

Abstract
Amino acids are essential for cellular metabolism, and it is important to understand how nutrient supply is coordinated with changing energy requirements during embryogenesis. Here, we show that the amino acid transporter Slc7a5/Lat1 is highly expressed in tissues undergoing morphogenesis and that Slc7a5‐null mouse embryos have profound neural and limb bud outgrowth defects. Slc7a5‐null neural tissue exhibited aberrant mTORC1 activity and cell proliferation; transcriptomics, protein phosphorylation and apoptosis analyses further indicated induction of the integrated stress response as a potential cause of observed defects. The pattern of stress response gene expression induced in Slc7a5‐null embryos was also detected at low level in wild‐type embryos and identified stress vulnerability specifically in tissues undergoing morphogenesis. The Slc7a5‐null phenotype is reminiscent of Wnt pathway mutants, and we show that Wnt/β‐catenin loss inhibits Slc7a5 expression and induces this stress response. Wnt signalling therefore normally supports the metabolic demands of morphogenesis and constrains cellular stress. Moreover, operation in the embryo of the integrated stress response, which is triggered by pathogen‐mediated as well as metabolic stress, may provide a mechanistic explanation for a range of developmental defects.
Keywords: amino acid transport, integrated stress response, mouse embryo morphogenesis, Slc7a5/Lat1, Wnt signalling
Subject Categories: Development & Differentiation, Membrane & Intracellular Transport, Signal Transduction
The amino acid transporter Slc7a5 sustains the metabolic demands of tissues undergoing morphogenesis during mouse embryogenesis and so constrains the activation of the integrated stress response.

Introduction
Embryonic development involves the patterned proliferation of emerging cell populations, with some tissues expanding more rapidly than others to generate new structures, including limb buds and regions of the developing nervous system. Such expansion is driven by the localised activity of key signalling pathways, including fibroblast growth factor and Wnt, e.g. 1, 2, 3, and there is growing evidence that links such signalling to the regulation of metabolic gene expression 4, 5. Morphogenesis of the embryo also depends on cell movement and shape changes, and all these cellular processes have high‐metabolic requirements that rely on efficient uptake of nutrients including amino acids. Indeed, amino acid supply is essential for survival of pre‐implantation mouse embryos 6, 7 and both under and over maternal nutrition can retard intrauterine growth and increase the risk of morphogenesis failures, including neural tube defects (reviewed in ref. 8). Amino acids are provided to cells via membrane‐localised transporters, which specialise in the import of specific types of amino acid. However, relatively little is known about the expression pattern, requirement and regulation of such transporters during embryogenesis.
The system L1 amino acid transporter isoforms Slc7a5 and Slc7a8 (aka Lat1 and Lat2, respectively) are the primary (although usually not exclusive) conduits for delivery of large neutral amino acids (LNAAs) to many mammalian cell and tissue types. These include the essential amino acids leucine, isoleucine, phenylalanine, tryptophan, valine and methionine. Slc7a5 and Slc7a8 are Na+‐independent amino acid antiporters, typically taking up essential LNAA in exchange for glutamine 9 or histidine 10. They are also able to transport phenylalanine/tyrosine derivatives such as thyroid hormones T3 and T4 11 as well as the neurotransmitter precursor L‐DOPA 12. Both Slc7a5 and Slc7a8 genes encode members of the heteromeric amino acid transporter family of proteins which require a regulatory glycoprotein subunit (in this case 4F2hc/CD98/Slc3a2) to function correctly 13, 14. Slc7a5‐mediated influx of essential LNAA in particular is recognised to promote net protein synthesis, cell growth and proliferation, reviewed in 15, by processes including activation of the mTORC1 signalling pathway 16, 17. Accordingly, Slc7a5 is over‐expressed in many cancers 18, 19 and induction of Slc7a5 is associated with periods of rapid cell growth and expansion during sustained activation of T lymphocytes 20. It also plays an important role in maintenance of critical amino acids in the brain 10.
Slc7a5 is clearly implicated in various processes essential for embryonic development such as protein synthesis, cell growth and proliferation, and we have discovered that Slc7a5‐null mice are embryonic lethal 16. Early embryos up to and including the blastocyst stage are able to take up LNAA effectively by system B0,+ Na+‐coupled transport 21. Slc7a5 therefore appears to be a good candidate gene for investigating the role and regulation of nutrient and hormone uptake during subsequent embryogenesis and to elucidate how gene regulatory mechanisms influence such environmental factors.
Here, we show that Slc7a5 expression is patterned in the mouse embryo and that Slc7a5‐null embryos exhibit profound neural and limb defects. We characterise this phenotype using key tissue and cell type‐specific markers and interrogate the Slc7a5‐null cell state. We detect modest effects on cell proliferation and mTORC1 activity in mutant embryos and uncover an early induction of the integrated stress response (ISR) 22, 23. The ISR is initially adaptive and acts to restore cell homeostasis, but ultimately leads to apoptosis, which is increased in Slc7a5‐null embryos. Moreover, we show that Wnt signalling is required for Slc7a5 expression and so prevents ISR induction, supports the elevated metabolic demands of tissue morphogenesis and protects against developmental defects.
Results
Slc7a5 is expressed in specific regions of the developing embryo
The spatial and temporal expression pattern of the LNAA transporter Slc7a5 was assessed by mRNA in situ hybridisation in whole mouse embryos from early primitive streak stages (Fig 1); probe specificity was assessed in Slc7a5‐null embryos, where no signal was detected (Appendix Fig S1). Slc7a5 mRNA was broadly detected in epiblast, primitive streak and emerging mesendoderm in the embryo at E7.0, as well as in extra‐embryonic epiblast and mesoderm 24 (Fig 1A, a1, a2, a2′). At E8.5 (Fig 1B, b1–b6), Slc7a5 was expressed in the open anterior (Fig 1B, b1, b2), and posterior neural plate, including preneural tube and the caudal lateral epiblast (Fig 1B, b5, b6), and dorsally in closed neural tube (which includes presumptive neural crest) and in somites (Fig 1B, b3, b4). At E9.5, Slc7a5 transcripts continued in all these domains, with high levels in forebrain and optic vesicle as well as in the otic vesicle and first brachial arch (Fig 1C, D, c1′–c1″), forming cranial ganglia (Fig 1D), dorsal hindbrain and spinal cord (Fig 1c2–c5) and in the progress zone of emerging limb buds (Fig 1E). At E10.5 Slc7a5 transcripts continued to be detected along the rostro‐caudal extent of the developing nervous system at varying levels (Fig EV1), including high expression in optic and otic vesicles, cranial ganglia (Fig EV1A, a1′–a2′), branchial arches (Fig EV1A, a2) and differentiating somites, neural crest derivatives and mesonephric duct (Fig EV1A, a3). Transcripts were detected more extensively in the limb bud (Fig EV1B, b1–b2). Notably, Slc7a5 mRNA was most strongly expressed in dorsal spinal cord (Fig EV1a3, a3′) and the forming neural tube arising from the tailbud (Fig EV1C, c1–c5). Slc7a5 is thus transcribed highly in neural and other tissues that undergo morphogenetic movements and/or proliferative expansion in the developing embryo.
Figure 1. Slc7a5 mRNA expression pattern in the early mouse embryo.
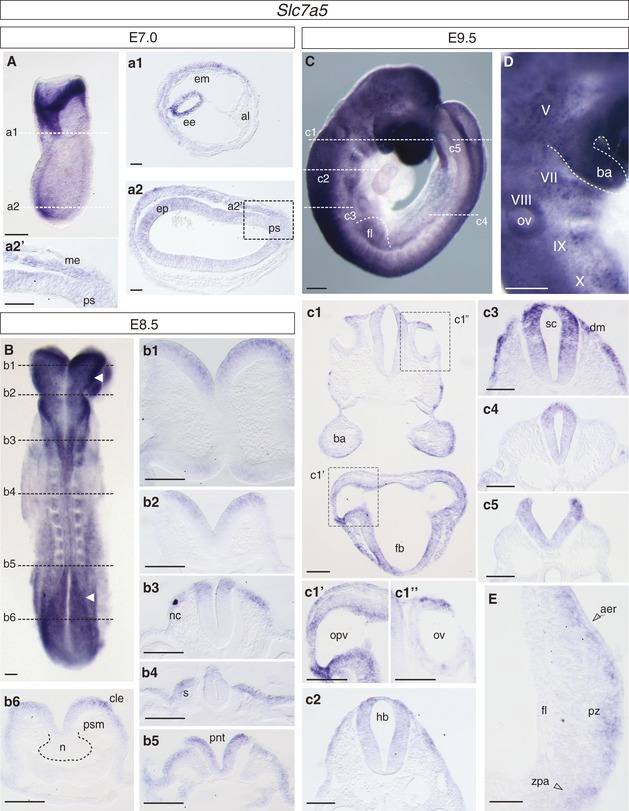
- E7.0 whole embryo and transverse section (TSs) (dashed lines) through (a1) extra‐embryonic tissue showing expression in a ring of extra‐embryonic ectoderm (ee) and extra‐embryonic mesoderm (em) and absence of transcripts in the allantois (al); (a2) revealing expression in epiblast (ep) and in mesendoderm (me) emerging from the primitive streak (ps), dashed box (a2′) defines higher magnification region shown in a2.
- E8.5 whole embryo dorsal view and TSs (dashed lines) showing expression in: (b1–b3) the open anterior neural plate (future fore‐, mid‐ and hindbrain) including (b3) neural crest (nc) emerging from hindbrain; (b4) in the dorsal neural tube (prospective neural crest) and dorsal somites (s); (b5) in closing preneural tube neural (pnt) and epithelising somites; (b6) in caudal lateral epiblast (cle) adjacent to the node (n) at the anterior tip of the primitive streak, but not in presomitic mesoderm (psm). White arrows indicate open anterior and posterior neural plate.
- E9.5 whole embryo side view and TSs (white dashed lines) showing expression in: (c1) forebrain (fb) and hindbrain, including optic vesicles (opv), (c1′) seen at high magnification (and indicated by box grey dashed line) and otic vesicles (ov) (c1″) seen at high magnification (box grey dashed line); (c2) high expression in dorsal hindbrain (hb) and (c3) spinal cord (sc) and dermamyotome (dm) of the differentiating somite; (c5) throughout open posterior neural plate.
- High magnification of E9.5 whole embryo showing expression in first branchial arch (ba) and forming cranial ganglia, which are just becoming apparent, (V), (VII), (VIII) (IX) and (X). OV, otic vesicle.
- Expression in mesenchyme cells underlying the apical ectodermal ridge (aer) in the progress zone (pz) and in the zone of polarising activity (zpa) of the forelimb (fl).
Figure EV1. Slc7a5 mRNA expression pattern in E10.5 mouse embryo.
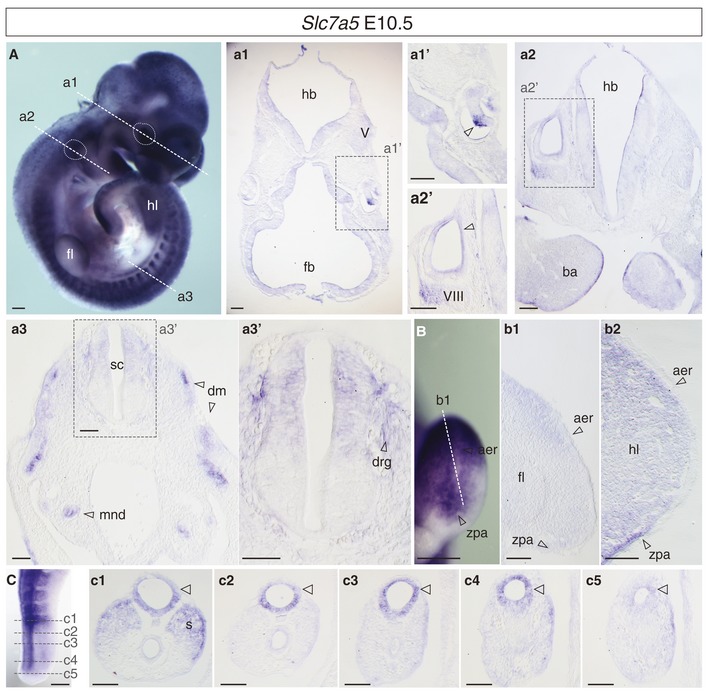
- E10.5 whole embryo side view and TSs (white dashed lines) showing expression in: (a1) forebrain (fb) and (a1′) optic vesicle, including regions of retina (open arrowhead) and lens; (a1, a2) hindbrain (hb), trigeminal ganglion (V) and (a2′) otic vesicle and vestibule–cochlear ganglion (VIII) and first branchial arch (ba); (a3, a3′) dorsal spinal cord (sc), dorsal root ganglia (drg), dermamyotome (dm), mesonephric duct (mnd).
- Forelimb bud (fl) (b1) and hindlimb bud (hl) (b2) in mesenchyme below the apical ectoderm ridge (aer) and in the zone of polarising activity (zpa).
- Forming somite (s) and secondary neural tube (white arrowhead).
Slc7a5‐null embryos exhibit morphological neural tube and limb defects
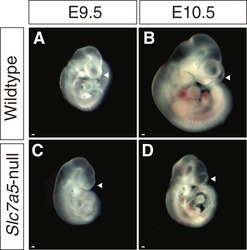
To determine the requirement for Slc7a5 during embryogenesis, null embryos were generated by inter‐crossing heterozygote Slc7a5 +/− mice (see Materials and Methods). An abnormal phenotype was first apparent at E9.5 (Fig 2A–H′). Mutant embryos appeared smaller than littermates and exhibited failure of zippering up along the fore‐ and midbrain and closure of the neural tube at site 2 (forebrain/midbrain boundary) (Fig 2B, F, F′) as well as a delay of posterior neuropore closure (Fig 2C, E′), while closure at site 1 (at the hindbrain/cervical spinal cord boundary) is completed 25. In addition, Slc7a5‐null embryos lacked optic vesicles and had small otic vesicles (Fig 2D, H, H′) and limb buds (Fig 2B and F). In a subset of Slc7a5‐null embryos, the forebrain formed but failed to expand (Fig EV2). This is similar to the “flat‐top” phenotype observed in mTORC1 mutants or rapamycin (mTORC1 complex inhibitor)‐treated embryos 26, 27. These overt morphological defects in neurulation and limb bud outgrowth correlate well with the regions in which Slc7a5 is highly expressed.
Figure 2. Slc7a5‐null embryos exhibit overt neural tube closure and limb bud outgrowth defects, reduced Fgf8 expression domains and aberrant neuronal and neural crest differentiation.

-
A–HLive wild‐type littermate and Slc7a5‐null embryos imaged shortly after dissection. (A–D) Wild‐type and (E–H) Slc7a5‐null E9.75 embryos from lateral (A, D, E, H), frontal (B, F) or dorsal (C, G) views; (e1–h1) higher magnification images of the Slc7a5‐null embryo. White arrowheads indicate the smaller limb bud (compare B and F), open/reduced forebrain (compare D and H), open neural tube at posterior (compare C and E, e1) and anterior (compare B and F, f1) regions. Black arrowheads indicate kinked neural tube (compare C and G, g1) and apparently missing optic and smaller otic vesicles (ov) (compare D and h1).
-
I–PmRNA in situ hybridisation and immunofluorescence in E9.5 or E10.5 wild‐type and Slc7a5‐null embryos for key marker genes. (I–L) Fgf8 mRNA transcripts were detected in wild‐type (I–j1) and in Slc7a5‐null (K–l1) E10.5 embryos (n = 4 each) (white arrowheads indicate limb buds in K and the isthmus in L) with (j1, l1) sections through isthmus at midbrain/hindbrain border and (i1, k1) lateral views of forelimb buds. (M, N) Wholemount tubulin‐β‐III (Tuj1) immunofluorescence performed on (M, m1) wild‐type and (N, n1) Slc7a5‐null E9.5–E10.5 embryos (n = 5 each condition). Dorsal root ganglia (DRG) and sympathetic chain ganglia (SCG) are indicated with open arrowheads together with cranial ganglia IX, X and XI. Images in (m1, n1) show higher magnification of the cranial ganglia V, VII/VIII (open arrowheads). (O, P) Neurog2 expression in wild‐type (O–o4) and Slc7a5‐null embryos (P–p4) (n = 5 and n = 6 each), showing reduced expression in forebrain (o2, p2), cranial ganglia (o1, o3, p1, p3, arrowheads) and spinal cord at level of forelimb (o4, p4, arrowheads indicate position of neural crest).
Figure EV2. A small subset of Slc7a5‐null embryos exhibits a “flat‐top‐like” phenotype.
-
A–DWild‐type littermate (A and B) and Slc7a5‐null embryos (C and D) were imaged at E9.5 (A and C) or E10.5 (B and D). A subset of Slc7a5‐null embryos (˜ 11%, 4/35 null embryos at E9.5 and 1/6 null embryos at E10.5) shows a “flat‐top”‐like phenotype with no expansion of the forebrain as described in 27 (see arrowheads). Scale bars 200 µm.
Tissue expansion, neurogenesis and neural crest defects in Slc7a5‐null embryos
Marker gene analysis of E9.5 embryos was undertaken next to elucidate these developmental defects. Fibroblast growth factor (Fgf) 8 locally regulates expansion of key embryonic tissues, including in the developing brain and limb 28, 29 and is co‐expressed in the rostral forebrain with FoxG1/BF1 30. These genes were both detected in normal (although reduced) domains in the forebrain in Slc7a5‐null embryos (Figs 2I, K, and EV3A, B, D, E) and Fgf8 was similarly present, but in reduced domains at the midbrain–hindbrain boundary and the apical ectodermal ridge which signals to the underlying proliferative progress zone of the limb bud (Fig 2I, J, i1, j1, K, L, k1, l1). As Fgf8 and FoxG1 are correctly localised, these data suggest that Slc7a5 loss does not disrupt overall tissue patterning, but attenuates expansion of cell populations in the developing brain and limb, which can compromise morphogenetic cell movements, such as those underlying neural tube closure 31.
Figure EV3. Slc7a5‐null embryos exhibit altered expression patterns of key neurogenesis genes.
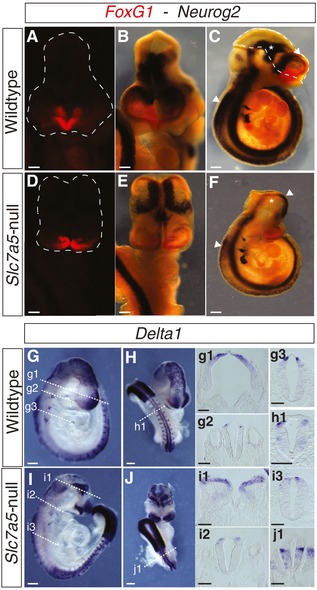
-
A–FmRNA in situ hybridisation in E9.5 wild‐type and Slc7a5‐null embryos for neurogenesis marker genes. FoxG1 and Neurog2 double in situ hybridisation in E9.5 (A–C) wild‐type and (D‐F) Slc7a5‐null embryos (n = 2 each for double FoxG1/Neurog2, a further n = 3 Slc7a5‐null and n = 2 wild‐type embryos were assessed for Neurog2 expression in wholemount and sections). Images in (A and D) show frontal view of FoxG1 mRNA detected with fast red (white dotted lines indicate shape of the head). Expression of FoxG1 in rostral‐most forebrain was detected in Slc7a5‐null embryos with similar intensity but in a reduced domain when compared to wild type. This suggests that Slc7a5 loss does not disrupt brain regionalisation, but leads to a reduction in the amount of tissue and/or failure of morphogenetic events underlying neural tube closure 31. (B–F) To determine whether neurogenesis was affected in Slc7a5‐null embryos, we assessed the expression pattern of Neurogenin‐2 (Neurog2), a neural progenitor marker 32. Widespread reduction in Neurog2 expression was apparent in whole Slc7a5‐null embryos (E, F); some Neurog2‐positive cell populations were represented in reduced domains (arrowheads in C, F), while others were absent (asterisk in C, F) (dotted line on the wild‐type embryo (C) indicates regions of fore‐ and midbrain not apparent in the Slc7a5‐null embryo).
-
G–JDelta1, a marker of newborn neurons 33, was detected in (G, H) wild type and (I, J) Slc7a5‐null E9.5 (n = 3 each) in fore‐ and midbrain but showed a distorted pattern in null embryos due to neurulation failure; TSs through (g1, i1) the forebrain, (g2, i2) at the level of the otic vesicles, (g3, i3) posterior hindbrain and (h1, j1) spinal cord show reduction of the dorsally located Delta1‐expressing cells in the Scl7a5‐null embryos. Scale bars 200 µm, except sections 100 µm.
To assess whether neuronal differentiation is affected in Slc7a5‐null embryos, we analysed expression of the neural progenitor marker Neurog2 32 and the neuronal marker Delta1 33. Defective neural tube closure at fore‐ and midbrain levels made expression patterns difficult to compare with littermate controls, but reduced Neurog2 expression in the brain and spinal cord were evident (Fig EV3B–F, G–i2), and analysis in the closed neural tube of the spinal cord revealed the lack of dorsally located Delta1 expressing cells 34, 35 in null embryos (Fig EV3g3, i3, h1, i4). Further, analysis of tubulin‐βIII (Tuj‐1) expression, which identifies neurons and their extending axons 36, revealed striking failure of axonal outgrowth in the brain and along the length of the neural tube in Slc7a5‐null embryos (Fig 2M–n1). This was particularly evident in neural crest‐derived cranial and spinal ganglia (Fig 2M, m1, N, n1), and this was consistent with reduced Neurog2 expression in these cell populations (Fig 2O–o4, P–p4) 37.
To dissect further the requirement for Slc7a5 in neural crest, Sox10 expression was used to identify migrating neural crest and its derivatives 38, 39. Sox10 expression domains were less extensive in Slc7a5‐null embryos (Fig 3A and B) including cranial as well as spinal ganglia (Fig 3a4, b4). We next assessed pre‐migratory neural crest cells within the dorsal neural tube itself. These cells express Pax3, which acts early in the neural crest gene regulatory network 40, 41. The domain of Pax3 expression in the dorsal neural tube was reduced in Slc7a5‐null embryos (Fig 3C and D, quantified in 3G), suggesting a role for Slc7a5 in the self‐renewal of the pre‐migratory pool of neural crest precursors 42. In contrast, expression of FoxA2 in the ventral neural tube and notochord appeared unaltered in Slc7a5‐null embryos (Fig 3E and F). These data identify specific defects in the expansion and differentiation of cells in the dorsal neural tube in Slc7a5‐null embryos. Overall, this marker gene analysis indicates a requirement for Slc7a5 in cell populations undergoing energetic activities, differentiation, extensive movement, cell shape change or expansive growth during the morphogenesis phase of embryonic development.
Figure 3. Slc7a5‐null embryos exhibit altered expression of neural crest genes.
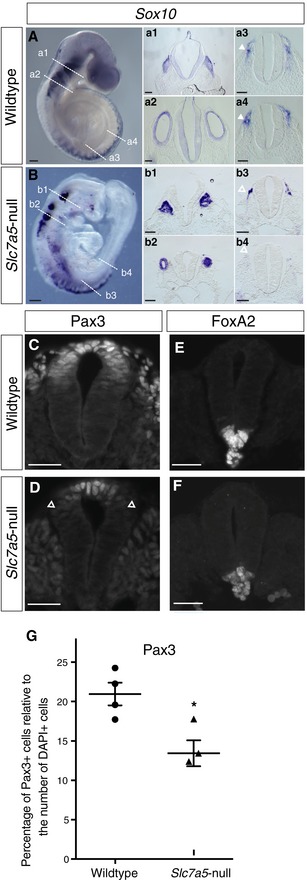
-
A, BSox10 in situ hybridisation in (A) wild‐type and (B) Slc7a5‐null E9.5 embryos (n = 4 each) with TSs of (a1, b1) the hindbrain at level of trigeminal ganglion V, (a2, b2) otic vesicles and the neural tube at more posterior levels (a3, a4, b3, b4). Scale bars 200 µm, except sections a1–b4, 100 µm. Closed white arrowheads in (a3, a4) indicate neural crest, and open white arrowheads indicate depleted neural crest in (b3) and position where neural crest should be in (b4).
-
C–FImmunofluorescence on TS caudal spinal cord of E9.5 wild‐type littermates and Slc7a5‐null embryos; Pax3 and FoxA2 were used as indicators for dorso‐ventral organisation in (C, E) wild type, n = 2 (for FoxA2) and n = 4 (For Pax3) and (D, F) Slc7a5‐null, n = 3 (for FoxA2) and n = 3 for Pax3) neural tube. Arrowheads indicate border of Pax3 expression domain. Scale bars 50 µm.
-
GPercentage of Pax3‐expressing cells was determined by counting these cells and all DAPI‐labelled nuclei in the neural tube and comparison made between wild type (four embryos, six sections each) and Slc7a5‐null (three embryos, six sections each), each dot represents the average for one embryo, unpaired t‐test, *P = 0.018 (see original source data). Error bars indicate SEM.
Source data are available online for this figure.
Slc7a5‐null embryos exhibit a mild cell proliferation defect and aberrant mTORC1 activity
To assess whether cells in Slc7a5‐null embryos exhibit reduced proliferation, we next used an antibody against phosphorylated histone 3 (phospho‐H3) to identify mitotic cells (Fig 4A–D). We first quantified phospho‐H3‐positive cells in the spinal cord (at the level of the forelimb) (Fig 4A and B). This revealed no difference in mitotic index in the absence of Slc7a5 (Fig 4E). The latter region was selected for analysis because mutant embryos complete neurulation in this region and exhibited minimal morphological defects and facilitating comparison with wild‐type embryos. We therefore next assessed the mitotic index in the forebrain, where Slc7a5‐null embryos exhibit profound defects, failing to form optic vesicles and undergo neurulation. Surprisingly, although the neuroepithelium appears depleted in Slc7a5‐null forebrain, the mitotic index was only just significantly reduced (Fig 4C, D and F). This modest effect in the forebrain and the lack of effect in the spinal cord suggest that reduced cell proliferation is unlikely to be the major explanation for the defects observed in Slc7a5‐null neural tube.
Figure 4. Slc7a5‐null neural tube exhibits slightly reduced cell proliferation and aberrant mTORC1 activity, and qPCR validation of significantly changed genes in Slc7a5‐null embryos.

-
A–DProliferation was assessed in the spinal cord (forelimb level) (A, B) using a phospho‐Histone 3 (phospho‐H3) antibody (green) to identify mitotic cells and DAPI (blue) to label nuclei; cells were counted in 3 wild‐type (18 sections) and 3 Slc7a5‐null embryos (18 sections) and mitotic index calculated; (C, D) proliferation was also assessed in the forebrain in three wild‐type embryos (17 sections) and three Slc7a5‐null embryos (16 sections). White dashed lines indicate midline.
-
E, FComparison of mitotic index in wild‐type and Slc7a5‐null (E) spinal cord, P = 0.872, and (F) forebrain *P = 0.049, each dot represents the average for one embryo, unpaired t‐test (see original source data).
-
G, HmTORC1 activity in the spinal cord (hindlimb level) was assessed by immunofluorescence using an antibody against the phospho‐ribosomal protein S6 (phospho‐S6 Ser 235/236).
-
ILabelling intensity was measured and plotted relative to DAPI intensity; six wild‐type embryos (18 sections) and five Slc7a5‐null embryos (19 sections). Each dot represents the average for one embryo, (*P = 0.0457) unpaired t‐test with Welch correction (see original source data).
-
JWestern blot of individual E9.5 wild‐type (n = 3) nd Slc7a5‐null (n = 4) embryo lysates immunoblotted using an antibody against phospho‐S6K (Thr389) and total P70S6K for loading control. Band intensities were measured with FIJI, and an unpaired t‐test was performed for statistical analysis (see original source data).
-
K, L(K) Validation by qPCR of the targets identified by RNA‐seq; qPCR was performed on individual E8.5 embryos of each genotype (wild type n = 5, heterozygous n = 9 and Slc7a5‐null n = 7). (L) qPCR was performed on individual E9.5 embryos (wild type n = 5, heterozygous n = 3 and Slc7a5‐null n = 4) using primers specific for genes associated with the integrated stress response. A one‐way ANOVA test with a Tukey post‐test was performed.
The resemblance of the forebrain phenotype in a subset of Slc7a5‐null embryos to the flat‐top phenotype resulting from reduced mTORC1 activity 26, 27, the requirement for Slc7a5 for full activation of mTORC1 in some other cellular contexts 16, 17, 20 along with the well‐established role of this pathway in integrating nutrient and mitogen signals 15, raised the possibility that altered mTORC1 signalling might underlie the Slc7a5‐null phenotype. We took two approaches to assess this possibility. By immunocytochemistry, the phosphorylated form of ribosomal protein S6 (S6 is a P70S6 kinase substrate and often used as surrogate reporter for mTORC1 signalling 43) was detected (using antibodies recognising phospho‐Ser235/236 (Fig 4G and H) or phospho‐Ser240/244 (Appendix Fig S2C and D). Strikingly, phospho‐S6 levels in neural tube sections varied markedly from cell to cell in Slc7a5‐null, but not in wild‐type embryos (Fig 4G and H, quantified in 4I), indicating a more heterogeneous pattern of mTORC1 activity in the loss‐of‐function condition. In contrast, Western blot for phospho‐Thr389 of P70S6 kinase (a key substrate of mTORC1) did not show a difference between wild‐type and Slc7a5‐null conditions in E9.5 embryo lysates (Fig 4J). This may reflect aberrant rather than absent mTORC1 signalling activity in a subset of cells, which may be beneath detection in whole embryo lysates. Together, these findings suggest that reduced mTORC1 activity is just beginning to be detectable at the time when profound morphological defects are manifest in Slc7a5‐null embryos.
Transcriptome analysis implicates the integrated stress response and discounts a role for thyroid hormone in the Slc7a5‐null phenotype
As we detected only a small effect on cell proliferation in Slc7a5‐null embryos, we next took an unbiased approach to uncover the earliest effects of Slc7a5 loss. This additionally allowed us to determine whether Slc7a5 transport of tyrosine derivatives (including thyroid hormones (TH) T3 and T4) as well as LNAAs might underlie the observed developmental defects. To this end, we carried out RNA‐seq on wild‐type and Slc7a5‐null embryos at E8.5, just prior to appearance of phenotypic defects and compared resulting transcriptomes (see Materials and Methods). Significant changes (FDR < 0.05) in the expression of only seven gene transcripts, one pseudogene and two lincRNAs were found in Slc7a5‐null embryos (Table 1, Datasets EV1 and EV2). These changes were confirmed by RT–qPCR for 6 genes: Slc7a5, Klhdc4 and Spire2 were downregulated and Chac1, Trib3 and Pck2 were upregulated (Fig 4K).
Table 1.
Seven genes significantly changed in E8.5 Slc7a5‐null embryos
| Gene_ID | Gene | logFC | P‐value | FDR | Description |
|---|---|---|---|---|---|
| ENSMUSG00000110631 | Gm42047 | −11.1 | 0 | 0 | lincRNA |
| ENSMUSG00000040010 | Slc7a5 | −4.95 | <10−16 | <10−16 | Solute carrier family 7 (cationic amino acid transporter, y+ system), member 5 |
| ENSMUSG00000074052 | BC048644 | −4.35 | 4.6 × 10−12 | 4.0 × 10−8 | cDNA sequence BC048644 |
| ENSMUSG00000112478 | AC153370.2 | −3.59 | <10−16 | 2.8 × 10−14 | lincRNA |
| ENSMUSG00000040263 | Klhdc4 | −1.14 | <10−16 | <10−16 | Kelch domain containing 4 |
| ENSMUSG00000010154 | Spire2 | −0.948 | 3.1 × 10−11 | 2.4 × 10−7 | Spire homolog 2 (Drosophila) |
| ENSMUSG00000032815 | Fanca | −0.387 | 7.2 × 10−6 | 0.038 | Fanconi anaemia, complementation group A |
| ENSMUSG00000040618 | Pck2 | 0.594 | 7.3 × 10−10 | 4.8 × 10−6 | Phosphoenolpyruvate carboxykinase 2 (mitochondrial) |
| ENSMUSG00000032715 | Trib3 | 1.851 | 6.9 × 10−8 | 0.00040 | Tribbles pseudokinase 3 |
| ENSMUSG00000027313 | Chac1 | 3.431 | 1.2 × 10−13 | 1.2 × 10−9 | ChaC, cation transport regulator 1 |
RNA‐seq data based on 5 wild‐type littermates and 5 Slc7a5‐null E8.5 embryos identified 7 genes showing significant change in expression (and see Appendix Table S1). Slc7a5 was downregulated with a log2 fold change of 4.948 (equivalent to > 99.9% reduction in mRNA expression (Fig 4H)). Klhdc4 (Kelch domain containing 4) and Spire2 (spire‐type actin nucleation factor 2) genes were both downregulated to a much lesser extent with log2 fold changes of ~ 1, confirmed by reductions in mRNA expression to ~ 50% of wild‐type level. Klhdc4 is a highly abundant transcript, and Spire2 is functionally redundant with Spire1 (Pfender et al, 2011; Fig 4H), so the observed changes in their expression levels seem unlikely to be functionally significant (and may be associated with their close proximity to the Slc7a5 locus on chromosome 8). Upregulated genes, Chac1, Trib3 and Pck2, are all metabolic stress‐related genes (see text). Correlation between samples was high (r > 0.98), and additional analysis without one WT outlier had a small effect on the significant gene list, adding several further genes associated with metabolic stress (Appendix Table S2).
These data confirm efficient knockout of Slc7a5, while Klhdc4 and Spire2 reduction are unlikely to be functionally significant, given the remaining level of expression in Slc7a5‐null embryos and the existence of other isoforms (see Table 1 and Fig 4K). Analysis in Slc7a5‐null embryos further confirmed that transcription and protein levels of Slc7a5 partner Slc3a2/CD98 are not reduced in this context (Appendix Fig S3). In contrast, two of the confirmed significantly upregulated genes Chac1 (cation transport regulator homolog‐1) and Trib3 (tribbles pseudokinase‐3, aka TRB3, NIPK or SKIP3) are implicated in endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) 45, 46, 47. Chac1 is distantly related to Botch, a negative regulator of Notch signalling 48; however, we found no evidence for change in Notch signalling in Slc7a5‐null embryos (Appendix Fig S4). Transcription of the third confirmed upregulated gene, Pck2 (phosphoenolpyruvate carboxykinase‐2), is activated by amino acid deficiency and ER stress and also plays a role in gluconeogenesis, a stress‐related metabolic pathway 45, 49. In addition, a further two upregulated genes just below significance, Aldh1l2 (aldehyde dehydrogenase 1 family, member L2) and Asns (asparagine synthetase) (Dataset EV1), are also well‐known cellular stress‐activated genes 50, 51. Multiple stress stimuli, including amino acid deprivation and ER stress, converge on induction of the integrated stress response (ISR) 22, 23. This is an adaptive response which acts to restore cellular homeostasis by decreasing global protein synthesis while promoting mRNA translation for selected proteins. These include the key mediator of the ISR, activating transcription factor (ATF) 4, and this leads to upregulation of ATF4 transcriptional targets, many of which alter cell metabolism so as to mitigate cellular stress 52. These findings therefore suggest that promotion of the ISR is an early consequence of Slc7a5 loss in developing embryos.
We further assessed whether altered transport of phenylalanine/tyrosine derivatives might contribute to the mutant phenotype. Indeed, TH signalling has been implicated in expansion of the cerebral cortex at later developmental stages 53. No changes in genes associated with L‐DOPA were found in the RNA‐seq data, and while TH target gene Dio3 was slightly reduced in an analysis in which one outlier data set was removed (Dataset EV2), this was not confirmed in Slc7a5‐null embryos assessed by qPCR (Appendix Fig S5A). Furthermore, Dio3 and thyroid hormone receptors THRβ and THRα were barely detectable until after E10.5 in wild‐type embryos (Appendix Fig S5B, D, E, F). These data strongly suggest that the dopamine and thyroid hormone systems do not operate during the period when the Slc7a5 phenotype is first manifest. Overall, these findings identify loss of Slc7a5 function as a transporter of LNAA as the likely underlying cause of these early neural developmental and limb defects.
Slc7a5‐null embryos exhibit localised integrated stress response
To investigate the activation of the ISR in Slc7a5‐null embryos, qPCR was carried out for key genes that mediate this response in stage E9.5 embryos (when the phenotype is now apparent). This included Chac1 and Trib3 and also ATF4 as well as C/EBP homologue protein (CHOP). CHOP is an effector of a pro‐apoptotic response which is triggered if cellular stress persists 52, 54. CHOP acts together with ATF4 to promote transcription of Chac1 and Trib3 which, along with other stress‐induced pathways, promote apoptosis 46, 47. Chac1, Trib3 and CHOP were significantly upregulated in Slc7a5‐null embryos (Fig 4L). ATF4 transcription showed a similar trend, but was not significantly increased, perhaps reflecting that ATF4 response to stress is primarily at the level of mRNA translation 55. In addition, Trib3 may act as a feedback inhibitor of ATF4 transcription 56.
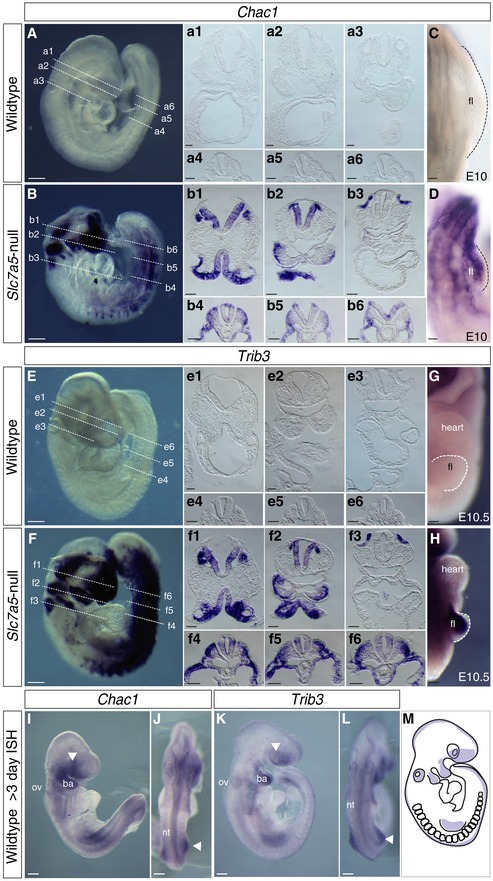
To localise this stress response within the developing Slc7a5‐null embryo, the expression patterns of Chac1 and Trib3 were next assessed by mRNA in situ hybridisation. Strikingly, transcripts were found at high levels in Slc7a5‐null embryos in regions that normally express Slc7a5 but were not detected in E9.5 wild‐type embryos processed in parallel (Fig 5A–H). This was particularly apparent in the brain, branchial arches and otic vesicles as well as dorsal neural tube and limb buds of mutant embryos (Fig 5B, b1–b6, D, F, f1–f6, H). To substantiate further Trib3 and Chac1 as markers of cellular stress in the embryo, we additionally analysed their expression following brief exposure of whole E8.5 wild‐type embryos to ER stress inducers, tunicamycin and thapsigargin, which also provoke the ISR 57. This revealed rapid and localised induction of both Trib3 and Chac1, particularly in the neural tube (Appendix Fig S6).
Figure 5. Slc7a5‐null embryos exhibit localised integrated stress response.
-
A–LmRNA in situ hybridisation was performed at E9.5 to detect Chac1 in (A–a6) wild‐type and (B–b6) Slc7a5‐null littermate embryos (n = 3 for each condition); Chac1 expression at E10 in emerging limb buds assessed in (C) wild‐type and (D) Slc7a5‐null littermate embryos (n = 2 for each condition); Trib3 mRNA detected in (E‐e6) wild‐type and (F–f6) Slc7a5‐null littermate embryos (n = 3 for each condition) and in emerging limb buds at E10.5 assessed in (G) wild‐type and (H) Slc7a5‐null littermate embryos (n = 2 for each condition). Chac1 and Trib3 transcripts were also detected in wild‐type E9.5 embryos after > 3 days in situ hybridisation reaction (n = 3 and n = 6, respectively). Lateral view (I and K), optic vesicles (white arrowheads), branchial arches (ba) and otic vesicles (ov). Dorsal view (J and L), neural tube (nt) and limb buds (white arrowheads). Scale bars 200 µm for whole embryo images and 100 µm for sections.
-
MSchematic indicating regions (purple) in which high levels of Slc7a5 (see Fig 1C–E) and ISR genes Trib3 and Chac1 are detected in wild‐type embryos.
Interestingly, Chac1 and Trib3 transcripts could also eventually be detected at low level in wild‐type embryos in a similar pattern to that in Slc7a5‐null, if the mRNA detection reaction was continued for > 3 days (Fig 5I–M and see Appendix Fig S7). These findings strongly suggest that loss of Slc7a5 promotes localised ISR in the embryo as it undergoes morphogenesis and, given detection of low‐level transcription of these ISR‐linked genes in these cell populations in wild‐type embryos, further suggest that this initially adaptive mechanism normally operates in dynamic cell populations to ensure cell homeostasis.
Slc7a5 loss triggers the integrated stress response via phosphorylation of GCN2 and eIF2α and induces apoptosis
The canonical ISR pathway is activated via phosphorylation, and so, inactivation of the translation initiation factor eIF2α by one or more of four eIF2α kinases and amino acid deprivation triggers activation of the kinase GCN2 23. Increased phosphorylation of GCN2 and eIF2α was detected in E9.5 Slc7a5‐null whole embryo lysates (Fig 6A and B). Moreover, we were able to detect localised induction of p‐eIF2α in E9.5 Slc7a5‐null embryos, extensively in the forebrain and locally in key tissues including the otic vesicle (Fig 6C–f2). These findings indicate that loss of Slc7a5 triggers activation of the kinase GCN2 and further substantiate the induction of ISR in Slc7a5‐null embryos.
Figure 6. Cell death is increased in Slc7a5‐null embryos, and exogenous ER stress induction in wild‐type embryos upregulates Chac1 and Trib3 .

-
A, BWestern blot of individual E9.5 wild‐type (n = 4) and Slc7a5‐null (n = 4) embryo lysates immunoblotted using an antibody against phospho‐GCN2 (Thr899) and total GCN2 (loading control), ***P = 0.0001; or against phospho‐eIF2α (Ser51) and total eIFα (loading control), *P = 0.0482, band intensities measured with FIJI and analysed with unpaired t‐test (see original source data).
-
C–FPhosphorylated eIFα was assessed by immunofluorescence in E9.5 embryos in the forebrain, wild type (C, c1) (n = 2 embryos, 0/14 sections) and Slc7a5‐null (D, d1) (n = 2 embryos 16/16 sections), and in hindbrain/anterior spinal cord, wild type (E–e2) (n = 4 embryos, 0/62 sections) and Slc7a5‐null (F–f2) (n = 4 embryos, 54/54 sections) including in the otic vesicle (compare e2, f2). Scale bars in (C–e1, D–f1) 100 µm and in (e2 and f2) 25 µm. Arrowheads indicate ventral midline.
-
GAn increase in Trib3 protein was detected by Western blot in Slc7a5‐null embryos; each lane represents an E9.5 embryo lysate (wild type n = 3, Slc7a5‐null, n = 4) with α‐tubulin loading control, unpaired t‐test *P = 0.0153 (see original source data).
-
H–LTUNEL assay to detect apoptotic cells in wild type (n = 3 embryos, 32 sections) or Slc7a5‐null (n = 5 embryos, 47 sections); (H, I) transverse sections through the head at level of the forebrain and (J, K) the spinal cord, (H–K) scale bars 50 µm, arrowheads indicate ventral midline. (L) Quantification of TUNEL‐positive cells within spinal cord sections was performed as stated in Materials and Methods. Each dot represents the average apoptosis cell count for a single embryo, Welch's correction unpaired t‐test, P = 0.250, F‐test to compare variances, P = 0.0003 (see original source data).
Induction of ATF4 targets Trib3, Chac1 and CHOP is indicative of a progressed cell stress response 46, 47, which if unresolved ultimately leads to apoptosis 52, 58. Here, we confirmed raised levels of Trib3 protein in Slc7a5‐null embryos (Fig 6G), which correlate with the localised high‐level transcription of this gene (Fig 5F–H). We therefore next assessed cell death in Slc7a5‐null embryos using a TUNEL assay. This revealed widespread apoptotic figures in the developing brain of E9.5 Slc7a5‐null embryos (Fig 6H–L); however, this varied between samples and the profound and varied morphological defects in this region in null embryos made it difficult to quantify with respect to wild type (Fig 6I). Analysis of TUNEL‐positive cells in sections of the dorsal spinal cord and caudal hindbrain also revealed variability between embryos (with an F‐test P = 0.0003), but could be quantified (Fig 6J and K, quantified in L). These findings indicate that most Slc7a5‐null embryos exhibit increased cell death. This is consistent with local triggering of apoptosis as stress levels rise on a cell‐by‐cell basis.
Slc7a5 expression depends on Wnt/β‐catenin signalling, and loss of this pathway induces the stress response gene Trib3
Finally, we noticed that the Slc7a5‐null phenotype is similar to that observed in Wnt pathway mutants, which also exhibit defects in the dorsal neural tube, involving neurogenesis and neural crest as well as in limb development 1, 2, 59. Moreover, there is a good correspondence between Wnt signalling and Slc7a5 expression in caudal epiblast/primitive streak and dorsal neural tube as well as in the limb bud 60 (Fig 1b3, b6, c3, E). Dorsal neural tube defects are dependent on canonical, β‐catenin‐mediated, Wnt signalling 61. This suggests that loss of such signalling might contribute to the Slc7a5‐null phenotype and/or that Slc7a5 transcription is normally promoted downstream of β‐catenin signalling.
To address whether canonical Wnt signalling is compromised in Slc7a5‐null embryos, we assessed expression of the β‐catenin transcriptional target Axin2. This revealed robust expression of Axin2 in both littermate (Fig 7A, a1–a3) and Slc7a5‐null embryos (Fig 7B, b1–b3) indicating that loss of Slc7a5 does not impact canonical Wnt signalling. We next investigated whether Slc7a5 is a potential canonical Wnt signalling target, first by analysing its promoter region for binding sites of relevant transcription factors using the eukaryotic promoter database and the MatInspector program 62. These analyses identified two putative LEF1 and TCF7L2 sites upstream of the mouse Slc7a5 transcription start site (TSS) and multiple potential MYC binding sites flanking the TSS (Fig EV4 and see Dataset EV3), supporting the possibility that Slc7a5 is a target of this pathway in the developing embryo.
Figure 7. Wnt β‐catenin signalling is unaffected in Slc7a5‐null embryos, but is required for Slc7a5 transcription and β‐catenin loss induces ISR gene Trib3 .
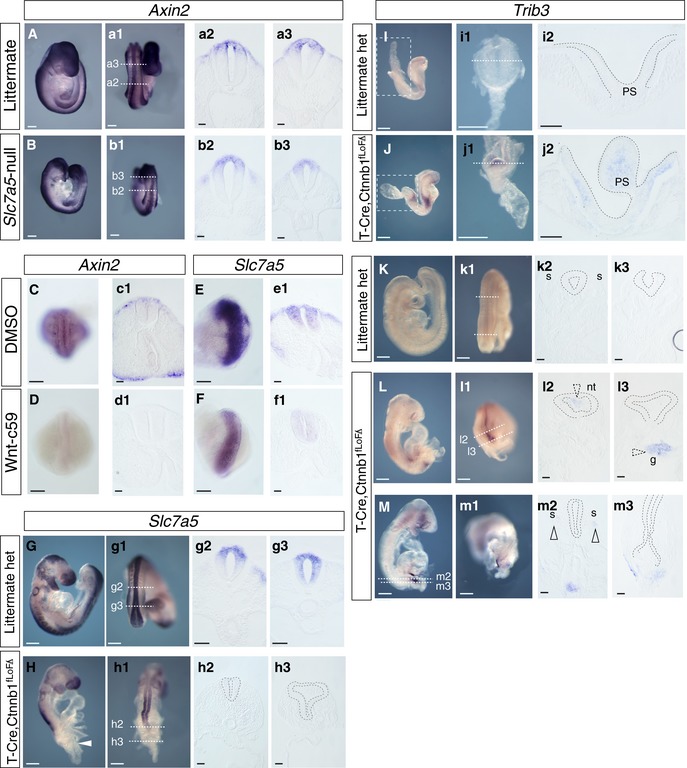
-
A, B(A, a1) Wnt β‐catenin target Axin2 strongly expressed in littermate, (a2, a3) in TS, and (B, b1) in Scl7a5‐null embryos (b2, b3) in TS (n = 2 littermates, n = 6/6 Slc7a5‐null embryos).
-
C, D(C, d1) Axin2 transcripts in embryo trunk explant exposed to DMSO (C, c1, n = 12/12) were reduced in Wnt‐c59‐exposed explants (D, d1, n = 14/14). Images in (c1, d1) show TS through explants (data from three independent embryo explant experiments).
-
E, FSlc7a5 transcripts in embryo trunk explant exposed to DMSO (E, e1, n = 9/10) or Wnt‐c59 (F, f1, n = 15/15). Images in (e1, f1) show TS through explants (data from 3 independent embryo explant experiments).
-
G, HSlc7a5 expression in control T‐Cre;Ctnnb1 flLOF/Δ heterozygous littermate embryo (G, g1) and in TS (g2, g3), and in T‐Cre;Ctnnb1 flLOF/Δ mutant embryo where Slc7a5 transcripts are absent in posterior neural tube (the region of T‐Cre recombination, indicated with white arrowhead in H) and shown in TS in (h2, h3) (n = 0/6 littermate and n = 6/6 mutant embryos).
-
I, JTrib3 is lacking in T‐Cre;Ctnnb1 flLOF/Δ heterozygous control embryos at E7.75 (I), caudal region dissected from the same embryo (white dashed box in I) (i1) and viewed in TS (i2); Trib3 detected in homozygous T‐Cre;Ctnnb1 flLOF/Δ embryo (J), in caudal region dissected from the same embryo (white dashed box in J) (j1) and shown in TS in caudal epiblast and remnant primitive streak (ps) (j2) (n = 3/3 T‐Cre;Ctnnb1 flLOF/Δ embryos and 14 littermate controls). Trib3 was also detected in the more rostral forming neural tube at this early stage (seen in J) and so in cells not directly experiencing β‐catenin loss; this may reflect the failure to form paraxial mesoderm, which provides signals that promote and support neurogenesis.
-
K–MAt E9.5, Trib3 was not detected in T‐Cre;Ctnnb1 flLOF/Δ heterozygous littermate embryos (K, k1) and in TS (k2, k3), but was detected in homozygous T‐Cre;Ctnnb1 flLOF/Δ mutant embryos (L‐m3) in patches of cells in neural tube (nt) (L‐l2), gut (g) (l3) and somites (s) (m2, arrowheads) (n = 9/9 T‐Cre;Ctnnb1 flLOF/Δ embryos and 12 littermate controls).
Figure EV4. Position and sequence of MYC and LEF/TCF binding sites in Slc7a5 promoter region.

Analysis of the −1,075 to +352 bp sequence of the Slc7a5 mouse gene with the MatInspector program 61 identified putative MYC binding sites at positions TSS −9/−25 and +30/+46 (with a matrix similarity comprised between 0.925 and 0.931). A further MYC/MAX binding site, similar to the MYC binding E‐box motif identified in human Slc7a5 promoter 19, was found at −TSS −154/−170. A potential LEF/TCF binding motif (sequence tagagataAAAGgccgc) at position −18/−34 from the transcription starting site. It showed a matrix similarity of 0.903 (see Dataset EV3). This motif was also found by analysing the −1,000 to +100 bp sequence of murine Slc7a5, using the eukaryotic promoter database (http://epd.vital-it.ch/) which is based on the JASPAR CORE 2018 vertebrate database 108. Another motif was found at position −505 with the cut‐off value set at P ≤ 0.001. Both sites were highlighted on the murine Slc7a5 gene sequence based on position and matrix similarity.
To test this regulatory relationship, embryo trunk explants were exposed to the Wnt secretion inhibitor Wnt‐c59, which inhibits Wnt palmitoylation 63. After 16 h, Axin2 expression was unchanged in DMSO vehicle‐only condition, but repressed by Wnt‐c59 (Fig 7C, c1, D, d1). Similarly, Slc7a5 was attenuated in the presence of Wnt‐c59 (Fig 7E, e1, F, f1), placing Slc7a5 transcription downstream of Wnt signalling. We then assessed Slc7a5 expression in mouse embryos lacking functional β‐catenin. As deletion of β‐catenin results in early embryonic lethality 64, we used a conditional approach, crossing mice harbouring floxed alleles of β‐catenin (Ctnnb1tm2Kem) 65 with a mouse line expressing Cre recombinase under the control of the T (Brachyury) promoter (T‐Cre) 66 and mutant embryos generated are referred to here as T‐Cre;Ctnnb1 flLOF/Δ. In these mice, the Cre recombinase is expressed in axial progenitors and their descendants which give rise to the posterior spinal cord and paraxial mesoderm 66, 67, 68. This cross‐generates embryos in which β‐catenin is lost from the developing body axis beginning in the caudal epiblast and primitive streak from ~ E7.5 and by E9.5 also in axial progenitor descendants. At E9.5, T‐Cre;Ctnnb1 flLOF/Δ embryos exhibit a truncated phenotype in which development of paraxial mesoderm is attenuated and remaining axial tissue forms neuroepithelium 68. Importantly, this caudal‐most neural tube lacked Slc7a5 transcripts compared to littermate controls (Fig 7G, g1–g3 and H, h1–h3), indicating that Scl7a5 transcription in the neural tube relies on β‐catenin.
As loss of β‐catenin leads to failure to form paraxial mesoderm, it is formally possible that the absence of other signals provided by this tissue underlies loss of Slc7a5 expression in the adjacent neural tube. If this were the case, we would expect Slc7a5 to be lost in embryos in which the β‐catenin downstream transcription factors Sp5 and Sp8 are mutated, as these embryos also fail to form paraxial mesoderm 69. However, as in littermate controls (Fig EV5A, a1–a3), Sp5/Sp8 compound mutant embryos continue to express Slc7a5 right to the caudal end (Fig EV5B, b1–b3). This finding indicates that Slc7a5 expression in the neural tube does not depend on signals from the newly formed paraxial mesoderm and further that Slc7a5 is regulated by β‐catenin downstream transcription factors other than Sp5 and Sp8. Indeed, available ChIP‐seq data show that while Sp5 and Sp8 bind to early mesodermal genes, they do not target Slc7a5 in differentiating mouse ESCs 70.
Figure EV5. Expression of Slc7a5 in Sp5/Sp8 double‐mutant embryos.
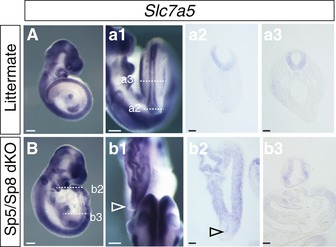
-
A, BSlc7a5 is expressed to the caudal tip of the Sp5/Sp8 double knockout embryos despite failure to form paraxial mesoderm. (A, a1) Slc7a5 expression in littermate control, (a2, a3) in TS, and (B, b1) in Sp5/Sp8 double knockout (dKO) embryo, (b2, b3) in TS (n = 5/5 mutant embryos and n = 3/3 littermate controls). Open arrowheads indicate posterior neural tube. Scale bars 200 µm, except sections 50 µm.
As Slc7a5 loss induces the ISR, we next assessed whether β‐catenin deletion also triggers this stress response. Analysis of early (E7.5–8.25) T‐Cre;Ctnnb1 flLOF/Δ embryos revealed mosaic induction of ISR gene Trib3 in the earliest T‐Cre domains, including posterior epiblast and primitive streak, but not in littermate controls (Fig 7I–j2). Later at E9.5, T‐Cre;Ctnnb1 flLOF/Δ embryos, but not littermate controls, exhibited patches of Trib3‐expressing cells in the region of the body axis truncation (Fig 7K–m3). This included groups of cells where somitic mesoderm should have formed (see open arrows Fig 7m2) and in gut endoderm (a known site of T‐Cre expression 66) (Fig 7l3) as well as cells in the lumen of the spinal cord (Fig 7l2); this pattern of later Trib3 induction may reflect mosaic Cre recombination and time required for the consequences of Slc7a5 loss to be manifest.
Together, these findings show that Wnt and β‐catenin signalling are required for Slc7a5 transcription in the forming neural tube and implicate canonical Wnt signalling in constraint of the integrated stress response.
Discussion
By elucidating the expression pattern, requirement and regulation of the amino acid transporter Slc7a5, this study uncovers a mechanism by which cell metabolism is regulated by developmental signalling in the mammalian embryo. Elevated expression of Slc7a5 in tissues undergoing morphogenesis revealed that amino acid transport is patterned in the embryo. The phenotype of Slc7a5‐null embryos indicated a requirement for increased amino acid transport for neural tube closure, neurogenesis and neural crest development as well as limb bud outgrowth. Aberrant mTORC1 activity and neural progenitor proliferation may underlie aspects of this phenotype. However, pre‐phenotypic transcriptomics and subsequent detection of ATF4 transcriptional targets as well as phosphorylated GCN2 and eIF2α indicated Slc7a5 loss rapidly triggers the ISR. Moreover, this correlated with elevated apoptosis, which is the likely cause of the observed developmental defects. The detection of low levels of ISR gene transcripts in wild‐type embryos further suggested that cells participating in morphogenesis are particularly vulnerable to cell stress. Finally, the requirement for Wnt signalling for Slc7a5 expression identified a new regulatory mechanism, in which Wnt promotes morphogenesis at least in part by inducing expression of this essential amino acid importer, thereby locally supporting metabolic demand and forestalling cell stress during this critical developmental period.
These data show that the effects of Slc7a 5 loss in the mouse embryo involve rapid induction of the ISR and are most likely due to intracellular LNAA deficiency activating the eIF2α kinase, GCN2. Indeed, ISR genes including Trib3, Aldh1l2 and Pck2 are among the top transcripts induced by leucine starvation in HEK293 cells 71; moreover, Slc7a5 loss in human cancer cell lines has been shown to induce this stress response 19, 72, 73; we detect aberrant activity of the amino acid sensor mTORC1; and transcriptomic analysis did not reveal changes in other potentially linked pathways, dopamine and thyroid hormone systems. While we detect elevated phosphorylation of GCN2 and eIf2α indicative of ISR induction in Slc7a5‐null embryos, progression of the ISR was indicated by induction of the pro‐apoptosis gene CHOP, which together with ATF4 directly promotes Trib3 47, 74 and Chac1 46. The correlation between Trib3 and Chac1 induction and increased apoptosis demonstrated that these genes are good reporters for ISR activity in the embryo and allowed identification of tissues particularly sensitive to cellular stresses. This included developing neural tissue, which we additionally confirmed in response to exogenous ER stressors in E8.5 embryos, but also limb buds which form later in E9.5 embryos and exhibit high‐level Chac1 and Trib3 in Slc7a5‐null embryos. Together, these data strongly suggest that rapid induction of apoptosis underlies the developmental defects generated by Slc7a5 loss and identify ISR induction as a potential upstream driver of this process. Interestingly, transcriptional targets for further cell death pathways identified in the context of ER stress, such as that mediated by IRE1 75, were not detected in our E8.5 RNA‐seq data, but studies at later stages are required to exclude such activity. To confirm ISR causality, rescue experiments are also required: although we note that inhibiting this stress response will not compensate for the underlying amino acid deficiency and so may attenuate but will not resolve this phenotype.
The appearance of a phenotype in Slc7a5‐null embryos only at E9.5 indicates that this gene is not critically required for some morphogenesis events that take place during early embryogenesis. This includes gastrulation and axial elongation and may reflect functional redundancy with the related gene, Slc7a8/Lat2 (although this is not fully reciprocal; the Slc7a8 knockout mouse has only a minor phenotype 76). Nonetheless, loss of amino acid import by both these transporters (a condition generated when their regulatory subunit, CD98/Slc3a2, is mutated 77, 78, 79) results in earlier lethality from E7.5 14. Death of Slc7a5‐null embryos at E10.5 may reflect onset of dependence on chorion–allantoic circulation and Slc7a5 expression in extra‐embryonic tissue and later placenta 80, 81.
Importantly, the detection of low‐level Trib3 and Chac1 transcripts in Slc7a5 domains of expression in wild‐type embryos suggested that adaptive ISR is normally ongoing in energetic cell populations in the mammalian embryo. This is consistent with a homeostatic role for elf2α phosphorylation in buffering protein synthesis in normal unstressed cells 82. Slc7a5 is one of a number of stress‐mitigating genes involved in amino acid transport reported to be upregulated by ER stress in cell lines 83, 84. Indeed, the Slc7a5 gene contains a cis‐acting ER stress response element (ERSE) that binds ATFs 83, 85. These findings suggest that Slc7a5 expression is normally regulated by positive feedback once the ISR is activated. Such action may contribute to elevated Slc7a5 expression during energetic activities and serve to ensure timely morphogenesis.
Although Slc7a5 may be upregulated by adaptive ISR in the embryo, this seemed unlikely to be its only regulator as mice lacking this stress pathway do not phenocopy the Slc7a5 deletion 82. The similarities between the phenotypes of Slc7a5‐null and Wnt pathway mutants are striking. Not only do they affect many of the same cell populations, with Wnt mutants also exhibiting defects in neurulation, neural crest 59 and neurogenesis 61 as well as limb bud outgrowth 1, 2, but they also share phenotypic detail. For example, in the dorsal neural tube, loss of Slc7a5 or Wnt reduces neuronal differentiation 61 and both Wnt3a/Wnt1 double‐mutant 59 and Slc7a5‐null embryos possess cell populations derived from the first emerging neural crest, but exhibit reduction of later forming dorsal root ganglia, consistent with a failure to expand the Pax3 expressing neural crest progenitor pool 42. Importantly, such dorsal neural tube defects have been shown to result specifically from loss of β‐catenin‐mediated signalling 61 and also Myc 42 and we show here that Slc7a5 transcription is lost following inhibition of Wnt secretion or deletion of β‐catenin in the forming neural tube. Furthermore, the presence of binding sites for known mediators of canonical Wnt signalling LEF/TCF and the direct Wnt target MYC upstream of Slc7a5, along with ChIP‐seq data, showing that β‐catenin binds the Slc7a5 promoter in the Xenopus embryo 86 and ChIP‐PCR data demonstrating that MYC directly binds the Slc7a5 promoter in human cancer cell lines and promotes Slc7a5 transcription 19 strongly support the possibility that Slc7a5 is a direct Wnt‐β‐catenin/MYC target in the mouse embryo. This conclusion is reinforced by screens which place Wnt upstream of Slc7a5 in other cellular contexts and point to conservation of this regulatory relationship 87, 88.
In addition, Wnts have other downstream pathways and these include promotion of mTORC1 activity in a variety of cell lines, independently of β‐catenin signalling 89, 90, 91. As loss of Slc7a5 and so local amino acid deficiency lead to aberrant mTORC1 activity, this suggests a further explanation for the similarity between mutations affecting Wnt ligands 59 and Slc7a5‐null embryos. Indeed, mTORC1 activity is required for normal neurogenesis in the chick neural tube 92 and for neural crest differentiation and survival 93. This also suggests operation of cross‐talk between mTORC1 and β‐catenin‐mediated Wnt pathways, which serves to maintain proteostasis. Such regulation may in turn be modulated by input from the adaptive ISR as it promotes expression of stress‐mitigating genes including Slc7a5. Moreover, Wnt promotion of metabolic gene expression is characteristic of many cancer cell states, for example, β‐catenin‐promoted cMyc expression upregulates glycolytic genes in such contexts 94, supporting the view that multiple Wnt downstream pathways orchestrate cellular metabolism. Our finding that the ISR gene Trib3 is induced in β‐catenin mutant embryos further connects Wnt signalling to the regulation of cellular stress.
Importantly, monitoring in Slc7a5‐null embryos for phospho‐S6 and also TUNEL‐positive cells underlined that the consequences of stress in tissues are manifest on a cell‐by‐cell basis and that these may differ according to the activity of individual cells during embryogenesis. Moreover, a critical mass of cells may need to be affected to generate a developmental defect and while adaptive stress may delay morphogenesis, chronic stress and so transition to apoptosis may be more likely to occur when multiple stresses are present. Indeed, the ISR integrates a range of cellular stresses which activate distinct eIF2α kinases; for example, amino acid deficiency activates GCN2 and ER stress kinase PERK (protein kinase R (PKR)‐like endoplasmic reticulum kinase), while viral or bacterial infection activates protein kinase R (PKR) and iron deficiency activates haem‐regulated kinase (HRI) by 23. It may be that small changes in, for example, nutrient supply could promote sustained cellular stress across a cell population in particular genetic backgrounds or when pathogens abound. Indeed, there are examples of gene–environment interactions which account for the sporadic nature of human diseases, such as Crohn's 95, neural tube 18, 96 and heart, skeletal and renal defects 97, as well as those induced by hypoxia 98, 99. Moreover, hypoxia‐induced reduction in FGFR1 protein leading to heart defects in mouse embryo has been linked to induction of p‐PERK and the ISR in the context of the unfolded protein response 100.
These findings make important new links between signalling, metabolism, cell stress and developmental defects in the mammalian embryo and suggest that Wnt activity normally locally promotes Slc7a5 to elevate amino acid supply in cells participating in morphogenesis. This identification of Wnt signalling as a key regulator of cell stress in embryonic tissues opens the way to wider investigation of how metabolic demand and the ISR are regulated in particular cell populations and how the level, timing and integration of cell stresses lead to specific developmental defects.
Materials and Methods
Mouse lines and embryo collection
Slc7a5 fl/fl mice harbouring two copies of the Slc7a5‐targeted allele (exon 1 of Slc7a5 flanked with two loxP sites) were crossed with a mouse line ubiquitously expressing Cre recombinase under the Bal1 promoter (Bal1‐cre) to obtain a global Slc7a5 knockout mouse line 16. Heterozygous Slc7a5 +/− C57Bl/6 mice were viable and fertile and were bred free of Bal1‐cre in subsequent generations 16; however, Slc7a5 +/− inter‐crosses did not produce any Slc7a5‐null live mice. Here, heterozygous Slc7a5 +/− mice were crossed to generate litters between E8.5 and E11.5. An abnormal phenotype was not found in E8.5‐null embryos (35/128 Slc7a5 −/− embryos from 18 litters), but was apparent at E9.5 (35/162 Slc7a5 −/− embryos from 19 litters) and the oldest live Slc7a5‐null embryos were found at E10.5 (6/28 Slc7a5 −/− embryos from three litters). Sex and genotype of embryos were determined by PCR.
To generate embryos lacking β‐catenin in axial progenitors, Ctnnb1tm2Kem mice 65 harbouring two copies of the floxed β‐catenin loss‐of‐function (LOF) allele (exons 2–6, including the ATG and domains essential for binding E‐cadherin and TCF/LEF flanked with two loxP sites), referred to as Ctnnb1 flLOF/flLOF, were crossed with a mouse line homozygous for the T‐Cre transgene (which expresses Cre recombinase under the control of the T/Bra promoter 66) and heterozygous at the Ctnnb1 locus deleted for exons 2–6 (T‐Cre tg/tg ; Ctnnb1 Δex2‐6/+). In this cross, both mutant (T‐Cre tg/+ ; Ctnnb Δex2‐6/flLOF) (referred to here as T‐Cre;Ctnnb1 flLOF/Δ) and control littermates (T‐Cre tg/+ ; Ctnnb1 flLOF/+) (referred to here as littermate het) are generated at 50% frequency. Sp5/Sp8 compound mutants were generated as described in 69.
Wild‐type CD1 embryos were collected between E7.0 and E9.5. Embryos were fixed in PFA 4% and processed for in situ hybridisation or immunofluorescence as described below. Mouse colonies were breed, mice sacrificed and embryos isolated following Home Office guidelines (PPL 60/04454 or PPL 60/3455 and/or 60/4118).
Mouse embryo and explant culture and exposure to small molecules
Live E8.5 CD1 embryos were dissected from the uterus and collected within yolk sacs in warm (37°C) culture media (F12 + Glutamax + FCS 10%). Hanging drop culture method 101 was then used with DMSO (1:1,000) or ER stress inducers: tunicamycin (1 μg/ml) and thapsigargin (1 μM). Embryos were cultured for 6 h at 37°C in 5% CO2, then dissected, fixed and processed for in situ hybridisation. E9.5 CD1 mouse embryos were dissected in ice‐cold medium, and explants of the trunk region were made, embedded in collagen and cultured in OptiMem/10%FBS/1×GlutaMAX/1× B27/1xPen strep (all from Gibco) in an incubator with 5% CO2 at 37°C, as described previously 102. Explants were cultured in either vehicle control DMSO or Wnt‐C59 (Tocris, Cat. No. 5148) at 4 μM (1/2,500) in DMSO for 16 h. Explants consisted of full thickness of the embryo at the level of the three most recently formed somites (caudal explant) or at the level of the three next rostral somites (rostral explant) (caudal explants were only compared with other caudal explants and rostral explants only compared with other rostral explants). Following fixation in ice‐cold 4% PFA, explants were processed for in situ hybridisation to detect mRNAs or for immunocytochemistry as described above.
In situ hybridisation for mRNA
In situ hybridisation experiments were performed on whole embryos to detect mRNA for Delta1, Neurog2, FoxG1, Fgf8, Hes5, Sox10, Slc7a5, Slc3a2, Chac1 and Trib3 following standard procedures. Primers used to clone Slc7a5, Slc3a2, Chac1 and Trib3 can be found in Appendix Table S1. Subset of embryos subjected to wholemount in situ hybridisation was embedded and cryo‐sectioned using standard procedures to localise mRNA at a cellular level.
Immunofluorescence
A standard protocol was used to embed and cryo‐section embryos at 20 μm for immunofluorescence. Primary antibodies were used at indicated concentrations: phospho‐H3 (S10), phospho‐S6 (S235/236) and phospho‐S6 (S240/244) (1:500—Cell Signaling Technology #9706, #5364, #2211), phospho‐eIf2α (1:500—Cell Signaling Technology #3398), tubulin‐βIII (Tuj1, 1:500—MMS‐435P, BioLegend), Pax3 (1:200—supernatant DSHB) and FoxA2 (1:200—ab108422, Abcam). Secondary antibodies goat anti‐rabbit IgG (H+L) cross‐adsorbed secondary antibody, Alexa Fluor 594 (A‐11012, Thermo Fisher) and donkey anti‐mouse IgG (H+L) secondary antibody, Alexa Fluor 488 conjugate (A‐21202, Thermo Fisher) were incubated for 30 min at 1:500 at room temperature. Nuclei were stained with DAPI.
TUNEL assay
Detection of cells with DNA‐strand breaks was performed on sections of E9.5 wild‐type or Slc7a5‐null embryos by the TUNEL labelling method, using an ApopTag kit (Millipore) according to the manufacturer's instructions. Images were taken with Leica DMRB microscope using a ×20 objective. To identify TUNEL‐positive cells, a colour threshold of 100 was set in ImageJ, and all cells above this threshold were counted in the dorsal 700 μm of the spinal cord. For quantification, see text, figure legends and original source data.
Microscopy
Whole embryo images (mRNA in situ hybridisation and Tuj‐1 immunofluorescence) were taken using a Micropublisher 3.3RT and Q‐Imaging on a Leica MZFLIII dissecting microscope. Representative sections were imaged using Leica DRB compound microscope. On sectioned tissues, immunofluorescence was imaged using either a personal DeltaVision imaging system (20× lens for Figs 3C–F, 4A, B, G, H and Appendix Fig S2A–D; 10× lens Fig 4C, D (nine images taken and stitched using ImageJ) or with SP8 Leica confocal microscope ×10 (Fig 6C–f1) ×60 (Fig 6e2, f2). To quantify the varied pattern of phospho‐S6 (Fig 4G, H, I), fluorescence intensity was measured using ImageJ and normalised relative to the intensity of DAPI fluorescence in the same area (see original source data). Figures were assembled using Adobe Photoshop and Illustrator software for composite figure construction.
Preparation of RNA‐seq samples
E8.5 embryos with 8–10 somites were dissected in cold PBS and lysed individually with TRIzol reagent. Embryos were stage matched with respect to somite number, and only male embryos were used to avoid potential sex‐related variations in transcriptome. Allantois was kept for genotyping and sexing the embryos by PCR 103. Total RNA was extracted using the RNeasy mini kit (Qiagen). Samples were DNase‐treated “on‐column” using RNase‐free DNase (Qiagen). Quality and quantity of total RNA were measured using a Qubit fluorometer (Invitrogen). Following quality control, 5 wild‐type and 5 Slc7a5‐null male embryos were selected as biological replicates. Samples started with 300 ng total RNA per embryo to which spike‐ins (ERCC, 2 μl of 1:100 dilution) were added. We used a TruSeq RNA v2 kit (Illumina RS‐122‐2001) to prepare the cDNA library following the manufacturer's instructions; polyA RNA selection was performed and fragment and random priming followed by cDNA synthesis, ligation and ×10 rounds of PCR amplification. RNA‐seq was performed by Illumina HiSeq 2000 with 100 bp paired ends and “dye‐swap” spike‐in of all samples. This gave paired‐end reads, length 101 bp, between 28 and 42 million reads per sample.
Computational analysis of RNA‐seq data
Reads were aligned to the Ensembl GRCm38 primary assembly genome (release 90) using STAR ver. 2.5.3a 104 with the following parameters –genomeLoad NoSharedMemory –outSAMstrandField intronMotif –outSAMmode Full –outFilterMultimapNmax 2 –outFilterMismatchNmax 5 –outFilterType BySJout –outSJfilterIntronMaxVsReadN 5000 10000 15000 20000 –readFilesCommand zcat –outSAMtype BAM SortedByCoordinate –outSAMunmapped Within –outReadsUnmapped Fastx –quantMode GeneCounts. Read counts per gene were calculated in the same STAR run. The gene annotation file (release 90) was downloaded from Ensembl. For differential expression, edgeR ver. 3.18.1 105, 106 was applied with its default normalisation, which resulted in a list of log2 fold changes, raw P‐values and Benjamini–Hochberg‐corrected P‐values to control the false discovery rate (FDR) (see Table 1, Appendix Tables S1 and S2).
Quantitative PCR
RNA from E8.5 or E9.5 embryos (respectively, 200 and 500 ng) was reverse‐transcribed using the qScript cDNA synthesis kit (Quanta Biosciences). A 1/10 dilution of the cDNA was used to perform qPCR on a Bio‐Rad iCycler with PerfeCTa SYBR Green FastMix (Quanta Biosciences). Primer pairs used can be found in Appendix Table S2. qPCRs for each embryo were run in triplicate, and the average of these technical replicates was taken to represent one independent experiment. Comparisons of such data from at least three embryos (see Figure legends) were made for each condition and each gene of interest. All data were determined using ΔΔCt method 107 and shown relative to GAPDH with error bars indicating ±SEM. A one‐way ANOVA with a Tukey post‐test was performed for statistical analyses (see original source data).
Preparation of whole embryo lysates and Western blotting
To prepare samples for SDS–PAGE electrophoresis, isolated whole mouse embryos were immediately transferred to a clean microtube containing ice‐cold lysis buffer [50 mM Tris–HCl, pH 7.5, 1% Triton X‐100, 1 mM EGTA, 1 mM EDTA, 150 mM NaCl, 0.27 M sucrose, 50 mM sodium fluoride, 10 mM sodium 2‐glycerophosphate, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM benzamidine, 1 mM PMSF and 0.1% 2‐mercaptoethanol, EDTA‐free protease inhibitor cocktail (Roche) (1 individual embryo per tube)]. Following sonication and vortexing, the resulting homogenate was centrifuged at 3,000 g for 10 min at 4°C to remove remaining tissue debris, and the resulting supernatant was used for Western blot analysis. Each lane was loaded with lysate from one embryo (this was 20 μg, except for the blot for Slc3a2 in which lanes were loaded with 25 μg). Loaded lysates were then subjected to SDS–polyacrylamide gel electrophoresis and immunoblotted as previously described 16 using primary antibodies (1:1,000) against (Cell Signaling, Beverly, MA, USA, phospho‐P70S6K (T389) #9205, P70S6K #2708, phospho‐eIF2α (S51) #3398 and eIF2α #9722, TRB3 (Proteintech, Chicago, IL, USA, #13300‐1‐AP) and α‐Tubulin (Sigma, St Louis, MO, USA, #T6074, used at 1:5000), pGCN2 (T899) (Abcam ab75386) and GCN2 (Abcam ab134053), Slc3a2 (Santa Cruz #sc‐20018), b‐Actin (Cell Signaling Technology cat#4970). Primary antibody detection was carried out with (Cell Signaling, Beverly, MA, USA) secondary antibodies, anti‐rabbit IgG‐HRP #7074 or anti‐mouse IgG‐HRP‐linked #7076 at (1:5,000) as appropriate by ECL. Resulting band intensities were quantified using FIJI software (NIH, Bethesda, MD), and unpaired t‐tests were performed for statistical analyses. In figures, solid black bars indicate where blots were cut and dashed lines separate wild‐type and Slc7a5‐null lanes. See original source data for complete blots.
Author contributions
Conception or design of the work or the acquisition, analysis or interpretation of data for the work: NP, MG, MF, CL, PAH, GAS, AD, TPY, PMT and KGS; Drafting the work or revising it critically for important intellectual content: NP, MG, MF, CL, PAH, TPY, PMT and KGS; Final approval of the version to be published: NP, MG, MF, CL, PAH, GAS, AD, Y‐BS, PMT and KGS; Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: KGS.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Appendix
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Acknowledgements
We thank members of the Storey Laboratory and colleagues Cheryll Tickle, Andy Copp, Linda Sinclair and David Ron for critical reading of versions of this manuscript. We are grateful to Dr Ravi Chalamalasetty for generation and collection of T‐Cre;Ctnnb1 flLOF/Δ and Sp5/Sp8 compound mutant embryos. This research was supported by a Wellcome Trust project grant to PT (WT094226) on which KGS was a named collaborator and NP was further supported for a short period by the ISSF (WT204816/Z/16). YBS is supported by NIH, and TPY is supported by NIH (project number 1ZIABC010345‐18). This study utilised microscopy resources supported by a Wellcome Trust multi‐user equipment grant for the development of tissue imaging approaches (WT101468). KGS is a Wellcome Trust Investigator (WT102817AIA).
EMBO Reports (2020) 21: e48469
Data availability
RNA‐seq data are available in ArrayExpress accession number E‐MTAB‐6336, https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6336/.
References
- 1. Galceran J, Farinas I, Depew MJ, Clevers H, Grosschedl R (1999) Wnt3a−/− like phenotype and limb deficiency in Lef1(−/−)Tcf1(−/−) mice. Genes Dev 13: 709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamaguchi TP, Takada S, Yoshikawa Y, Wu N, McMahon AP (1999) T (Brachyury) is a direct target of Wnt3a during paraxial mesoderm specification. Genes Dev 13: 3185–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crossley PH, Martinez S, Martin GR (1996) Midbrain development induced by FGF8 in the chick embryo. Nature 380: 66–68 [DOI] [PubMed] [Google Scholar]
- 4. Oginuma M, Moncuquet P, Xiong F, Karoly E, Chal J, Guevorkian K, Pourquie O (2017) A gradient of glycolytic activity coordinates FGF and Wnt signaling during elongation of the body axis in amniote embryos. Dev Cell 40: 342–353 e310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miyazawa H, Aulehla A (2018) Revisiting the role of metabolism during development. Development 145: dev131110 [DOI] [PubMed] [Google Scholar]
- 6. Gardner DK, Lane M (1993) Amino acids and ammonium regulate mouse embryo development in culture. Biol Reprod 48: 377–385 [DOI] [PubMed] [Google Scholar]
- 7. Devreker F, Hardy K, Van den Bergh M, Vannin AS, Emiliani S, Englert Y (2001) Amino acids promote human blastocyst development in vitro . Hum Reprod 16: 749–756 [DOI] [PubMed] [Google Scholar]
- 8. Wu G, Bazer FW, Cudd TA, Meininger CJ, Spencer TE (2004) Maternal nutrition and fetal development. J Nutr 134: 2169–2172 [DOI] [PubMed] [Google Scholar]
- 9. Kanai Y, Segawa H, Miyamoto K, Uchino H, Takeda E, Endou H (1998) Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J Biol Chem 273: 23629–23632 [DOI] [PubMed] [Google Scholar]
- 10. Tarlungeanu DC, Deliu E, Dotter CP, Kara M, Janiesch PC, Scalise M, Galluccio M, Tesulov M, Morelli E, Sonmez FM et al (2016) Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell 167: 1481–1494 e1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Friesema EC, Docter R, Moerings EP, Verrey F, Krenning EP, Hennemann G, Visser TJ (2001) Thyroid hormone transport by the heterodimeric human system L amino acid transporter. Endocrinology 142: 4339–4348 [DOI] [PubMed] [Google Scholar]
- 12. Kageyama T, Nakamura M, Matsuo A, Yamasaki Y, Takakura Y, Hashida M, Kanai Y, Naito M, Tsuruo T, Minato N et al (2000) The 4F2hc/LAT1 complex transports L‐DOPA across the blood‐brain barrier. Brain Res 879: 115–121 [DOI] [PubMed] [Google Scholar]
- 13. Mastroberardino L, Spindler B, Pfeiffer R, Skelly PJ, Loffing J, Shoemaker CB, Verrey F (1998) Amino‐acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature 395: 288–291 [DOI] [PubMed] [Google Scholar]
- 14. Sato Y, Heimeier RA, Li C, Deng C, Shi YB (2011) Extracellular domain of CD98hc is required for early murine development. Cell Biosci 1: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laplante M, Sabatini DM (2009) mTOR signaling at a glance. J Cell Sci 122: 3589–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poncet N, Mitchell FE, Ibrahim AF, McGuire VA, English G, Arthur JS, Shi YB, Taylor PM (2014) The catalytic subunit of the system L1 amino acid transporter (slc7a5) facilitates nutrient signalling in mouse skeletal muscle. PLoS One 9: e89547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C et al (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136: 521–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang F, Wu Y, Gu H, Reece EA, Fang S, Gabbay‐Benziv R, Aberdeen G, Yang P (2015) Ask1 gene deletion blocks maternal diabetes‐induced endoplasmic reticulum stress in the developing embryo by disrupting the unfolded protein response signalosome. Diabetes 64: 973–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yue M, Jiang J, Gao P, Liu H, Qing G (2017) Oncogenic MYC activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis. Cell Rep 21: 3819–3832 [DOI] [PubMed] [Google Scholar]
- 20. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA (2013) Control of amino‐acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 14: 500–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Winkle LJ, Tesch JK, Shah A, Campione AL (2006) System B0,+ amino acid transport regulates the penetration stage of blastocyst implantation with possible long‐term developmental consequences through adulthood. Hum Reprod Update 12: 145–157 [DOI] [PubMed] [Google Scholar]
- 22. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R et al (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633 [DOI] [PubMed] [Google Scholar]
- 23. Pakos‐Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM (2016) The integrated stress response. EMBO Rep 17: 1374–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pereira PN, Dobreva MP, Graham L, Huylebroeck D, Lawson KA, Zwijsen AN (2011) Amnion formation in the mouse embryo: the single amniochorionic fold model. BMC Dev Biol 11: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Copp AJ, Greene ND (2013) Neural tube defects–disorders of neurulation and related embryonic processes. Wiley Interdiscip Rev Dev Biol 2: 213–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hentges K, Thompson K, Peterson A (1999) The flat‐top gene is required for the expansion and regionalization of the telencephalic primordium. Development 126: 1601–1609 [DOI] [PubMed] [Google Scholar]
- 27. Hentges K, Sirry B, Gingeras A‐C, Sarbassov D, Sonenberg N, Sabatini DM, Peterson A (2001) FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proc Natl Acad Sci USA 98: 13796–13801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Okada T, Okumura Y, Motoyama J, Ogawa M (2008) FGF8 signaling patterns the telencephalic midline by regulating putative key factors of midline development. Dev Biol 320: 92–101 [DOI] [PubMed] [Google Scholar]
- 29. Storm EE, Rubenstein JL, Martin GR (2003) Dosage of Fgf8 determines whether cell survival is positively or negatively regulated in the developing forebrain. Proc Natl Acad Sci USA 100: 1757–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shimamura K, Rubenstein JL (1997) Inductive interactions direct early regionalization of the mouse forebrain. Development 124: 2709–2718 [DOI] [PubMed] [Google Scholar]
- 31. Nikolopoulou E, Galea GL, Rolo A, Greene ND, Copp AJ (2017) Neural tube closure: cellular, molecular and biomechanical mechanisms. Development 144: 552–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gradwohl G, Fode C, Guillemot F (1996) Restricted expression of a novel murine atonal‐related bHLH protein in undifferentiated neural precursors. Dev Biol 180: 227–241 [DOI] [PubMed] [Google Scholar]
- 33. Henrique D, Adam J, Myat A, Chitnis A, Lewis J, Ish‐Horowicz D (1995) Expression of a Delta homologue in prospective neurons in the chick. Nature 375: 787–790 [DOI] [PubMed] [Google Scholar]
- 34. Scardigli R, Schuurmans C, Gradwohl G, Guillemot F (2001) Crossregulation between Neurogenin2 and pathways specifying neuronal identity in the spinal cord. Neuron 31: 203–217 [DOI] [PubMed] [Google Scholar]
- 35. Helms AW, Johnson JE (2003) Specification of dorsal spinal cord interneurons. Curr Opin Neurobiol 13: 42–49 [DOI] [PubMed] [Google Scholar]
- 36. Lee MK, Tuttle JB, Rebhun LI, Cleveland DW, Frankfurter A (1990) The expression and posttranslational modification of a neuron‐specific beta‐tubulin isotype during chick embryogenesis. Cell Motil Cytoskeleton 17: 118–132 [DOI] [PubMed] [Google Scholar]
- 37. Dupin E, Coelho‐Aguiar JM (2013) Isolation and differentiation properties of neural crest stem cells. Cytometry A 83: 38–47 [DOI] [PubMed] [Google Scholar]
- 38. Southard‐Smith EM, Kos L, Pavan WJ (1998) Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat Genet 18: 60–64 [DOI] [PubMed] [Google Scholar]
- 39. Kuhlbrodt K, Herbarth B, Sock E, Hermans‐Borgmeyer I, Wegner M (1998) Sox10, a novel transcriptional modulator in glial cells. J Neurosci 18: 237–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sato T, Sasai N, Sasai Y (2005) Neural crest determination by co‐activation of Pax3 and Zic1 genes in Xenopus ectoderm. Development 132: 2355–2363 [DOI] [PubMed] [Google Scholar]
- 41. Betancur P, Bronner‐Fraser M, Sauka‐Spengler T (2010) Assembling neural crest regulatory circuits into a gene regulatory network. Annu Rev Cell Dev Biol 26: 581–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kerosuo L, Bronner ME (2016) cMyc regulates the size of the premigratory neural crest stem cell pool. Cell Rep 17: 2648–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18: 1926–1945 [DOI] [PubMed] [Google Scholar]
- 44. Pfender S, Kuznetsov V, Pleiser S, Kerkhoff E, Schuh M (2011) Spire‐type actin nucleators cooperate with formin‐2 to drive asymmetric oocyte division. Curr Biol 21: 955–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mendez‐Lucas A, Hyrossova P, Novellasdemunt L, Vinals F, Perales JC (2014) Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK‐M) is a pro‐survival, endoplasmic reticulum (ER) stress response gene involved in tumor cell adaptation to nutrient availability. J Biol Chem 289: 22090–22102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mungrue IN, Pagnon J, Kohannim O, Gargalovic PS, Lusis AJ (2009) CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4‐ATF3‐CHOP cascade. J Immunol 182: 466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H (2005) TRB3, a novel ER stress‐inducible gene, is induced via ATF4‐CHOP pathway and is involved in cell death. EMBO J 24: 1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chi Z, Zhang J, Tokunaga A, Harraz MM, Byrne ST, Dolinko A, Xu J, Blackshaw S, Gaiano N, Dawson TM et al (2012) Botch promotes neurogenesis by antagonizing Notch. Dev Cell 22: 707–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stark R, Guebre‐Egziabher F, Zhao X, Feriod C, Dong J, Alves TC, Ioja S, Pongratz RL, Bhanot S, Roden M et al (2014) A role for mitochondrial phosphoenolpyruvate carboxykinase (PEPCK‐M) in the regulation of hepatic gluconeogenesis. J Biol Chem 289: 7257–7263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Quiros PM, Prado MA, Zamboni N, D'Amico D, Williams RW, Finley D, Gygi SP, Auwerx J (2017) Multi‐omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216: 2027–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lomelino CL, Andring JT, McKenna R, Kilberg MS (2017) Asparagine synthetase: function, structure, and role in disease. J Biol Chem 292: 19952–19958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13: 89–102 [DOI] [PubMed] [Google Scholar]
- 53. Stenzel D, Wilsch‐Brauninger M, Wong FK, Heuer H, Huttner WB (2014) Integrin alphavbeta3 and thyroid hormones promote expansion of progenitors in embryonic neocortex. Development 141: 795–806 [DOI] [PubMed] [Google Scholar]
- 54. Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ (2008) Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 118: 3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC (2010) Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem 285: 33165–33174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jousse C, Deval C, Maurin AC, Parry L, Cherasse Y, Chaveroux C, Lefloch R, Lenormand P, Bruhat A, Fafournoux P (2007) TRB3 inhibits the transcriptional activation of stress‐regulated genes by a negative feedback on the ATF4 pathway. J Biol Chem 282: 15851–15861 [DOI] [PubMed] [Google Scholar]
- 57. Oslowski CM, Urano F (2011) Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol 490: 71–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tabas I, Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13: 184–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ikeya M, Lee SM, Johnson JE, McMahon AP, Takada S (1997) Wnt signalling required for expansion of neural crest and CNS progenitors. Nature 389: 966–970 [DOI] [PubMed] [Google Scholar]
- 60. Ferrer‐Vaquer A, Piliszek A, Tian G, Aho RJ, Dufort D, Hadjantonakis AK (2010) A sensitive and bright single‐cell resolution live imaging reporter of Wnt/ss‐catenin signaling in the mouse. BMC Dev Biol 10: 121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Valenta T, Gay M, Steiner S, Draganova K, Zemke M, Hoffmans R, Cinelli P, Aguet M, Sommer L, Basler K (2011) Probing transcription‐specific outputs of beta‐catenin in vivo . Genes Dev 25: 2631–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T (2005) MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21: 2933–2942 [DOI] [PubMed] [Google Scholar]
- 63. Proffitt KD, Madan B, Ke Z, Pendharkar V, Ding L, Lee MA, Hannoush RN, Virshup DM (2013) Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT‐driven mammary cancer. Cancer Res 73: 502–507 [DOI] [PubMed] [Google Scholar]
- 64. Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R (1995) Lack of beta‐catenin affects mouse development at gastrulation. Development 121: 3529–3537 [DOI] [PubMed] [Google Scholar]
- 65. Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R (2001) Inactivation of the beta‐catenin gene by Wnt1‐Cre‐mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128: 1253–1264 [DOI] [PubMed] [Google Scholar]
- 66. Perantoni AO, Timofeeva O, Naillat F, Richman C, Pajni‐Underwood S, Wilson C, Vainio S, Dove LF, Lewandoski M (2005) Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development 132: 3859–3871 [DOI] [PubMed] [Google Scholar]
- 67. Dunty WC Jr, Biris KK, Chalamalasetty RB, Taketo MM, Lewandoski M, Yamaguchi TP (2008) Wnt3a/beta‐catenin signaling controls posterior body development by coordinating mesoderm formation and segmentation. Development 135: 85–94 [DOI] [PubMed] [Google Scholar]
- 68. Garriock RJ, Chalamalasetty RB, Kennedy MW, Canizales LC, Lewandoski M, Yamaguchi TP (2015) Lineage tracing of neuromesodermal progenitors reveals novel Wnt‐dependent roles in trunk progenitor cell maintenance and differentiation. Development 142: 1628–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dunty WC Jr, Kennedy MW, Chalamalasetty RB, Campbell K, Yamaguchi TP (2014) Transcriptional profiling of Wnt3a mutants identifies Sp transcription factors as essential effectors of the Wnt/beta‐catenin pathway in neuromesodermal stem cells. PLoS One 9: e87018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kennedy MW, Chalamalasetty RB, Thomas S, Garriock RJ, Jailwala P, Yamaguchi TP (2016) Sp5 and Sp8 recruit beta‐catenin and Tcf1‐Lef1 to select enhancers to activate Wnt target gene transcription. Proc Natl Acad Sci USA 113: 3545–3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hensen SM, Heldens L, van Enckevort CM, van Genesen ST, Pruijn GJ, Lubsen NH (2012) Heat shock factor 1 is inactivated by amino acid deprivation. Cell Stress Chaperones 17: 743–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rosilio C, Nebout M, Imbert V, Griessinger E, Neffati Z, Benadiba J, Hagenbeek T, Spits H, Reverso J, Ambrosetti D et al (2015) L‐type amino‐acid transporter 1 (LAT1): a therapeutic target supporting growth and survival of T‐cell lymphoblastic lymphoma/T‐cell acute lymphoblastic leukemia. Leukemia 29: 1253–1266 [DOI] [PubMed] [Google Scholar]
- 73. Cormerais Y, Giuliano S, LeFloch R, Front B, Durivault J, Tambutte E, Massard PA, de la Ballina LR, Endou H, Wempe MF et al (2016) Genetic disruption of the multifunctional CD98/LAT1 complex demonstrates the key role of essential amino acid transport in the control of mTORC1 and tumor growth. Cancer Res 76: 4481–4492 [DOI] [PubMed] [Google Scholar]
- 74. Carraro V, Maurin AC, Lambert‐Langlais S, Averous J, Chaveroux C, Parry L, Jousse C, Ord D, Ord T, Fafournoux P et al (2010) Amino acid availability controls TRB3 transcription in liver through the GCN2/eIF2alpha/ATF4 pathway. PLoS One 5: e15716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A et al (2012) IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase‐2. Science 338: 818–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Braun D, Wirth EK, Wohlgemuth F, Reix N, Klein MO, Gruters A, Kohrle J, Schweizer U (2011) Aminoaciduria, but normal thyroid hormone levels and signalling, in mice lacking the amino acid and thyroid hormone transporter Slc7a8. Biochem J 439: 249–255 [DOI] [PubMed] [Google Scholar]
- 77. Broer A, Friedrich B, Wagner CA, Fillon S, Ganapathy V, Lang F, Broer S (2001) Association of 4F2hc with light chains LAT1, LAT2 or y+LAT2 requires different domains. Biochem J 355: 725–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nakamura E, Sato M, Yang H, Miyagawa F, Harasaki M, Tomita K, Matsuoka S, Noma A, Iwai K, Minato N (1999) 4F2 (CD98) heavy chain is associated covalently with an amino acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J Biol Chem 274: 3009–3016 [DOI] [PubMed] [Google Scholar]
- 79. Fenczik CA, Zent R, Dellos M, Calderwood DA, Satriano J, Kelly C, Ginsberg MH (2001) Distinct domains of CD98hc regulate integrins and amino acid transport. J Biol Chem 276: 8746–8752 [DOI] [PubMed] [Google Scholar]
- 80. Chrostowski MK, McGonnigal BG, Stabila JP, Padbury JF (2009) LAT‐1 expression in pre‐ and post‐implantation embryos and placenta. Placenta 30: 270–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ritchie JW, Taylor PM (2001) Role of the System L permease LAT1 in amino acid and iodothyronine transport in placenta. Biochem J 356: 719–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner‐Weir S, Kaufman RJ (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7: 1165–1176 [DOI] [PubMed] [Google Scholar]
- 83. Dombroski BA, Nayak RR, Ewens KG, Ankener W, Cheung VG, Spielman RS (2010) Gene expression and genetic variation in response to endoplasmic reticulum stress in human cells. Am J Hum Genet 86: 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Krokowski D, Han J, Saikia M, Majumder M, Yuan CL, Guan BJ, Bevilacqua E, Bussolati O, Broer S, Arvan P et al (2013) A self‐defeating anabolic program leads to beta‐cell apoptosis in endoplasmic reticulum stress‐induced diabetes via regulation of amino acid flux. J Biol Chem 288: 17202–17213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yoshida H, Haze K, Yanagi H, Yura T, Mori K (1998) Identification of the cis‐acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose‐regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem 273: 33741–33749 [DOI] [PubMed] [Google Scholar]
- 86. Nakamura Y, de Paiva Alves E, Veenstra GJ, Hoppler S (2016) Tissue‐ and stage‐specific Wnt target gene expression is controlled subsequent to beta‐catenin recruitment to cis‐regulatory modules. Development 143: 1914–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA (2009) Wnt/beta‐catenin signaling is required for CNS, but not non‐CNS, angiogenesis. Proc Natl Acad Sci USA 106: 641–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rossol‐Allison J, Stemmle LN, Swenson‐Fields KI, Kelly P, Fields PE, McCall SJ, Casey PJ, Fields TA (2009) Rho GTPase activity modulates Wnt3a/beta‐catenin signaling. Cell Signal 21: 1559–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K et al (2006) TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126: 955–968 [DOI] [PubMed] [Google Scholar]
- 90. Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS (2009) mTOR mediates Wnt‐induced epidermal stem cell exhaustion and aging. Cell Stem Cell 5: 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chen J, Tu X, Esen E, Joeng KS, Lin C, Arbeit JM, Ruegg MA, Hall MN, Ma L, Long F (2014) WNT7B promotes bone formation in part through mTORC1. PLoS Genet 10: e1004145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fishwick KJ, Li RA, Halley P, Deng P, Storey KG (2010) Initiation of neuronal differentiation requires PI3‐kinase/TOR signalling in the vertebrate neural tube. Dev Biol 338: 215–225 [DOI] [PubMed] [Google Scholar]
- 93. Nie X, Zheng J, Ricupero CL, He L, Jiao K, Mao JJ (2018) mTOR acts as a pivotal signaling hub for neural crest cells during craniofacial development. PLoS Genet 14: e1007491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sherwood V (2015) WNT signaling: an emerging mediator of cancer cell metabolism? Mol Cell Biol 35: 2–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW (2010) Virus‐plus‐susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell 141: 1135–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Greene ND, Copp AJ (2014) Neural tube defects. Annu Rev Neurosci 37: 221–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shi H, Enriquez A, Rapadas M, Martin E, Wang R, Moreau J, Lim CK, Szot JO, Ip E, Hughes JN et al (2017) NAD deficiency, congenital malformations, and niacin supplementation. N Engl J Med 377: 544–552 [DOI] [PubMed] [Google Scholar]
- 98. Sparrow DB, Chapman G, Smith AJ, Mattar MZ, Major JA, O'Reilly VC, Saga Y, Zackai EH, Dormans JP, Alman BA et al (2012) A mechanism for gene‐environment interaction in the etiology of congenital scoliosis. Cell 149: 295–306 [DOI] [PubMed] [Google Scholar]
- 99. Moreau JLM, Kesteven S, Martin E, Lau KS, Yam MX, O'Reilly VC, Del Monte‐Nieto G, Baldini A, Feneley MP, Moon AM et al (2019) Gene‐environment interaction impacts on heart development and embryo survival. Development 146: dev172957 [DOI] [PubMed] [Google Scholar]
- 100. Shi H, O'Reilly VC, Moreau JL, Bewes TR, Yam MX, Chapman BE, Grieve SM, Stocker R, Graham RM, Chapman G et al (2016) Gestational stress induces the unfolded protein response, resulting in heart defects. Development 143: 2561–2572 [DOI] [PubMed] [Google Scholar]
- 101. Potter SW, Morris JE (1985) Development of mouse embryos in hanging drop culture. Anat Rec 211: 48–56 [DOI] [PubMed] [Google Scholar]
- 102. Placzek M, Dale K (1999) Tissue recombinations in collagen gels. Methods Mol Biol 97: 293–304 [DOI] [PubMed] [Google Scholar]
- 103. McFarlane L, Truong V, Palmer JS, Wilhelm D (2013) Novel PCR assay for determining the genetic sex of mice. Sex Dev 7: 207–211 [DOI] [PubMed] [Google Scholar]
- 104. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. McCarthy DJ, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res 40: 4288–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- 108. Khan A, Fornes O, Stigliani A, Gheorghe M, Castro‐Mondragon JA, van der Lee R, Bessy A, Cheneby J, Kulkarni SR, Tan G et al (2018) JASPAR 2018: update of the open‐access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 46: D260–D266 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Appendix
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Data Availability Statement
RNA‐seq data are available in ArrayExpress accession number E‐MTAB‐6336, https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6336/.