Woodroof et al., 2011). Thus compounds that enhance PINK1 kinase activity have been identified and tested as a therapeutic strategy for diseases where mitochondrial quality control is deficient, including ATP analog kinetin triphosphate KTP, kinetin riboside and its ProTides (Hertz et al., 2013; Osgerby et al., 2017), as well as mitochondrial uncoupler niclosamide and analogs (Barini et al., 2018). Although these compounds activate PINK1, the pleiotropic effects of these compounds limit their use in enhancing mitochondrial quality control as treatment strategies.
Targeting DJ-1 has also been achieved with compounds which have been shown to attenuate its oxidative modification at C106 (Miyazaki et al., 2008). Similarly, a DJ-1 based peptide has been shown to attenuate dopaminergic degeneration in ischemia injury, neurotoxin Parkinson’s disease and multiple system atrophy mouse models (Glat et al., 2016; Lev et al., 2015; Molcho et al., 2018). On the outer membrane, the mitochondrial cholesterol transporter, translocator protein (18kDa) (TSPO), has been used as a biomarker in brain injury and inflammation (Rupprecht et al., 2010). TSPO imaging ligands are being tested in animal models and clinical trials of several neurological and psychiatric diseases including Multiple Sclerosis (Rupprecht et al., 2010). One potential mechanism for the detrimental effect of high abundance of TSPO is that it has been shown to bind VDAC, and inhibits mitophagy downstream of PINK1-Parkin function and prevents p62 mitochondrial translocation (Gatliff et al., 2014).
Natural compounds with mitophagy-inducing properties include spermidine and urolithin A. Spermidine can be provided from aged cheese, mushrooms, soy products, legumes and whole grains, and has been shown to extends mouse lifespan, improves cardiac diastolic function, and delay cardiac aging, this is associated with increased complex I activity in mitochondria isolated from aged mice and enhanced autophagic and mitophagic flux in cardiomyocytes (Eisenberg et al., 2016). Urolithin A is a metabolite from the gut bacteria and with precursors in plants including pomegranates, strawberries, raspberries, and walnuts. It, too, has been found to extend lifespan in an AMPK- and mitophagy-dependent manner in C.elegans (Ryu et al., 2016). Recent studies found that C. elegans expressing human Aβ exhibited decreased basal and reserve capacity oxygen consumption rate, and APOE/E4 iPSC-derived neurons exhibited decreased maximal and reserve capacity oxygen consumption rate. Both Aβ worms and APOE/E4 cells exhibited decreased mitophagy. Urolithin A supplementation stimulated mitophagy, likely due to their ability to increase PINK1 and Parkin, and the decreased in oxygen consumption rates in Aβ worms and APOE/E4 cells were partially restored (Fang et al., 2019).
Pharmacological agents that enhance general autophagy are also potentially useful for enhancement of mitophagy, thus improving mitochondrial quality control. One of the pharmacological agents discovered is rapamycin, which was originally isolated from soil sample from Easter Island (Li et al., 2014). Rapamycin was first used as an anti-fungal drug, and then found to be an immunosuppressant via binding to FKBP (Li et al., 2014). In the late 1980s and early 1990s, target of rapamycin (TOR) was identified first in yeast and then in mammalian cells (Li et al., 2014). Then inhibition of mTOR by rapamycin was found to upregulate autophagy, and in animal models promote longevity (Allison et al., 2014; Kennedy et al., 2014; Saxton et al., 2017). Rapamycin has been shown to protect against rotenone induced cell death in SH-SY5Y cells and primary neurons (Giordano et al., 2014; Pan et al., 2009). Interestingly, rapamycin decreased maximum and reserve capacity oxygen consumption rate in intact primary neurons, and further decreased rotenone-induced decrease of reserve capacity oxygen consumption rate (Giordano et al., 2014). However, the maximal complex I and II substrate linked oxygen consumption rate in permeabilized primary neurons were unchanged by rapamycin (Dodson et al., 2017), suggesting that rapamycin changes nutrient utilization. Rapamycin was not able to ameliorate mitochondrial bioenergetics or brain injuries in a mouse model of CoQ deficiency in a recent study, signaling its likely limitations in attenuating mitochondrial deficit-related disease (Barriocanal-Casado et al., 2019).
Another established drug that has been shown to also impact mitochondrial function and mitochondrial quality control is metformin. Metformin was first used as an antidiabetic drug in in 1950s although the use of the goat’s-rue or French Lilac plant which produces a biguanide metformin precursor was documented in medieval Europe as a treatment for diabetes (Bailey et al., 1996). Metformin improves insulin sensitivity by increasing peripheral uptake of glucose, decreasing output by the liver, and decreasing fatty acid oxidation (Bailey et al., 1996). Recent studies also suggested its potential beneficial effects in cardiovascular disease, Alzheimer’s disease, cancer and improving healthspan (Foretz et al., 2014; Martin-Montalvo et al., 2013). The effect of metformin on mitochondrial complex I activity was demonstrated first in 2000 in permeabilized hepatocytes and isolated mitochondria (El-Mir et al., 2000; Owen et al., 2000) and later reported in many other cell types, although high concentrations have been used in these studies; while a lower concentration was found to be required to activate AMPK and inhibit mitochondrial glycerophosphate dehydrogenase (He et al., 2015). The effect of metformin in activating autophagy has also been reported, through AMPK activation or the SIRT1 pathway (He et al., 2015; Ren et al., 2018). In a bleomycin model, metformin reverses established lung fibrosis, and this effect is associated with activation of AMPK and increased mitochondrial biogenesis (Rangarajan et al., 2018). A recent study demonstrated that phenformin and inhibition of complex I inhibit autophagic and mitophagic flux (Thomas et al., 2018). Taken together, although metformin is able to inhibit complex I, its effect on mitochondrial quality control and mitophagy is more likely dependent on AMPK inhibition and independent of inhibition of complex I.
Similarly, a cell permeable tricyclin pyrone CP2 has been identified to inhibit intracellular Aβ oligomeric accumulation and cell death (Maezawa et al., 2006). It was later found to inhibit complex I activity when added to isolated mitochondria from mouse brains. Primary neurons exposed to CP2 exhibited decreased basal, ATP linked, leak linked and non-mitochondrial OCR while reserve capacity, AMPA activity, and survival on exposure to H2O2 are increased. In vivo, CP2 decreases amyloid beta in 5xFAD Alzheimer’s disease mice, and attenuates cognitive decline in APP/PS1 Alzheimer’s disease mice (Zhang et al., 2015). It appears that CP2 resembles the complex I inhibition and AMPA activity effects of metformin. Whether CP2 affects autophagy/mitophagy needs to be determined. As mitophagy and mitochondrial biogenesis must work together to ensure mitochondrial quality control, small molecules that stimulate mitochondrial biogenesis has been examined. For example, AICAR activates AMPK and SIRT1 thereby activating PGC1α (Canto et al., 2009), as well as promoting mitochondrial fission thereby stimulating mitophagy (Toyama et al., 2016). In this category, thiazolidinedione drugs also target peroxisome proliferator-activated receptors, including PGC1α.
Emerging studies also strongly support the concept that many of the mitochondrial metabolites are important modulators for health and diseases. Early pioneer work has demonstrated a key role of mitochondrial complex II substrate, TCA cycle intermediate succinate, in reperfusion injury (Chouchani et al., 2014). Based on this and follow-up studies, reversible inhibition of complex II has been explored as a treatment strategy. For example, malonate has been shown to be cardioprotective in the isolated mouse heart (Boylston et al., 2015; Valls-Lacalle et al., 2016). Succinate is not the only metabolite in the mitochondrial matrix that exerts roles in inflammation and injury. Itaconate has been shown to directly modify Keap1 and thereby enabling Nrf2 activation (Mills et al., 2018). Cell permeable itaconate derivative dimethyl itaconate and 4-octyl itaconate, have then been tested for their anti-inflammatory activities (Mills et al., 2018). As Nrf2 has been shown to regulate p62 expression (Dodson et al., 2015), it is likely that itaconate can affect mitochondrial quality control by regulating mitophagy. Dimethyl fumarate, a derivative of the TCA cycle intermediate fumarate, has also been shown to modulate Keap1 and thereby affect Nrf2 activity; however, while a structurally distinct inhibitor of Nrf2-Keap1 interaction could promote mitochondrial respiration, and superoxide-activated mitophagy in mouse embryonic fibroblasts, in the same setting, dimethyl fumarate does not affect mitophagy (Georgakopoulos et al., 2017). Because they have diverse celllular targets, the effects of mitochondrial metabolites on bioenergetics and mitophagy are likely to be multifaceted.
Summary
In this overview, we have outlined how circulating cells, and specifically platelets, can be utilized not only as a marker of bioenergetic health and disease, but can be exploited to understand the bioenergetic and metabolite responses to physiologic factors and pathologic stressors. Interestingly, it also suggests that exposure to environmental toxins results in modification of bioenergetic programs and this can be detected in platelets. These data can be integrated together to define a bioenergetic-metabolite interactome which has the capacity to quantitatively assign regulatory functions to not only human metabolites but compounds present in the environment and produced by the microbiome. Although metabolomics is already on the forefront of precision medicine, numerous challenges remain (Clish, 2015; Jang et al., 2018; Wishart, 2016; Zamboni et al., 2015). The methodology itself presents a considerable analytical challenge because it involves diverse strategies for sample preparation, chromatography, mass spectrometry, metabolite identification, and data interpretation. There remain few standard workflows across laboratories, which makes data comparison difficult. Moreover, interpretation of stable isotope labeling patterns often requires formal analysis and modeling (Antoniewicz, 2013; Crown et al., 2013; Sauer, 2006). Nevertheless, as metabolomics expertise continues to build, we expect the field will overcome these challenges and gain even deeper insights into the role of metabolism in health and disease. A better understanding of the bioenergetic-metabolic interaction will also aid the development of metabolotherapies.
Understanding the conditions and mechanisms that lead to mitochondrial transfer is important to elucidate the role of mitochondria in intracellular signaling. However, evidence that cells can incorporate exogenous healthy mitochondria into their bioenergetic network raises the possibility to utilize this phenomenon for therapeutics. Indeed, these types of therapeutic pre-clinical studies have been initiated and show early promise (Cowan et al., 2016; Emani et al., 2018; McCully et al., 2009). Thus, it is clear that mitochondrial transfer will remain an active area of translational research for years to come. Taken together mitochondrial health is now emerging as a central concept in precision medicine.
Acknowledgements:
The authors are grateful for support from the UAB-UCSD O’Brien Center P30 DK079337 (VDU), UAB Blue Sky program (VDU) and UAB Nathan Shock Center P30 G050886 (VDU, JZ, SB), NIH R01 HL133003-01A1 (SS), R01HL142216 (JZ), VA I01 BX004251-01A1 (JZ), and the Hemophilia Center of Western Pennsylvania (SS), (: HL130174, ES028268, HL078825; (BGH) the American Diabetes Association Pathway to Stop Diabetes Grant (BH: 1-16-JDF-041).
REFERENCES
- Abu Bakar MH, Sarmidi MR, Cheng KK, Ali Khan A., Suan CL, Zaman Huri H. and Yaakob H. (2015). Metabolomics - the complementary field in systems biology: a review on obesity and type 2 diabetes. Mol Biosyst. 11, 1742–1774. [DOI] [PubMed] [Google Scholar]
- Allis CD and Jenuwein T. (2016). The molecular hallmarks of epigenetic control. Nat Rev Genet. 17, 487–500. [DOI] [PubMed] [Google Scholar]
- Allison DB, Antoine LH, Ballinger SW, Bamman MM, Biga P., Darley-Usmar VM, Fisher G., Gohlke JM, Halade GV, Hartman JL, Hunter GR, Messina JL, Nagy TR, Plaisance EP, Powell ML, Roth KA, Sandel MW, Schwartz TS, Smith DL, Sweatt JD, Tollefsbol TO, Watts SA, Yang Y., Zhang J. and Austad SN (2014). Aging and energetics’ ‘Top 40’ future research opportunities 2010–2013. F1000Res. 3, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Dolado M., Pardal R., Garcia-Verdugo JM, Fike JR, Lee HO, Pfeffer K., Lois C., Morrison SJ and Alvarez-Buylla A. (2003). Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 425, 968–973. [DOI] [PubMed] [Google Scholar]
- Antoniewicz MR (2013). 13C metabolic flux analysis: optimal design of isotopic labeling experiments. Curr Opin Biotechnol. 24, 1116–1121. [DOI] [PubMed] [Google Scholar]
- Avila C., Huang RJ, Stevens MV, Aponte AM, Tripodi D., Kim KY and Sack MN (2012). Platelet mitochondrial dysfunction is evident in type 2 diabetes in association with modifications of mitochondrial anti-oxidant stress proteins. Experimental and clinical endocrinology & diabetes : official journal, German Society of Endocrinology [and] German Diabetes Association. 120, 248–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CJ and Turner RC (1996). Metformin. N Engl J Med. 334, 574–579. [DOI] [PubMed] [Google Scholar]
- Ballinger SW (2013). Beyond retrograde and anterograde signalling: mitochondrial-nuclear interactions as a means for evolutionary adaptation and contemporary disease susceptibility. Biochem. Soc. Trans. 41, 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banoth B. and Cassel SL (2018). Mitochondria in innate immune signaling. Transl Res. 202, 52–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barini E., Miccoli A., Tinarelli F., Mulholland K., Kadri H., Khanim F., Stojanovski L., Read KD, Burness K., Blow JJ, Mehellou Y. and Muqit MMK (2018). The Anthelmintic Drug Niclosamide and Its Analogues Activate the Parkinson’s Disease Associated Protein Kinase PINK1. Chembiochem. 19, 425–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriocanal-Casado E., Hidalgo-Gutierrez A., Raimundo N., Gonzalez-Garcia P., Acuna-Castroviejo D., Escames G. and Lopez LC (2019). Rapamycin administration is not a valid therapeutic strategy for every case of mitochondrial disease. EBioMedicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellizzi D., D’Aquila P., Scafone T., Giordano M., Riso V., Riccio A. and Passarino G. (2013). The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 20, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero E., Maack C. and O’Rourke B. (2018). Mitochondrial transplantation in humans: “magical” cure or cause for concern? J Clin Invest. 128, 5191–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt AM, King AL, Fetterman JL, Millender-Swain T., Finley RD, Oliva CR, Crowe DR, Ballinger SW and Bailey SM (2014). Mitochondrial-nuclear genome interactions in nonalcoholic fatty liver disease in mice. Biochem. J. 46, 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordt EA, Clerc P., Roelofs BA, Saladino AJ, Tretter L., Adam-Vizi V., Cherok E., Khalil A., Yadava N., Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T., Uppuluri S., Miller AM, Itoh K., Karbowski M., Sesaki H., Hill RB and Polster BM (2017). The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev Cell. 40, 583–594 e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau LH, Duchez AC, Cloutier N., Soulet D., Martin N., Bollinger J., Pare A., Rousseau M., Naika GS, Levesque T., Laflamme C., Marcoux G., Lambeau G., Farndale RW, Pouliot M., Hamzeh-Cognasse H., Cognasse F., Garraud O., Nigrovic PA, Guderley H., Lacroix S., Thibault L., Semple JW, Gelb MH and Boilard E. (2014). Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 124, 2173–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylston JA, Sun J., Chen Y., Gucek M., Sack MN and Murphy E. (2015). Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J Mol Cell Cardiol. 88, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray AW and Ballinger SW (2017). Mitochondrial DNA mutations and cardiovascular disease. Curr Opin Cardiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronstein JM, Paul K., Yang L., Haas RH, Shults CW, Le T. and Ritz B. (2015). Platelet mitochondrial activity and pesticide exposure in early Parkinson’s disease. Mov Disord. 30, 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, Cleland JG, Colucci WS, Butler J., Voors AA, Anker SD, Pitt B., Pieske B., Filippatos G., Greene SJ and Gheorghiade M. (2017). Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 14, 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher JM, Antoniewicz MR, Boros LG, Burgess SC, Brunengraber H., Clish CB, DeBerardinis RJ, Feron O., Frezza C., Ghesquiere B., Gottlieb E., Hiller K., Jones RG, Kamphorst JJ, Kibbey RG, Kimmelman AC, Locasale JW, Lunt SY, Maddocks OD, Malloy C., Metallo CM, Meuillet EJ, Munger J., Noh K., Rabinowitz JD, Ralser M., Sauer U., Stephanopoulos G., St-Pierre J., Tennant DA, Wittmann C., Vander Heiden MG, Vazquez A., Vousden K., Young JD, Zamboni N. and Fendt SM (2015). A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol. 34, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C., Gerhart-Hines Z., Feige JN, Lagouge M., Noriega L., Milne JC, Elliott PJ, Puigserver P. and Auwerx J. (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 458, 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenes N., Corey C., Geary L., Jain S., Zharikov S., Barge S., Novelli EM and Shiva S. (2014). Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood. 123, 2864–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko BK, Kramer PA, Ravi S., Benavides GA, Mitchell T., Dranka BP, Ferrick D., Singal AK, Ballinger SW, Bailey SM, Hardy RW, Zhang J., Zhi D. and Darley-Usmar VM (2014). The Bioenergetic Health Index: a new concept in mitochondrial translational research. Clin Sci (Lond). 127, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko BK, Kramer PA, Ravi S., Johnson MS, Hardy RW, Ballinger SW and Darley-Usmar VM (2013). Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Laboratory investigation; a journal of technical methods and pathology. 93, 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko BK, Smith MR, Johnson MS, Benavides G., Culp ML, Pilli J., Shiva S., Uppal K., Go YM, Jones DP and Darley-Usmar VM (2019). Mitochondria in precision medicine; linking bioenergetics and metabolomics in platelets. Redox Biol. 22, 101165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko BK, Zhi D., Darley-Usmar VM and Mitchell T. (2016). The Bioenergetic Health Index is a sensitive measure of oxidative stress in human monocytes. Redox Biol. 8, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B. and Williams GR (1955). Respiratory enzymes in oxidative phosphorylation. II. Difference spectra. J Biol Chem. 217, 395–407. [PubMed] [Google Scholar]
- Chance B. and Williams GR (1956). Respiratory enzymes in oxidative phosphorylation. VI. The effects of adenosine diphosphate on azide-treated mitochondria. J Biol Chem. 221, 477–489. [PubMed] [Google Scholar]
- Cho YM, Kim JH, Kim M., Park SJ, Koh SH, Ahn HS, Kang GH, Lee JB, Park KS and Lee HK (2012). Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS One. 7, e32778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E., Aksentijevic D., Sundier SY, Robb EL, Logan A., Nadtochiy SM, Ord EN, Smith AC, Eyassu F., Shirley R., Hu CH, Dare AJ, James AM, Rogatti S., Hartley RC, Eaton S., Costa AS, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K., Shattock MJ, Robinson AJ, Work LM, Frezza C., Krieg T. and Murphy MP (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 515, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SF and Foster JR (2012). A history of blood glucose meters and their role in self-monitoring of diabetes mellitus. Br J Biomed Sci. 69, 83–93. [PubMed] [Google Scholar]
- Clish CB (2015). Metabolomics: an emerging but powerful tool for precision medicine. Cold Spring Harb Mol Case Stud. 1, a000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RRJ, Patel K., Putnam WC, Kapur P. and Rakheja D. (2017). Oncometabolites: A New Paradigm for Oncology, Metabolism, and the Clinical Laboratory. Clin Chem. 63, 1812–1820. [DOI] [PubMed] [Google Scholar]
- Cortassa S., Caceres V., Bell LN, O’Rourke B., Paolocci N. and Aon MA (2015). From metabolomics to fluxomics: a computational procedure to translate metabolite profiles into metabolic fluxes. Biophys J. 108, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortopassi GA and Arnheim N. (1990). Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Research. 18, 6927–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan DB, Yao R., Akurathi V., Snay ER, Thedsanamoorthy JK, Zurakowski D., Ericsson M., Friehs I., Wu Y., Levitsky S., Del Nido PJ, Packard AB and McCully JD (2016). Intracoronary Delivery of Mitochondria to the Ischemic Heart for Cardioprotection. PLoS One. 11, e0160889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown SB and Antoniewicz MR (2013). Parallel labeling experiments and metabolic flux analysis: Past, present and future methodologies. Metab Eng. 16, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czajka A., Ajaz S., Gnudi L., Parsade CK, Jones P., Reid F. and Malik AN Altered Mitochondrial Function, Mitochondrial DNA and Reduced Metabolic Flexibility in Patients With Diabetic Nephropathy. EBioMedicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta G., Kramer PA, Johnson MS, Sawada H., Smythies LE, Crossman DK, Chacko B., Ballinger SW, Westbrook DG, Mayakonda P., Anantharamaiah GM, Darley-Usmar VM and White CR (2015). Bioenergetic programming of macrophages by the apolipoprotein A-I mimetic peptide 4F. Biochem J. 467, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv. 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dela Cruz CS and Kang MJ (2018). Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion. 41, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarest TG and McCarthy MM (2015). Sex differences in mitochondrial (dys)function: Implications for neuroprotection. J Bioenerg Biomembr. 47, 173–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X. and Mochly-Rosen D. (2013). Acute Inhibition of Excessive Mitochondrial Fission After Myocardial Infarction Prevents Long-term Cardiac Dysfunction. J Am Heart Assoc. 2, e000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson M., Redmann M., Rajasekaran NS, Darley-Usmar V. and Zhang J. (2015). KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem J. 469, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson M., Wani WY, Redmann M., Benavides GA, Johnson M., Ouyang X., Cofield SS, Mitra K., Darley-Usmar V. and Zhang J. (2017). Regulation of autophagy, mitochondrial dynamics and cellular bioenergetics by 4-hydroxynonenal in primary neurons. Autophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Benavides GA, Diers AR, Giordano S., Zelickson BR, Reily C., Zou L., Chatham JC, Hill BG, Zhang J., Landar A. and Darley-Usmar VM (2011). Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free radical biology & medicine. 51, 1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Benavides GA, Diers AR, Giordano S., Zelickson BR, Reily C., Zou LY, Chatham JC, Hill BG, Zhang JH, Landar A. and Darley-Usmar VM (2011). Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radical Biology and Medicine. 51, 1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Hill BG and Darley-Usmar VM (2010). Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radical Biology and Medicine. 48, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek J. (2017). Role of Cardiolipin in Mitochondrial Signaling Pathways. Front Cell Dev Biol. 5, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham-Snary KJ and Ballinger SW (2015). GENETICS. Mitochondrial-nuclear DNA mismatch matters. Science. 349, 1449–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham-Snary KJ, Sandel MW, Sammy MJ, Westbrook DG, Xiao R., McMonigle RJ, Ratcliffe WF, Penn A., Young ME and Ballinger SW (2018). Mitochondrial - nuclear genetic interaction modulates whole body metabolism, adiposity and gene expression in vivo. EBioMedicine. 36, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehinger JK, Morota S., Hansson MJ, Paul G. and Elmer E. (2016). Mitochondrial Respiratory Function in Peripheral Blood Cells from Huntington’s Disease Patients. Mov Disord Clin Pract. 3, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T., Abdellatif M., Schroeder S., Primessnig U., Stekovic S., Pendl T., Harger A., Schipke J., Zimmermann A., Schmidt A., Tong M., Ruckenstuhl C., Dammbrueck C., Gross AS, Herbst V., Magnes C., Trausinger G., Narath S., Meinitzer A., Hu Z., Kirsch A., Eller K., Carmona-Gutierrez D., Buttner S., Pietrocola F., Knittelfelder O., Schrepfer E., Rockenfeller P., Simonini C., Rahn A., Horsch M., Moreth K., Beckers J., Fuchs H., Gailus-Durner V., Neff F., Janik D., Rathkolb B., Rozman J., de Angelis MH, Moustafa T., Haemmerle G., Mayr M., Willeit P., von Frieling-Salewsky M., Pieske B., Scorrano L., Pieber T., Pechlaner R., Willeit J., Sigrist SJ, Linke WA, Muhlfeld C., Sadoshima J., Dengjel J., Kiechl S., Kroemer G., Sedej S. and Madeo F. (2016). Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 22, 1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mir MY, Nogueira V., Fontaine E., Averet N., Rigoulet M. and Leverve X. (2000). Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 275, 223–228. [DOI] [PubMed] [Google Scholar]
- Emani SM and McCully JD (2018). Mitochondrial transplantation: applications for pediatric patients with congenital heart disease. Transl Pediatr. 7, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emani SM, Piekarski BL, Harrild D., Del Nido PJ and McCully JD (2017). Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 154, 286–289. [DOI] [PubMed] [Google Scholar]
- Fan J., Ye J., Kamphorst JJ, Shlomi T., Thompson CB and Rabinowitz JD (2014). Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 510, 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D., Wang Y., Zhang Z., Du H., Yan S., Sun Q., Zhong C., Wu L., Vangavaragu JR, Yan S., Hu G., Guo L., Rabinowitz M., Glaser E., Arancio O., Sosunov AA, McKhann GM, Chen JX and Yan SS (2015). Increased neuronal PreP activity reduces Abeta accumulation, attenuates neuroinflammation and improves mitochondrial and synaptic function in Alzheimer disease’s mouse model. Hum Mol Genet. 24, 5198–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Hou Y., Palikaras K., Adriaanse BA, Kerr JS, Yang B., Lautrup S., Hasan-Olive MM, Caponio D., Dan X., Rocktaschel P., Croteau DL, Akbari M., Greig NH, Fladby T., Nilsen H., Cader MZ, Mattson MP, Tavernarakis N. and Bohr VA (2019). Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 22, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeley KP, Bray AW, Westbrook DG, Johnson LW, Kesterson RA, Ballinger SW and Welch DR (2015). Mitochondrial Genetics Regulate Breast Cancer Tumorigenicity and Metastatic Potential. Cancer Res. 75, 4429–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vizarra E., Enriquez JA, Perez-Martos A., Montoya J. and Fernandez-Silva P. (2011). Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion. 11, 207–213. [DOI] [PubMed] [Google Scholar]
- Fessel JP and Oldham WM (2018). Pyridine Dinucleotides from Molecules to Man. Antioxid Redox Signal. 28, 180–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetterman JL, Sammy MJ and Ballinger SW (2017). Mitochondrial toxicity of tobacco smoke and air pollution. Toxicology. 391, 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetterman JL, Zelickson BR, Johnson LW, Moellering DR, Westbrook DG, Pompilius M., Sammy MJ, Johnson M., Dunham-Snary KJ, Cao X., Bradley WE, Zhang J., Wei CC, Chacko B., Schurr TG, Kesterson RA, Dell’Italia LJ, rley-Usmar VM, Welch DR and Ballinger SW (2013). Mitochondrial genetic background modulates bioenergetics and susceptibility to acute cardiac volume overload. Biochem. J. 455, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M., Guigas B., Bertrand L., Pollak M. and Viollet B. (2014). Metformin: from mechanisms of action to therapies. Cell Metab. 20, 953–966. [DOI] [PubMed] [Google Scholar]
- Foryst-Ludwig A., Kreissl MC, Benz V., Brix S., Smeir E., Ban Z., Januszewicz E., Salatzki J., Grune J., Schwanstecher AK, Blumrich A., Schirbel A., Klopfleisch R., Rothe M., Blume K., Halle M., Wolfarth B., Kershaw EE and Kintscher U. (2015). Adipose Tissue Lipolysis Promotes Exercise-induced Cardiac Hypertrophy Involving the Lipokine C16:1n7-Palmitoleate. J Biol Chem. 290, 23603–23615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami MH and Salganicoff L. (1973). Isolation and properties of human platelet mitochondria. Blood. 42, 913–918. [PubMed] [Google Scholar]
- Fulghum K. and Hill BG (2018). Metabolic Mechanisms of Exercise-Induced Cardiac Remodeling. Front Cardiovasc Med. 5, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatliff J., East D., Crosby J., Abeti R., Harvey R., Craigen W., Parker P. and Campanella M. (2014). TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy. 10, 2279–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakopoulos ND, Frison M., Alvarez MS, Bertrand H., Wells G. and Campanella M. (2017). Reversible Keap1 inhibitors are preferential pharmacological tools to modulate cellular mitophagy. Sci Rep. 7, 10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb AA, Epstein PN, Uchida S., Zheng Y., McNally LA, Obal D., Katragadda K., Trainor P., Conklin DJ, Brittian KR, Tseng MT, Wang J., Jones SP, Bhatnagar A. and Hill BG (2017). Exercise-Induced Changes in Glucose Metabolism Promote Physiological Cardiac Growth. Circulation. 136, 2144–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb AA and Hill BG (2018). Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ Res. 123, 107–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb AA, Lorkiewicz PK, Zheng YT, Zhang X., Bhatnagar A., Jones SP and Hill BG (2017). Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem J. 474, 2785–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S., Darley-Usmar V. and Zhang J. (2014). Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox. Biol. 2, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S., Dodson M., Ravi S., Redmann M., Ouyang X., Darley-Usmar VM and Zhang J. (2014). Bioenergetic adaptation in response to autophagy regulators during rotenone exposure. J. Neurochem. 131, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glat MJ, Ben-Zur T., Barhum Y. and Offen D. (2016). Neuroprotective Effect of a DJ-1 Based Peptide in a Toxin Induced Mouse Model of Multiple System Atrophy. PLoS One. 11, e0148170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory JC, Buffa JA, Org E., Wang Z., Levison BS, Zhu W., Wagner MA, Bennett BJ, Li L., DiDonato JA, Lusis AJ and Hazen SL (2015). Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem. 290, 5647–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohm J., Kim SW, Mamrak U., Tobaben S., Cassidy-Stone A., Nunnari J., Plesnila N. and Culmsee C. (2012). Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death. Differ. 19, 1446–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Disatnik MH, Monbureau M., Shamloo M., Mochly-Rosen D. and Qi X. (2013). Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J Clin Invest. 123, 5371–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie R. and Susi A. (1963). A Simple Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics. 32, 338–343. [PubMed] [Google Scholar]
- Hauser CJ and Otterbein LE (2018). Danger signals from mitochondrial DAMPS in trauma and post-injury sepsis. Eur J Trauma Emerg Surg. 44, 317–324. [DOI] [PubMed] [Google Scholar]
- Hayakawa K., Esposito E., Wang X., Terasaki Y., Liu Y., Xing C., Ji X. and Lo EH (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 535, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L. and Wondisford FE (2015). Metformin action: concentrations matter. Cell Metab. 21, 159–162. [DOI] [PubMed] [Google Scholar]
- Herst PM, Dawson RH and Berridge MV (2018). Intercellular Communication in Tumor Biology: A Role for Mitochondrial Transfer. Front Oncol. 8, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz NT, Berthet A., Sos ML, Thorn KS, Burlingame AL, Nakamura K. and Shokat KM (2013). A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase PINK1. Cell. 154, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill BG, Benavides GA, Lancaster JR, Ballinger S., Dell’italia L., Zhang J. and Darley-Usmar VM (2012). Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biological chemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill BG, Benavides GA, Lancaster JR Jr., Ballinger S., Dell’Italia L., Jianhua Z. and Darley-Usmar VM (2012). Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biological chemistry. 393, 1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill BG, Dranka BP, Zou L., Chatham JC and Darley-Usmar VM (2009). Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem J. 424, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN and Daly MJ (2005). Genome-wide association studies for common diseases and complex traits. Nature Reviews Genetics. 6, 95–108. [DOI] [PubMed] [Google Scholar]
- Holmsen H., Day HJ and Storm E. (1969). Adenine nucleotide metabolism of blood platelets. VI. Subcellular localization of nucleotide pools with different functions in the platelet release reaction. Biochim Biophys Acta. 186, 254–266. [DOI] [PubMed] [Google Scholar]
- Hong EE, Okitsu CY, Smith AD and Hsieh CL (2013). Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol Cell Biol. 33, 2683–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough KP, Trevor JL, Strenkowski JG, Wang Y., Chacko BK, Tousif S., Chanda D., Steele C., Antony VB, Dokland T., Ouyang X., Zhang J., Duncan SR, Thannickal VJ, Darley-Usmar VM and Deshane JS (2018). Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to T cells. Redox Biol. 18, 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Chandler JD, Park S., Liu K., Fernandes J., Orr M., Smith MR, Ma C., Kang SM, Uppal K., Jones DP and Go YM (2019). Low-dose cadmium disrupts mitochondrial citric acid cycle and lipid metabolism in mouse lung. Free Radic Biol Med. 131, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlimann HC, Laloo B., Simon-Kayser B., Saint-Marc C., Coulpier F., Lemoine S., Daignan-Fornier B. and Pinson B. (2011). Physiological and toxic effects of purine intermediate 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR) in yeast. J Biol Chem. 286, 30994–31002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husted AS, Trauelsen M., Rudenko O., Hjorth SA and Schwartz TW (2017). GPCR-Mediated Signaling of Metabolites. Cell Metab. 25, 777–796. [DOI] [PubMed] [Google Scholar]
- Iacobazzi V., Castegna A., Infantino V. and Andria G. (2013). Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab. 110, 25–34. [DOI] [PubMed] [Google Scholar]
- Ishikawa K., Takenaga K., Akimoto M., Koshikawa N., Yamaguchi A., Imanishi H., Nakada K., Honma Y. and Hayashi J. (2008). ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 320, 661–664. [DOI] [PubMed] [Google Scholar]
- Jang C., Chen L. and Rabinowitz JD (2018). Metabolomics and Isotope Tracing. Cell. 173, 822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Japiassu AM, Santiago AP, d’Avila JC, Garcia-Souza LF, Galina A., Castro Faria-Neto HC, Bozza FA and Oliveira MF (2011). Bioenergetic failure of human peripheral blood monocytes in patients with septic shock is mediated by reduced F1Fo adenosine-5’-triphosphate synthase activity. Critical care medicine. 39, 1056–1063. [DOI] [PubMed] [Google Scholar]
- Jin SM and Youle RJ (2012). PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 125, 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP and Sies H. (2015). The Redox Code. Antioxid Redox Signal. 23, 734–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AU, Ebert AE, Haileselassie B. and Mochly-Rosen D. (2019). Drp1/Fis1-mediated mitochondrial fragmentation leads to lysosomal dysfunction in cardiac models of Huntington’s disease. J Mol Cell Cardiol. 127, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AU, Saw NL, Shamloo M. and Mochly-Rosen D. (2018). Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget. 9, 6128–6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AU, Saw NL, Vogel H., Cunnigham AD, Shamloo M. and Mochly-Rosen D. (2018). Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol Med. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlstaedt A., Zhang X., Vitrac H., Harmancey R., Vasquez H., Wang JH, Goodell MA and Taegtmeyer H. (2016). Oncometabolite d-2-hydroxyglutarate impairs alpha-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc Natl Acad Sci U S A. 113, 10436–10441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK and Pennypacker JK (2014). Drugs that modulate aging: the promising yet difficult path ahead. Transl. Res. 163, 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizhakekuttu TJ and Widlansky ME (2010). Natural antioxidants and hypertension: promise and challenges. Cardiovascular therapeutics. 28, e20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Wang Z., Levison BS, Buffa JA, Org E., Sheehy BT, Britt EB, Fu X., Wu Y., Li L., Smith JD, DiDonato JA, Chen J., Li H., Wu GD, Lewis JD, Warrier M., Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ and Hazen SL (2013). Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 19, 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PA, Chacko BK, George DJ, Zhi D., Wei CC, Dell’Italia LJ, Melby SJ, George JF and Darley-Usmar VM (2015). Decreased Bioenergetic Health Index in monocytes isolated from the pericardial fluid and blood of post-operative cardiac surgery patients. Biosci Rep. 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PA, Ravi S., Chacko B., Johnson MS and Darley-Usmar VM (2014). A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic biomarkers. Redox Biology. 2, 206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs HA and Johnson WA (1937). The role of citric acid in intermediate metabolism in animal tissues. Enzymologia. 4, 148–156. [DOI] [PubMed] [Google Scholar]
- Krzywanski DM, Moellering DR, Fetterman JL, Dunham-Snary KJ, Sammy MJ and Ballinger SW (2011). The mitochondrial paradigm for cardiovascular disease susceptibility and cellular function: a complementary concept to Mendelian genetics. Lab Invest. 91, 1122–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu BK, Zhong M., Sen S., Davogustto G., Keller SR and Taegtmeyer H. (2015). Remodeling of glucose metabolism precedes pressure overload-induced left ventricular hypertrophy: review of a hypothesis. Cardiology. 130, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L., Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J., Kapoor K., Koves TR, Stevens R., Ilkayeva OR, Vega RB, Attie AD, Muoio DM and Kelly DP (2014). Energy Metabolic Re-Programming in the Hypertrophied and Early Stage Failing Heart: A Multi-systems Approach. Circ Heart Fail. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardy HA and Wellman H. (1952). Oxidative phosphorylations; role of inorganic phosphate and acceptor systems in control of metabolic rates. J Biol Chem. 195, 215–224. [PubMed] [Google Scholar]
- Le A., Lane AN, Hamaker M., Bose S., Gouw A., Barbi J., Tsukamoto T., Rojas CJ, Slusher BS, Zhang H., Zimmerman LJ, Liebler DC, Slebos RJ, Lorkiewicz PK, Higashi RM, Fan TW and Dang CV (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. (2016). Mitochondrial drug targets in neurodegenerative diseases. Bioorg Med Chem Lett. 26, 714–720. [DOI] [PubMed] [Google Scholar]
- Lee J., Giordano S. and Zhang J. (2012). Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem. J. 441, 523–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev N., Barhum Y., Ben-Zur T., Aharony I., Trifonov L., Regev N., Melamed E., Gruzman A. and Offen D. (2015). A DJ-1 Based Peptide Attenuates Dopaminergic Degeneration in Mice Models of Parkinson’s Disease via Enhancing Nrf2. PLoS One. 10, e0127549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Pin S., Zeng Z., Wang MM, Andreasson KA and McCullough LD (2005). Sex differences in cell death. Ann Neurol. 58, 317–321. [DOI] [PubMed] [Google Scholar]
- Li J., Kim SG and Blenis J. (2014). Rapamycin: one drug, many effects. Cell Metab. 19, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan A. and Murphy MP (2017). Using chemical biology to assess and modulate mitochondria: progress and challenges. Interface Focus. 7, 20160151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lood C., Blanco LP, Purmalek MM, Carmona-Rivera C., De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB and Kaplan MJ (2016). Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 22, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C., Galluzzi L., Freije JM, Madeo F. and Kroemer G. (2016). Metabolic Control of Longevity. Cell. 166, 802–821. [DOI] [PubMed] [Google Scholar]
- Lou E., Fujisawa S., Morozov A., Barlas A., Romin Y., Dogan Y., Gholami S., Moreira AL, Manova-Todorova K. and Moore MA (2012). Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS One. 7, e33093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY and Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 27, 441–464. [DOI] [PubMed] [Google Scholar]
- Lusk G. (1905). Theories of Metabolism. Science. 22, 6–12. [DOI] [PubMed] [Google Scholar]
- Maezawa I., Hong HS, Wu HC, Battina SK, Rana S., Iwamoto T., Radke GA, Pettersson E., Martin GM, Hua DH and Jin LW (2006). A novel tricyclic pyrone compound ameliorates cell death associated with intracellular amyloid-beta oligomeric complexes. J Neurochem. 98, 57–67. [DOI] [PubMed] [Google Scholar]
- Manczak M., Mao P., Calkins MJ, Cornea A., Reddy AP, Murphy MP, Szeto HH, Park B. and Reddy PH (2010). Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. Journal of Alzheimer’s disease : JAD. 20 Suppl 2, S609–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Montalvo A., Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M., Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M., Pollak M., Zhang Y., Yu Y., Becker KG, Bohr VA, Ingram DK, Sinclair DA, Wolf NS, Spindler SR, Bernier M. and de Cabo R. (2013). Metformin improves healthspan and lifespan in mice. Nat Commun. 4, 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuzawa A., Black KM, Pacak CA, Ericsson M., Barnett RJ, Drumm C., Seth P., Bloch DB, Levitsky S., Cowan DB and McCully JD (2013). Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 304, H966–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilainen O., Quiros PM and Auwerx J. (2017). Mitochondria and Epigenetics - Crosstalk in Homeostasis and Stress. Trends Cell Biol. 27, 453–463. [DOI] [PubMed] [Google Scholar]
- McCully JD, Cowan DB, Pacak CA, Toumpoulis IK, Dayalan H. and Levitsky S. (2009). Injection of isolated mitochondria during early reperfusion for cardioprotection. Am J Physiol Heart Circ Physiol. 296, H94–H105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Ryan DG, Prag HA, Dikovskaya D., Menon D., Zaslona Z., Jedrychowski MP, Costa ASH, Higgins M., Hams E., Szpyt J., Runtsch MC, King MS, McGouran JF, Fischer R., Kessler BM, McGettrick AF, Hughes MM, Carroll RG, Booty LM, Knatko EV, Meakin PJ, Ashford MLJ, Modis LK, Brunori G., Sevin DC, Fallon PG, Caldwell ST, Kunji ERS, Chouchani ET, Frezza C., Dinkova-Kostova AT, Hartley RC, Murphy MP and O’Neill LA (2018). Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 556, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 191, 144–148. [DOI] [PubMed] [Google Scholar]
- Mitchell T., Chacko B., Ballinger SW, Bailey SM, Zhang J. and Darley-Usmar V. (2013). Convergent mechanisms for dysregulation of mitochondrial quality control in metabolic disease: implications for mitochondrial therapeutics. Biochem. Soc. Trans. 41, 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki S., Yanagida T., Nunome K., Ishikawa S., Inden M., Kitamura Y., Nakagawa S., Taira T., Hirota K., Niwa M., Iguchi-Ariga SM and Ariga H. (2008). DJ-1-binding compounds prevent oxidative stress-induced cell death and movement defect in Parkinson’s disease model rats. J. Neurochem. 105, 2418–2434. [DOI] [PubMed] [Google Scholar]
- Molcho L., Ben-Zur T., Barhum Y. and Offen D. (2018). DJ-1 based peptide, ND-13, promote functional recovery in mouse model of focal ischemic injury. PLoS One. 13, e0192954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna P., Sacquegna T., Martinelli P., Cortelli P., Bresolin N., Moggio M., Baldrati A., Riva R. and Lugaresi E. (1988). Mitochondrial abnormalities in migraine. Preliminary findings. Headache. 28, 477–480. [DOI] [PubMed] [Google Scholar]
- Montal ED, Dewi R., Bhalla K., Ou L., Hwang BJ, Ropell AE, Gordon C., Liu WJ, DeBerardinis RJ, Sudderth J., Twaddel W., Boros LG, Shroyer KR, Duraisamy S., Drapkin R., Powers RS, Rohde JM, Boxer MB, Wong KK and Girnun GD (2015). PEPCK Coordinates the Regulation of Central Carbon Metabolism to Promote Cancer Cell Growth. Mol Cell. 60, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moras M., Lefevre SD and Ostuni MA (2017). From Erythroblasts to Mature Red Blood Cells: Organelle Clearance in Mammals. Front Physiol. 8, 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschoi R., Imbert V., Nebout M., Chiche J., Mary D., Prebet T., Saland E., Castellano R., Pouyet L., Collette Y., Vey N., Chabannon C., Recher C., Sarry JE, Alcor D., Peyron JF and Griessinger E. (2016). Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood. 128, 253–264. [DOI] [PubMed] [Google Scholar]
- Murray LMA and Krasnodembskaya AD (2019). Concise Review: Intercellular Communication Via Organelle Transfer in the Biology and Therapeutic Applications of Stem Cells. Stem Cells. 37, 14–25. [DOI] [PubMed] [Google Scholar]
- Nadtochiy SM, Schafer X., Fu D., Nehrke K., Munger J. and Brookes PS (2016). Acidic pH is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadtochiy SM, Urciuoli W., Zhang J., Schafer X., Munger J. and Brookes PS (2015). Metabolomic profiling of the heart during acute ischemic preconditioning reveals a role for SIRT1 in rapid cardioprotective metabolic adaptation. J Mol Cell Cardiol. 88, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M., Bhatnagar A. and Sadoshima J. (2012). Overview of Pyridine Nucleotides Review Series. Circulation Research. 111, 604–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H. and Otsu K. (2018). Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem J. 475, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen QL, Corey C., White P., Watson A., Gladwin MT, Simon MA and Shiva S. (2017). Platelets from pulmonary hypertension patients show increased mitochondrial reserve capacity. JCI Insight. 2, e91415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeso JA, Stamelou M., Goetz CG, Poewe W., Lang AE, Weintraub D., Burn D., Halliday GM, Bezard E., Przedborski S., Lehericy S., Brooks DJ, Rothwell JC, Hallett M., DeLong MR, Marras C., Tanner CM, Ross GW, Langston JW, Klein C., Bonifati V., Jankovic J., Lozano AM, Deuschl G., Bergman H., Tolosa E., Rodriguez-Violante M., Fahn S., Postuma RB, Berg D., Marek K., Standaert DG, Surmeier DJ, Olanow CW, Kordower JH, Calabresi P., Schapira AHV and Stoessl AJ (2017). Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. 32, 1264–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H., Goto S., Sato K., Fujibuchi W., Bono H. and Kanehisa M. (1999). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H., Bradfute SB, Gallardo TD, Nakamura T., Gaussin V., Mishina Y., Pocius J., Michael LH, Behringer RR, Garry DJ, Entman ML and Schneider MD (2003). Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci U S A. 100, 12313–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SB, Subrayan S., Lim SY, Yellon DM, Davidson SM and Hausenloy DJ (2010). Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 121, 2012–2022. [DOI] [PubMed] [Google Scholar]
- Osgerby L., Lai YC, Thornton PJ, Amalfitano J., Le Duff CS, Jabeen I., Kadri H., Miccoli A., Tucker JHR, Muqit MMK and Mehellou Y. (2017). Kinetin Riboside and Its ProTides Activate the Parkinson’s Disease Associated PTEN-Induced Putative Kinase 1 (PINK1) Independent of Mitochondrial Depolarization. J Med Chem. 60, 3518–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MR, Doran E. and Halestrap AP (2000). Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 348 Pt 3, 607–614. [PMC free article] [PubMed] [Google Scholar]
- Palikaras K., Daskalaki I., Markaki M. and Tavernarakis N. (2017). Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol Ther. 178, 157–174. [DOI] [PubMed] [Google Scholar]
- Pan T., Rawal P., Wu Y., Xie W., Jankovic J. and Le W. (2009). Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. 164, 541–551. [DOI] [PubMed] [Google Scholar]
- Parker WD Jr., Boyson SJ and Parks JK (1989). Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol. 26, 719–723. [DOI] [PubMed] [Google Scholar]
- Petersen MC and Shulman GI (2018). Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev. 98, 2133–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros JA, Baumann AK, Ruiz-Pesini E., Amin MB, Sun CQ, Hall J., Lim S., Issa MM, Flanders WD, Hosseini SH, Marshall FF and Wallace DC (2005). mtDNA mutations increase tumorigenicity in prostate cancer. Proceedings Of The National Academy Of Sciences Of The United States Of America. 102, 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phinney DG, Di Giuseppe M., Njah J., Sala E., Shiva S., St Croix CM, Stolz DB, Watkins SC, Di YP, Leikauf GD, Kolls J., Riches DW, Deiuliis G., Kaminski N., Boregowda SV, McKenna DH and Ortiz LA (2015). Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun. 6, 8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picca A., Lezza AMS, Leeuwenburgh C., Pesce V., Calvani R., Landi F., Bernabei R. and Marzetti E. (2017). Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int J Mol Sci. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prezant TR, Agapian JV, Bohlman MC, Bu X., Oztas S., Qiu WQ, Arnos KS, Cortopassi GA, Jaber L., Rotter JI and et al. (1993). Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 4, 289–294. [DOI] [PubMed] [Google Scholar]
- Qi X., Qvit N., Su YC and Mochly-Rosen D. (2013). A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 126, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Langer T. and Lopez-Otin C. (2015). New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 16, 345–359. [DOI] [PubMed] [Google Scholar]
- Rangarajan S., Bone NB, Zmijewska AA, Jiang S., Park DW, Bernard K., Locy ML, Ravi S., Deshane J., Mannon RB, Abraham E., Darley-Usmar V., Thannickal VJ and Zmijewski JW (2018). Metformin reverses established lung fibrosis in a bleomycin model. Nat Med. 24, 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappold PM, Cui M., Grima JC, Fan RZ, de Mesy-Bentley KL, Chen L., Zhuang X., Bowers WJ and Tieu K. (2014). Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat Commun. 5, 5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi S., Chacko B., Sawada H., Kramer PA, Johnson MS, Benavides GA, O’Donnell V., Marques MB and Darley-Usmar VM (2015). Metabolic plasticity in resting and thrombin activated platelets. PLoS One. 10, e0123597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi S., Johnson MS, Chacko BK, Kramer PA, Sawada H., Locy ML, Wilson LS, Barnes S., Marques MB and Darley-Usmar VM (2016). Modification of platelet proteins by 4-hydroxynonenal: Potential Mechanisms for inhibition of aggregation and metabolism. Free Radic Biol Med. 91, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmann M., Dodson M., Boyer-Guittaut M., Darley-Usmar V. and Zhang J. (2014). Mitophagy mechanisms and role in human diseases. Int. J. Biochem. Cell Biol. 53, 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J. and Zhang Y. (2018). Targeting Autophagy in Aging and Aging-Related Cardiovascular Diseases. Trends Pharmacol Sci. 39, 1064–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M., Phoon CK and Schlame M. (2014). Metabolism and function of mitochondrial cardiolipin. Prog Lipid Res. 55, 1–16. [DOI] [PubMed] [Google Scholar]
- Roberts DJ, Tan-Sah VP, Ding EY, Smith JM and Miyamoto S. (2014). Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol Cell. 53, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A., Jackson R., Harman CC, Li T., West AP, de Zoete MR, Wu Y., Yordy B., Lakhani SA, Kuan CY, Taniguchi T., Shadel GS, Chen ZJ, Iwasaki A. and Flavell RA (2014). Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 159, 1563–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubertoux PL, Sluyter F., Carlier M., Marcet B., Maarouf-Veray F., Cherif C., Marican C., Arrechi P., Godin F., Jamon M., Verrier B. and Cohen-Salmon C. (2003). Mitochondrial DNA modifies cognition in interaction with the nuclear genome and age in mice. Nature Genetics. 35, 65–69. [DOI] [PubMed] [Google Scholar]
- Rupprecht R., Papadopoulos V., Rammes G., Baghai TC, Fan J., Akula N., Groyer G., Adams D. and Schumacher M. (2010). Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 9, 971–988. [DOI] [PubMed] [Google Scholar]
- Russo E., Napoli E. and Borlongan CV (2018). Healthy mitochondria for stroke cells. Brain Circ. 4, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D., Mouchiroud L., Andreux PA, Katsyuba E., Moullan N., Nicolet-Dit-Felix AA, Williams EG, Jha P., Lo Sasso G., Huzard D., Aebischer P., Sandi C., Rinsch C. and Auwerx J. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 22, 879–888. [DOI] [PubMed] [Google Scholar]
- Rzem R., Achouri Y., Marbaix E., Schakman O., Wiame E., Marie S., Gailly P., Vincent MF, Veiga-da-Cunha M. and Van Schaftingen E. (2015). A mouse model of L-2-hydroxyglutaric aciduria, a disorder of metabolite repair. PLoS One. 10, e0119540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salabei JK, Lorkiewicz PK, Mehra P., Gibb AA, Haberzettl P., Hong KU, Wei X., Zhang X., Li Q. and Wysoczynski M. (2016). Type 2 diabetes dysregulates glucose metabolism in cardiac progenitor cells. Journal of Biological Chemistry. 291, 13634–13648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansbury BE, DeMartino AM, Xie Z., Brooks AC, Brainard RE, Watson LJ, DeFilippis AP, Cummins TD, Harbeson MA, Brittian KR, Prabhu SD, Bhatnagar A., Jones SP and Hill BG (2014). Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail. 7, 634–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansbury BE, Jones SP, Riggs DW, Darley-Usmar VM and Hill BG (2011). Bioenergetic function in cardiovascular cells: the importance of the reserve capacity and its biological regulation. Chem Biol Interact. 191, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer U. (2006). Metabolic networks in motion: 13C-based flux analysis. Mol Syst Biol. 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell. 169, 361–371. [DOI] [PubMed] [Google Scholar]
- Schmitt K., Grimm A., Dallmann R., Oettinghaus B., Restelli LM, Witzig M., Ishihara N., Mihara K., Ripperger JA, Albrecht U., Frank S., Brown SA and Eckert A. (2018). Circadian Control of DRP1 Activity Regulates Mitochondrial Dynamics and Bioenergetics. Cell Metab. 27, 657–666 e655. [DOI] [PubMed] [Google Scholar]
- Schneider WC (1948). Intracellular distribution of enzymes; the oxidation of octanoic acid by rat liver fractions. J Biol Chem. 176, 259–266. [PubMed] [Google Scholar]
- Sciacovelli M. and Frezza C. (2016). Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic Biol Med. 100, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott GR, Schulte PM, Egginton S., Scott AL, Richards JG and Milsom WK (2011). Molecular evolution of cytochrome C oxidase underlies high-altitude adaptation in the bar-headed goose. Mol. Biol. Evol. 28, 351–363. [DOI] [PubMed] [Google Scholar]
- Sellers K., Fox MP, Bousamra M 2nd, Slone SP, Higashi RM, Miller DM, Wang Y., Yan J., Yuneva MO, Deshpande R., Lane AN and Fan TW (2015). Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest. 125, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S., Kundu BK, Wu HC, Hashmi SS, Guthrie P., Locke LW, Roy RJ, Matherne GP, Berr SS, Terwelp M., Scott B., Carranza S., Frazier OH, Glover DK, Dillmann WH, Gambello MJ, Entman ML and Taegtmeyer H. (2013). Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc. 2, e004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao D., Villet O., Zhang Z., Choi SW, Yan J., Ritterhoff J., Gu H., Djukovic D., Christodoulou D., Kolwicz SC Jr., Raftery D. and Tian R. (2018). Glucose promotes cell growth by suppressing branched-chain amino acid degradation. Nat Commun. 9, 2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpley MS, Marciniak C., Eckel-Mahan K., McManus M., Crimi M., Waymire K., Lin CS, Masubuchi S., Friend N., Koike M., Chalkia D., MacGregor G., Sassone-Corsi P. and Wallace DC (2012). Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell. 151, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z., Ye C., McCain K. and Greenberg ML (2015). The Role of Cardiolipin in Cardiovascular Health. Biomed Res Int. 2015, 891707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C., Guo K., Yew DT, Yao Z., Forster EL, Wang H. and Xu J. (2008). Effects of ageing and Alzheimer’s disease on mitochondrial function of human platelets. Exp Gerontol. 43, 589–594. [DOI] [PubMed] [Google Scholar]
- Shi X., Zhao M., Fu C. and Fu A. (2017). Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion. 34, 91–100. [DOI] [PubMed] [Google Scholar]
- Shock LS, Thakkar PV, Peterson EJ, Moran RG and Taylor SM (2011). DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A. 108, 3630–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AC, Almeida S., Laco M., Duarte AI, Domingues J., Oliveira CR, Januario C. and Rego AC (2013). Mitochondrial respiratory chain complex activity and bioenergetic alterations in human platelets derived from pre-symptomatic and symptomatic Huntington’s disease carriers. Mitochondrion. 13, 801–809. [DOI] [PubMed] [Google Scholar]
- Spees JL, Olson SD, Whitney MJ and Prockop DJ (2006). Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A. 103, 1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer AC and Spremulli LL (2004). Interaction of mitochondrial initiation factor 2 with mitochondrial fMet-tRNA. Nucleic Acids Res. 32, 5464–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreekumar A., Poisson LM, Rajendiran TM, Khan AP, Cao Q., Yu J., Laxman B., Mehra R., Lonigro RJ, Li Y., Nyati MK, Ahsan A., Kalyana-Sundaram S., Han B., Cao X., Byun J., Omenn GS, Ghosh D., Pennathur S., Alexander DC, Berger A., Shuster JR, Wei JT, Varambally S., Beecher C. and Chinnaiyan AM (2009). Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 457, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Sullivan JE, Brocklehurst KJ, Marley AE, Carey F., Carling D. and Beri RK (1994). Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 353, 33–36. [DOI] [PubMed] [Google Scholar]
- Sun H., Olson KC, Gao C., Prosdocimo DA, Zhou M., Wang Z., Jeyaraj D., Youn JY, Ren S., Liu Y., Rau CD, Shah S., Ilkayeva O., Gui WJ, William NS, Wynn RM, Newgard CB, Cai H., Xiao X., Chuang DT, Schulze PC, Lynch C., Jain MK and Wang Y. (2016). Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation. 133, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun RC, Fan TW, Deng P., Higashi RM, Lane AN, Le AT, Scott TL, Sun Q., Warmoes MO and Yang Y. (2017). Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat Commun. 8, 1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH and Birk AV (2014). Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther. 96, 672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AS, Baty JW, Dong LF, Bezawork-Geleta A., Endaya B., Goodwin J., Bajzikova M., Kovarova J., Peterka M., Yan B., Pesdar EA, Sobol M., Filimonenko A., Stuart S., Vondrusova M., Kluckova K., Sachaphibulkij K., Rohlena J., Hozak P., Truksa J., Eccles D., Haupt LM, Griffiths LR, Neuzil J. and Berridge MV (2015). Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 21, 81–94. [DOI] [PubMed] [Google Scholar]
- Thiele I., Swainston N., Fleming RM, Hoppe A., Sahoo S., Aurich MK, Haraldsdottir H., Mo ML, Rolfsson O., Stobbe MD, Thorleifsson SG, Agren R., Bolling C., Bordel S., Chavali AK, Dobson P., Dunn WB, Endler L., Hala D., Hucka M., Hull D., Jameson D., Jamshidi N., Jonsson JJ, Juty N., Keating S., Nookaew I., Le Novere N., Malys N., Mazein A., Papin JA, Price ND, Selkov E Sr., Sigurdsson MI, Simeonidis E., Sonnenschein N., Smallbone K., Sorokin A., van Beek JH, Weichart D., Goryanin I., Nielsen J., Westerhoff HV, Kell DB, Mendes P. and Palsson BO (2013). A community-driven global reconstruction of human metabolism. Nat Biotechnol. 31, 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas HE, Zhang Y., Stefely JA, Veiga SR, Thomas G., Kozma SC and Mercer CA (2018). Mitochondrial Complex I Activity Is Required for Maximal Autophagy. Cell Rep. 24, 2404–2417 e2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torralba D., Baixauli F. and Sanchez-Madrid F. (2016). Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front Cell Dev Biol. 4, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S., Courchet J., Lewis TL Jr., Loson OC, Hellberg K., Young NP, Chen H., Polleux F., Chan DC and Shaw RJ (2016). Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 351, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trounce I., Byrne E. and Marzuki S. (1989). Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet. 1, 44–45. [DOI] [PubMed] [Google Scholar]
- Tyrrell DJ, Bharadwaj MS, Jorgensen MJ, Register TC and Molina AJ (2016). Blood cell respirometry is associated with skeletal and cardiac muscle bioenergetics: Implications for a minimally invasive biomarker of mitochondrial health. Redox Biol. 10, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell DJ, Bharadwaj MS, Jorgensen MJ, Register TC, Shively C., Andrews RN, Neth B., Dirk Keene C., Mintz A., Craft S. and Molina AJA (2017). Blood-Based Bioenergetic Profiling Reflects Differences in Brain Bioenergetics and Metabolism. Oxid Med Cell Longev. 2017, 7317251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell DJ, Bharadwaj MS, Van Horn CG, Kritchevsky SB, Nicklas BJ and Molina AJ (2015). Respirometric Profiling of Muscle Mitochondria and Blood Cells Are Associated With Differences in Gait Speed Among Community-Dwelling Older Adults. J Gerontol A Biol Sci Med Sci. 70, 1394–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valls-Lacalle L., Barba I., Miro-Casas E., Alburquerque-Bejar JJ, Ruiz-Meana M., Fuertes-Agudo M., Rodriguez-Sinovas A. and Garcia-Dorado D. (2016). Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc Res. 109, 374–384. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangavaragu JR, Valasani KR, Gan X. and Yan SS (2014). Identification of human presequence protease (hPreP) agonists for the treatment of Alzheimer’s disease. Eur J Med Chem. 76, 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent EE, Sergushichev A., Griss T., Gingras MC, Samborska B., Ntimbane T., Coelho PP, Blagih J., Raissi TC, Choiniere L., Bridon G., Loginicheva E., Flynn BR, Thomas EC, Tavare JM, Avizonis D., Pause A., Elder DJ, Artyomov MN and Jones RG (2015). Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol Cell. 60, 195–207. [DOI] [PubMed] [Google Scholar]
- Wallace DC (2015). Mitochondrial DNA variation in human radiation and disease. Cell. 163, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual review of genetics. 2005/11/16, 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Shoffner JM, Trounce I., Brown MD, Ballinger SW, Corral-Debrinski M., Horton T., Jun AS and Lott MT (1995). Mitochondrial DNA mutations in human degenerative diseases and aging. Biochim. Biophys. Acta. 1271, 141–151. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Singh G., Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ and Nikoskelainen EK (1988). Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 242, 1427–1430. [DOI] [PubMed] [Google Scholar]
- Wang TJ, Ngo D., Psychogios N., Dejam A., Larson MG, Vasan RS, Ghorbani A., O’Sullivan J., Cheng S., Rhee EP, Sinha S., McCabe E., Fox CS, O’Donnell CJ, Ho JE, Florez JC, Magnusson M., Pierce KA, Souza AL, Yu Y., Carter C., Light PE, Melander O., Clish CB and Gerszten RE (2013). 2-Aminoadipic acid is a biomarker for diabetes risk. J Clin Invest. 123, 4309–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Cui J., Sun X. and Zhang Y. (2011). Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 18, 732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Klipfell E., Bennett BJ, Koeth R., Levison BS, Dugar B., Feldstein AE, Britt EB, Fu X., Chung YM, Wu Y., Schauer P., Smith JD, Allayee H., Tang WH, DiDonato JA, Lusis AJ and Hazen SL (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 472, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Tang WH, Buffa JA, Fu X., Britt EB, Koeth RA, Levison BS, Fan Y., Wu Y. and Hazen SL (2014). Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur Heart J. 35, 904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Patel J., Wise DR, Abdel-Wahab O., Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M., Su SM, Sharp KA, Levine RL and Thompson CB (2010). The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 17, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W., Staron M., Tal MC, Pineda CM, Lang SM, Bestwick M., Duguay BA, Raimundo N., MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A. and Shadel GS (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP and Shadel GS (2017). Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 17, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. (2013). Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 27, 2065–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJ, McArthur K., Metcalf D., Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S., Lessene G., Ritchie ME, Huang DC and Kile BT (2014). Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 159, 1549–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins HM, Koppel SJ, Bothwell R., Mahnken J., Burns JM and Swerdlow RH (2017). Platelet cytochrome oxidase and citrate synthase activities in APOE epsilon4 carrier and non-carrier Alzheimer’s disease patients. Redox Biol. 12, 828–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins HM, Weidling IW, Ji Y. and Swerdlow RH (2017). Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front Immunol. 8, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willig AL, Kramer PA, Chacko BK, Darley-Usmar VM, Heath SL and Overton ET (2017). Monocyte bioenergetic function is associated with body composition in virologically suppressed HIV-infected women. Redox Biol. 12, 648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnica D., Corey C., Mullett S., Reynolds M., Hill G., Wendell S., Que L., Holguin F. and Shiva S. (2019). Bioenergetic Differences in the Airway Epithelium of Lean Versus Obese Asthmatics Are Driven by Nitric Oxide and Reflected in Circulating Platelets. Antioxid Redox Signal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS (2016). Emerging applications of metabolomics in drug discovery and precision medicine. Nat Rev Drug Discov. 15, 473–484. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Jewison T., Guo AC, Wilson M., Knox C., Liu Y., Djoumbou Y., Mandal R., Aziat F., Dong E., Bouatra S., Sinelnikov I., Arndt D., Xia J., Liu P., Yallou F., Bjorndahl T., Perez-Pineiro R., Eisner R., Allen F., Neveu V., Greiner R. and Scalbert A. (2013). HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res. 41, D801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodroof HI, Pogson JH, Begley M., Cantley LC, Deak M., Campbell DG, van Aalten DM, Whitworth AJ, Alessi DR and Muqit MM (2011). Discovery of catalytically active orthologues of the Parkinson’s disease kinase PINK1: analysis of substrate specificity and impact of mutations. Open Biol. 1, 110012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q., Xia SX, Li QQ, Gao Y., Shen X., Ma L., Zhang MY, Wang T., Li YS, Wang ZF, Luo CL and Tao LY (2016). Mitochondrial division inhibitor 1 (Mdivi-1) offers neuroprotection through diminishing cell death and improving functional outcome in a mouse model of traumatic brain injury. Brain Res. 1630, 134–143. [DOI] [PubMed] [Google Scholar]
- Wurtz P., Soininen P., Kangas AJ, Ronnemaa T., Lehtimaki T., Kahonen M., Viikari JS, Raitakari OT and Ala-Korpela M. (2013). Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care. 36, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W., Wang RS, Handy DE and Loscalzo J. (2018). NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid Redox Signal. 28, 251–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Cardenes N., Corey C., Erzurum SC and Shiva S. (2015). Platelets from Asthmatic Individuals Show Less Reliance on Glycolysis. PLoS One. 10, e0132007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni N., Saghatelian A. and Patti GJ (2015). Defining the metabolome: size, flux, and regulation. Mol Cell. 58, 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE and Henikoff S. (2013). Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 20, 259–266. [DOI] [PubMed] [Google Scholar]
- Zhang L., Zhang S., Maezawa I., Trushin S., Minhas P., Pinto M., Jin LW, Prasain K., Nguyen TD, Yamazaki Y., Kanekiyo T., Bu G., Gateno B., Chang KO, Nath KA, Nemutlu E., Dzeja P., Pang YP, Hua DH and Trushina E. (2015). Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s Disease. EBioMedicine. 2, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K. and Hauser CJ (2010). Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 464, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Sun X., Hu D., Prosdocimo DA, Hoppel C., Jain MK, Ramachandran R. and Qi X. (2019). ATAD3A oligomerization causes neurodegeneration by coupling mitochondrial fragmentation and bioenergetics defects. Nat Commun. 10, 1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zharikov S. and Shiva S. (2013). Platelet mitochondrial function: from regulation of thrombosis to biomarker of disease. Biochemical Society transactions. 41, 118–123. [DOI] [PubMed] [Google Scholar]