

Abstract
Objective
To characterize the time course of ventricular volume expansion in genetic frontotemporal dementia (FTD) and identify the onset time and rates of ventricular expansion in presymptomatic FTD mutation carriers.
Methods
Participants included patients with a mutation in MAPT, PGRN, or C9orf72, or first-degree relatives of mutation carriers from the GENFI study with MRI scans at study baseline and at 1 year follow-up. Ventricular volumes were obtained from MRI scans using FreeSurfer, with manual editing of segmentation and comparison to fully automated segmentation to establish reliability. Linear mixed models were used to identify differences in ventricular volume and in expansion rates as a function of time to expected disease onset between presymptomatic carriers and noncarriers.
Results
A total of 123 participants met the inclusion criteria and were included in the analysis (18 symptomatic carriers, 46 presymptomatic mutation carriers, and 56 noncarriers). Ventricular volume differences were observed 4 years prior to symptom disease onset for presymptomatic carriers compared to noncarriers. Annualized rates of ventricular volume expansion were greater in presymptomatic carriers relative to noncarriers. Importantly, time-intensive manually edited and fully automated ventricular volume resulted in similar findings.
Conclusions
Ventricular volume differences are detectable in presymptomatic genetic FTD. Concordance of results from time-intensive manual editing and fully automatic segmentation approaches support its value as a measure of disease onset and progression in future studies in both presymptomatic and symptomatic genetic FTD.
Frontotemporal dementia (FTD) is a heritable neurodegenerative disorder characterized clinically by behavioral or language deficits and atrophy within the frontal and temporal lobes. Approximately 30% of patients with FTD present with an autosomal dominant family history, with mutations in MAPT, PGRN, and C9Forf72 each presenting in 5%–25% of familial FTD cases.1 Advances have been made in developing disease-modifying treatments that target the underlying pathology of FTD.2 As the initiation of FTD treatment is anticipated to be necessary during the presymptomatic or prodromal stages of the disease, biomarkers sensitive to these disease periods are needed. Brain volumetric measurements may be a promising candidate measure of disease onset and progression, as atrophy in regions of the frontal and temporal lobes may appear as early as 5–10 years before anticipated disease onset in genetic FTD.3
Changes in ventricular volume represent a particularly attractive candidate index of neuronal survival in FTD. Ventricular expansion is seen across the heterogeneous clinical, molecular, and genetic subtypes of FTD at the symptomatic stage.4,5 Additional advantages include reduced image distortion from gradient nonlinearities due to the ventricles’ proximity to the magnet’s isocenter and high contrast in intensity between ventricles and tissue, which facilitates automated segmentation techniques and implementation in large clinical trials.6,7
Ventricular expansion during the presymptomatic stages of genetic FTD has not yet been characterized. The objective of the present study was to examine ventricular volume expansion cross-sectionally and over a 1-year interval in carriers of an FTD-causing genetic mutation and biologically related noncarriers to determine its utility as a measure of early or presymptomatic disease in genetic FTD.
Methods
Participants
The data used in this study were obtained from the multicenter international Genetic Frontotemporal Dementia Initiative (GENFI). Participants were recruited from 12 research sites across Canada, Sweden, Italy, the United Kingdom, and the Netherlands and were either a known symptomatic carrier of a pathogenic FTD-causing mutation in MAPT, PGRN, or C9orf72 or a first-degree relative of a known symptomatic mutation carrier. As described previously,3 all participants completed clinical interviews and standardized neuropsychological testing at baseline and at 1-year follow-up. At-risk first-degree relatives underwent genetic testing to determine mutation carrier status.
Therefore, the sample was composed of symptomatic mutation carriers and biologically related presymptomatic mutation carriers and noncarriers.
Imaging and ventricular volume processing
Volumetric T1-weighted scans were acquired from a 3T Philips (Best, the Netherlands), Siemens (Munich, Germany) Trio, Siemens Skyra, or GE (Chicago, IL) machine; 1.5 Siemens or GE scanners were utilized if a 3T was not available. Scanning protocols were designed to accommodate the different scanners and field strengths.4 Longitudinal scans collected approximately 1 year after baseline, using the same scanner and protocol as the baseline scan, were included in the analysis.
The default longitudinal stream in FreeSurfer, version 5.1 (surfer.nmr.mgh.harvard.edu/), was used for ventricular volume processing.8 DICOM images were converted into NIfTI format using the mri_convert command available in FreeSurfer. An unbiased within-subject template was created using inverse consistent registration.9 In addition, utilizing information from the within-subject template, processing steps including skull stripping, Talairach transformation, atlas registration, spherical surface maps, and parcellations were initialized to increase reliability and statistical power.8 Ventricular segmentations of the lateral, inferior, third, and fourth ventricles were visually checked and manually edited by TPT, while blinded to familial mutation group membership, mutation status (carrier vs noncarrier), and study period (baseline or follow-up). Volumes of the left and right lateral and inferior ventricles, third and fourth ventricles, and total intracranial were extracted from aseg.txt longitudinal output files.
Statistical analyses
Total ventricular volume was calculated as the sum of the left and right lateral (including inferior), third, and fourth ventricles and was expressed as a percentage of the individual's total intracranial volume. Main analyses compared ventricular volume in presymptomatic carriers vs noncarriers, collapsed across the mutation types to maximize sample size. Exploratory analysis included genetic mutation type (C9orf72, PGRN, MAPT) to compare ventricular changes across the 3 genes.
Linear mixed models were used to examine differences in baseline ventricular volumes and change over 1 year. Presymptomatic carriers were compared to noncarriers to examine whether differences were detectable between asymptomatic carriers vs noncarriers. Predictor variables in these analyses included random effects (family membership [variance components covariance structure] and each participant nested within the family [unstructured covariance structure], and a random intercept for each) and fixed effects (visit [baseline, follow-up], genetic status [GS; carrier, noncarriers], years from expected symptom onset, and an interaction term for GS and years from expected symptom onset). Years from expected age at symptom onset were calculated by subtracting the mean age at symptom onset within the family from the participant's current age at the time of baseline scan and follow-up scan.3 As ventricular volume was predicted to change in a nonlinear fashion over time as individuals approached the time of expected symptom onset, a quadratic term for time from expected symptom onset and its interaction with genetic mutation status were included. Thus, using ventricular volume at time 1 and 2 within individuals, the model could evaluate whether a linear or quadratic change in ventricular volume was present across individuals as a function of years to expected symptom onset. In order to create parsimonious models, nonsignificant interaction terms were removed. The visit by GS interaction was examined in a separate model without the time to expected symptom onset by GS interaction due to multicollinearity. Residual and influence analyses were conducted to examine model quality and to identify potential outliers. Studentized and conditional residuals were examined, along with several influence diagnostic measures including Cook D, COVRATIO, Restricted Likelihood Distance, the PRESS statistic, and MDFFIT. Given assumptions made in using the expected years to symptom onset based on the average age at symptom onset in the family, we also conducted a confirmatory analysis using the final model, substituting years to expected symptom onset with the participant’s age.
To examine differences in ventricular volume across the presymptomatic, prodromal, and affected stages of the disease, similar models were included to compare all mutation carriers (symptomatic and presymptomatic mutation carriers and progressors) relative to noncarriers. This model allows ventricular volume to be examined across the continuum of the disease and allowed an opportunity to examine whether and how the model might change if symptomatic patients were included with presymptomatic carriers, as has been done in some prior studies.
Significant interactions between years from expected symptom onset and GS were followed up with t tests to assess potential differences between the genetic carriers and noncarriers in the years prior to and after expected symptom onset, across the baseline and follow-up periods. Significant results were also followed up with analysis of regional ventricular volumes (i.e., left and right). Given reported gray matter asymmetries in PGRN mutation carriers,10–12 we also computed and examined a laterality index, defined as the absolute difference between the left and right ventricular volumes divided by the total ventricular volume.3 All statistical analyses were conducted using SAS (version 9.4).
Standard protocol approvals, registrations, and patient consents
Local ethics committees at each site approved the study and all participants provided written informed consent at enrollment.
Data availability
The data for this study were obtained from the GENFI data freeze 2. Further details on the GENFI protocol, cohorts, and data policies can be found at http://genfi.org.uk/. Due to the sensitive nature of the genetic data in the GENFI cohort, and consistent with practices adopted by the DIAN study of genetic Alzheimer disease, individual data points are not presented to preserve participant blinding to GS and confidentiality. Supplemental analysis as described below has been deposited in the open data repository linked to this article (additional results, e-tables 1–9, figure e-1, doi.org/10.5061/dryad.1qj7630).
Results
Participants
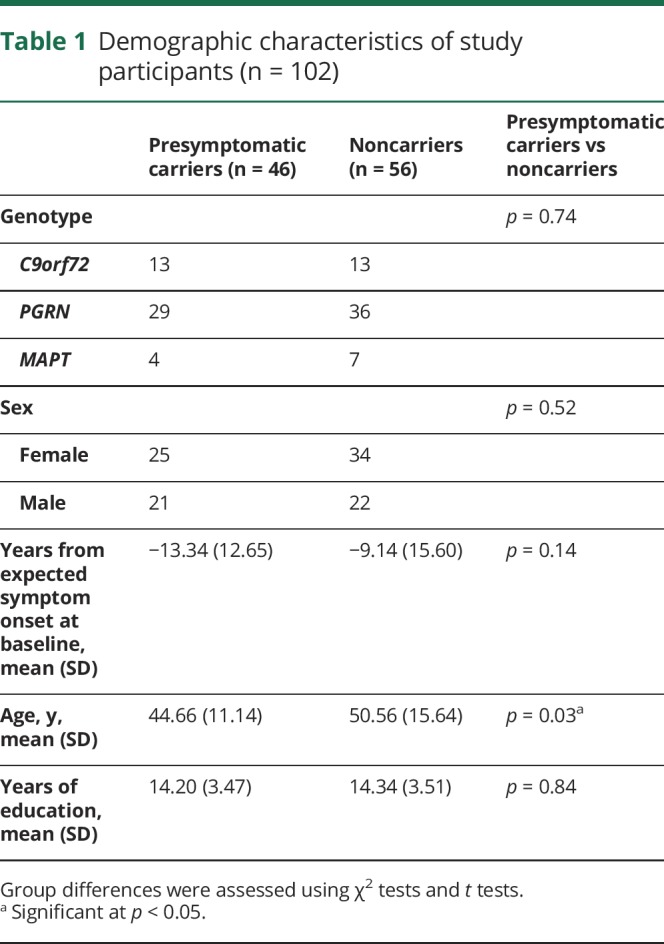
A total of 106 participants met the inclusion criteria. After processing in FreeSurfer, 4 participants were removed prior to statistical analysis: 1 due to scaling errors, 1 with extensive segmentation errors, and 2 found to be extreme outliers (1 carrier and 1 noncarrier from PGRN families; mean volumes >3 SD), leaving 102 participants from 43 family cohorts entered into the statistical model (table 1).
Table 1.
Demographic characteristics of study participants (n = 102)
Presymptomatic carriers vs noncarriers
The final model included the GS by time to symptom onset (linear) interaction, which illustrates differences between the genetic groups at differing points of time to symptom onset. A diagnostic analysis identified 2 high-influential participants (noncarriers) who were subsequently removed (table e-8, doi.org/10.5061/dryad.1qj7630), resulting in a main effect of GS and a GS by time from symptom onset interaction. In addition, visual inspection of the scatterplot indicated an extreme case (noncarrier). Table 2 shows the model estimates, p values, and confidence intervals for the main effects (visit, GS, time from symptom onset [linear and quadratic term]), and the interaction between time from symptom onset and GS. Unadjusted post hoc t tests demonstrated differences in total ventricular volume between presymptomatic carriers and noncarriers beginning 2 years prior to expected symptom onset, or beginning at 4 years prior to expected symptom onset when one extreme case (noncarrier) was removed from the model (figure 1). Table 3 shows the model estimates for the total ventricular volume and total left and right volumes at specific years prior to expected symptom onset (−25 years to 10 years). To create parsimonious models, we excluded sites (n = 11) as random effects and only accounted for family membership. In a confirmatory analysis, we included site as a random effect in the final model to examine the potential influence of data collection from multiple sites. Confirmatory analysis supported that site was not significant (p = 0.32). In the supportive analysis, with age used instead of expected years to symptom onset, a significant genetic group by age interaction was found. Post hoc tests demonstrated that in comparison to noncarriers, presymptomatic carriers showed greater ventricular volume beginning at age 49.
Table 2.
Total ventricular volume estimates for presymptomatic (n = 46) and noncarriers (n = 53), with no influential cases (n = 2) or extreme case (n = 1)
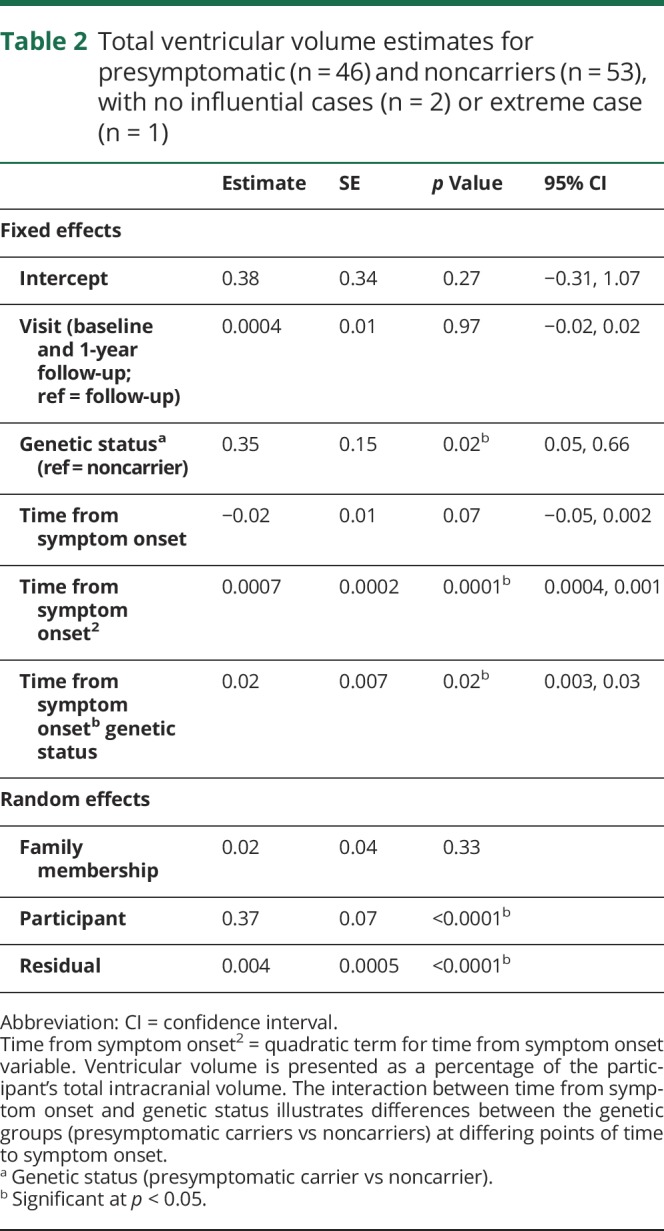
Figure 1. Total ventricular volume by years from expected symptom onset in presymptomatic carriers vs noncarriers.

Total ventricular volume by estimated years from expected symptom onset in presymptomatic carriers (blue, n = 46) and noncarriers (red, n = 53). Ventricular volume is expressed as a percentage of intracranial volume. To prevent disclosure of genetic status, individual data points are not plotted. Differences are noted beginning at 4 years prior to symptom onset as indicated by the dashed line (p = 0.04).
Table 3.
Total ventricle volume estimates from post hoc test between presymptomatic mutation carriers (n = 46) and noncarriers (n = 53) by time to expected symptom onset with no influential cases (n = 2) or extreme case (n = 1)

Manually edited vs fully automated ventricular volumes
Manual edits to the ventricular segmentations performed in FreeSurfer were made on all study participants for each time point (mean differences in edited vs unedited volumes are reported in the e-supplement). Substitution of the fully automated ventricular volumes produced by FreeSurfer into the final models resulted in similar findings, demonstrating that for presymptomatic carriers vs noncarriers, significant differences were observed at 4 years prior to symptom onset (table e-9, a and b, doi.org/10.5061/dryad.1qj7630). See table e-6 (doi.org/10.5061/dryad.1qj7630) for annualized change of unedited ventricular volume.
Total ventricular expansion over 1 year
To assess potential differences in ventricular expansion over the 1-year interval, an additional model was comprised that included the same family and participant random effects as above and the following fixed effects: visit, years to symptom onset (linear and quadratic terms), GS, and an interaction between visit and GS. Significant visit by GS interaction was followed up by simple effects estimation. There was a significant time by GS interaction (p = 0.03); however, follow-up tests did not reach significance (all ps > 0.18). Annual rates of change of total ventricular volume are presented in table e-7 (doi.org/10.5061/dryad.1qj7630), as a function of GS and years to expected symptom onset.
Mutation type
Given previously reported differences in atrophy patterns across the different genotypes, we conducted an exploratory analysis to assess potential differences in total ventricular volume and in the laterality index between the genotypes. Specifically, utilizing the final models from previous analysis (with the extreme case and 2 influential cases), we included genotype (C9orf72, PGRN, MAPT) and the interaction between genotype and GS as fixed effects in the model. There was no significant interaction between genotype and GS (p = 0.10). There was no significant interaction between GS and genotype for the laterality index (p = 0.63).
Discussion
In this multicenter cohort of individuals from families with genetic FTD, we found that ventricular volume enlargement is detectable in the presymptomatic period, on average 4 years prior to the anticipated onset of symptoms. We also provide the first estimates of annualized rates of ventricular expansion in presymptomatic gene carriers compared to biologically related noncarriers.
We also examined ventricular volume changes across the presymptomatic and prodromal to symptomatic stages of the disease, which offers a unique opportunity to explore ventricular volume changes throughout the disease continuum. This method has also been employed by other studies3 delineating cross-sectional gray matter volumes in a genetic cohort. When all mutation carriers (symptomatic and presymptomatic) were included in the model, ventricular volume changes emerged 12 years prior to symptom onset. This is in contrast to the model including only presymptomatic participants. Importantly, the presymptomatic model allows the examination of subtle changes that emerge a few years prior to disease symptom onset, unbiased by the increased rate of change that may occur during the symptomatic period. Thus, the different model estimates and the earlier detection of ventricular volume changes in the combined model of symptomatic patients with the presymptomatic individuals likely reflects the increased the rate of change of ventricular volume as the disease progresses. We suggest that the presymptomatic carrier vs noncarrier model identifying measurable differences 4 years prior to anticipated disease symptom onset offers the more accurate depiction of ventricular volume changes throughout the presymptomatic period.
Ventricular volume expansion has been well-documented in patients with symptomatic sporadic and genetic forms of FTD.13–15 The annual mean rates of expansion for symptomatic mutation carriers in this GENFI cohort range from 6% to 11% and are in line with those reported in a series of 6 symptomatic C9orf72 carriers (mean annualized rate of ventricular expansion of ∼9%)14 and in 21 MAPT symptomatic carriers (∼9%).16 The expansion rate is slightly lower than that reported previously in a cohort of patients with sporadic FTD (mean 11%–14%)13 despite similarities in the mean age of symptomatic participants. Prior small series of presymptomatic mutation carriers have not detected significant differences in ventricular expansion rates in 7 C9orf72 presymptomatic carriers (mean age 41) over a 6-month interval,17 or in 9 presymptomatic MAPT carriers (1-year interval).16
The varied and dramatic atrophy patterns observed in FTD can introduce difficulties for automated segmentation programs.18 We focused our analysis on total ventricular volume in particular, as in the presymptomatic state, the laterality and exact brain regions that may display the earliest signs of atrophy are not certain, even within a genetic mutation. Such advantages of whole brain measurements, such as total ventricular volume, for tracking outcomes have been described.13,19,20 Despite theoretical concerns about lesser sensitivity due to averaging across brain regions, this study supports the potential for total ventricular volume measurements to provide an unbiased approach to capture accelerated rates of atrophy in presymptomatic and early symptomatic stages of disease. Our comparison of time-intensive, detailed manual editing of all scans included in the study relative to the fully automated segmentation with no editing produced remarkably similar results, further supporting the feasibility of total ventricular volume as a practical measure of disease onset and progression in multicenter clinical trials in FTD.
Despite the relatively large sample for a cohort of presymptomatic FTD mutation carriers, subgrouping by mutation type and years to expected symptom onset resulted in significant variability in some estimates. While significant variability in rates of atrophy has been reported within mutation groups and even within families,21 in the current study, the variability may potentially be due at least in part to the sample size of subgroups. Due to subgroup sample size, the examination of differences between carriers of C9orf72, PRGN, and MAPT was exploratory, and did not reach significance. Examination of subregions of ventricular expansion identified the third ventricle as one of the earliest markers in symptomatic patients (e-Results, doi.org/10.5061/dryad.1qj7630). While we did not identify a significant interaction between GS and time to expected symptom onset for the third ventricle in presymptomatic carriers, inspection of the post hoc tests indicate early expansion of the third ventricle (∼14 years prior to expected symptom onset) in this cohort as well. Together these findings suggest that enlargement of the third ventricle in presymptomatic C9orf72 carriers may be one of the first neuroimaging derived markers, due to early thalamic atrophy.22
An additional potential limitation of this study is the use of the estimated age at symptom onset, calculated by subtracting the mean age at symptom onset within the family from the participant’s age at the time of testing. While previous work has demonstrated a strong association between patient age at symptom onset and mean familial age at symptom onset,3 it has been observed that age at symptom onset within families is particularly variable in GRN mutations and somewhat variable in C9ORF72. Although we found similar results when current age or actual age at symptom onset was substituted into the models, we cannot yet confirm how accurately the anticipated age at symptom onset represents the actual age in the majority of individuals in the presymptomatic cohort. Data anticipated from collaborations across large FTD cohorts including LEFTDS, ARTFL, GENFI, and DINAD examining individual age at symptom onset with family age at symptom onset, parent age at symptom onset, and other potential mediator factors will be helpful in the future to improve such models. Furthermore, differences between the presymptomatic carriers and noncarriers were based on groupwise estimates and could not yet be applied on an individual basis. Data collection of additional longitudinal timepoints continues for the GENFI cohort, which will inform future estimates of individual rates of change in ventricular volumes.
Overall, the present study shows ventricular volume differences during the presymptomatic period in genetic FTD pathophysiology and supports the potential of application of ventricular volume as one index of disease symptom onset in the prodromal stages of FTD. Future longitudinal follow-up of this GENFI cohort, as well as comparison with anticipated results from other familial FTD cohorts such as Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (memory.ucsf.edu/lefftds), and with complementary measures such as rates of change in total brain volume will enable further modeling according to specific genotype and confirm the rates of change.
Glossary
- FTD
frontotemporal dementia
- GENFI
Genetic Frontotemporal Dementia Initiative
- GS
genetic status
Author contributions
T.P. Tavares: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, statistical analysis. D. Mitchell: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval. K. Coleman: data acquisition, accepts responsibility for conduct of research and final approval, acquisition of data, study supervision. C. Shoesmith: data acquisition, accepts responsibility for conduct of research and final approval. R. Bartha: study concept or design, accepts responsibility for conduct of research and final approval. D.M. Cash: data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval. K.M. Moore: data acquisition, accepts responsibility for conduct of research and final approval, acquisition of data, study supervision. J. van Swieten: drafting/revising the manuscript, data acquisition, study concept or design, accepts responsibility for conduct of research and final approval, acquisition of data. B. Boroni: data acquisition, accepts responsibility for conduct of research and final approval, acquisition of data. D. Galimberti: drafting/revising the manuscript, accepts responsibility for conduct of research and final approval, contribution of vital reagents/tools/patients. M.C. Tartaglia: drafting/revising the manuscript, accepts responsibility for conduct of research and final approval. J. Rowe: drafting/revising the manuscript, data acquisition, study concept or design, accepts responsibility for conduct of research and final approval, acquisition of data. C. Graff: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, contribution of vital reagents/tools/patients, acquisition of data, study supervision, obtaining funding. F. Tagliavini: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval. G. Frisoni: data acquisition, accepts responsibility for conduct of research and final approval, obtaining funding. S. Cappa: data acquisition, accepts responsibility for conduct of research and final approval. R. Laforce: data acquisition, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, obtaining funding. A. de Mendonça: drafting/revising the manuscript, data acquisition, accepts responsibility for conduct of research and final approval. S. Sorbi: data acquisition, accepts responsibility for conduct of research and final approval, acquisition of data. G. Wallstrom: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, statistical analysis. M. Masellis: drafting/revising the manuscript, data acquisition, accepts responsibility for conduct of research and final approval. J.D. Rohrer: drafting/revising the manuscript, data acquisition, study concept or design, accepts responsibility for conduct of research and final approval, study supervision, obtaining funding. E. Finger: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data, statistical analysis, study supervision, obtaining funding.
Study funding
This work was funded by the UK Medical Research Council, the Italian Ministry of Health and the Canadian Institutes of Health Research as part of a Centres of Excellence in Neurodegeneration grant, and also a Canadian Institutes of Health Research operating grant (MOP 327387) and funding from the Weston Brain Institute to M.M and E.F. J.D.R., D.C. and K.M.M. are supported by the NIHR Queen Square Dementia Biomedical Research Unit, the NIHR UCL/H Biomedical Research Centre and the Leonard Wolfson Experimental Neurology Centre (LWENC) Clinical Research Facility. J.D.R. is supported by an MRC Clinician Scientist Fellowship (MR/M008525/1) and has received funding from the NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH), the MRC UK GENFI grant (MR/M023664/1) and The Bluefield Project. K.M.M. is supported by an Alzheimer's Society PhD Studentship (AS-PhD-2015-005). J. Rowe is supported by the Medical Research Council, Wellcome Trust (103848) and NIHR Cambridge Biomedical Research Centre. F.T. is supported by the Italian Ministry of Health (Grant NET-2011-02346784). L.C.J and J.V.S are supported by the Association for frontotemporal Dementias Research Grant 2009, ZonMw Memorabel project number 733050103 and 733050813, and the Bluefield project. The Swedish contributors C.G., L.O. and C.A. were supported by grants from JPND Prefrontals Swedish Research Council (VR) 529-2014-7504, Swedish Research Council (VR) 2015-02926, Swedish Research Council (VR) 2018-02754, Swedish FTD Initiative- Schörling Foundation, Swedish Brain Foundation, Swedish Alzheimer Foundation, Stockholm County Council ALF, Karolinska Institutet Doctoral Funding and StratNeuro, Swedish Demensfonden, during the conduct of the study.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 2012;8:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai RM, Boxer AL. Therapy and clinical trials in frontotemporal dementia: past, present, and future. J Neurochem 2016;138(suppl 1):211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rohrer JD, Nicholas JM, Cash DM, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal Dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol 2015;14:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rohrer JD. Structural brain imaging in frontotemporal dementia. Biochim Biophys Acta 2012;1822:325–332. [DOI] [PubMed] [Google Scholar]
- 5.Whitwell JL, Xu J, Mandrekar J, et al. Frontal asymmetry in behavioral variant frontotemporal dementia: clinicoimaging and pathogenetic correlates. Neurobiol Aging 2013;34:636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitwell JL, Jack CR Jr, Pankratz VS, et al. Rates of brain atrophy over time in autopsy-proven frontotemporal dementia and Alzheimer disease. Neuroimage 2008;39:1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nestor SM, Rupsingh R, Borrie M, et al. Ventricular enlargement as a possible measure of Alzheimer's disease progression validated using the Alzheimer's Disease Neuroimaging Initiative database. Brain 2008;131:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reuter M, Schmansky NJ, Rosas HD, Fischl B. Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage 2012;61:1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reuter M, Rosas HD, Fischl B. Highly accurate inverse consistent registration: a robust approach. Neuroimage 2010;53:1181–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whitwell JL, Weigand SD, Boeve BF, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 2012;135:794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohrer JD, Ridgway GR, Modat M, et al. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 2010;53:1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irwin DJ, McMillan CT, Xie SX, et al. Asymmetry of post-mortem neuropathology in behavioural-variant frontotemporal dementia. Brain 2018;141:288–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knopman DS, Jack CR Jr, Kramer JH, et al. Brain and ventricular volumetric changes in frontotemporal lobar degeneration over 1 year. Neurology 2009;72:1843–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahoney CJ, Downey LE, Ridgway GR, et al. Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimers Res Ther 2012;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitwell JL, Boeve BF, Weigand SD, et al. Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur J Neurol 2015;22:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones DT, Weig S, Przybelski S, et al. Longitudinal changes in brain MRI and neuropsychological measures in asymptomatic and symptomatic familial frontotemporal lobar degeneration with mutations in MAPT. Alzheimers Dis Demen 2014;10(suppl):P6. [Google Scholar]
- 17.Floeter MK, Bageac D, Danielian LE, Braun LE, Traynor BJ, Kwan JY. Longitudinal imaging in C9orf72 mutation carriers: relationship to phenotype. Neuroimage Clin 2016;12:1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avants BB, Epstein CL, Grossman M, Gee JC. Symmetric diffeomorphic image registration with cross-correlation: evaluating automated labeling of elderly and neurodegenerative brain. Med Image Anal 2008;12:26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitwell JL. Biomarkers in randomized clinical trials: magnetic resonance imaging. Front Neurol Neurosci 2016;39:101–108. [DOI] [PubMed] [Google Scholar]
- 20.McMillan CT. Neurodegenerative disease: MRI biomarkers: a precision medicine tool in neurology? Nat Rev Neurol 2016;12:323–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boeve BF, Tremont-Lukats IW, Waclawik AJ, et al. Longitudinal characterization of two siblings with frontotemporal dementia and parkinsonism linked to chromosome 17 associated with the S305N tau mutation. Brain 2005;128:752–772. [DOI] [PubMed] [Google Scholar]
- 22.Cash DM, Bocchetta M, Thomas DL, et al. Patterns of gray matter atrophy in genetic frontotemporal dementia: results from the GENFI study. Neurobiol Aging 2018;62:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data for this study were obtained from the GENFI data freeze 2. Further details on the GENFI protocol, cohorts, and data policies can be found at http://genfi.org.uk/. Due to the sensitive nature of the genetic data in the GENFI cohort, and consistent with practices adopted by the DIAN study of genetic Alzheimer disease, individual data points are not presented to preserve participant blinding to GS and confidentiality. Supplemental analysis as described below has been deposited in the open data repository linked to this article (additional results, e-tables 1–9, figure e-1, doi.org/10.5061/dryad.1qj7630).