

Abstract
Cathepsin K (Cat K) is expressed in cancer cells, but the effect of Cat K on apoptosis is still elusive. Here, we showed that inhibition of Cat K sensitized the human carcinoma cells to anti-cancer drug through up-regulation of Bim. Inhibition of Cat K increased USP27x expression, and knock down of USP27x markedly blocked Cat K-induced up-regulation of Bim expression. Furthermore, inhibition of Cat K induced proteasome-dependent degradation of regulatory associated protein of mammalian target of rapamycin (Raptor). Down-regulation of Raptor expression increased mitochondrial ROS production, and mitochondria specific superoxide scavengers prevented USP27x-mediated stabilization of Bim by inhibition of Cat K. Moreover, combined treatment with Cat K inhibitor (odanacatib) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) reduced tumor growth and induced cell death in a xenograft model. Our results demonstrate that Cat K inhibition enhances anti-cancer drug sensitivity through USP27x-mediated the up-regulation of Bim via the down-regulation of Raptor.
Keywords: Apoptosis, Bim, Cathepsin K, USP27x, Mitochondria, Raptor
Highlights
-
•
Inhibition of Cat K sensitizes cancer cells to anti-cancer drugs.
-
•
Reduction of Raptor by inhibition of Cat K induces mitochondria dysfunction.
-
•
Mitochondrial ROS induction by inhibition of Cat K induces USP27X expression.
-
•
Up-regulation of USP27X by inhibition of Cat K stabilizes Bim protein.
1. Introduction
Cathepsin (Cat) K belongs to the papain-like cysteine proteases and is mainly located in the lysosomes [1]. Cat K is originally considered to regulate bone homeostasis in osteoclasts [2]. Cat K is also detected in other cells, such as fibroblasts, chondrocytes, neurons, and glia [[3], [4], [5], [6]], and Cat K has been shown to be highly expressed in cancer cells [7]. Increased expression of Cats makes cancer cells more sensitive to cathepsin inhibitors. For example, inhibition of Cat S induces reactive oxygen species (ROS)-mediated inhibition of the phosphoinositide 3-kinases (PI3K)/Akt signaling and activation of the JNK signaling pathway in human glioblastoma cells [8], and an inhibitor of cysteine cathepsins (Z-Phenylalanine-Glycine–NHO–Bz) induces caspase- and p53-independent cell death in multiple human cancer cells [9]. Similarly, a dual inhibitor of Cat B and L (Fmoc-Tyr-Ala-CHN2) increases cell death in neuroblastoma [10] and an inhibitor of Cats B, L, S and V (VBY-825) decreases tumor burden and tumor number in a RIP1-Tag2 mouse model of pancreatic neuroendocrine cancer [11]. In other cancers, inhibition of Cats alone has no effect on apoptosis but increases cell sensitivity to anti-cancer drugs. Inhibition of Cat S and Cat G enhances TRAIL-mediated apoptosis through the modulation of the expression of anti-apoptotic proteins [12,13], and inhibition of Cat L increases the rate of irradiation-mediated apoptosis [14,15]. In addition, inhibition of Cats enhances anti-cancer effects, including induction of apoptosis and inhibition of invasion and cell growth [[16], [17], [18], [19], [20], [21]]. Although Cat K modulates invasion and metastasis through degradation of matrix proteins in melanoma [22], it is still unclear whether Cat K plays additional roles in cancer cells.
mTOR signaling is highly activated in cancer cells. mTOR forms two distinct multiprotein complexes, mTORC1 and mTORC2 [23,24]. mTORC1 is composed of mTOR, regulatory associated protein of mammalian target of rapamycin (Raptor), G protein beta protein subunit-like (GβL), DEP domain-containing mTOR-interacting protein (DEPTOR), and proline-rich Akt substrate of 40 kDa (PRAS40), and it regulates phosphorylation of p70 ribosomal protein S6 kinase 1 (S6K) and eukaryotic initiation factor 4E binding protein 1 (4EBP1). mTORC2 includes mTOR, GβL, rapamycin-insensitive companion of mammalian target of rapamycin (Rictor), Sin1, proline-rich protein 5 (PRR5)/PRR5-like (L), and DEPTOR and modulates phosphorylation of Akt and protein kinase C (PKC) [23,24]. Activation of mTORC1/2 regulates cell growth, migration, invasion, and metastasis in cancer cells [25]. The importance of the mTOR signaling in cancer cells is emphasized by the fact that its activation is critical for drug resistance. Inhibitors of RAF or MEK are used for the treatment of BRAF-mutant melanoma; however, their successful action is dependent on the inhibition of TORC1/2 activity. Sustained TORC1 activity induces drug resistance [26], and inhibition of mTORC1/2 sensitizes BRAF-mutant melanoma to MEK inhibitor-induced apoptosis [27]. Up-regulation of Raptor also contributes to resistance to the treatment with a PI3K-mTOR inhibitor in renal cancer cells [28]. In addition, inhibition of the PI3K/Akt/mTOR signaling overcomes resistance to trastuzumab-treated HER2-targeted therapy in breast cancer cells [29] and to everolimus in pancreatic neuroendocrine tumor cells [30]. Thus, modulation of the mTOR signaling can be a targeted treatment with a dual effect in cancer cells: it can negatively interfere with the onset of cancer and suppress anti-cancer resistance.
In view of this evidence, in this study, we investigated the effect of Cat K inhibition on cell death, and the related molecular mechanisms were evaluated in human renal carcinoma Caki cells.
2. Materials and methods
2.1. Cells
Caki (ATCC HTB-46), ACHN (ATCC CRL-1611), A498 (ATCC CRL-7908), Hela (ATCC CCL-2), and TCMK-1 (ATCC CCL-139) cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and normal human mesangial cells (NHMCs) were purchased from Lonza (CC-2559, Basel, Switzerland). The human head and neck cancer cells, AMC-HN4, were obtained from the Asan Medical Center (Seoul, Korea). All cells were cultured in appropriate medium containing 10% Fetal Bovine Serum (FBS; Welgene, Gyeongsan, Korea), 1% penicillin-streptomycin and 100 μg/mL gentamycin (Thermo Fisher Scientific, Waltham, MA, USA). All cell lines tested negative for mycoplasma contamination. The lines were authenticated by standard morphologic examination using microscopy. The information of used materials and plasmids in this study are described in Supplementary Table 1.
2.2. Generation of human cathepsin K KO cell lines using CRISPR-Cas9 system
Two small guide RNAs (sgRNAs) were designed to target human cathepsin K using the CRISPR designing tool [31]. The sequences are as follows: oligomer1 5′-CAC CGA AAT CTC TCG GCG TTT AAT T-3′ and oligomer2 5′-AAA CAA TTA AAC GCC GAG AGA TTT C-3’. The sgRNAs were cloned into the pSpCas9 (BB)-2A-Puro (PX459) (Addgene, Watertown, MA, USA), and transiently transfected into Caki cells using Lipofactor-pMAX (Aptabio, Yongin, Korea). After 48 h, transfected cells were selected by 0.5 μg/mL puromycin for 2–3 week. Single-cell clones were randomly isolated and screened for knockout efficiency of cathepsin K using western blotting.
2.3. Knockdown of gene by siRNA
The siRNA transfected into cells using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA, USA).
2.4. Analysis of cell cycle and apoptosis using flow cytometry
Cells were fixed with 100% ethanol for 2 h, incubated in 1.12% sodium citrate buffer containing 50 μg/mL RNase for 30 min at 37 °C, added to 50 μg/mL propidium iodide, and measured using the BD Accuri™ C6 Cytometer (BD Biosciences, San Jose, CA, USA).
2.5. Western blotting
Western blotting was performed according to methods described in our previous study [32]. In brief, the lysates were collected, boiled with 5X sample buffer, and separated by SDS-PAGE. Proteins on membrane were probed with specific antibodies, and the antibodies were detected by enhanced chemiluminescence (ECL) solution (EMD Millipore, Darmstadt, Germany).
2.6. Detection of apoptosis by DNA fragmentation and DEVDase activity assay
Caki cells were treated with ODN alone, TRAIL alone or combinations of ODN plus TRAIL for 24 h. To check the change of cellular nuclei, cells were fixed with 1% paraformaldehyde and added to 4′,6′-diamidino-2-phenylindole solution for 5 min (Roche, Basel, Switzerland). The change of cell morphology and condensation of the nucleus were examined by fluorescence microscope (Carl Zeiss, Jena, Germany). DNA fragmentation assay was used for cell death detection ELISA plus kit (Roche, Basel, Switzerland). For DEVDase activity assay, cells were harvested and incubated with reaction buffer containing acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) substrate.
2.7. Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated using the TriZol (Life Technologies; Gaithersburg, MD, USA) [33] and cDNA was obtained using M-MLV reverse transcriptase (Gibco-BRL; Gaithersburg, MD, USA). For PCR, we used Blend Taq DNA polymerase (Toyobo, Osaka, Japan) with primers targeting raptor, Bim and actin. The used primers were consulted in previous studies [20,34].
2.8. Ubiquitination assay
This assay was described using the tagged-ubiquitin plasmid and pretreatment of MG132 as previously report [35]. Briefly, cells were harvested, washed with PBS containing 10 mM N-ethylmaleimide (NEM) (EMD Millipore, Darmstadt, Germany), resuspended in 90 μL PBS/NEM containing 1% SDS, and boiled for 10 min at 95 °C. Lysates were added to lysis buffer involving 1 mM PMSF and 5 mM NEM, dissolved using l mL syringe for 3–4 times and centrifuged at 13,000×g for 10 min at 4 °C. The supernatants were incubated with primary antibody of target protein for overnight and reacted by adding protein G agarose bead for 2 h. After centrifuging, the supernatants were removed, washed with lysis buffer containing 1 mM PMSF and 5 mM NEM at 2 times and boiled using 2X sample buffer for 10 min. Ubiquitinated Raptor and Bim were detected using HRP-conjugated anti-Ub.
2.9. Construct of stable cell lines by transfection
To construct the stable cell lines, pDsRed2-Mito vector plasmids transiently transfected into Caki cells using Lipofactor-pMAX (Aptabio, Yongin, Korea). After 2 days, cells were replaced with fresh media and selected by the G418 (700 μg/mL) (Invitrogen, Carlsbad, CA, USA). After 3 weeks, red fluorescence of labeling of mitochondria was detected by fluorescence microscope.
2.10. Analysis of mitochondrial lengths
Caki/pDsRed2-Mito cells were treated with 2 μM ODN for 6 h. Fluorescence images of mitochondrial morphology was analyzed by Confocal Laser Microscope (Carl Zeiss, Jena, Germany). Mitochondrial lengths were measured using LSM 5 Image Browser. In 3 independent experiments, average lengths of at least five mitochondria were analyzed from randomly selected areas for each data. Showing images were obtained from differences between six individual images.
2.11. Detection of mitochondrial damage
For mitochondrial damage, Caki cells were stained to MitoTracker Deep Red and MitoTracker Green dye (Molecular Probes Inc., Eugene, OR, USA) for 15 min after treatment of ODN for 6 h. Cells were trypsinized and resuspended 300 μL of PBS. Mitochondrial damage was measured using the FACSCanto™ flow cytometer (BD Biosciences, San Jose, CA, USA).
2.12. ATP production assay
Detection of ATP levels were analyzed using ATP determination kit (A22066, Thermo Fisher Scientific, Waltham, MA, USA). Caki cells were treated with ODN for 6 h. After, cells were harvested, washed with cold PBS and lysed in provided lysis buffers in the kits according to the manufacturer's instructions. After centrifuging, the supernatants were mixed with standard reaction buffer in 96 well microplates and incubated for 15 min at room temperature. ATP production was measured by luminescence using Infinite® 200 PRO microplate reader (Tecan, Männedorf, Switzerland).
2.13. Measurement of mitochondrial ROS
To measure mitochondrial ROS production, cells were determined using the MitoSOX Red mitochondrial superoxide indicator (Thermo Fisher scientific, Waltham, MA, USA). Before the harvest of lysate, cells were stained with the MitoSOX Red dye for 10 min. And then cells were trypsinized and resuspended in PBS, and mitochondrial ROS production was measured by red fluorescence using the BD Accuri™ C6 Cytometer (BD Biosciences, San Jose, CA, USA).
2.14. Animal
Male BALB/c-nude mice, aged 5 weeks, were purchased from the Central Lab Animal Inc. (Seoul, Korea). All the mice were allowed 1 week to acclimatize to the surroundings before the experiments, and were kept at 25 ± 2 °C, with a relative humidity of 55 ± 5% and a 12 h light–dark cycle. The study protocol was approved by the IRB Keimyung University Ethics Committee.
2.15. In vivo xenograft model and detection of TUNEL assay
Development of xenograft models were previously described in our previous study [36]. Experiment grope were divided by vehicle alone, 5 mg/kg ODN (20% DMSO + PBS) alone, 3 mg/kg GST-TRAIL alone, and in combinations of ODN and GST-TRAIL for 24 days. For apoptosis in vivo, TUNEL assay was performed according to methods described in our previous study [36].
2.16. Statistical analysis
We repeated experiments in our studies at least three times, and all data are represented as the means. Statistical analysis was performed by a one-way ANOVA and post hoc comparisons (Student-Newman–Keuls) using the SPSS (Statistical Package for the Social Sciences, version 22.0) (SPSS Inc.; Chicago, IL). We decide the sample size on the basis of the minimum effects we wish to measure. The p-values <0.05 were considered significant.
3. Results
3.1. Knockout and knockdown of Cat K sensitize the cancer cells to anti-cancer drugs
Since it has been known that Cats are highly expressed in cancer cells compared with normal cells [7], we investigated the effect of Cat K inhibition on cancer cell death. To evaluate the effects of Cat K genomic deletion in human renal carcinoma Caki cells, we used the CRISPR-Cas9 genome editing system [31]. The effect of Cat K knockout (KO) on apoptosis was minimal until five days (Caki/Cat K KO; Fig. 1A). Therefore, we investigated whether inhibition of Cat K sensitizes the cells to anti-cancer drugs. The Cat K inhibitor odanacatib (ODN) alone had no effect on apoptosis, but the combined treatment with ODN and a sub-lethal dose of anti-cancer drugs markedly induced apoptosis (Fig. 1B). We also confirmed the chemosensitizing effect of Cat K inhibition using Caki/Cat K KO cells. Sub-lethal doses of TRAIL, a cancer cell-specific inducer of apoptosis, had no effect on the apoptosis in Caki cells, but apoptosis and cleaved PARP (being PARP a substrate of caspase-3) were markedly increased in TRAIL-treated Caki/Cat K KO cells (Fig. 1C). In addition, sub-lethal doses of other cancer drugs (doxorubicin, etoposide, 5FU, cisplatin, and oxaliplatin) also induced apoptosis in Cat K KO cells (Fig. 1D). Using Cat K RNA interference in other cancer cells (human renal carcinoma ACHN, human head and neck carcinoma AMC-HN4, and human cervical carcinoma Hela cells), we found that the sensitizing effect of Cat K inhibition to TRAIL-mediated apoptosis was a common response (Fig. 1E). Therefore, inhibition of Cat K sensitizes cancer cells to anti-cancer drugs.
Fig. 1.

Inhibition of Cat K overcomes anti-cancer drugs resistance. (A) The cell cycle of Caki WT and Cat K KO cells was assessed by flow cytometry. (B) Caki cells were treated with the combination of 50 ng/mL TRAIL, 5 ng/mL TNF-α plus 2.5 μg/mL cycloheximide (CHX), 500 ng/mL anti-Fas, 1 μM doxorubicin, 3 μg/mL etoposide, 250 μM 5FU, 30 μM cisplatin, 25 μM oxaliplatin, 0.1 μM gefitinib and 5 μM sorafenib in the presence or absence of 2 μM ODN for 24 h. (C and D) Caki WT and three different Cat K KO cell lines were treated with 50 ng/mL TRAIL, 1 μM doxorubicin, 3 μg/mL etoposide, 250 μM 5FU, 30 μM cisplatin, and 25 μM oxaliplatin for 24 h. (E) The indicated cancer cell lines were transfected with control siRNA (siCont) or Cat K siRNA (siCat K) and treated with 50 ng/mL TRAIL for 24 h. Apoptosis and protein expression were measured by flow cytometry and western blotting. The values in the graphs (A–E) represent the mean ± SD of three independent experiments. *p < 0.01 compared to the control.
3.2. Cat K inhibitors enhance TRAIL-induced apoptosis via caspase-3 activation
Next, we tested whether Cat K inhibitors could mimic anti-cancer effect of cathepsin K knockdown (KD) or knockout (KO). Two Cat K inhibitors, ODN and balicatib, dose-dependently induced apoptosis and PARP cleavage in TRAIL-treated Caki cells (Fig. 2A), and combined treatment with ODN and TRAIL induced apoptosis-related morphological changes, nuclear chromatin damage, and DNA fragmentation (Fig. 2B–D). Furthermore, combined treatment markedly activated caspase-3 (Fig. 2E). The pan-caspase inhibitor (z-VAD) completely blocked apoptosis (Fig. 2F). Cat K inhibitors also sensitized the cells to TRAIL.
Fig. 2.

Combined treatment with ODN and TRAIL induces apoptosis. (A) Caki cells were treated with 50 ng/mL TRAIL in the presence or absence of the indicated concentrations of Cat K inhibitors (ODN and balicatib) for 24 h. (B–E) Caki cells were treated with 2 μM ODN, 50 ng/mL TRAIL or ODN plus TRAIL for 24 h. Cell morphology (B) and nuclear condensation (C) were assesses using a microscope. Quantification of DNA fragments was determined using a DNA fragmentation assay kit (D). Detection of caspase activity was measured using a DEVDase colorimetric assay kit (E). (F) Caki cells were treated with 2 μM ODN and 50 ng/mL TRAIL in the presence or absence of a pan-caspase inhibitor, z-VAD-fmk (20 μM), for 24 h. Apoptosis and protein expression were measured by flow cytometry, and western blotting, respectively. The values in the graphs (A and D-F) represent the mean ± SD of three independent experiments. *p < 0.01 compared to the control. #p < 0.01 compared to the combinations of ODN and TRAIL.
3.3. Up-regulation of Bim expression plays a critical role in the enhancement of the cytotoxicity of anti-cancer drugs
Next, we wondered which molecular mechanisms were associated with the increased sensitivity to anti-cancer drugs upon inhibition of Cat K. When we screened for apoptosis-related proteins whose levels changed upon ODN treatment, Bim and Bax expression was increased, whereas other proteins were not altered (Fig. 3A and B). Unlike ODN, balicatib induced down-regulation of c-FLIP and Mcl-1 expression, and up-regulation of Bim and Bax expression (Fig. 3A and B). Since KD and KO of Cat K did not reduce expression of c-FLIP and Mcl-1, but increased Bim and Bax expression (Fig. 3C and D). Down-regulation of c-FLIP and Mcl-1 by balicatib might be independent of inhibition of Cat K. To validate the role of Bim up-regulation by ODN, cells were transfected with Bim siRNA. Knockdown of Bim decreased ODN plus TRAIL-induced apoptosis and PARP cleavage (Fig. 3E), and balicatib plus TRAIL-induced apoptosis is also inhibited by knockdown of Bim (Supplementary Fig. S1). For further studies, we focused on up-regulation of Bim using ODN. These results suggested that up-regulation of Bim expression plays a critical role in the enhancement of the cytotoxicity of TRAIL by Cat K inhibition.
Fig. 3.

Inhibition of Cat K induces Bim protein expression. (A–B) Caki cells were treated with various concentrations of ODN or balicatib for 24 h. Band intensity was analyzed using ImageJ (B). (C and D) We used Cat K KD (C) or KO Caki cells (D). (E) Caki cells were transfected with siCont or Bim siRNA (siBim) and treated with/without 2 μM ODN plus 50 ng/mL TRAIL for 24 h. Apoptosis and protein expression were measured by flow cytometry and western blotting, respectively. The values in the graphs (B and E) represent the mean ± SD of three independent experiments. *p < 0.01 compared to the control. †p < 0.01 compared to the combinations of ODN and TRAIL in siCont.
3.4. USP27x stabilizes Bim
Within 6 h from the treatment, ODN increased Bim expression at the protein, but not at the mRNA level (Fig. 4A). Therefore, we investigated whether ODN modulates Bim expression at the post-translational level. Caki cells were treated with or without ODN in the presence of cycloheximide (CHX; inhibitor of de novo protein synthesis) for various periods. ODN significantly enhanced Bim stability compared with CHX alone (Fig. 4B). Next, to identify the molecular mechanism, which modulates protein stability of Bim, we investigated the expression of E3 ligases (TRIM2, β-TrCP, and Cdc20) and deubiquitinases (USP9x and USP27x) of Bim [[37], [38], [39], [40], [41]]. ODN did not effect on expression levels of TRIM2, β-TrCP, Cdc20 and USP9x (Fig. 4C and D). However, ODN only increased USP27x expression within 3 h (Fig. 4D), and KD or KO of Cat K also induced USP27x expression (Fig. 4E and F). ODN had no effect on USP27x mRNA expression (Supplementary Fig. S2). Furthermore, we found that ODN-induced up-regulation of USP27x expression is related with Bim stabilization (Fig. 4G). In addition, ODN dramatically inhibited ubiquitination of Bim, but KD of USP27x blocked ODN-induced deubiquitination of Bim (Fig. 4H). These data indicated that inhibition of Cat K increases Bim stabilization via up-regulation of USP27x expression.
Fig. 4.

ODN stabilize Bim via up-regulation of USP27x. (A) Caki cells were treated with 2 μM ODN for the indicated time. (B) Caki cells were treated with 20 μg/mL CHX in the presence or absence of 2 μM ODN for the indicated time. (C and D) Caki cells were treated with 2 μM ODN for the indicated time. (E and F) We used Cat K KD (E) or KO Caki cells (F). (G) Caki cells were transfected with siCont or USP27x siRNA (siUSP27x) and treated with/without 2 μM ODN for 24 h. (H) To analyze the ubiquitination of endogenous Bim, Caki cells were co-transfected with siCont or siUSP27x and HA-ubiquitin (HA-Ub) and treated with 2 μM ODN for 12 h. Immunoprecipitation was performed using an anti-Bim antibody. Protein and mRNA expression were measured by western blotting and RT-PCR, respectively. The values in the graphs (B) represent the mean ± SD of three independent experiments. *p < 0.01 compared to the CHX.
3.5. Down-regulation of raptor is a critical role for Bim stabilization in ODN -treated cells
Next, we elucidated the molecular mechanisms underlying the stabilization of Bim upon Cat K inhibition. We investigated modulation of various kinases phosphorylation to find signaling pathways controlled by ODN, and we found ODN inhibited mTORC1 signaling, which is known to inhibit the expression of Bim [42]. As shown in Fig. 5A, ODN significantly reduced the phosphorylation of mTOR at S2448 (Fig. 5A), which is related with mTORC1 signaling [43]. In addition, the expression of Raptor and PRAS40, and the phosphorylation of S6K and 4EBP1 (substrates of mTORC1) were also markedly reduced by ODN (Fig. 5A). In contrast, the expression and phosphorylation of mTORC2-related proteins, and the expression of mTORC1/2 common proteins did not significantly change (Fig. 5A). Therefore, mTORC1 signaling pathway may regulate Bim expression through inhibition of Cat K. Since Raptor is a critical for the binding of mTORC1 substrates [44] and dissociation of Raptor from mTORC1 inhibits mTOR signaling [45], we first checked the role of Raptor. Overexpression of Raptor reversed ODN-mediated dephosphorylation of mTOR (S2448), S6K, 4EBP1 (Fig. 5B), and inhibited the stabilization of Bim and resulted in inhibition of ODN plus TRAIL-induced apoptosis and PARP cleavage (Fig. 5C). PRAS40, which has an inhibitory effect of mTORC1 kinase activity [46], was down-regulated by ODN (Fig. 5A). However, PRAS40 overexpression did not inhibit ODN plus TRAIL-induced apoptosis and stabilization of Bim (Fig. 5D).
Fig. 5.

Cat K decreases Raptor protein stability. (A) Caki cells were treated with 2 μM ODN for the indicated time points. (B) Caki cells were transfected with pRK5 (Vec) or pRK5-myc-Raptor (Raptor) and treated with 2 μM ODN for 12 h. (C) Caki cells were transfected with pRK5 (Vec) or pRK5-myc-Raptor (Raptor) and treated with 2 μM ODN and 50 ng/mL TRAIL for 24 h. (D) Flow cytometry and western blotting analysis in Caki cells transfected with pRK5 (Vec) or pRK5-Flag-PRAS40 (PRAS40 WT) and treated with 2 μM ODN and 50 ng/mL TRAIL for 24 h. (E) Caki cells were treated with 2 μM ODN for the indicated time. mRNA levels were assessed by reverse transcription PCR. (F) Caki cells were pretreated with 0.5 μM MG132 for 30 min and then treated with 2 μM ODN for 24 h. (G) Caki cells were treated with 20 μg/mL CHX in the presence or absence of 2 μM ODN for the indicated time. (H) To analyze the ubiquitination of endogenous Raptor, Caki cells were transfected with HA-ubiquitin (HA-Ub) and treated with 2 μM ODN in the presence of 0.5 μM MG132 for 12 h. Immunoprecipitation was performed using an anti-Raptor antibody. (I) Indicated cells were treated with the various concentrations of ODN for 24 h. (J) The indicated cancer cell lines were transfected with control siRNA (siCont) or Cat K siRNA (siCat K) for 24 h. Apoptosis and protein expression were measured by flow cytometry and western blotting, respectively. The values in the graphs (C, D and G) represent the mean ± SD of three independent experiments. *p < 0.01 compared to the combinations of ODN and TRAIL in Caki/Vec. #p < 0.01 compared to the CHX.
To investigate further the molecular mechanisms underlying the down-regulation of Raptor upon Cat K inhibition, we analyzed the transcriptional regulation of Raptor. The expression levels of Raptor mRNA did not change in ODN-treated cells (Fig. 5E). We then investigated whether ODN regulated Raptor at the post-translational level. Treatment with a proteasome inhibitor (MG132) rescued ODN-induced down-regulation of Raptor expression (Fig. 5F), and the combined treatment with CHX and ODN markedly reduced Raptor stability, compared with the treatment with CHX alone (Fig. 5G). Since the levels of Raptor are regulated through ubiquitination [47,48], we tested the effect of ODN on the ubiquitination of Raptor. ODN markedly increased polyubiquitination of Raptor (Fig. 5H). Therefore, our data suggested that ODN decreases the stability of Raptor at the protein level. We also detected up-regulation of Bim and USP27x expression and down-regulation of Raptor by pharmacological Cat K inhibitor (ODN) and Cat K siRNA in other cancer cell lines (Fig. 5I and J), Therefore, these data suggested that Cat K inhibition induces stabilization of Bim through down-regulation of Raptor.
3.6. Down-regulation of raptor inhibits mitochondrial fission
We investigated how down-regulation of Raptor by ODN sensitizes the cells to anti-cancer drugs. Since mTORC1 signaling is a key regulator of mitochondrial dynamics [49], we first focused on this process. ODN increased the number of elongated mitochondria (>5 μm), whereas the number of fragmented (<2 μm) and intermediate (2–5 μm) mitochondria decreased (Fig. 6A). ODN inhibited phosphorylation of Drp1 at S616, related with pro-fission activity (Fig. 6B) [[50], [51], [52]]. Furthermore, using two dyes that distinguish respiring mitochondria (MitoTtracker Deep Red) and total mitochondria (MitoTracker Green) [53], we found that damaged mitochondria increased (Fig. 6C). ODN-induced mitochondrial damage decreased ATP production and increased mitochondrial ROS production (Fig. 6D and E). We then investigated whether mitochondrial ROS are involved in ODN plus TRAIL-induced apoptosis. A blocker of mitochondrial ROS (Mito-TEMPO) significantly inhibited apoptosis and PARP cleavage (Fig. 6F). Up-regulation of Bim expression was also reversed by Mito-TEMPO treatment, while Mito-TEMPO failed to reverse the expression of Raptor in ODN-treated cells (Fig. 6G), indicating that Raptor is an upstream signaling molecule for mitochondrial ROS. To prove this hypothesis, we overexpressed Raptor in Caki cells. Overexpression of Raptor markedly inhibited mitochondrial ROS production, and reversed dephosphorylation of Drp1 at S616 in ODN-treated cells (Fig. 6H and I). Furthermore, overexpression of Raptor also reduced the number of elongated mitochondria (>5 μm) in ODN-treated cells (Fig. 6J). Next, we investigated whether mitochondrial ROS is related with mitochondrial fusion. Blockers of mitochondrial ROS (Mito-TEMPO and MnTMPyP) inhibited ODN-induced dephosphorylation of Drp1 (Fig. 6K). In addition, overexpression of Raptor and blockers of mitochondrial ROS blocked ODN-induced USP27x expression (Fig. 6L and M). These data suggested that Raptor downregulation-mediated mitochondrial ROS production is critical for the enhancement of ODN-induced anti-cancer drugs sensitivity via USP27x-mediated Bim stabilization.
Fig. 6.

Downregulation of Raptor expression induces mitochondrial ROS production. (A) Representative confocal images obtained on cells stably transfected with a pDsRed2-mito (red) plasmid labeling mitochondria after treatment with 2 μM ODN for 6 h. The nuclei were stained with DAPI (blue), and the length of the mitochondria was measured using LSM 5 Image Browser. (B–E) Caki cells were treated with 2 μM ODN for the indicated time (B and E) or 6 h (C and D) and analyzed for indicated proteins by western blotting (B). Flow cytometry was used to detect fluorescence intensity for mitochondrial damage (C) and ATP production was measured using an ATP determination kit (D). Flow cytometry was used to detect fluorescence intensity for mitochondrial ROS (E). (F and G) Caki cells were pretreated with different concentrations of Mito-TEMPO and treated with 2 μM ODN and 50 ng/mL TRAIL (F) or 2 μM ODN (G) for 24 h, and apoptosis and protein expression were measured by flow cytometry and western blotting, respectively. (H and I) Caki cells were transfected with pRK5 (Caki/Vec) or pRK5-Myc-Raptor (Caki/Raptor) and treated with 2 μM ODN for 6 h (H) or 24 h (I). Mitochondrial ROS production was analyzed by MitoSOX Red reagent staining and flow cytometry. (J) Representative confocal images obtained on Caki/pDsRed2-mito cells transfected with pRK5 (Vec) or pRK5-Myc-Raptor (Raptor) after treatment with 2 μM ODN for 6 h. The nuclei were stained with DAPI, and the length of the mitochondria was measured using LSM 5 Image Browser. (K) Caki cells were pretreated with different concentrations of Mito-TEMPO or MnTMPyP and treated with 2 μM ODN for 24 h. (L) Caki cells were transfected with pRK5 (Vec) or pRK5-myc-Raptor (Raptor) and treated with 2 μM ODN for 24 h. (M) Caki cells were pretreated with different concentrations of Mito-TEMPO or MnTMPyP and treated with 2 μM ODN for 24 h. Apoptosis and protein expression were measured by flow cytometry and western blotting, respectively. The values in graph (A, D-F, H and J) represent the mean ± SD of three independent experiments. *p < 0.01 compared to the control. #p < 0.01 compared to the combinations of ODN and TRAIL. &p < 0.01 compared to the ODN treatment in Caki/Vec. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.7. Combined treatment with ODN and TRAIL reduces tumor growth in vivo
Finally, we examined the effect of the combined treatment with ODN and TRAIL in xenograft model. ODN plus TRAIL markedly suppressed tumor growth, compared with the other conditions (Fig. 7A and B), and increased TUNEL-positive staining (Fig. 7C). Furthermore, ODN also induced up-regulation of Bim and down-regulation of Raptor expression (Fig. 7D). In addition, combined treatment with ODN and TRAIL had no effect on cell death in normal mouse kidney cells (TCMK-1) and normal human mesangial cells (MC) (Fig. 7E). ODN did not induce up-regulation of Bim expression and down-regulation of Raptor expression in mesangial cells, while Raptor and Bim proteins were undetected in TCMK-1 cells (Fig. 7F). These results suggested that inhibition of Cat K sensitizes the cells to anti-cancer drug-induced apoptosis, and up-regulation of Bim expression is involved in the enhancement of the cytotoxicity of anti-cancer drugs.
Fig. 7.

Inhibition of Cat K overcomes anti-cancer drugs resistance in vivo. (A–D) Mice bearing Caki xenograft tumors were treated with 5 mg/kg ODN, 3 mg/kg GST-TRAIL, ODN and GST-TRAIL or vehicle for 24 days. Tumor volume (A) and tumor size (B) were then measured. TUNEL assays were used to measure apoptosis in vivo (C). Protein expression was measured by western blotting (D). (E) Caki, mesangial cells (MC) and TCMK-1 cells were treated with 2 μM ODN, 50 ng/mL TRAIL, ODN plus TRAIL (E), or the indicated concentrations of ODN (F) for 24 h. Cell morphology was assessed using a microscope (E). Apoptosis (E) and protein expression (F) were measured by flow cytometry and western blotting, respectively. (positive control, p.c: Caki cell lysates). The values in the graph (A and E) represent the mean ± SD of three independent experiments. *p < 0.01 compared to the vehicle.
4. Discussion
In the present study, we demonstrated that inhibition of Cat K enhanced the chemosensitivity of cancer cells. Down-regulation of Raptor by ODN increased mitochondrial ROS levels, and inhibited mitochondria fission via regulation of Drp1 phosphorylation at S616. Furthermore, ODN-mediated mitochondrial ROS production induced USP27x expression. Up-regulation of USP27x by ODN is associated with the up-regulation of Bim expression, which is critical for ODN-induced chemosensitivity of cancer cells (Fig. 8). We also revealed that combined treatment with ODN and TRAIL reduced tumor size and increased apoptosis in a xenograft model (Fig. 7A–C).
Fig. 8.
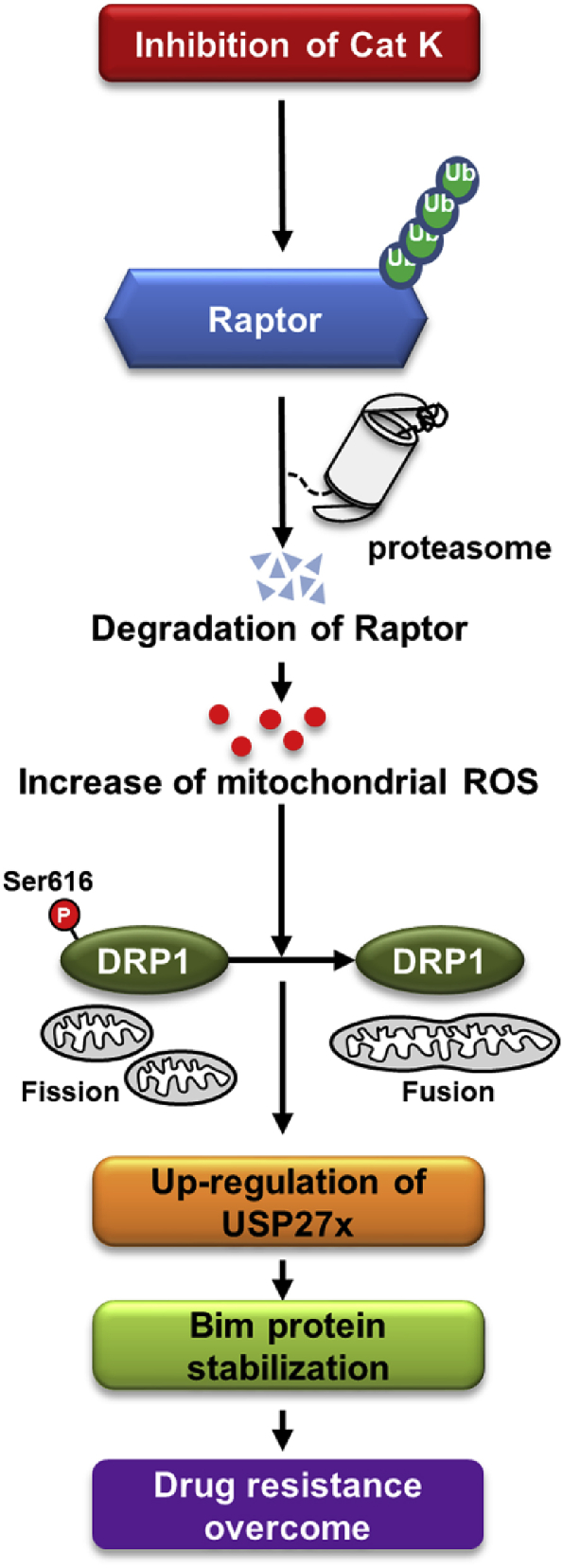
Scheme indicating the mechanism to overcome resistance to anti-cancer drugs through inhibition of Cat K.
There are many evidences about the links between levels of ROS and mitochondrial dynamics. In general, ROS induces mitochondrial fission via multiple pathways [54]. However, ODN-induced ROS promoted mitochondria fusion via dephosphorylation of Drp1 at S616 (Fig. 6K). Morita et al. reported that mitochondrial fission process protein 1 (MTFP1) plays a critical role in Drp1 phosphorylation at S616 during mTORC1 inhibition [49]. Inhibition of mTOR reduces MTFP1 expression, resulting in inhibition of Drp1 phosphorylation at S616. However, they did not find how MTFP1 regulates DRP1 phosphorylation at S616. We thought that ROS are one of regulators, which induces dephosphorylation of Drp1 at S616 during mTORC1 inhibition, and there is a possibility that MTFP1 will be involved in ODN-induced dephosphorylation of Drp1 at S616. However, we need further study to identify how ROS modulates mitochondrial dynamic via modulation of Drp1 phosphorylation at S616.
Inhibition of Cat K increased Bim expression in multiple cancer cells lines (Fig. 3, Fig. 5J). Up-regulation of Bim expression by ODN was not detected at the transcriptional level (Fig. 4A), and stability of Bim protein was increased in ODN-treated cells (Fig. 4B). Stability of Bim protein is modulated via ubiquitin-proteasome pathway. For examples, Cdc20 induces Bim ubiquitination, resulting in degradation of Bim [39]. Tripartite motif protein 2 (TRIM2) and β-TrCP also induced ubiquitination of Bim in an ERK MAPK- and Rsk1/2-dependent manner, respectively [37,38]. We examined whether ODN modulates E3 ligases of Bim in Caki cells, but ODN did not change the expression of E3 ligases, such as TRIM2, β-TrCP, and Cdc20 (Fig. 4C). Therefore, we focused on DUBs of Bim. As shown in Fig. 4D, ODN markedly increased USP27x expression, but not USP9x. Furthermore, KD of USP27x blocked ODN-induced Bim expression and deubiquitination of Bim (Fig. 4G and H). Weber et al. reported that USP27x stabilizes Bim and induces apoptosis [41]. Phosphorylation of Bim by Raf‐ERK pathway increases ubiquitination in a β-TrCP dependent manner, resulting in degradation of Bim. USP27x had no affect the phosphorylation of Bim, but increases stability of Bim by inhibition of ubiquitination [41]. In addition, USP27x is DUB of Snail1 [55]. Snail1 is a critical transcription factor to modulate epithelial-to-mesenchymal transition (EMT)-related genes and is related with resistance to chemotherapy. USP27x is increased TGF-β, which is induces EMT, and stabilized Snail1 [55]. Furthermore, USP27x interacts with cyclin E, resulting in stabilization [56]. Treatment with 5-fluorouracil (5-FU) decreases USP27x expression, and then induces degradation of cyclin E [56]. In our study, ODN also increased USP27x expression (Fig. 4D). The regulation of USP27x expression might be important on modulation of stability of substrates. Therefore, modulatory mechanism of USP27x expression also need further detailed investigation.
In case of Raptor, inhibition of Cat K also markedly induced down-regulation of Raptor expression (Fig. 5A). As shown in Fig. 5E–G, down-regulation of Raptor was not detected at the transcriptional level, and inhibition of Cat K decreased the stability of Raptor in a proteasome-dependent manner. There are few reports on the regulation of Raptor expression at the post-translational level. Bridges et al. reported that USP9x directly binds to Raptor and inhibits its proteasomal degradation, resulting in activation of the mTORC1 signaling in neural progenitor cells [48]. However, in our system, ODN had no effect on USP9x expression and KD of USP9x did not inhibit Raptor expression (Fig. 4D and Negative data; Data not shown). In addition, UCHL1 inhibits the ubiquitination of Raptor [47]. However, KD of UCHL1 inhibits ubiquitination of Raptor by DDB1-CUL4, resulting in instability of mTORC1. Deubiquitination of Raptor by UCHL1 did not change stability of Raptor. Antagonizing DDB1-CUL4-mediated ubiquitination of Raptor by UCHL1 decreases mTORC1 signaling, but activates mTORC2 signaling. Therefore, we could rule out the UCHL1 as a DUB of Raptor. We have identified several possibilities to determine the regulatory mechanisms of the protein stability of Bim and Raptor by inhibition of Cat K, but further research needs to validate. For renal cell carcinoma patients, inhibitors of multi-targeted tyrosine kinase, VEGF receptor, and mTOR are treated to improve the survival [57]. However, resistance to these drugs alleviates the anti-cancer effects. Interestingly, Earwaker et al. studied how renal carcinoma cells acquire resistance to PI3K/mTOR inhibitor. They suggested that drug resistance is correlated with Raptor expression [28]. In our study, inhibition of Cat K sensitized cancer cells to multiple anti-cancer drugs (Fig. 1B), but overexpression of Raptor completely blocked ODN-induced sensitization cancer cells to TRAIL (Fig. 5C). Previous studies reported that mTOR signaling pathway is associated with mitochondria functions. For examples, complex formation of mTOR and raptor is correlated with mitochondrial oxygen consumption and oxidative capacity [58], and mTOR controls mitochondrial oxygen consumption through the complex formation with a yin-yang 1 (YY1), peroxisome-proliferator activated receptor coactivator (PGC)-1α, and mTORC1 [59]. Furthermore, MTFP1 has been known to regulate mTORC1-mediated modulation of mitochondria dynamic [49]. In our study, inhibition of Cat K also induced mitochondrial dysfunction and ROS production (Fig. 6C–E), and overexpression of Raptor prevented mitochondrial damage by inhibition of Cat K (Fig. 6H and I). Inhibition of mTORC1 by inhibitors is transient [60], thus degradation of Raptor by ODN might have more effective inhibitory function on mTORC1, because raptor is critical for activation of mTORC1 signaling as a scaffold protein. Therefore, down-regulation of raptor by inhibition of Cat K could be effective strategy for anti-cancer drug resistant cancer cells therapy.
Taken together, we showed that Cat K inhibition sensitizes the cells to anti-cancer drugs through the up-regulation of USP27x-mediated Bim expression by down-regulation of Raptor. These observations suggest that inhibition of Cat K sensitizes cancer cells to anti-cancer drugs.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgements
We appreciate critical reading of the manuscript by Jae Man Lee (Kyungpook National University), Gang Min Hur (Chungnam National University) and Dr. Jaewhan Song (Yonsei University). This work was supported by an NRF grant funded by the Korea Government (MSIP) (2014R1A5A2010008 and NRF-2019R1A2C2005921).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101422.
Contributor Information
Kyoung-jin Min, Email: kjmin@dgmif.re.kr.
Taeg Kyu Kwon, Email: kwontk@dsmc.or.kr.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Brix K., Dunkhorst A., Mayer K., Jordans S. Cysteine cathepsins: cellular roadmap to different functions. Biochimie. 2008;90:194–207. doi: 10.1016/j.biochi.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Costa A.G., Cusano N.E., Silva B.C., Cremers S., Bilezikian J.P. Cathepsin K: its skeletal actions and role as a therapeutic target in osteoporosis. Nat. Rev. Rheumatol. 2011;7:447–456. doi: 10.1038/nrrheum.2011.77. [DOI] [PubMed] [Google Scholar]
- 3.Quintanilla-Dieck M.J., Codriansky K., Keady M., Bhawan J., Runger T.M. Expression and regulation of cathepsin K in skin fibroblasts. Exp. Dermatol. 2009;18:596–602. doi: 10.1111/j.1600-0625.2009.00855.x. [DOI] [PubMed] [Google Scholar]
- 4.Morko J.P., Soderstrom M., Saamanen A.M., Salminen H.J., Vuorio E.I. Up regulation of cathepsin K expression in articular chondrocytes in a transgenic mouse model for osteoarthritis. Ann. Rheum. Dis. 2004;63:649–655. doi: 10.1136/ard.2002.004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dauth S., Sirbulescu R.F., Jordans S., Rehders M., Avena L., Oswald J., Lerchl A., Saftig P., Brix K. Cathepsin K deficiency in mice induces structural and metabolic changes in the central nervous system that are associated with learning and memory deficits. BMC Neurosci. 2011;12:74. doi: 10.1186/1471-2202-12-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lendeckel U., Kahne T., Ten Have S., Bukowska A., Wolke C., Bogerts B., Keilhoff G., Bernstein H.G. Cathepsin K generates enkephalin from beta-endorphin: a new mechanism with possible relevance for schizophrenia. Neurochem. Int. 2009;54:410–417. doi: 10.1016/j.neuint.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 7.Verbovsek U., Van Noorden C.J., Lah T.T. Complexity of cancer protease biology: cathepsin K expression and function in cancer progression. Semin. Cancer Biol. 2015;35:71–84. doi: 10.1016/j.semcancer.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L., Wang H., Xu J., Zhu J., Ding K. Inhibition of cathepsin S induces autophagy and apoptosis in human glioblastoma cell lines through ROS-mediated PI3K/AKT/mTOR/p70S6K and JNK signaling pathways. Toxicol. Lett. 2014;228:248–259. doi: 10.1016/j.toxlet.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 9.Zhu D.M., Uckun F.M., Phe-Gly-NHO-Bz Z.- An inhibitor of cysteine cathepsins, induces apoptosis in human cancer cells. Clin. Cancer Res. 2000;6:2064–2069. [PubMed] [Google Scholar]
- 10.Cartledge D.M., Colella R., Glazewski L., Lu G., Mason R.W. Inhibitors of cathepsins B and L induce autophagy and cell death in neuroblastoma cells. Investig. New Drugs. 2013;31:20–29. doi: 10.1007/s10637-012-9826-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elie B.T., Gocheva V., Shree T., Dalrymple S.A., Holsinger L.J., Joyce J.A. Identification and pre-clinical testing of a reversible cathepsin protease inhibitor reveals anti-tumor efficacy in a pancreatic cancer model. Biochimie. 2010;92:1618–1624. doi: 10.1016/j.biochi.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seo B.R., Min K.J., Woo S.M., Choe M., Choi K.S., Lee Y.K., Yoon G., Kwon T.K. Inhibition of cathepsin S induces mitochondrial ROS that sensitizes TRAIL-mediated apoptosis through p53-mediated downregulation of Bcl-2 and c-FLIP. Antioxidants Redox Signal. 2017;27:215–233. doi: 10.1089/ars.2016.6749. [DOI] [PubMed] [Google Scholar]
- 13.Woo S.M., Min K.J., Seo S.U., Kim S., Park J.W., Song D.K., Lee H.S., Kim S.H., Kwon T.K. Up-regulation of 5-lipoxygenase by inhibition of cathepsin G enhances TRAIL-induced apoptosis through down-regulation of survivin. Oncotarget. 2017;8:106672–106684. doi: 10.18632/oncotarget.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Q.Q., Wang W.J., Li J., Yang N., Chen G., Wang Z., Liang Z.Q. Cathepsin L suppression increases the radiosensitivity of human glioma U251 cells via G2/M cell cycle arrest and DNA damage. Acta Pharmacol. Sin. 2015;36:1113–1125. doi: 10.1038/aps.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W., Long L., Wang L., Tan C., Fei X., Chen L., Huang Q., Liang Z. Knockdown of Cathepsin L promotes radiosensitivity of glioma stem cells both in vivo and in vitro. Cancer Lett. 2016;371:274–284. doi: 10.1016/j.canlet.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 16.Hsieh M.J., Lin C.W., Chen M.K., Chien S.Y., Lo Y.S., Chuang Y.C., Hsi Y.T., Lin C.C., Chen J.C., Yang S.F. Inhibition of cathepsin S confers sensitivity to methyl protodioscin in oral cancer cells via activation of p38 MAPK/JNK signaling pathways. Sci. Rep. 2017;7:45039–45049. doi: 10.1038/srep45039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Primon M., Huszthy P.C., Motaln H., Talasila K.M., Torkar A., Bjerkvig R., Lah Turnsek T. Cathepsin L silencing enhances arsenic trioxide mediated in vitro cytotoxicity and apoptosis in glioblastoma U87MG spheroids. Exp. Cell Res. 2013;319:2637–2648. doi: 10.1016/j.yexcr.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 18.Primon M., Huszthy P.C., Motaln H., Talasila K.M., Miletic H., Atai N.A., Bjerkvig R., Lah Turnsek T. Cathepsin L silencing increases As2O3 toxicity in malignantly transformed pilocytic astrocytoma MPA58 cells by activating caspases 3/7. Exp. Cell Res. 2017;356:64–73. doi: 10.1016/j.yexcr.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 19.Hah Y.S., Noh H.S., Ha J.H., Ahn J.S., Hahm J.R., Cho H.Y., Kim D.R. Cathepsin D inhibits oxidative stress-induced cell death via activation of autophagy in cancer cells. Cancer Lett. 2012;323:208–214. doi: 10.1016/j.canlet.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Seo S.U., Woo S.M., Min K.J., Kwon T.K., FL-COCHO Z.- a cathepsin S inhibitor, enhances oxaliplatin-induced apoptosis through upregulation of Bim expression. Biochem. Biophys. Res. Commun. 2018;498:849–854. doi: 10.1016/j.bbrc.2018.03.068. [DOI] [PubMed] [Google Scholar]
- 21.Bell-McGuinn K.M., Garfall A.L., Bogyo M., Hanahan D., Joyce J.A. Inhibition of cysteine cathepsin protease activity enhances chemotherapy regimens by decreasing tumor growth and invasiveness in a mouse model of multistage cancer. Cancer Res. 2007;67:7378–7385. doi: 10.1158/0008-5472.CAN-07-0602. [DOI] [PubMed] [Google Scholar]
- 22.Quintanilla-Dieck M.J., Codriansky K., Keady M., Bhawan J., Runger T.M. Cathepsin K in melanoma invasion. J. Investig. Dermatol. 2008;128:2281–2288. doi: 10.1038/jid.2008.63. [DOI] [PubMed] [Google Scholar]
- 23.Wullschleger S., Loewith R., Hall M.N. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guri Y., Hall M.N. mTOR signaling confers resistance to targeted cancer drugs. Trends Cancer. 2016;2:688–697. doi: 10.1016/j.trecan.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Corcoran R.B., Rothenberg S.M., Hata A.N., Faber A.C., Piris A., Nazarian R.M., Brown R.D., Godfrey J.T., Winokur D., Walsh J., Mino-Kenudson M., Maheswaran S., Settleman J., Wargo J.A., Flaherty K.T., Haber D.A., Engelman J.A. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci. Transl. Med. 2013;5:196ra198. doi: 10.1126/scitranslmed.3005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gopal Y.N., Rizos H., Chen G., Deng W., Frederick D.T., Cooper Z.A., Scolyer R.A., Pupo G., Komurov K., Sehgal V., Zhang J., Patel L., Pereira C.G., Broom B.M., Mills G.B., Ram P., Smith P.D., Wargo J.A., Long G.V., Davies M.A. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1alpha and oxidative phosphorylation in melanoma. Cancer Res. 2014;74:7037–7047. doi: 10.1158/0008-5472.CAN-14-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Earwaker P., Anderson C., Willenbrock F., Harris A.L., Protheroe A.S., Macaulay V.M. RAPTOR up-regulation contributes to resistance of renal cancer cells to PI3K-mTOR inhibition. PLoS One. 2018;13 doi: 10.1371/journal.pone.0191890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brien N.A., McDonald K., Tong L., von Euw E., Kalous O., Conklin D., Hurvitz S.A., di Tomaso E., Schnell C., Linnartz R., Finn R.S., Hirawat S., Slamon D.J. Targeting PI3K/mTOR overcomes resistance to HER2-targeted therapy independent of feedback activation of AKT. Clin. Cancer Res. 2014;20:3507. doi: 10.1158/1078-0432.CCR-13-2769. [DOI] [PubMed] [Google Scholar]
- 30.Vandamme T., Beyens M., de Beeck K.O., Dogan F., van Koetsveld P.M., Pauwels P., Mortier G., Vangestel C., de Herder W., Van Camp G., Peeters M., Hofland L.J. Long-term acquired everolimus resistance in pancreatic neuroendocrine tumours can be overcome with novel PI3K-AKT-mTOR inhibitors. Br. J. Canc. 2016;114:650–658. doi: 10.1038/bjc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woo S.M., Min K.J., Seo B.R., Kwon T.K. YM155 sensitizes TRAIL-induced apoptosis through cathepsin S-dependent down-regulation of Mcl-1 and NF-kappaB-mediated down-regulation of c-FLIP expression in human renal carcinoma Caki cells. Oncotarget. 2016;7:61520–61532. doi: 10.18632/oncotarget.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park K.-H., Kim J., Jung S., Sung K.-h., Son Y.-K., Bae J.M., Kim B.-H. Alleviation of ultraviolet B-induced photoaging by 7-MEGATM 500 in hairless mouse skin. Toxicol. Res. 2019;35:353–359. doi: 10.5487/TR.2019.35.4.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han K., Xu X., Xu Z., Chen G., Zeng Y., Zhang Z., Cao B., Kong Y., Tang X., Mao X. SC06, a novel small molecule compound, displays preclinical activity against multiple myeloma by disrupting the mTOR signaling pathway. Sci. Rep. 2015;5:12809. doi: 10.1038/srep12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seo J., Lee E.W., Shin J., Seong D., Nam Y.W., Jeong M., Lee S.H., Lee C., Song J. K6 linked polyubiquitylation of FADD by CHIP prevents death inducing signaling complex formation suppressing cell death. Oncogene. 2018;37:4994–5006. doi: 10.1038/s41388-018-0323-z. [DOI] [PubMed] [Google Scholar]
- 36.Woo S.M., Min K.J., Seo B.R., Nam J.O., Choi K.S., Yoo Y.H., Kwon T.K. Cafestol overcomes ABT-737 resistance in Mcl-1-overexpressed renal carcinoma Caki cells through downregulation of Mcl-1 expression and upregulation of Bim expression. Cell Death Dis. 2014;5:e1514. doi: 10.1038/cddis.2014.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson S., Pearson A.N., Ashley M.D., Jessick V., Murphy B.M., Gafken P., Henshall D.C., Morris K.T., Simon R.P., Meller R. Identification of a novel Bcl-2-interacting mediator of cell death (Bim) E3 ligase, tripartite motif-containing protein 2 (TRIM2), and its role in rapid ischemic tolerance-induced neuroprotection. J. Biol. Chem. 2011;286:19331–19339. doi: 10.1074/jbc.M110.197707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dehan E., Bassermann F., Guardavaccaro D., Vasiliver-Shamis G., Cohen M., Lowes K.N., Dustin M., Huang D.C., Taunton J., Pagano M. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol. Cell. 2009;33:109–116. doi: 10.1016/j.molcel.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan L., Tan M., Yang J., Inuzuka H., Dai X., Wu T., Liu J., Shaik S., Chen G., Deng J., Malumbres M., Letai A., Kirschner M.W., Sun Y., Wei W. APC(Cdc20) suppresses apoptosis through targeting Bim for ubiquitination and destruction. Dev. Cell. 2014;29:377–391. doi: 10.1016/j.devcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trivigno D., Essmann F., Huber S.M., Rudner J. Deubiquitinase USP9x confers radioresistance through stabilization of Mcl-1. Neoplasia. 2012;14:893–904. doi: 10.1593/neo.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber A., Heinlein M., Dengjel J., Alber C., Singh P.K., Hacker G. The deubiquitinase Usp27x stabilizes the BH3-only protein Bim and enhances apoptosis. EMBO Rep. 2016;17:724–738. doi: 10.15252/embr.201541392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugatani T., Hruska K.A. Akt1/Akt2 and mammalian target of rapamycin/Bim play critical roles in osteoclast differentiation and survival, respectively, whereas Akt is dispensable for cell survival in isolated osteoclast precursors. J. Biol. Chem. 2005;280:3583–3589. doi: 10.1074/jbc.M410480200. [DOI] [PubMed] [Google Scholar]
- 43.Copp J., Manning G., Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009;69:1821–1827. doi: 10.1158/0008-5472.CAN-08-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nojima H., Tokunaga C., Eguchi S., Oshiro N., Hidayat S., Yoshino K., Hara K., Tanaka N., Avruch J., Yonezawa K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 2003;278:15461–15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 45.Oshiro N., Yoshino K., Hidayat S., Tokunaga C., Hara K., Eguchi S., Avruch J., Yonezawa K. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells. 2004;9:359–366. doi: 10.1111/j.1356-9597.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- 46.Wang L., Harris T.E., Roth R.A., Lawrence J.C. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007;282:20036–20044. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 47.Hussain S., Feldman A.L., Das C., Ziesmer S.C., Ansell S.M., Galardy P.J. Ubiquitin hydrolase UCH-L1 destabilizes mTOR complex 1 by antagonizing DDB1-CUL4-mediated ubiquitination of raptor. Mol. Cell. Biol. 2013;33:1188–1197. doi: 10.1128/MCB.01389-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bridges C.R., Tan M.-C., Premarathne S., Nanayakkara D., Bellette B., Zencak D., Domingo D., Gecz J., Murtaza M., Jolly L.A., Wood S.A. USP9X deubiquitylating enzyme maintains RAPTOR protein levels, mTORC1 signalling and proliferation in neural progenitors. Sci. Rep. 2017;7:391. doi: 10.1038/s41598-017-00149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morita M., Prudent J., Basu K., Goyon V., Katsumura S., Hulea L., Pearl D., Siddiqui N., Strack S., McGuirk S., St-Pierre J., Larsson O., Topisirovic I., Vali H., McBride H.M., Bergeron J.J., Sonenberg N. mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol. Cell. 2017;67:922–935. doi: 10.1016/j.molcel.2017.08.013. e925. [DOI] [PubMed] [Google Scholar]
- 50.Cereghetti G.M., Stangherlin A., Martins de Brito O., Chang C.R., Blackstone C., Bernardi P., Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U. S. A. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang C.R., Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007;282:21583–21587. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 52.Qi X., Disatnik M.H., Shen N., Sobel R.A., Mochly-Rosen D. Aberrant mitochondrial fission in neurons induced by protein kinase Cδ under oxidative stress conditions in vivo. Mol. Biol. Cell. 2011;22:256–265. doi: 10.1091/mbc.E10-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 54.Jezek J., Cooper K.F., Strich R. Reactive oxygen species and mitochondrial dynamics: the Yin and Yang of mitochondrial dysfunction and cancer progression. Antioxidants. 2018;7:E13. doi: 10.3390/antiox7010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lambies G., Miceli M., Martinez-Guillamon C., Olivera-Salguero R., Pena R., Frias C.P., Calderon I., Atanassov B.S., Dent S.Y.R., Arribas J., Garcia de Herreros A., Diaz V.M. TGFbeta-activated USP27X deubiquitinase regulates cell migration and chemoresistance via stabilization of Snail1. Cancer Res. 2019;79:33–46. doi: 10.1158/0008-5472.CAN-18-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dong L., Yu L., Bai C., Liu L., Long H., Shi L., Lin Z. USP27-mediated Cyclin E stabilization drives cell cycle progression and hepatocellular tumorigenesis. Oncogene. 2018;37:2702–2713. doi: 10.1038/s41388-018-0137-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Calvo E., Schmidinger M., Heng D.Y., Grunwald V., Escudier B. Improvement in survival end points of patients with metastatic renal cell carcinoma through sequential targeted therapy. Cancer Treat Rev. 2016;50:109–117. doi: 10.1016/j.ctrv.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 58.Schieke S.M., Phillips D., McCoy J.P., Jr., Aponte A.M., Shen R.F., Balaban R.S., Finkel T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J. Biol. Chem. 2006;281:27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- 59.Cunningham J.T., Rodgers J.T., Arlow D.H., Vazquez F., Mootha V.K., Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 60.Rodrik-Outmezguine V.S., Chandarlapaty S., Pagano N.C., Poulikakos P.I., Scaltriti M., Moskatel E., Baselga J., Guichard S., Rosen N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011;1:248–259. doi: 10.1158/2159-8290.CD-11-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.