

SUMMARY
In eukaryotes, cellular respiration is driven by mitochondrial cytochrome c oxidase (CcO), an enzyme complex that requires copper cofactors for its catalytic activity. Insertion of copper into its catalytically active subunits, including COX2, is a complex process that requires metallochaperones and redox proteins including SCO1, SCO2, and COA6, a recently discovered protein whose molecular function is unknown. To uncover the molecular mechanism by which COA6 and SCO proteins mediate copper delivery to COX2, we have solved the solution structure of COA6, which reveals a coiled-coil-helix-coiled-coil-helix domain typical of redox-active proteins found in the mitochondrial inter-membrane space. Accordingly, we demonstrate that COA6 can reduce the copper-coordinating disulfides of its client proteins, SCO1 and COX2, allowing for copper binding. Finally, our determination of the interaction surfaces and reduction potentials of COA6 and its client proteins provides a mechanism of how metallochaperone and disulfide reductase activities are coordinated to deliver copper to CcO.
Graphical Abstract
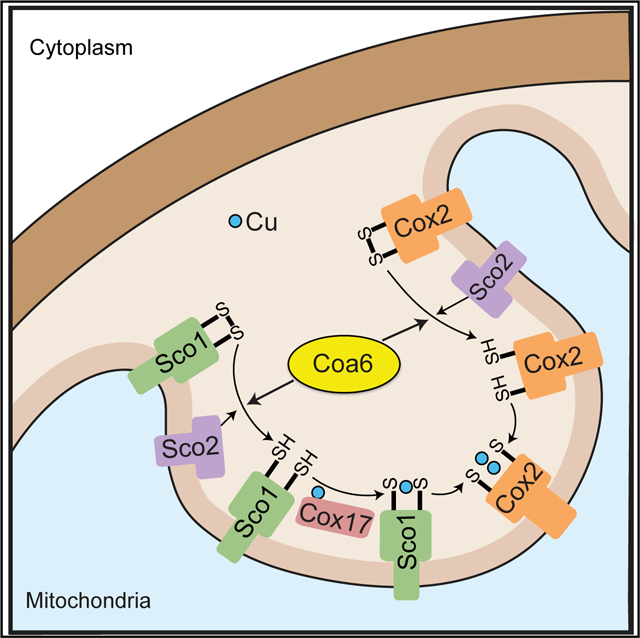
In Brief
Soma et al. reports the solution structure of cytochrome c oxidase assembly factor COA6 and establishes that it functions as a thiol-disulfide oxidoreductase in a relay system that delivers copper to COX2, a copper-containing subunit of the mitochondrial cytochrome c oxidase.
INTRODUCTION
Copper (Cu) is a redox-active transition metal that plays essential roles in cellular physiology by acting as a catalytic cofactor in numerous enzymatic reactions (Kim et al., 2008). One of the most critical Cu-containing enzymes is cytochrome c oxidase (CcO), a multimeric heme-Cu oxidase that is integral to the inner mitochondrial membrane and is the main site of cellular respiration (Ferguson-Miller and Babcock, 1996). CcO activity and its assembly depend on the formation of two Cu centers—CuA and CuB—on the COX2 and COX1 subunits, respectively (Tsukihara et al., 1995). These centers provide a molecular conduit for the transfer of electrons from reduced cytochrome c to molecular oxygen, the terminal step in mitochondrial respiration.
The high chemical reactivity and thiophilic nature of Cu ions present a challenge for its transport and insertion into target enzymes. Aqueous Cu ions can drive the production of deleterious hydroxyl radicals (Halliwell and Gutteridge, 1984) and can displace other metal ions from proteins, impairing their proper function and preventing its own delivery to cuproproteins (Foster et al., 2014; Macomber and Imlay, 2009). Moreover, the reduced oxidation state of cysteine ligands in cuproproteins, which coordinate Cu via their sulfhydryl groups, is necessary for Cu binding (Abriata et al., 2008). Not surprisingly, metallation of CcO is a complex process that requires several evolutionarily conserved proteins with distinct roles in Cu delivery and insertion (Soto et al., 2012; Timón-Gómez et al., 2018). For example, delivery of Cu to the cysteine-bridged binuclear CuA site requires at least four proteins– COX17, SCO1, SCO2, and COA6; all of which are either localized to the mitochondrial inter-membrane space (IMS) or anchored to the inner mitochondrial membrane with their functional domains facing the IMS (Vögtle et al., 2017). These proteins are part of a relay system, where Cu ions are sequentially transferred from COX17 to SCO1 and then to the CuA site of the COX2 subunit (Horng et al., 2004; Banci et al., 2008; Morgada et al., 2015). Structural and functional analyses of the yeast and human COX17 and SCO1 proteins have shown that they both coordinate Cu through their cysteinyl residues (Abajian et al., 2004; Abajian and Rosenzweig 2006; Banci et al., 2006, 2008) and act as metallochaperones (Banci et al., 2008; Horng et al., 2004; Morgada et al., 2015). Human SCO2 acts as a thiol-disulfide oxidoreductase of SCO1 in vivo and COX2 in vitro (Leary et al., 2009; Morgada et al., 2015). These findings show that in addition to the metallochaperone activity of COX17 and SCO1, a thiol-disulfide oxidoreductase activity is essential for inserting Cu into the CuA site.
Recently, we reported that the poorly characterized CcO assembly factor COA6 is a novel member of the Cu delivery pathway to CcO (Ghosh et al., 2014). Subsequent biochemical and genetic interaction studies showed that COA6 interacts with SCO1, SCO2, and COX2, and that it has an overlapping function with SCO2 in the biogenesis of the CuA site (Ghosh et al., 2016; Pacheu-Grau et al., 2015; Stroud et al., 2015). Loss of COA6 in many organisms results in a severe CcO deficiency, demonstrating its conserved role in this pathway (Ghosh et al., 2014). Loss-of-function mutations in COA6 in human mitochondrial disease patients result in fatal infantile cardiomyopathies, further highlighting its critical requirement in cellular respiration (Baertling et al., 2015; Calvo et al., 2012). Pathogenic mutations have also been reported in the COA6-interacting partners SCO1 and SCO2, and SCO patients present with similar, early onset clinical syndromes owing to an isolated CcO deficiency (Papadopoulou et al., 1999; Stiburek et al., 2009). Although these observations emphasize the centrality of this pathway to CcO biogenesis, the precise biochemical function of COA6 and the nature of its interactions with various partners during CuA site maturation are still largely unknown.
Here, we address these unknowns by a combination of a nuclear magnetic resonance (NMR)-based structural determination and in vitro and in vivo mechanistic biochemistry. We show that COA6 adopts a coiled-coil-helix-coiled-coil-helix (CHCH) domain, preferentially interacts with SCO1, and exhibits thiol-disulfide oxidoreductase activity both in vitro and in vivo, with SCO1 and COX2 being its client proteins. We further demonstrate that COA6 is not a Cu-binding protein under physiological conditions and that its enzymatic activity is independent of metalation state. Collectively, our data suggest a mechanistic picture in which COA6 and SCO2 act as disulfide reductases during the stepwise transfer of Cu from Cox17 to the CuA site.
RESULTS
Human COA6 Is a Helical Protein with a CHCH Domain
To uncover the molecular function of COA6, we first solved the solution structure of human COA6 isoform 3, hereafter referred to as COA6 (Figure 1A). We chose isoform 3 for our structural studies because it encompasses the most well-conserved region of the full-length protein and it contains the primary structure that functionally complements yeast coa6Δ cells (Ghosh et al., 2016). Human COA6 was expressed as a glutathione S-transferase (GST) fusion protein and the GST tag was cleaved with thrombin protease to release free COA6 (Figure 1B). Subsequent gel-filtration analysis suggests that COA6 exists predominantly as an ~18-kDa dimer (Figure 1C). Isotope-enriched COA6 samples were then prepared and the structure determined using distance and angle restraints obtained from 2D and 3D heteronuclear NMR spectra. The 1H-15N heteronuclear signal quantum correlation (HSQC) spectrum of COA6 shows well-dispersed resonances, indicative of a folded protein as well as parts lacking stable structure (Figure 1D; PDB: 6NL3). The unstructured nature of N- and C-terminal residues is evident from 1H-15N NOE data (Figures S1A–S1C). Sixty-seven out of the expected 75 15N backbone amide resonances were detected and approximately 90% of all detectable carbon, nitrogen, and hydrogen nuclei were assigned.
Figure 1. Solution Structure of Human COA6.

(A) Primary structure of human COA6. No NMR assignments were achieved for the residues colored in red.
(B) SDS-PAGE analysis of the samples obtained during COA6 purification steps: lane 1, supernatant of lysate from E. coli expressing the COA6-GST fusion protein; lane 2, eluate containing COA6-GST from the GST binding column; lane 3, COA6-GST after cleaving the GST tag with thrombin; and lane 4, purified COA6 sample from gel filtration chromatography.
(C) Elution profile of purified COA6 from the Superdex 75 gel-filtration column.
(D) 1H-15N HSQC spectrum of human COA6.
(E) Ensemble of the 20 NMR solution structures of COA6 by backbone representation.
(F) Ribbon representation of human COA6 based on the lowest energy conformer of solution structures. Human COA6 has four helixes labeled as H1, H2, H3, and H4, and the two disulfide bonds between H1 and H2 are shown in yellow.
To determine if dimerization of COA6 is affected by its concentration, we acquired the 1H-15N HSQC spectrum of COA6 at varying protein concentrations (Figure S1D). Analysis of the relative chemical shift perturbations at the lowest compared to the highest COA6 concentration (Figure S1E) indicates that the residues in helix 3 of COA6 are likely involved in dimerization. Comparable gel-filtration elution profiles in the presence and absence of tris(2-carboxyethyl)phosphine (TCEP) argue that the dimerization of COA6 is unaffected by a strong reducing agent (Figure S1F). This finding suggests that dimerization is not due to inter-disulfide bonding between cysteine residues of each COA6 monomer.
We obtained an ensemble of 20 COA6 structures in excellent agreement with experimental restraints (Table S1). This ensemble indicates that the N and C termini of COA6 (residues 1–8 and 65–79, respectively) are unstructured (Figure 1E). The lowest energy conformer of COA6 shows that the compact core, encompassing residues 9–64, is entirely helical and is composed of four helices, with the first two, H1 and H2, stabilized by a pair of disulfide bonds (Figure 1F). The CB chemical shifts of each cysteine (C12, 37.566 ppm; C22, 32.450 ppm; C33, 32.006 ppm; and C44, 45.634 ppm) were interpreted alongside NOESY patterns to deduce disulfide pairing (Sharma and Rajarathnam, 2000). Addition of TCEP to purified COA6 produced only localized changes in the HSQC spectrum, including splitting of the W20 side chain peak (Figures S1G and S1H). Since W20 is proximal to the C22-C33 disulfide, this phenomenon is likely due to the reduction of this disulfide bond. COA6 exhibits a CHCH domain reminiscent of many mitochondrial IMS proteins with proposed roles in redox chemistry (Banci et al., 2009b). To further determine if the structural features of COA6 are conserved in yeast Coa6, we analyzed the 1H-15N HSQC spectrum and chemical shift index (CSI) of recombinant yeast Coa6 and found that it is also a helical protein (Figures S2A and S2B).
Mapping COA6 Patient Mutations onto the COA6 Structure Uncovers the Structural Basis of Pathogenicity
Mutations in COA6 have been reported in two unrelated human mitochondrial disease patients: one with compound heterozygous mutations (W59C and E87X) (Calvo et al., 2012) and the other with a homozygous missense mutation (W66R) (Baertling et al., 2015). These residues are highly conserved, suggesting their essential function (Figure 2A). To understand how these mutations affect COA6 function, we mapped the mutated residues onto the COA6 structure. The truncation mutation (E87X) clearly disrupts the CHCH domain by removing a large portion of the protein from helix 2 onward (Figures 2A and 2B). The other two mutations, W59C and W66R, are found within the first helix of COA6, where the side chains of each tryptophan face the bulk solvent, suggesting that these residues may facilitate interactions with their client proteins (Figure 2B).
Figure 2. Mapping COA6 Patient Mutations onto the COA6 Structure.

(A) Sequence alignment of the conserved region of COA6 across indicated model organisms. Horizontal lines above Cx9C and Cx10C residues show the conserved Cx9CxnCx10C motif. Schematic representation of the yeast-human hybrid COA6 (hyCOA6) protein, where the sequences from yeast Coa6 and human COA6 are shown in cyan and blue, respectively. Arrows indicate amino acid residues that were mutated in human COA6 patients and the corresponding residues in the hyCOA6 protein.
(B) Ribbon representation of COA6 patient mutations mapped onto the COA6 structure in red.
(C) Wild-type (WT) and coa6Δ cells transformed with empty vector (EV) or vector-expressing hyCOA6 or hyCOA6 harboring patient mutations (W26C, W33R, and E54X) were serially diluted and seeded on YP glycerol-ethanol (YPGE) plates. Cells were grown at 30°C and 37°C for 5 days.
(D and E) SDS-PAGE/western blot analysis of whole cell (D) and mitochondrial protein (E) extracts from coa6Δ cells expressing wild-type or mutant hyCOA6 proteins. Por1 and Pgk1 were used as loading controls.
(F) Human control and COA6 patient cell lines were transduced with cDNAs expressing either WT or pathogenic variants of COA6 followed by measurement of CcO activity. Data are represented as mean ± SEM, n = 3.
(G) SDS-PAGE/western blot depicting COA6, COX2, and GAPDH (loading control) levels in protein lysate from control and COA6 cell lines as indicated in (D). The data shown are representative of at least three independent replicates.
To further test the effect of these two missense mutations on COA6 function, we introduced patient mutations into a human-yeast chimera (hyCOA6) that consists of the bulk of the human protein and the N-terminal 24 amino acid residues of yeast Coa6 to facilitate mitochondrial targeting (Figure 2A). As expected based on our previous study, the respiratory growth of coa6Δ cells was restored by hyCOA6 (Ghosh et al., 2016) but not by either of the tryptophan variants, suggesting that these missense mutations disrupt COA6 function or expression (Figure 2C). To distinguish between these two possibilities, we measured the expression of COA6 mutants and found that while hyCOA6W33R and the truncated protein hyCOA6E54X are undetectable, hyCOA6W26C is expressed and localizes to mitochondria (Figures 2D and 2E). These data suggest that the W26C mutation perturbs COA6 function. To further study the effect of these mutations within the context of human cells, we overexpressed the wild-type (WT) and mutant alleles of COA6 in control and COA6 patient fibroblasts and found that the W66R variant fails to rescue CcO activity (Figure 2F). In contrast, expression of the W59C mutant leads to a partial recovery of CcO activity and COX2 levels (Figures 2D and 2E), consistent with a previous report (Stroud et al., 2015). Curiously, COA6 abundance in patient cells overexpressing the W59C and W66R mutants was comparable and at the lower limits of detection (Figure 2G). It seems that in most cell types residual levels of the partially functional W59C allele are not sufficient to support CcO assembly and mitochondrial respiration because coa6Δ cells expressing the W59C variant do not exhibit respiratory growth (Figure 2C) and the human patient with the W59C mutation exhibits a severe CcO deficiency in cardiac tissue (Calvo et al., 2012). Taken together, these data strongly suggest that the conserved tryptophans are critical for COA6 stability and possibly for their interactions with client proteins.
COA6 Preferentially Interacts with SCO1 over SCO2
Previous studies from yeast and human cell lines have shown that COA6 interacts with COX2 and SCO proteins in vivo (Ghosh et al., 2016; Pacheu-Grau et al., 2015; Stroud et al., 2015). Genetic and physical interaction studies demonstrate that yeast Coa6 interacts with both Sco1 and Sco2 (Ghosh et al., 2016). However, studies of human COA6 are less conclusive, with one group reporting that COA6 physically interacts only with SCO2 (Pacheu-Grau et al., 2015) and another group reporting interactions only with SCO1 (Stroud et al., 2015).
Given the uncertainty surrounding the relative affinity of COA6 for its interacting partner(s), we decided to further investigate COA6 interactions with the SCO proteins by conducting competitive binding analysis using size-exclusion chromatography (SEC). First, we determined the elution profiles of each protein in isolation. The elution profile for COA6 shows a right-tailing-effect (Figure 3A), suggesting equilibrium between monomeric and dimeric forms (~18 kDa) of the protein. The soluble domains of SCO1 and SCO2 behave as monomeric 20- and 17.6-kDa proteins, respectively (Figures 3B and 3C).
Figure 3. Human COA6 Exhibits a Stronger Interaction with SCO1 than SCO2.

(A–C) Size exclusion chromatography elution profiles of (A) COA6, (B) SCO1, and (C) SCO2.
(D–H) Elution profiles of the indicated mixtures of proteins (solid lines), compared to simulations of the indicated individual proteins or mixtures of two or more proteins (dotted lines).
(I) SDS-PAGE/western blot analysis of different fractions from size exclusion chromatogram shown as the solid black line in (F)–(H).
We then obtained elution profiles for different mixtures of these proteins. An equimolar mixture of COA6 and SCO1 led to the formation of a complex of ~27 kDa, and the absence of a peak corresponding to COA6 alone, indicating that all of the COA6 is found within the higher molecular weight complex (Figure 3D). Excess SCO1 appears as a shoulder on the chromatogram (Figure 3D). Based on the apparent molecular weight of the complex, COA6 appears to interact with SCO1 as a monomer and with relatively high affinity. Next, we analyzed an equimolar mixture of COA6 and SCO2 and observed the formation of higher molecular weight complexes, suggesting that SCO2 can also interact with COA6 (Figure 3E).
To better probe the competitive binding of SCO1 and SCO2 with COA6, a mixture of all three proteins was prepared and analyzed by SEC. Three distinct peaks were identified in the elution profile (Figure 3F). Through curve fitting, we compared this elution profile with traces of each individual protein (Figure 3G) and with traces of the mixtures of two proteins (Figure 3H), and found that the high molecular weight peak corresponds to the SCO1:COA6 complex and the remaining two peaks correspond to SCO1 and SCO2 alone. To confirm the constituents of the elution profile, we performed an immunoblot analysis of each eluted fraction to detect both SCO proteins and COA6. We found that the majority of COA6 co-elutes with SCO1 (Figure 3I), strongly suggesting that at least in vitro SCO1 is the dominant interacting partner of COA6.
COA6 Uses a Largely Non-polar Interface to Interact with SCO1
To identify the surfaces that facilitate interactions between COA6 and SCO1, we compared HSQC spectra of 15N-labeled COA6 obtained in the presence and absence of unlabeled SCO1. A comparison of the two spectra identified pronounced chemical shift perturbations in residues 49–55 present in helix 3 and residues 65–69 that form a β-hairpin turn of COA6 (Figures 4A and 4B). Most of these COA6 residues are non-polar, suggesting hydrophobic interactions between COA6 and SCO1. Notably, we did not detect perturbations in the N-terminal residues of COA6, around the patient mutations corresponding to W20, suggesting that these residues are not critical for the interaction with SCO1 (Figures 4A and 4B).
Figure 4. Mapping the Surfaces That Mediate Interactions between COA6 and SCO1.

(A) Average chemical shift perturbation of 15N-labeled COA6 upon addition of equimolar unlabeled SCO1. Horizontal solid line, mean chemical shift perturbation; dashed line, mean + SD of chemical shift perturbation for all residues; solid circles, unassigned residues; and open circles, residues with broadened peaks upon complex formation.
(B) COA6 residues with significant chemical shift perturbations are colored in red on a ribbon representation of COA6.
(C) Chemical shift perturbation of 15N-labeled SCO1 residues upon addition of equimolar unlabeled COA6, as displayed in (A).
(D) SCO1 residues with significant chemical shift perturbations are colored in red on a ribbon representation of SCO1.
(E) A HADDOCK docking model of the COA6:SCO1 complex generated using chemical shift perturbation (CSP) data. The side chain of the COA6 tryptophan (W48) and the indicated cysteine residues of COA6 and SCO1 are shown in stick representations in red.
(F) WT and coa6Δ cells transformed with empty vector (EV) or vector expressing either full-length or truncated hyCOA6. Transformants were serially diluted on agar plates and grown for 5 days.
(G) SDS-PAGE/western blot analysis of protein extracts from coa6Δ cells expressing wild-type or truncated mutants of hyCOA6. Pgk1 was used as loading control.
To obtain complementary information about the interaction surface on SCO1, we performed the reciprocal experiment where HSQC spectra of 15N-labeled SCO1 were obtained in the presence and absence of unlabeled COA6. This led to the identification of a number of non-polar residues present on the same face of the SCO1 surface (Figures 4C and 4D). Interestingly, the surface that SCO1 uses to interact with COA6 is also highly conserved in SCO2 (Figure S3A), suggesting that COA6 is capable of interacting with each SCO protein via a common surface (Figure S3). Using the chemical shift perturbation data, we generated a hypothetical docking model for the SCO1:COA6 complex using High Ambiguity Driven protein-protein Docking (HADDOCK) (de Vries et al., 2010). This model shows that the Cu-binding cysteines of SCO1 (Cys169 and Cys173) are located in a U-shaped turn region that makes direct intermolecular contact with the H3 helix of COA6 (Figure 4E). In particular, the indole side chain of W48 from COA6 (highlighted red in Figure 4E) is within 3Å of the sulfhydryl side chain of Cys173 from SCO1 (Figure 4E). The indole side chain of W48 is also located halfway between Cys173 of SCO1 and Cys44 of COA6, suggesting that W48 may bridge or otherwise relay electrons from the Cys12/ Cys44 disulfide bridge of COA6 to the Cu-binding cysteines of SCO1 (Cys169/Cys173). The COA6 and SCO1 intermolecular structural interaction is largely composed of hydrophobic residues localized to the H3 helix and the C-terminal tail of COA6 (Figure 4E). To test the validity of the interaction model, we generated two truncated forms of COA6 by deleting residues 64–79 (COA6Δ64–79) and residues 72–79 (COA6Δ72–79) from the C terminus. We confirmed that both the truncated forms of COA6 are expressed but are not functional, likely due to loss of their interaction with SCO1 (Figures 4F and 4G). Superimposition of the known missense mutations in SCO1 and SCO2 onto their ribbon diagrams further suggests that at least some of the pathogenic variants (e.g., SCO1 P174L, SCO2, and E140K) are close to the mapped interacting residues in COA6 (Figures S3B and S3C). Taken together, interaction mapping and genetic complementation data suggest that H3 and C-terminal residues of COA6 are critical for its interaction with SCO1.
COA6 Function Can Be Bypassed in a Reducing Environment
Although previous work has shown that human COA6 binds Cu with high affinity in vitro (Stroud et al., 2015), suggesting a possible metallochaperone function, Cu was not detected in the purified COA6 samples that we used for our structural studies. We therefore sought to definitively determine the Cu-binding properties of COA6. To do this, we first utilized a custom-built online liquid chromatography-inductively coupled plasma mass spectrometry (LC-ICP-MS) system, which allows simultaneous detection of Cu and protein in elution fractions obtained from SEC. When purified COA6 was applied to this LC-ICP-MS system, we detected a major peak at A280 corresponding to the COA6 dimer, although the peak was asymmetric and exhibited a right-tailing-effect, suggesting minor contributions from a monomeric species (Figure S4A). We did not detect any Cu peaks in the chromatogram, indicating that recombinant COA6 does not bind endogenous Cu from E. coli (Figure S4A). When purified COA6 was reconstituted with CuSO4 and reduced glutathione as described previously (Stroud et al., 2015), we detected two major A280 peaks corresponding to a dimer and a low-intensity higher-molecular-weight species that likely corresponds to an aggregate of COA6 and Cu (Figure S4B). Indeed, the Cu peak was detected in the same fractions as COA6, with excess Cu found in the high-molecular-weight COA6 aggregates (Figure S4B). Thus, upon reconstitution in the presence of reduced glutathione, COA6 can bind Cu; however, we suspected that this was not the physiological form of the protein.
To address this issue, we tested whether Cu can bind Coa6 in vivo by analyzing yeast mitochondrial fractions. Lysates from anaerobically purified WT and coa6Δ mitochondria were subjected to LC-ICPMS to identify Cu-specific peaks using a method we recently described (Nguyen et al., 2019). In WT mitochondria, we detected four Cu peaks, including one at the void volume and the others associated with masses of circa 30, 20, and 10 kDa (Figure 5A). coa6Δ samples exhibited nearly identical Cu peaks in the region associated with the same masses (Figure 5B). Notably, none of these Cu peaks corresponds to the fractions containing Coa6 (Figures 5C and 5D). The lack of any difference in the Cu peaks in the WT and coa6Δ mitochondrial lysates suggests that Coa6 does not bind Cu under physiological conditions in yeast cells and is therefore unlikely to act as a Cu metallochaperone.
Figure 5. A Reducing Environment Rescues the Respiratory Growth of coa6Δ Cells.

(A and B) Cu traces from online LC-ICP-MS of solubilized mitochondrial extracts from WT (A) or coa6Δ (B) cells. The dotted lines indicate simulated curves generated by peak fitting.
(C and D) SDS-PAGE/western blot depicting Cox2 and Coa6 protein levels in the different LC-ICP-MS fractions of WT (C) and coa6Δ (D) soluble mitochondrial extracts.
(E) Serially diluted WT, coa6Δ, sco1Δ, sco2Δ, cox17Δ, cox11Δ, and coa6Δsco2Δ cells were spotted on YP dextrose (YPD) and YP glycerol-ethanol (YPGE) plates and grown under normoxic or hypoxic (4% O2) conditions.
(F) Western blot of Cox2 in the indicated yeast strains. Por1 was used as a loading control.
(G) WT and coa6Δ cells were cultured in YPGE medium at 37°C in the presence or absence of reduced glutathione (GSH) at the indicated concentrations. The cell density was measured spectrophotometrically at the indicated time points. These data are representative of at least three independent replicates.
The presence of a CHCH domain in the COA6 structure is consistent with a redox role for the protein in CuA site maturation, because the CHCH domain has been proposed to represent the minimal oxidoreductase domain, and a number of mitochondrial IMS proteins with this domain are indeed redox active (Banci et al., 2009a, 2009b). To begin investigating the potential redox role for COA6 in Cu delivery to CcO in vivo, we utilized hypoxic (4% O2) conditions (Bihlmaier et al., 2007) to counter the oxidizing environment of the mitochondrial IMS (Hu et al., 2008). coa6Δ and other mutants involved in Cu delivery to CcO were cultured under normoxic or hypoxic conditions in both fermentable and respiratory media. The respiratory growth of coa6Δ was almost fully rescued in hypoxic yeast, while that of yeast strains lacking proteins with established Cu metallochaperone activity (sco1Δ, cox17Δ, and cox11Δ) was not (Figure 5E). This finding suggests that hypoxia cannot replace a metallochaperone function and is likely more specific to rescuing the functional deficiency in redox-related proteins (Figure 5E). Consistent with this idea, we also observed a partial rescue of coa6Δ yeast that also lacks Sco2, a protein with an established disulfide reductase activity in Cu delivery to CcO (Figure 5E). We were unable to test the effect of hypoxia on a sco2Δ strain directly, because yeasts lacking Sco2 do not have a pronounced respiratory growth-related phenotype (Figure 5E).
The respiratory growth defect of coa6Δ cells has been attributed to a decrease in the levels of Cox2, which degrades rapidly when Cu delivery to CcO is impeded (Ghosh et al., 2014; Pacheu-Grau et al., 2015). Therefore, to test whether Cu delivery to Cox2 is restored under hypoxic conditions, we measured Cox2 abundance in coa6Δ cells and found that it was indeed restored to the WT levels (Figure 5F). Moreover, exogenous supplementation of a normoxic culture with reduced glutathione (GSH) also partially rescued respiratory growth of coa6Δ cells (Figure 5G), further supporting a redox role for Coa6 in the Cu delivery process. Taken together, these results strongly suggest that Coa6 has a redox as opposed to a metallochaperone function in Cu delivery to Cox2.
COA6 Acts as a Disulfide Reductase of SCO and COX2 Proteins
Previous work on the biogenesis of the prokaryotic CuA center of heme-Cu oxidases established the requirement for both Cu metallochaperones and disulfide reductases (Abriata et al., 2008). The presence of a CHCH domain, its redundancy in a hypoxic environment, and its overlapping function with SCO2 (Ghosh et al., 2016) all argue that COA6 harbors disulfide reductase activity. However, the redox potential of COA6 is unknown. To determine whether reduction of COX2 and SCO disulfides by COA6 is thermodynamically favorable, we measured the reduction potential of the ox/red COA6 couple using the DTTox:DTTred redox couple as a calibrant, and found that at −330 mV (pH 7.0) it is lower than that of SCO1 (−280 mV) and COX2 (−290 mV) (Figures 6A and 6B). The reduction potential of SCO2 has previously been shown to be < −300 mV (Banci et al., 2007). These reduction potential values suggest that both COA6 and SCO2 can reduce SCO1 and COX2 in a thermodynamically favorable manner.
Figure 6. COA6 Acts as a Thiol-Disulfide Reductase of SCO and COX2 Proteins.

(A) Determination of the redox potential of recombinant human COA6 by subjecting it to increasing dithiothreitol (DTT) concentrations followed by treatment with the alkylating agent AMS to irreversibly modify cysteines containing reduced thiols. Oxidized and reduced species of COA6 were resolved by non-reducing SDS-PAGE.
(B) Schematic representation of the redox potential of COA6 relative to the previously determined redox potentials of COX2, SCO1, and the mitochondrial inter-membrane space (IMS).
(C) A schematic of how AMS reacts with reduced protein thiols resulting in protein-AMS adducts of increased mass and with retarded migration in SDS-PAGE gels under non-reducing conditions.
(D and E) Migration patterns of purified recombinant human COA6, SCO1, and SCO2 (D) and COX2 (E) proteins with and without AMS treatment on non-reducing SDS-PAGE. 1-h co-incubation of reduced COA6 with oxidized SCO1, SCO2, or COX2 was followed by AMS treatment and SDS-PAGE. SCO and COA6 proteins were used at 1:1 and 1:2 stoichiometries. COX2 and COA6 proteins were used at a 1:1 stoichiometry.
(F) Isolated human mitochondria from control (control 1 and control 2) and COA6 patient fibroblasts were incubated in the presence or absence of the reductant dithiothreitol (DTT), followed by treatment with the alkylating agents iodoacetamide (IAM) or AMS to irreversibly modify cysteines containing reduced thiols. Species of SCO1 and SCO2 containing oxidized (ox) cysteines were then resolved from those with reduced (red) cysteines by non-reducing SDS-PAGE and detected by immunoblotting with the indicated antibodies. Red* refers to reduced thiols that were modified in the presence of AMS. These data are representative of three independent replicates.
We then tested whether COA6 can effectively reduce the disulfide bonds of Cu-coordinating cysteines of its interacting partners. To do this, we used a thiol-alkylating reagent 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS), which selectively reacts with free thiols on proteins to form a protein-AMS adduct. This modification increases the protein mass by ~ 0.5 kDa per free thiol. The additional mass leads to an upward shift on an SDS-PAGE gel (Figure 6C). Utilizing this assay, we observed that a 1-h co-incubation of reduced COA6 with either of the oxidized SCO proteins led to the appearance of two SCO-specific bands in the AMS-treated samples (Figure 6D). This observation suggests that COA6 can reduce the disulfides of SCO proteins, generating free sulfhydryl groups that then react with AMS. Similarly, we found that COA6 can also reduce the cysteines of COX2 (Figure 6E).
Finally, to corroborate these in vitro findings, we tested the redox state of the cysteinyl thiols of SCO proteins in vivo in COA6 patient fibroblasts using AMS and a second alkylating agent iodoacetamide (IAM). As shown previously, the cysteinyl sulfurs of SCO1 and SCO2 proteins exist as a mixed redox population in control fibroblasts that is composed of both oxidized and reduced thiols (Figure 6F) (Leary et al., 2009). Strikingly, mutations in COA6 significantly skew the relative ratio of reduced to oxidized cysteinyl sulfurs of SCO1, with the oxidized species predominating (Figure 6F). In contrast, the ratio of oxidized-to-reduced sulfur species of SCO2 was comparable in control and COA6 patient fibroblasts (Figure 6F). Taken together, these findings provide further evidence that COA6 acts as a disulfide reductase in vivo and suggest that SCO1 is one of its substrates.
To determine how the redox function of COA6 affects the synthesis and stability of COX2, we performed pulse-chase labeling of mitochondrial DNA-encoded proteins in control, COA6, SCO1, and SCO2 patient fibroblasts. Consistent with our previous studies, we found that COX2 is synthesized normally in SCO1–1 cells but is rapidly turned over during the chase (Cobine et al., 2006), while its synthesis is attenuated in SCO2 cells yet it accumulates during the chase (Leary et al., 2009) (Figure S5). The profile of COX2 synthesis and degradation in COA6 patient fibroblasts most closely mirrors that observed for SCO2 cells (Figure S5), a result that would be expected if COA6 has a similar biochemical function to that of SCO2, which has previously been shown to have disulfide reductase activity (Leary et al., 2009; Morgada et al., 2015). Consistent with the idea that COA6 potentiates SCO1 function and functionally overlaps with SCO2 to some degree, we found that overexpressing COA6 in SCO1 and SCO2 patient cell lines partially rescues CcO activity and COX2 levels (Figures S6A and S6B).
DISCUSSION
A large body of data has established that Cu insertion in the evolutionarily conserved binuclear CuA center of CcO occurs in the mitochondrial IMS (Baker et al., 2017; Timón-Gómez et al., 2018). Catalyzing the necessary biochemical reactions in this sub-mitochondrial compartment poses two challenges. First, the Cu-coordinating cysteines of COX2 must be re-reduced to the dithiol form if they become oxidized to disulfides in the oxidizing environment of the IMS. Second, the Cu metallochaperone responsible for delivering Cu to the CuA site must be present in the IMS with its Cu-coordinating thiols kept in the reduced state in order to bind Cu ions. Thus, CuA assembly is necessarily a two-step process that involves reducing the disulfides of newly synthesized apoCOX2 followed by Cu transfer from a metallochaperone. Here, we show that COA6 is a CHCH domain-containing protein that facilitates CuA site maturation by exhibiting disulfide reductase activity toward apo- COX2 and SCO1, the metallochaperone that delivers Cu to reduced apoCOX2. Thus, COA6 likely plays a dual role in CuA site maturation by maintaining the Cu-coordinating cysteines of COX2 and its metallochaperone SCO1 in the thiol/thiolate oxidation state to allow for Cu binding.
We have recently shown that metallation of the CuA site can be achieved in vitro by the combined action of SCO1 and SCO2 proteins, where SCO1 acts as a metallochaperone to transfer two Cu(I) equivalents to COX2 after SCO2 has reduced its cysteine residues to render it competent for Cu binding (Morgada et al., 2015). While the two orthologous SCO proteins thus are sufficient to form the CuA center in vitro, genetic and protein-protein interaction data all argue that the final step in the metallation of CuA site requires the combined activities of at least three proteins: SCO1, SCO2, and COA6 (Ghosh et al., 2016; Pacheu-Grau et al., 2015; Stroud et al., 2015). To address the discrepancy between the in vitro and in vivo data, we decided to focus on determining the function of COA6, the most recently discovered member of this Cu delivery pathway.
The fact that human COA6 is a small, soluble protein makes it highly amenable to NMR-based structure determination. Thus, we utilized NMR to solve the solution structure of COA6 and identify a CHCH domain (Figure 1F). Consistent with our studies, Maghool et al. (2019) recently published the X-ray crystallographic structure of a COA6 dimer while we were revising our article, showing that each COA6 monomer has a CHCH fold with the first two helices being stabilized by disulfide bonds. The presence of COA6 as a dimer both in vitro (Figure 1C) and in vivo (Figures 5A and 5C) suggests that dimerization may be physiologically significant. Based on our observations that the COA6 dimer is readily disrupted in the presence of either SCO protein and binds to SCO proteins as a monomer (Figure 3), we speculate that COA6 dimerization in vivo may prevent its non-specific activity against other mitochondrial intermembrane space proteins. The CHCH domain identified in COA6 has been proposed to be the minimal domain require for thiol-disulfide oxidoreductase activity (Banci et al., 2009a). We found that COA6 function can be bypassed under reducing or hypoxic conditions (Figures 5E–5G), suggesting that it may exhibit disulfide reductase activity. Indeed, our in vitro and in vivo experiments further support this notion and identified SCO1 and COX2 as COA6 client proteins (Figures 3 and 6). Having two client proteins for a disulfide reductase is not without precedent; the bacterial periplasmic protein thioredoxin TlpA acts as a specific reductant for both of the metallochaperones SCO1 and COXB, the bacterial equivalent of COX2 (Abicht et al., 2014; Mohorko et al., 2012). Thus, COA6 appears to be a functional homolog of bacterial TlpA; however, it likely represents a product of convergent evolution, as there is no sequence homology between the two proteins. We propose that the client-specific disulfide reductases in the bacterial periplasmic space and the mitochondrial IMS are required to counter the oxidizing environment of these compartments, which would prevent Cu coordination by promoting disulfide bond formation in the Cu-binding proteins (Herrmann et al., 2009).
Our findings beg the question why three proteins—COA6, SCO1, and SCO2—are needed in vivo when SCO1 and SCO2 are sufficient to form a CuA center in vitro. The redox states of SCO1 and SCO2 in vitro were set to favor Cu delivery to the CuA site (Morgada et al., 2015). In vivo, however, the reduced states of these proteins must be continuously regenerated for the proteins to function. Thus, after delivering Cu to the CuA site, the cysteines of SCO1 are likely to be more prone to oxidation given the oxidizing nature of the mitochondrial IMS, necessitating their subsequent reduction to bind Cu for the next round of Cu delivery to apoCOX2. In this setting, COA6 function may be critical. Previously, it was shown that the metallochaperone COX17 is capable of transferring both reducing equivalents and Cu to SCO1 in vitro, although the redox potential of the S-S/2SH redox couple of COX17 (−198 mV) is not thermodynamically favored to reduce the disulfide of SCO1 (−277 mV) (Banci et al., 2008) but a Cu-bound COX17 is able to overcome unfavorable thermodynamic driving force in vitro. Based on these observations and our findings, it appears that COA6 is a more likely electron donor for SCO1 in vivo.
By combining both in vivo and in vitro approaches, our study demonstrates that the mechanism of mitochondrial CuA site biogenesis requires the combined activities of COA6, SCO1, and SCO2, with COA6 facilitating Cu delivery to the CuA site by keeping the Cu-binding cysteines of SCO1 and COX2 in their reduced states (Figure 7). This model raises an obvious question regarding the ultimate source of the reducing power. Although at present this remains unknown, our model is consistent with the previously documented synthetic lethal interaction between coa6Δ and sco2Δ yeast mutants (Ghosh et al., 2016), in that both COA6 and SCO2 exhibit disulfide reductase activities with some overlapping substrate specificity in SCO1 and/ or COX2 (Figure 7). Our structural model of the COA6:SCO1 complex (Figure 4E) suggests a possible redox pathway for shuttling electrons from COA6 to the Cu-binding cysteines of SCO1 (Cys 169 and Cys173). Both Cys169 and Cys173 are in close proximity to the indole ring of W48 from COA6, which is also within 5Å of the Cys12/Cys44 disulfide bridge of COA6. We propose that the polarizable π electrons of the W48 indole ring could help relay the transfer of electrons between Cys12/Cys44 (of COA6) and Cys169/Cys173 (of SCO1) as a possible mechanism of the observed COA6 disulfide reductase activity. We further suggest that the other COA6 disulfide bridge (Cys22/Cys33) may similarly help catalyze the reduction of disulfides in COX2. In this scenario, a ternary complex of COA6 bound to both SCO1 and COX2 could simultaneously promote Cu binding to both SCO1 and COX2. This model is supported by our previous study, which showed that a pathogenic mutation in the first tryptophan residue of COA6 disrupts its ability to interact with COX2 but not with SCO1, indicating that COA6 uses distinct surfaces to interact with each protein (Ghosh et al., 2016). Additional support for our model comes from a recent report, which identified a SCO1-COA6-COX2 assembly module in yeast CcO biogenesis (Franco et al., 2018). In summary, our work provides structural, biochemical, and in vivo data supporting the role of COA6 as a disulfide reductase in the IMS and advances the mechanistic model of CuA site assembly within the complex picture of CcO biogenesis.
Figure 7. A Proposed Model of CuA Site Biogenesis.

CuA site formation on the Cox2 subunit of CcO involves sequential transfer of Cu from Cox17 to Sco1 to Cox2. This process occurs in the mitochondrial inter-membrane space, an oxidizing environment that would promote the formation of disulfide bonds in the Cu-coordinating cysteines of apo-Cox2 and apo-Sco1 proteins and prevent Cu binding. The thioldisulfide oxidoreductase activities of Coa6 and Sco2 overcome this problem by reducing the disulfides of apo-Cox2 and apo-Sco1 to dithiols, thereby allowing for Cu binding. Previous studies and the work described here suggest that Coa6 has dual substrates in Sco1 and Cox2 and that it exhibits overlapping substrate specificity with Sco2.
STAR✶METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vishal M. Gohil (vgohil@tamu.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restrictions.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast Strains and Culture Conditions
All strains used in this study are listed in the Key Resources Table, and all the primers used in this study are listed in Table S2. Yeast cells were grown in YP (1% yeast extract, 2% bactopeptone) medium with 2% dextrose (YPD) or 3% glycerol + 1% ethanol (YPGE) as carbon sources. Synthetic media was prepared with 0.17% yeast nitrogen base, 0.5% ammonium sulfate, 0.2% dropout amino acid mix and 2% dextrose as a carbon source. Solid media was prepared by further adding 2% agar. Growth was measured spectrophotometrically at 600 nm in liquid medium or by spotting on solid plates. For hypoxic growth conditions on solid media, indicated plates were incubated in a hypoxia incubator chamber (STEMCELL Technologies) purged with a certified gas mixture (4% oxygen, 5% carbon dioxide and 91% nitrogen). For hypoxic growth conditions in liquid media, synthetic complete raffinose liquid media, which constitutes 2% raffinose and 1x TEM (0.5% Tween80, 2.4 μg/ml ergosterol, 55.6 μg/ml methionine), was kept in a glove box with nitrogen gas for 24 hr prior to culturing the indicated strains at 30°C while shaking at 225 rpm.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Por11 | Abcam | Cat# ab110326; RRID:AB_10865182 |
| Mouse monoclonal anti-Cox2 | Abcam | Cat# ab110271; RRID:AB_10858117 |
| Rabbit polyclonal anti-Coa6 | Gohil Lab | N/A |
| Rabbit polyclonal anti- SCO1 | Bourens et al., 2014 | N/A |
| Rabbit polyclonal anti- SCO2 | Fitzgerald | Cat# 70R-20118 |
| Rabbit Polyclonal anti- COA6 | Proteintech | Cat# 24209–1-AP |
| Mouse monoclonal anti- COX2 | Mitoscience | Cat# MS405; RRID:AB_1618186 |
| Rabbit monoclonal anti- GAPDH | Cell signaling | Cat# 14C10–2118 |
| hFAB Rhodamine anti-Actin | Bio-Rad | Cat# 12004163 |
| Bacterial and Virus Strains | ||
| E. coli strain DH5α | NEB | Cat# C2987I |
| E. coli BL21(DE3) | NEB | Cat# C2527I |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ammonium-15N chloride | Sigma-Aldrich | Cat# 299251 |
| BME Vitamins 100x solution | Sigma-Aldrich | Cat# B6891 |
| D-(+)-Raffinose pentahydrate | Sigma-Aldrich | Cat# R0250 |
| D-Glucose-13C6 | Sigma-Aldrich | Cat# 389374 |
| DL-Dithiothreitol | Sigma-Aldrich | Cat# D0632 |
| Ergosterol | Sigma-Aldrich | Cat# 45480 |
| Isopropyl β-D-thiogalactoside | Sigma-Aldrich | Cat# I6758 |
| Trans-4,5-Dihydroxy-1,2-dithiane (Oxidized DTT) | Sigma-Aldrich | Cat# D3511 |
| Deposited Data | ||
| Human COA6 structure | This Paper | PDB: 6NL3 |
| Experimental Models: Cell Lines | ||
| COA6 patient cell line | Baertling et al., 2015 | N/A |
| SCO1–1 (R149X/P174L) patient cell line | Valnot et al., 2000 | N/A |
| SCO1–2 (V93X/M294V) patient cell line | Leary et al., 2013 | N/A |
| SCO2–9 patient cell line | Leary et al., 2013 | N/A |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: Strain background: BY4741 WT - MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 | Miriam L. Greenberg | N/A |
| S. cerevisiae: Strain background: BY4741 coa6Δ - MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 coa6Δ::kanMX4 | Open Biosystems | N/A |
| S. cerevisiae: Strain background: BY4741 sco1Δ - MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 sco1Δ::kanMX4 | Open Biosystems | N/A |
| S. cerevisiae: Strain background: BY4741 sco2Δ - MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 sco2Δ::kanMX4 | Open Biosystems | N/A |
| S. cerevisiae: Strain background: BY4741 cox11Δ - MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 cox11Δ::kanMX4 | Open Biosystems | N/A |
| S. cerevisiae: Strain background: BY4741 cox17Δ - MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 cox17Δ::kanMX4 | Open Biosystems | N/A |
| S. cerevisiae: Strain background: STY10 sco2Δcoa6Δ - MATa, his3Δ1, leu2Δ0, ura3Δ0, lys2Δ0, sco2Δ::KanMX4, coa6Δ::NatMX4 | Ghosh et al., 2016 | N/A |
| Recombinant DNA | ||
| pGEX4t-1- COA6 isoform3 | This paper | N/A |
| pET28a- Coa6 | This paper | N/A |
| pETG-30A- SCO1 | Banci et al., 2006 | N/A |
| pETG-30A- SCO2 | Banci et al., 2007 | N/A |
| pET9a-COX2-chimeric protein | Morgada et al., 2015 | N/A |
| pRS416 -hyCOA6 | Ghosh et al., 2016 | N/A |
| pRS416 -hyCOA6w26C | Soma et al., 2018 | N/A |
| pRS416 -hyCOA6w33R | Soma et al., 2018 | N/A |
| pRS416 -hyCOA6E54X | Soma et al., 2018 | N/A |
| pRS416 -hyCOA6(Δ65–79) | This paper | N/A |
| pRS416 -hyCOA6(Δ73–79) | This paper | N/A |
| Software and Algorithms | ||
| The PyMOL Molecular Graphics System, v2.0 | Schrödinger LLC | https://pymol.org/2/ |
| HADDOCK2.2 | van Zundert and Bonvin, 2014 | http://haddock.science.uu.nl/services/HADDOCK2.2/ |
| Topspin, version 3.2 | Bruker, Germany | N/A |
| Sparky, version 3.112 | UCSF | https://www.cgl.ucsf.edu/home/sparky/distrib-3.112/download.html |
| CYANA software package | Güntert and Buchner, 2015 | https://www.las.jp/english/products/cyana.html |
| TALOS+ | Shen et al., 2009 | https://spin.niddk.nih.gov/bax/nmrserver/talos/ |
| Xplor NIH version 2.50 | Schwieters et al., 2003 | https://nmr.cit.nih.gov/xplor-nih/ |
| Protein Structure Validation Software suite 1.5 | Bhattacharya et al., 2007 | https://psvs-1_5-dev.nesg.org/ |
Mammalian Cell Culture
Primary fibroblasts from control, COA6 (Baertling et al., 2015), SCO1–1 (R149X/P174L; Valnot et al., 2000), SCO1–2 (V93X/M294V; Leary et al., 2013) and SCO2–9 (Leary et al., 2013) were immortalized as described (Leary et al., 2004). Fibroblasts were transduced at 40%–60% confluency with retrovirus produced by the Phoenix amphotrophic packaging cell line, and selected in media containing hygromycin or puromycin to yield stable overexpression cell lines (Leary et al., 2004). Both fibroblasts and the packaging cells were grown in high-glucose DMEM containing 10% bovine growth serum (ThermoFisher) at 37°C in 5% CO2 and were tested to ensure that they were Mycoplasma-free (Lonza MycoAlert) before harvesting.
METHOD DETAILS
Cloning and Expression
E.coli codon optimized human COA6 isoform3 was cloned into the pGEX4t-1 vector using BamHI and XhoI restriction endonucleases to generate N-terminal, GST-fused COA6. E.coli codon optimized yeast COA6 was cloned into the pET28a plasmid using XhoI and EcoRI restriction endonucleases to generate N-terminal, His-GFP-fused COA6. The resultant plasmids were then transformed into E.coli BL 21(DE3) cells. Human COA6 and yeast Coa6 expression was induced by the addition of 0.5 mM isopropyl-D-thiogalactopyranoside (IPTG) at an optical density of 600 nm (OD600) of 0.6, and the cells were grown at 37°C and 16°C for 4 hr and 12 hr respectively. 15N/13C labeled proteins for NMR structural characterization were expressed in cells grown in minimal medium, which contained 3 g/liter KH2PO4, 6.8 g/liter Na2HPO4, 0.1 mM CaCl2, 2 mM MgCl2, 1x Basal Medium Eagle (BME) vitamin solution (Sigma), 1 g/liter 15N-ammonium chloride and 4 g/liter 13C-D-glucose (Sigma).
Protein Purification
To purify human COA6, cells were resuspended in lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1 mM PMSF and 0.1% Triton X-100) and lysed by repeated sonication on ice for a total of 4 min. The lysate was clarified by centrifugation at 40,000 g for 30 min at 4°C and the supernatant was applied to a GST binding column pre-equilibrated with a buffer containing 50 mM Tris (pH 7.4) and 150 mM NaCl. GST-COA6 was eluted in equilibration buffer supplemented with 10 mM reduced glutathione. Eluted GST-COA6 was subjected to thrombin digestion (20 units of thrombin for 100 mg of GST-COA6 protein) for 12 hr at room temperature. COA6 from the flow-through was then injected into a HiPrep Sephacryl S-100 column pre-equilibrated with a 20 mM Tris (pH 7.4) and 150 mM NaCl solution.
To purify yeast Coa6, cells were resuspended in lysis buffer containing 20 mM Tris (pH 8), 500 mM NaCl, 1 mM PMSF and 6 mM imidazole and sonicated on ice for 4 min. The lysate was clarified by centrifugation at 40,000 g for 30 min at 4°C and the supernatant was applied to a HisTrap HP column pre-equilibrated with 20 mM Tris (pH 8), 500 mM NaCl, and 6mM imidazole. His-GFP-Coa6 was eluted using 20 mM Tris (pH 8), 500 mM NaCl, and 400 mM imidazole. Eluted His-GFP-Coa6 was subjected to TEV digestion for 12 hr at 4°C. Yeast Coa6 was further purified using a Superdex 200 column pre-equilibrated with 20mM Tris (pH 7.4) and 500 mM NaCl. Human SCO1 and SCO2 were purified as described (Banci et al., 2006, 2007).
NMR Spectroscopy
NMR data were acquired in Shigemi tubes on 0.015 – 0.947 mM COA6 samples buffered in 20mM Tris (pH 7.4), 150 mM NaCl and 10% (v/v) D2O. All NMR experiments used for the structure calculation were carried out at 25°C on the Bruker Avance 800 MHz, 600 MHz, or 500 MHz NMR spectrometers equipped with a 5 mm triple-resonance cryoprobe and single-axis pulsed field gradient by using standard pulse programs provided by the instrument manufacturer. NMR data were acquired and processed by using the software Topspin, version 3.2 (Bruker, Germany), and were further analyzed in Sparky, version 3.112 (University of California, San Francisco).
1H, 13C, and 15N resonance assignments for Coa6 were indirectly referenced according to the IUPAC recommendations (Markley et al., 1998). The protein backbone resonance assignments were achieved through analysis of the 3D-triple resonance experiments and the aliphatic side-chain resonances were primarily assigned using 3D-HCCH-COSY and 15N-edited TOCSY-HSQC acquired with 60 ms mixing time. Aromatic side-chains were assigned using 2D-homo-nuclear TOCSY and NOESY spectra and verified with 2D-experiments 13C-HMQC and (HB)CB(CGCD)HD. NOE distance restraints were based on 3D-experiments, 13C-edited NOESYHSQC and 15N-edited NOESY HSQC, each acquired with a 150 ms mixing time. Semi-automated NOE cross-peaks were assigned using the CYANA software package (Güntert and Buchner, 2015) and were manually checked for correctness in an iterative manner. Dihedral angle restraints were predicted from the backbone chemical shifts using the program TALOS+ (Shen et al., 2009). Sixteen slowly exchanging backbone amide protons identified in the H/D exchange experiments were predicted to belong in a-helical regions by the chemical shift index analysis (Wishart and Sykes, 1994) and the NOESY patterns. Thus 32 corresponding distance restraints for HN-O and N-O atoms were imposed in the final structure calculation to depict the H-bonds (Williamson et al., 1985). Isotope-filtered NOESY experiments (3D-13C,15N-filtered/edited NOESY HSQC) were performed on a sample prepared by mixing a 1:1 ratio of unlabeled and 13C,15N-labeled sample to identify inter-subunit contacts between COA6 dimer. These experiments failed to reveal any cross peaks due to the weak dimerization constant and thus were not used in further analysis.
COA6 subunit structure calculation was performed using CYANA, version 3.96, following a torsion angle molecular dynamics protocol (Güntert et al., 1997). Two hundred random conformers were annealed in 15,000 steps for 7 cycles, and 20 structures with the lowest target functions after the final cycle were selected for water refinement, which was performed using the software X-plor NIH version, 2.50 (Schwieters et al., 2003). The structure ensemble was validated with the Protein Structure Validation Software suite 1.5 (Bhattacharya et al., 2007).
Relaxation Measurements
15N relaxation experiments were performed on 15N-labeled COA6 samples at 25°C on the 600 MHz spectrometer. Ten 15N longitudinal relaxation rate constant (R1) experiments were performed in random order, with relaxation delays of 10 (duplicate), 90, 190, 320, 470 (duplicate), 660, 936, 1500 ms. Similarly, the transverse relaxation rate constant (R2) experiments were performed with relaxation delays of 0 (duplicate), 16.9, 33.9, 50.9 (duplicate), 67.8, 84.8, 118.7, 186.6 ms. Both rate constants were calculated using the program Curvefit, assuming a monoexponential decay of the peak intensities. The errors in the peak intensities were calculated from the two duplicate experiments. The steady-state heteronuclear [1H]-15N NOE experiment was carried out in an interleaved manner, with and without proton saturation and repeated thrice. The NOE effect was calculated as an average ratio of the peak intensities.
Online Liquid Chromatography-Inductively Coupled Plasma-Mass Spectrometry (LC-ICP-MS)
Cu binding to purified COA6 and COA6 within yeast mitochondrial extracts was measured using an online LC-ICP-MS system. A bioinert HPLC system (Agilent Technologies, 1260) located inside a chilled and refrigerated glove box (MBraun, labmaster 130) was connected to an ICP-MS (Agilent Technologies, 7700x) via PEEK tubing coming from the diode array of the LC system in the glove box. The HPLC system was composed of a pump (G5611A), diode array (G4212B), fraction collector (G5664A) and manual injector. The ICP-MS was interfaced with the LC system via a micromist nebulizer attached to a Scott-type spray chamber. The ICP-MS was tuned daily prior to analyzing samples. Samples were analyzed in helium collision mode. The instrument was tuned as described (Dziuba et al., 2018).
Samples were analyzed either with two Superdex Peptide 10/300 GL (GEHealthcare) columns connected in series or with a Bio SEC-3 (Agilent Technologies) column. Samples were passed through either a Titan regenerated cellulose 0.2 mm filter (Thermo scientific) or a 0.2 mm cellulose acetate filter (VWR). The dual peptide columns used a mobile phase of 20 mM Tris (pH 7.4) and 10 mM NaCl at a flow rate of 0.350 mL/min for a total of 160 min per run and a loop volume of 300 μL. The Bio SEC-3 column used a mobile phase of 20 mM Tris (pH 7.4) and 150 mM NaCl at a flow rate of 0.4 mL/min for a total of 10 min per run and a loop of 20 μL. The diode array was set for detection and data collection at 280 nm. Molecular mass standards were cyanocobalamin (1.3 kDa) from Fisher BioReagents and cytochrome c (12 kDa), carbonic anhydrase (30 kDa), albumin (66 kDa) and apoferritin (443 kDa) all from Sigma.
Gel-Filtration Chromatography
The apparent molecular mass of COA6 was determined using gel filtration chromatography. COA6 (100 μM) was loaded onto a Superdex 75 10/300 GL column equilibrated with 20 mM Tris (pH 7.4) and 150 mM NaCl at a flow rate of 0.3 ml/min. Aprotinin (6.5 kDa), cytochrome c (12.4 kDa), carbonic anhydrase (29 kDa) and albumin (66 kDa) were used as molecular weight references (Sigma, MWGF70).
Haddock Docking Calculation of the COA6:SCO1 Complex
The molecular docking of COA6 and SCO1 was performed using the Haddock d-level 2.2 web server as described (van Zundert and Bonvin, 2014). Chemical shift perturbation data were used as structural restraints (referred to as ambiguous interaction restraints (AIR)). The docking calculation used the NMR structure of COA6 determined in this study and the crystal structure of SCO1 (PDB: 2GVP) as input structures for the docking calculation. A total of 26 AIR restraints were used based on chemical shift perturbation data for COA6 (Figure 4A) and SCO1 (Figure 4C). Initial docking calculations generated 200 structures (with the lowest energy) that were then used for subsequent simulated annealing and water refinement. The final docked structures (10 lowest energy structures) had an overall RMSD of 1.8Å.
SDS-PAGE and Western Blotting
Yeast mitochondrial proteins (20 μg) were separated by SDS-PAGE and blotted onto a polyvinylidene difluoride membrane. Membranes were treated for 1 hr in blocking buffer containing 5% nonfat milk dissolved in Tris-buffered saline with 0.1% Tween 20 (TBST-milk), followed by overnight incubation with primary antibody in TBST-milk at 4°C. Primary antibodies were used at the following dilutions: Cox2, 1:100,000 (Abcam 110271); Coa6, 1:5000 and Porin, 1:50,000 (Abcam 110326).
For the experiments with human cell lines, whole cells were homogenized on ice in STE buffer (250 mM sucrose, 10 mM Tris-HCl, pH 7.4, and 1 mM EDTA) supplemented with a 1 × cOmplete protease inhibitor cocktail (Roche, Indianapolis, IN) and 0.5 mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich) and centrifuged twice at 4°C for 10 min at 600 × g to obtain a postnuclear supernatant. A crude mitochondrial fraction was then obtained by centrifugation at 4°C for 10 min at 8000 × g and used for non-reducing SDS– PAGE analyses as described (Leary and Sasarman, 2009). Alternatively, whole cells were extracted in a RIPA extraction buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA and 1x Complete protease inhibitor cocktail), adjusted to a final concentration of 5 mg/ml, incubated on ice for 30 min and then centrifuged at 4 °C for 10 min at 14 000 × g and used for reducing SDS-PAGE analyses (Boulet et al., 2018).
For both reducing and nonreducing SDS–PAGE gels, equal amounts of protein were separated on precast gels (BioRad), transferred to nitrocellulose membrane, blocked for 2 hr in Tris buffered saline supplemented with 0.05% Tween 20 (TBST; 25 mM Tris (pH 7.4), 137 mM NaCl, 2.5 mM KCl and 0.05% Tween 20) containing 5% milk, and incubated overnight at 4 °C in primary antibody. Membranes were then washed six times for 5 min in TBST, incubated for 60 min in the appropriate horseradish peroxidase conjugated secondary antibody (BioRad, 1: 2500) in TBST containing 5% milk, and washed again in TBST as above. Primary antibodies were used at the following dilutions: SCO1, 1:1000 (Bourens et al., 2014); SCO2, 1:1000 (Fitzgerald 70R-20118); COA6, 1:1000 (Proteintech 24209–1-AP); COX2, 1:1000 (Mitoscience MS405); GAPDH, 1:1000 (Cell signaling 14C10–2118) and Actin, 1:5000 (Bio-Rad 12004163). Membranes were developed using a homemade luminol-enhanced chemiluminescence solution, and visualized with the BioRad ChemiDoc MP Imaging System.
CcO Activity
Cco activity was measured as described (Capaldi et al., 1995). Briefly, we used triethanolamine/EDTA buffer to make a cell suspension, which is then added to a 96-well plate and the oxidation rate of cytochrome c was measured at 550nm. The activity assay was performed in potassium phosphate buffer (50mM pH 7) that contained cytochrome c and the mild detergent dodecylmaltoside.
Redox State Determination
The in vitro redox state of the cysteine residues in protein and protein mixtures were investigated by nonreducing SDS-PAGE after reaction of the samples with and without AMS as described previously (Morgada et al., 2015). Reduced and oxidized samples and protein mixtures (20–40 μM) were treated in an anaerobic environment with 1% (wt/vol) SDS and 10 mM AMS for 1 hr at 37°C, and resolved on 17% SDS-PAGE gels under nonreducing conditions.
Reduction Potential Determination
The midpoint reduction potential of the cysteine residues of COA6 was estimated by monitoring the redox state of thiols in COA6, using AMS and non-reducing SDS-PAGE after incubation in redox buffers. Samples of fully oxidized Coa6 (25 mM) were incubated for 24 hr in 50 mM phosphate pH 7 buffer with different ratios of oxidized and reduced DTT. Reduced DTT and Oxidized DTT solutions were prepared by dissolving the solid form of each (sigma Aldrich D0632 and D3511 respectively) in 50 mM nitrogen-fluxed potassium phosphate buffer (pH 7) under anaerobic conditions. The redox potential of the buffers (EBuff) was estimated using the Nernst equation: EBuff = E0 + 29.4log ([DTTox]/[DTT]2) with E0 being −332 mV for the DTT redox couple at pH 7.0 using with a 1 mM being the maximal concentration of reduced DTT. After incubation, reactions were quenched by addition of 5 mM AMS (Thermo Fisher A485), 2% SDS and incubated at 37°C for 1 hr. Non-reducing protein loading buffer was added to the samples and the redox state of the proteins was followed by SDS-Gel shift assay in 16% polyacrylamide gel. The reduced fractions were quantified using the ImageJ software.
In Vitro Labeling of Mitochondrial Translation Products
Cells were labeled in 6 cm dishes as described in detail elsewhere (Leary and Sasarman, 2009). Briefly, cells were incubated in chloramphenicol (Sigma) prior to labeling to inhibit mitochondrial translation and allow for the accumulation of nuclear-encoded respiratory chain subunits. 400 mCi of a [35S]-methionine and cysteine mixture (Easy Tag EXPRESS) was added for 60 min to cells in methionine- and cysteine-free DMEM containing 10% dialyzed fetal bovine serum (GIBCO) and anisomycin (Sigma), to reversibly inhibit cytoplasmic translation. Total cellular protein (50 mg) was resuspended in sample loading buffer containing β-mercaptoethanol, sonicated and run on a 12%–20% gradient gel. Gels were subsequently transferred to nitrocellulose under semi-dry conditions and the [35S]-labeled mitochondrial translation products were detected through digital autoradiography.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are shown as mean ± standard deviation (SD) or mean ± standard error of the mean (SEM), and the number of replicates is indicated in the figure legends. Values of p < 0.05 were considered significant. Statistical analysis was performed using GraphPad Prism version 8 (GraphPad Software, La Jolla California USA, https://www.graphpad.com).
DATA AND CODE AVAILABILITY
The solution structure of human COA6 isoform 3 has been deposited in the Protein Data Bank with the accession code PDB: 6NL3.
Supplementary Material
Highlights.
COA6 is a coiled-coil-helix-coiled-coil-helix domain containing protein
COA6 preferentially interacts with SCO1 over SCO2
COA6 acts as a disulfide reductase of SCO1 and COX2
COA6 function can be bypassed under hypoxic conditions
ACKNOWLEDGMENTS
Research reported in this publication was supported by NIH awards R01GM111672 to V.M.G., R35GM127021 to P.A.L., and R01EY012347 to J.B.A. This work was further supported by Welch Foundation grant A-1810 to V.M.G., a T3 grant from Texas A&M University to V.M.G. and P.A.L., and a Canadian Institutes of Health Research Operating grant (MOP 133562) to S.C.L. A.J.V. is a CONICET staff member and M.N.M. is a recipient of a postdoctoral fellowship from CONICET. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.054.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following reference appears in the Supplemental Information: Rebelo et al., 2018.
REFERENCES
- Abajian C, and Rosenzweig AC (2006). Crystal structure of yeast Sco1. J. Biol. Inorg. Chem 11, 459–466. [DOI] [PubMed] [Google Scholar]
- Abajian C, Yatsunyk LA, Ramirez BE, and Rosenzweig AC (2004). Yeast cox17 solution structure and Copper(I) binding. J. Biol. Chem 279, 53584–53592. [DOI] [PubMed] [Google Scholar]
- Abicht HK, Schärer MA, Quade N, Ledermann R, Mohorko E, Capitani G, Hennecke H, and Glockshuber R (2014). How periplasmic thioredoxin TlpA reduces bacterial copper chaperone ScoI and cytochrome oxidase subunit II (CoxB) prior to metallation. J. Biol. Chem 289, 32431–32444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abriata LA, Banci L, Bertini I, Ciofi-Baffoni S, Gkazonis P, Spyroulias GA, Vila AJ, and Wang S (2008). Mechanism of Cu(A) assembly. Nat. Chem. Biol 4, 599–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baertling F, A M van den Brand M, Hertecant JL, Al-Shamsi A, P van den Heuvel L, Distelmaier F, Mayatepek E, Smeitink JA, Nijtmans LG, and Rodenburg RJ (2015). Mutations in COA6 cause cytochrome c oxidase deficiency and neonatal hypertrophic cardiomyopathy. Hum. Mutat 36, 34–38. [DOI] [PubMed] [Google Scholar]
- Baker ZN, Cobine PA, and Leary SC (2017). The mitochondrion: a central architect of copper homeostasis. Metallomics 9, 1501–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L, Bertini I, Calderone V, Ciofi-Baffoni S, Mangani S, Martinelli M, Palumaa P, and Wang S (2006). A hint for the function of human Sco1 from different structures. Proc. Natl. Acad. Sci. USA 103, 8595–8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L, Bertini I, Ciofi-Baffoni S, Gerothanassis IP, Leontari I, Martinelli M, and Wang S (2007). A structural characterization of human SCO2. Structure 15, 1132–1140. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Ciofi-Baffoni S, Hadjiloi T, Martinelli M, and Palumaa P (2008). Mitochondrial copper(I) transfer from Cox17 to Sco1 is coupled to electron transfer. Proc. Natl. Acad. Sci. USA 105, 6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L, Bertini I, Cefaro C, Ciofi-Baffoni S, Gallo A, Martinelli M, Sideris DP, Katrakili N, and Tokatlidis K (2009a). MIA40 is an oxidoreductase that catalyzes oxidative protein folding in mitochondria. Nat. Struct. Mol. Biol 16, 198–206. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Ciofi-Baffoni S, and Tokatlidis K (2009b). The coiled coil-helix-coiled coil-helix proteins may be redox proteins. FEBS Lett. 583, 1699–1702. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Tejero R, and Montelione GT (2007). Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795. [DOI] [PubMed] [Google Scholar]
- Bihlmaier K, Mesecke N, Terziyska N, Bien M, Hell K, and Herrmann JM (2007). The disulfide relay system of mitochondria is connected to the respiratory chain. J. Cell Biol 179, 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulet A, Vest KE, Maynard MK, Gammon MG, Russell AC, Mathews AT, Cole SE, Zhu X, Phillips CB, Kwong JQ, et al. (2018). The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J. Biol. Chem 293, 1887–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourens M, Boulet A, Leary SC, and Barrientos A (2014). Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum. Mol. Genet 23, 2901–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, Tucker EJ, Laskowski A, Garone C, Liu S, Jaffe DB, et al. (2012). Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med 4, 118ra10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaldi RA, Marusich MF, and Taanman JW (1995). Mammalian cytochrome-c oxidase: characterization of enzyme and immunological detection of subunits in tissue extracts and whole cells. Methods Enzymol. 260, 117–132. [DOI] [PubMed] [Google Scholar]
- Cobine PA, Pierrel F, Leary SC, Sasarman F, Horng YC, Shoubridge EA, and Winge DR (2006). The P174L mutation in human Sco1 severely compromises Cox17-dependent metallation but does not impair copper binding. J. Biol. Chem 281, 12270–12276. [DOI] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk M, and Bonvin AM (2010). The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc 5, 883–897. [DOI] [PubMed] [Google Scholar]
- Dziuba N, Hardy J, and Lindahl PA (2018). Low-molecular-mass iron in healthy blood plasma is not predominately ferric citrate. Metallomics 10, 802–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson-Miller S, and Babcock GT (1996). Heme/Copper Terminal Oxidases. Chem. Rev 96, 2889–2908. [DOI] [PubMed] [Google Scholar]
- Foster AW, Osman D, and Robinson NJ (2014). Metal preferences and metallation. J. Biol. Chem 289, 28095–28103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco LVR, Su CH, McStay GP, Yu GJ, and Tzagoloff A (2018). Cox2p of yeast cytochrome oxidase assembles as a stand-alone subunit with the Cox1p and Cox3p modules. J. Biol. Chem 293, 16899–16911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Trivedi PP, Timbalia SA, Griffin AT, Rahn JJ, Chan SS, and Gohil VM (2014). Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of COA6 deficiency. Hum. Mol. Genet 23, 3596–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Pratt AT, Soma S, Theriault SG, Griffin AT, Trivedi PP, and Gohil VM (2016). Mitochondrial disease genes COA6, COX6B and SCO2 have overlapping roles in COX2 biogenesis. Hum. Mol. Genet 25, 660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güntert P, and Buchner L (2015). Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 62, 453–471. [DOI] [PubMed] [Google Scholar]
- Güntert P, Mumenthaler C, and Wüthrich K (1997). Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol 273, 283–298. [DOI] [PubMed] [Google Scholar]
- Halliwell B, and Gutteridge JM (1984). Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J 219, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JM, Kauff F, and Neuhaus HE (2009). Thiol oxidation in bacteria, mitochondria and chloroplasts: common principles but three unrelated machineries? Biochim. Biophys. Acta 1793, 71–77. [DOI] [PubMed] [Google Scholar]
- Horng YC, Cobine PA, Maxfield AB, Carr HS, and Winge DR (2004). Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J. Biol. Chem 279, 35334–35340. [DOI] [PubMed] [Google Scholar]
- Hu J, Dong L, and Outten CE (2008). The redox environment in the mitochondrial intermembrane space is maintained separately from the cytosol and matrix. J. Biol. Chem 283, 29126–29134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BE, Nevitt T, and Thiele DJ (2008). Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol 4, 176–185. [DOI] [PubMed] [Google Scholar]
- Leary SC, and Sasarman F (2009). Oxidative phosphorylation: synthesis of mitochondrially encoded proteins and assembly of individual structural subunits into functional holoenzyme complexes. Methods Mol. Biol 554, 143–162. [DOI] [PubMed] [Google Scholar]
- Leary SC, Kaufman BA, Pellecchia G, Guercin GH, Mattman A, Jaksch M, and Shoubridge EA (2004). Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum. Mol. Genet 13, 1839–1848. [DOI] [PubMed] [Google Scholar]
- Leary SC, Sasarman F, Nishimura T, and Shoubridge EA (2009). Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet 18, 2230–2240. [DOI] [PubMed] [Google Scholar]
- Leary SC, Antonicka H, Sasarman F, Weraarpachai W, Cobine PA, Pan M, Brown GK, Brown R, Majewski J, Ha KC, et al. (2013). Novel mutations in SCO1 as a cause of fatal infantile encephalopathy and lactic acidosis. Hum. Mutat 34, 1366–1370. [DOI] [PubMed] [Google Scholar]
- Macomber L, and Imlay JA (2009). The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci. USA 106, 8344–8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghool S, Cooray NDG, Stroud DA, Aragão D, Ryan MT, and Maher MJ (2019). Structural and functional characterization of the mitochondrial complex IV assembly factor Coa6. Life Sci. Alliance 2, e201900458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, and Wüthrich K; IUPAC-IUBMB-IUPAB Inter-Union Task Group on the Standardization of Data Bases of Protein and Nucleic Acid Structures Determined by NMR Spectroscopy (1998). Recommendations for the presentation of NMR structures of proteins and nucleic acids. J. Biomol. NMR 12, 1–23. [DOI] [PubMed] [Google Scholar]
- Mohorko E, Abicht HK, Bühler D, Glockshuber R, Hennecke H, and Fischer HM (2012). Thioredoxin-like protein TlpA from Bradyrhizobium japonicum is a reductant for the copper metallochaperone ScoI. FEBS Lett. 586, 4094–4099. [DOI] [PubMed] [Google Scholar]
- Morgada MN, Abriata LA, Cefaro C, Gajda K, Banci L, and Vila AJ (2015). Loop recognition and copper-mediated disulfide reduction underpin metal site assembly of CuA in human cytochrome oxidase. Proc. Natl. Acad. Sci. USA 112, 11771–11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TQ, Dziuba N, and Lindahl PA (2019). Isolated Saccharomyces cerevisiae vacuoles contain low-molecular-mass transition-metal polyphosphate complexes. Metallomics 11, 1298–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheu-Grau D, Bareth B, Dudek J, Juris L, Vögtle FN, Wissel M, Leary SC, Dennerlein S, Rehling P, and Deckers M (2015). Cooperation between COA6 and SCO2 in COX2 maturation during cytochrome c oxidase assembly links two mitochondrial cardiomyopathies. Cell Metab. 21, 823–833. [DOI] [PubMed] [Google Scholar]
- Papadopoulou LC, Sue CM, Davidson MM, Tanji K, Nishino I, Sadlock JE, Krishna S, Walker W, Selby J, Glerum DM, et al. (1999). Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet 23, 333–337. [DOI] [PubMed] [Google Scholar]
- Rebelo AP, Saade D, Pereira CV, Farooq A, Huff TC, Abreu L, Moraes CT, Mnatsakanova D, Mathews K, Yang H, et al. (2018). SCO2 mutations cause early-onset axonal Charcot-Marie-Tooth disease associated with cellular copper deficiency. Brain 141, 662–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, and Clore GM (2003). The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson 160, 65–73. [DOI] [PubMed] [Google Scholar]
- Sharma D, and Rajarathnam K (2000). 13C NMR chemical shifts can predict disulfide bond formation. J. Biomol. NMR 18, 165–171. [DOI] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, and Bax A (2009). TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma S, Latimer AJ, Chun H, Vicary AC, Timbalia SA, Boulet A, Rahn JJ, Chan SSL, Leary SC, Kim BE, et al. (2018). Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc. Natl. Acad. Sci. USA 115, 8161–8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto IC, Fontanesi F, Liu J, and Barrientos A (2012). Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta 1817, 883–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiburek L, Vesela K, Hansikova H, Hulkova H, and Zeman J (2009). Loss of function of Sco1 and its interaction with cytochrome c oxidase. Am. J. Physiol. Cell Physiol 296, C1218–C1226. [DOI] [PubMed] [Google Scholar]
- Stroud DA, Maher MJ, Lindau C, Vögtle FN, Frazier AE, Surgenor E, Mountford H, Singh AP, Bonas M, Oeljeklaus S, et al. (2015). COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet 24, 5404–5415. [DOI] [PubMed] [Google Scholar]
- Timón-Gómez A, Nývltová E, Abriata LA, Vila AJ, Hosler J, and Barrientos A (2018). Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol 76, 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, and Yoshikawa S (1995). Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 A. Science 269, 1069–1074. [DOI] [PubMed] [Google Scholar]
- Valnot I, Osmond S, Gigarel N, Mehaye B, Amiel J, Cormier-Daire V, Munnich A, Bonnefont JP, Rustin P, and Rötig A (2000). Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet 67, 1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert GC, and Bonvin AM (2014). Modeling protein-protein complexes using the HADDOCK webserver “modeling protein complexes with HADDOCK”. Methods Mol. Biol 1137, 163–179. [DOI] [PubMed] [Google Scholar]
- Vögtle FN, Burkhart JM, Gonczarowska-Jorge H, Kücükköse C, Taskin AA, Kopczynski D, Ahrends R, Mossmann D, Sickmann A, Zahedi RP, and Meisinger C (2017). Landscape of submitochondrial protein distribution. Nat. Commun 8, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson MP, Havel TF, and Wüthrich K (1985). Solution conformation of proteinase inhibitor IIA from bull seminal plasma by 1H nuclear magnetic resonance and distance geometry. J. Mol. Biol 182, 295–315. [DOI] [PubMed] [Google Scholar]
- Wishart DS, and Sykes BD (1994). The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The solution structure of human COA6 isoform 3 has been deposited in the Protein Data Bank with the accession code PDB: 6NL3.