

β-catenin regulates the cross-talk among mature adipocytes, macrophages, and preadipocytes under overnutrition.
Abstract
Overnutrition results in adiposity and chronic inflammation with expansion of white adipose tissue (WAT). However, genetic factors controlling fat mass and adiposity remain largely undetermined. We applied whole-exome sequencing in young obese subjects and identified rare gain-of-function mutations in CTNNB1/β-catenin associated with increased obesity risk. Specific ablation of β-catenin in mature adipocytes attenuated high-fat diet–induced obesity and reduced sWAT mass expansion with less proliferated Pdgfrα+ preadipocytes and less mature adipocytes. Mechanistically, β-catenin regulated the transcription of serum amyloid A3 (Saa3), an adipocyte-derived chemokine, through β-catenin–TCF (T-Cell-Specific Transcription Factor) complex in mature adipocytes, and Saa3 activated macrophages to secrete several factors, including Pdgf-aa, which further promoted the proliferation of preadipocytes, suggesting that β-catenin/Saa3/macrophages may mediate mature adipocyte-preadipocyte cross-talk and fat expansion in sWAT. The identification of β-catenin as a key regulator in fat expansion and human adiposity provides the basis for developing drugs targeting Wnt/β-catenin pathway to combat obesity.
INTRODUCTION
Obesity has become a worldwide noncommunicable epidemic with rising prevalence in past decades, and causes a severe health burden as a consequence of its notorious complications, such as type 2 diabetes, cardiovascular diseases, and colorectal cancers (1, 2). In the developed and developing countries, overnutrition results in excess calorie intake, fat accumulation, and further adiposity, which is characterized by chronic inflammation and expansion of subcutaneous and visceral white adipose tissues (WATs) (3–5).
WATs are composite of mature adipocytes, preadipocytes, vessel fractions, and other infiltrated inflammatory cells, e.g., macrophages and T cells (6). Stromal vascular fractions (SVFs) of adipose tissues were identified to be the origin of adipocyte progenitors, giving rise to new white adipocytes in subcutaneous WAT (sWAT) and visceral WAT (vWAT) (7–10). Macrophages also provide a favorable microenvironment influencing the apoptosis, proliferation, and differentiation of adipocyte progenitors (8, 11, 12). The expansion of WAT in obesity involves both the increased cell size of existing adipocytes (hypertrophy) and the generation of new adipocytes from the proliferation and differentiation of adipocyte progenitors/preadipocytes (hyperplasia) during fat development or under overnutrition such as high-fat diet (HFD) feeding (4, 13). It was previously believed that the total number of adipocytes has been set in early life; however, evidence suggests that adipocyte turnover also occurs throughout the adult stage and is especially enhanced during excess fat accumulation (14), identifying the important role of adipocyte hyperplasia in human obesity (15). Of note, chronic macrophage infiltration accompanies the development of obesity, not only contributing to insulin resistance but also participating adipose tissue remodeling (3, 12). Both healthy and pathological adipose tissue expansion need the participation of macrophages. However, genetic factors that control macrophage recruitment, fat expansion, and adiposity are still largely undetermined. The interplay between different components of WAT, e.g., mature adipocyte, macrophages, and preadipocyte, in regulating fat mass has also not been fully unveiled.
Human genetic and animal experimental studies have revealed that canonical Wnt/β-catenin signaling plays a pivotal role in body fat distribution and adipose tissue development (16). Gain-of-function LRP5 and LGR4, two Wnt signaling amplifiers, promote more fat storage in sWAT of humans (17, 18). Common variants in ZNRF3 and RSPO3, two other Wnt signaling modulators, are associated with increased waist-to-hip ratio (WHR) in several large-scale genome-wide association studies (GWAS) (19, 20). Although constitutive activation of Wnt signaling by active β-catenin mutant at an early stage inhibits adipogenesis in vitro (21), the activation of β-catenin in adipocyte progenitor cells driven by PPARγ-tTA;TRE-Cre results in fibrotic replacement in sWAT and lower vWAT fat mass with enlarged adipocytes in mutant mice (22). These findings illustrate the crucial but discrepant effects of activated Wnt signaling on adipogenesis in vitro and in vivo and the development of different WAT pads.
Identifying genetic mutations in CTNNB1/β-catenin, the hub gene of canonical Wnt signaling, is of importance to understanding the physiological function of Wnt signaling in obesity and adipocyte function. In the present study, we sequenced the whole coding regions of CTNNB1 in a young obese cohort and identified that rare gain-of-function mutations in CTNNB1 were associated with human obesity risk and body fat distribution. With the Cre/Loxp system to conditionally knock out CTNNB1/β-catenin in adipocytes with aP2-cre (termed as APBKO) and adiponectin-cre (termed as ABKO), respectively, we observed the crucial roles of β-catenin in fat expansion and obesity. We further revealed that the Wnt/β-catenin/Saa3 pathway mediated the cross-talk among the mature adipocyte-macrophage-preadipocyte circuit that controlled WAT expansion and adiposity, providing a promising drug target for the intervention of obesity.
RESULTS
Rare gain-of-function mutations in CTNNB1 are associated with human obesity
Our and others’ findings have reported the pathogenic roles of Wnt signaling mutations in human obesity (18, 23, 24). To further explore the potential association between genetic variants in CTNNB1, the core gene of the canonical Wnt signaling pathway and obesity, we first applied a large-scale GWAS database in Genetic Investigation of ANthropometric Traits (GIANT) and UK Biobank Meta-analysis (25), including 795,640 subjects in the Type 2 Diabetes Knowledge Portal (http://www.type2diabetesgenetics.org/), and found that the common noncoding variants in and around CTNNB1 were significantly associated with body mass index (BMI) (the most significant variant, rs9814633, β = 0.012, P = 2.10 × 10−11) (fig. S1 and table S1). Next, we screened the low-frequency/rare variants with minor allele frequency (MAF) less than 5% in the CTNNB1 gene in our in-home database of whole-exome sequencing (WES) data consisting of 1408 young, severely obese cases (age, 23.8 ± 7.3 years; BMI, 35.2 ± 4.7 kg/m2) and the published exome sequencing data containing 1455 ethnically matched nonobese controls (fig. S2A) (26). A total of seven rare missense mutations (MAF < 1%) were identified and verified by Sanger sequencing (table S2 and fig. S2B), which were conserved among different species (fig. S2C) without hotspot mutation features (fig. S2D). We obtained gene-based mutation prevalence of 0.71 versus 0.14% in obese cases and control subjects, respectively, which presented a significant enrichment of germline CTNNB1 mutations in obesity (odds ratio, 5.20; 95% confidence interval, 1.14 to 23.77; P = 0.02) (Fig. 1A).
Fig. 1. Genetic mutations in the CTNNB1 gene are associated with human obesity.
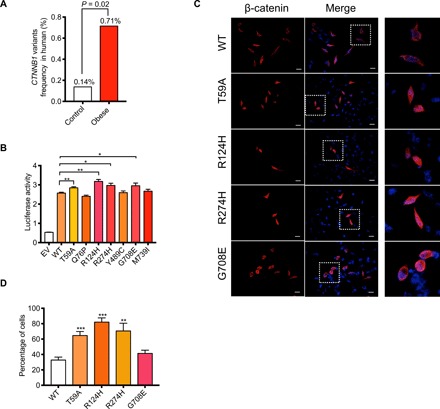
(A) Comparison of the low-frequency CTNNB1 mutations in control and obese subjects. (B) Luciferase reporter assay performed in human embryonic kidney (HEK) 293T cells 48 hours after transfection with the indicated plasmids. pRL-TK (expressing Renilla luciferase) was used as the normalized control. WT, wild type. (C) Representative images of β-catenin staining in HeLa cells that were overexpressed with indicated plasmids. Scale bars, 20 μm. The right panels were the amplified images of those in the corresponding squares in the middle panel. (D) Quantification of the percentage of the cells with β-catenin accumulated in the nucleus relative to all cells transfected with wild-type or four mutant plasmids. EV, empty vector; WT, wild-type. Data are shown as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
To further explore whether the seven rare missense mutations affected the function of β-catenin protein, we constructed plasmids expressing the mutations and examined their transcriptional activities through the TOP-Flash system, which is used to evaluate the canonical Wnt pathway activation by a luciferase reporter. p.T59A, p.R124H, p.R274H, and p.G708E mutants showed higher transcriptional activities than wild-type β-catenin (Fig. 1B), which were not found in the gnomAD_database of 8624 East Asians (table S2). To verify whether the higher transcriptional activity may be due to an increase in β-catenin translocation from cytoplasm to nucleus, we overexpressed these mutants into HeLa cells and calculated the percentage of cells with β-catenin accumulating in the nucleus. We found that three of four mutants except p.G708E had a higher accumulation in the nucleus than in wild-type β-catenin (Fig. 1, C and D). These results together suggested that these mutations conferred higher functional activity for β-catenin protein. Previous studies demonstrated the determinant roles of canonical Wnt signaling in body fat distribution (17). We next evaluated the obesity and metabolic features of these gain-of-function CTNNB1 mutation carriers. Four young obese female subjects carrying p.T59A, p.R124, and p.R274H mutations were included and received physical examination, abdominal computed tomography scanning, and biochemical analysis, while age-, sex-, ethnic-, and geography-matched obese subjects without CTNNB1 mutations were applied as general obese controls. Of note, the visceral fat content and liver enzymes including ALT (alanine aminotransferase), AST (aspartate aminotransferase), and GGT (gamma-glutamyl transferase), which are usually referred as the nonspecific indicators of lipid accumulation in liver in obesity development, were much lower in CTNNB1 mutation carriers than in matched obese controls (table S3). Other clinical parameters such as waist circumference and WHR did not show an obvious difference between carriers and noncarriers despite a trend to decrease in carriers (table S3). There was no difference in blood glucose, glucose tolerance, and serum lipids between groups (table S3). These results suggested that rare gain-of-function mutation in CTNNB1 confers high risk for obesity coupled with unique clinical features, at least, in these young female carriers.
Wnt/β-catenin signaling is induced in sWAT by overnutrition
To further examine the potential roles of β-catenin in the development of obesity, we first examined β-catenin expression in adipose tissues between lean and obese humans and mice. β-Catenin expression was significantly higher in sWAT of obese subjects than in normal weight controls (Fig. 2, A and B). Moreover, β-catenin expression was significantly higher in inguinal WAT (iWAT) and epididymal WAT (eWAT) of leptin-deficient (ob/ob) mice (Fig. 2, C and D). We next examined β-catenin expression in different fractions of WAT in mice. β-Catenin expression was significantly higher in mature adipocytes than SVFs in both WATs, consistent with the change in mature adipocyte markers such as leptin and adiponectin (Fig. 2, E and F), indicating that β-catenin might be involved in the function of mature adipocytes. We further examined the time course change of the canonical Wnt pathway in HFD-induced obese C57BL/6 J mice and revealed that β-catenin and Wisp2, a canonical Wnt/β-catenin pathway target gene, were gradually induced in iWAT in parallel with leptin increase (Fig. 2, G to I); β-catenin expression also showed positive correlation with leptin levels (Fig. 2J). However, these changes were not observed in eWAT of HFD mice, indicating much stronger activation of Wnt signaling in iWAT than in eWAT by overnutrition (Fig. 2, K to N). These results also supported the potential involvement of canonical Wnt/β-catenin activation in adipose tissue expansion and obesity.
Fig. 2. β-Catenin was induced in the development of obesity in humans and mice.
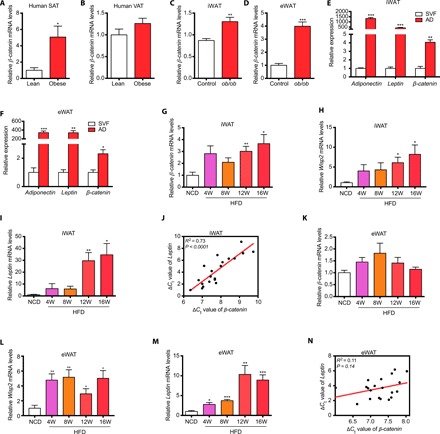
(A) β-catenin expression was significantly higher in subcutaneous adipose tissue of obese subjects than in lean subjects. (B) β-catenin expression was unaltered in visceral adipose tissue of obese subjects compared with that of lean subjects. (For A and B, n = 11 for lean subjects, n = 21 for obese subjects.) (C and D) The expression of β-catenin was higher in inguinal WAT (iWAT) (C) and epididymal WAT (eWAT) (D) of ob/ob mice than in wild-type mice (n = 10 for each group). (E and F) The expression of β-catenin was highly enriched in mature adipocytes than in SVF cells in iWAT (E) and eWAT (F) (n = 3 for each group). Leptin and adiponectin were used as the markers of mature adipocytes. (G to I) β-catenin expression was induced in iWAT during long-term HFD feeding (G) along with high expression of Wisp2 (H) and WAT marker Leptin (I). (J) β-catenin expression was associated with Leptin expression in iWAT. ΔCt values of β-catenin and Leptin were used to examine the association in linear regression analysis. (K to M) β-catenin expression was not significantly induced in eWAT during HFD feeding (K) despite the high expression of Wsip2 (L) and WAT marker Leptin (M). (N) β-catenin expression was not significantly associated with Leptin expression in eWAT. For (G) to (N), n = 4 for each group. AD, adipocyte; SAT, subcutaneous adipose tissue; VAT, visceral adipose tissue; NCD, normal chow diet; HFD, high-fat diet; iWAT, inguinal white adipose tissue; eWAT, epididymal white adipose tissue; SVF, stromal vascular fraction. Data are shown as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Ablation of β-catenin in mature adipocyte resists HFD-induced obesity
We next examined the effects of β-catenin ablation in adipocytes by crossing β-cateninflox/flox mice (27) with aP2-cre mice (APBKO) (Fig. 3A and fig. S3A). β-Catenin was significantly reduced in the adipose tissues; however, we also observed that several other tissues, including brain, hypothalamus, bone, and muscle, showed an obvious decrease in β-catenin mRNA levels (fig. S3B). Possibly because of this nonspecific leakage of β-catenin deletion using aP2-cre, APBKO mice showed a lower birth rate than expected according to Mendelian law of inheritance (fig. S3C) and severe neonatal death (fig. S3D). Despite this defect, a small portion of APBKO mice avoided the fetal lethality of global β-catenin knockout mice and survived to 8 weeks old. Phenotypically, survived APBKO mice showed reduced body weight and body size (Fig. 3, B and C). Moreover, iWAT mass of APBKO mice was markedly decreased, and eWAT was notably absent (fig. S3, E and F). Histologically, the adipocytes in iWAT of APBKO mice lost typical white adipocyte features showing an underdeveloped structure without droplet accumulation (fig. S3G). Brown adipocytes of APBKO mice also showed abnormal signs of piled nuclei compared with control mice (fig. S3G). Genes related to adipogenesis such as PPARγ2 and C/EBPα were also reduced in brown adipose tissues (BATs) of APBKO mice (fig. S3H). Despite nonspecific leakage of gene deletion, these results indicated that β-catenin may be involved in the development and maturation of adipocytes.
Fig. 3. Ablation of β-catenin in mature adipocyte resists long-term HFD–induced obesity.
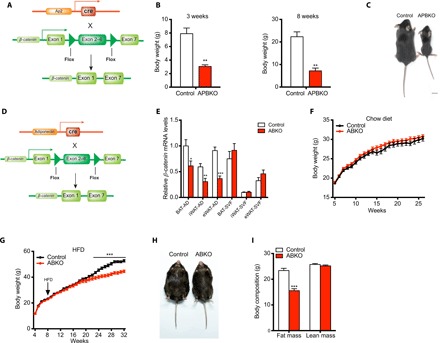
(A) Schematic of conditional β-catenin deletion in mature adipocytes with aP2-cre. (B) Body weight of control and APBKO mice at 3 and 8 weeks (n = 5 for each group at 3 weeks, n = 3 for each group at 8 weeks). (C) A representative photograph of 8-week-old control and APBKO mice. (D) Schematic of conditional β-catenin deletion in mature adipocytes with adiponectin-cre. (E) Comparison of β-catenin expression in mature adipocytes and SVF cells from brown adipose tissues (BAT), iWAT, and eWAT between chow diet–fed control and ABKO mice (n = 7 for each group). (F and G) Body weight curve of control and ABKO mice under chow diet (F) and HFD (G) (n = 15 to 19 for chow diet, n = 13 for HFD). (H) A representative photograph of 24-week HFD-fed control and ABKO mice. (I) Fat mass and lean mass in 24-week HFD-fed control and ABKO mice (n = 12 for each group). AD, adipocyte; ABKO, Adiponectin-cre;β-cateninflox/flox; APBKO, aP2-cre;β-cateninflox/flox; HFD, high-fat diet; SVF, stromal vascular fraction; BAT, brown adipose tissue; iWAT, inguinal white adipose tissue; eWAT, epididymal white adipose tissue. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
However, the leakage of β-catenin deletion in nonfat tissues and early death added complexity to the exploration of its roles in adipocytes and obesity. We next crossed β-cateninflox/flox mice with adipocyte-specific adiponectin-cre mice (ABKO) (Fig. 3D) and found that β-catenin was specifically and effectively deleted in mature adipocytes but not in SVFs, which contain adipocyte progenitors (Fig. 3E), or in other tissues, e.g., hypothalamus, liver, and muscle (fig. S4A). Thus, β-catenin ablation was restricted in the mature adipocytes in ABKO mice. Next, ABKO mice fed chow diet showed no difference in body weight (Fig. 3F), slight decrease in fat mass (fig. S4, B and C), unaltered plasma lipid levels (fig. S4, D to H), and glucose tolerance (fig. S4I) compared with control mice. There was no change in these parameters until 18 months old (fig. S4, J to L), indicating that β-catenin ablation in mature adipocyte did not affect age-induced body weight gain. As obesity also develops under overnutrition, we subsequently fed both genotypes with HFD from 8 weeks of age and found that ABKO mice began to gain less weight compared with the control group from 22 weeks of age. At the end of the 24-week HFD (32 weeks of age), ABKO mice showed 9.0-g body weight reduction compared with control mice (about 16.9% body weight of control mice) (Fig. 3, G and H). Furthermore, fat content analysis showed significantly reduced fat mass (Δ = 7.8 g, P < 0.001) in ABKO mice and similar lean mass between the two groups (Δ = 0.5 g, P = 0.28) (Fig. 3I). Collectively, these results suggested that specific ablation of β-catenin in mature adipocytes resisted long-term HFD–induced adipose tissue expansion and obesity, and fat mass reduction contributed largely to body weight loss.
Ablation of β-catenin in mature adipocytes improves metabolic homeostasis
With such a significant decrease in body weight and adiposity, we postulated that metabolic disturbances under HFD could also be greatly improved in ABKO mice. As expected, fasted ABKO mice showed reduced blood glucose levels compared with control mice (Fig. 4A), which was much more evident when we challenged the mice with d-glucose, as reflected by the improved glucose tolerance (Fig. 4, A and B). Furthermore, insulin sensitivity was also improved in ABKO mice (Fig. 4, C and D), and plasma insulin levels were consistently decreased (Fig. 4E). This was further supported by enhanced insulin signaling pathway with enhanced phosphorylation of Akt in iWAT (Fig. 4F). In addition, serum lipid levels were reduced in ABKO mice (Fig. 4, G and H), and HFD-induced ectopic fat deposition in the liver was also markedly attenuated (Fig. 4, I and J). HFD-induced macrophage infiltration was reduced in iWAT of ABKO mice compared with controls, as indicated by the reduced expression of macrophage markers such as Cd68, F4/80, and Cd11c and the reduced proportion of F4/80+Cd11b+ macrophages (Fig. 4, K and L, and fig. S4, M and N). However, we did not find any changes in macrophage number in the BAT or eWAT of HFD-fed ABKO mice (fig. S4, M to O). Furthermore, we found that the markers of classically activated macrophages (M1), such as interleukin-1β (IL-1β), tumor necrosis factor–α (TNFα), and C-C motif chemokine ligand 2 (Ccl2), were also significantly reduced in the iWAT of ABKO mice (Fig. 4M), while the markers of alternatively activated macrophages (M2), such as Il-4 and Il-13, were unaltered (fig. S4P). In line with the reduced adiposity and improved iWAT inflammation, ABKO mice also showed improved systemic inflammatory reaction, indicated by lower plasma levels of Leptin, Resistin, and Pai-1 when compared with control mice (Fig. 4, N to P). These results together demonstrated that ablation of β-catenin in mature adipocytes improved global metabolic homeostasis under long-term overnutrition.
Fig. 4. Ablation of β-catenin in mature adipocyte improves metabolic homeostasis.
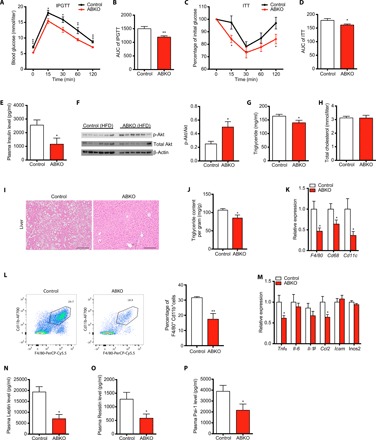
(A) Glucose tolerance test after 16-hour fasting in 24-week HFD-fed control and ABKO mice (n = 13 for each group). (B) Quantification of area under the curve (AUC) for glucose tolerance test. (C) Insulin tolerance test after 6-hour fasting in 24-week HFD-fed control and ABKO mice (n = 13 for each group). (D) Quantification of AUC for insulin tolerance test. (E) Fasting plasma Insulin levels of 26-week HFD-fed control and ABKO mice (n = 8 for each group). (F) Left: Western blot of p-Akt and total Akt (t-Akt) in iWAT of 26-week HFD-fed control and ABKO mice fasted for 16 hours. Right: Quantification of p-Akt/t-Akt ratio (n = 6 for each group). (G and H) Plasma triglyceride (G) and total cholesterol levels (H) in 26-week HFD-fed control and ABKO mice (n = 13 for each group). (I) Representative images of hematoxylin and eosin (HE) staining of liver in 26-week HFD-fed control and ABKO mice. Scale bars, 100 μm. (J) Liver triglyceride content quantification (n = 13 for each group). (K) The mRNA expression of macrophage markers F4/80, Cd68, and Cd11c in iWAT of 26-week HFD-fed control and ABKO mice (n = 13 for each group). (L) Left: Representative cell sorting of F4/80+Cd11b+ cells from iWAT SVF of 26-week HFD-fed control and ABKO mice by fluorescence-activated cell sorting (FACS). Right: Quantification of the percentage of F4/80+Cd11b+ cells to Cd45+ cells in SVF of iWAT (n = 4 for each group). (M) mRNA expression of the indicated M1 macrophage markers in iWAT of 26-week HFD-fed control and ABKO mice (n = 13 for each group). (N to P) Plasma levels of Leptin (N), Resistin (O), and Pai-1 (P) in 26-week HFD-fed control and ABKO mice (n = 8 for each group). IPGTT, intraperitoneal glucose tolerance test; ITT, insulin tolerance test; ABKO, Adiponectin-cre;β-cateninflox/flox; HFD, high-fat diet. Data are shown as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
To fully evaluate the energy balance in ABKO mice, food intake, fecal energy excretion, physical activity, and energy expenditure, represented by O2 consumption and CO2 production, were systemically measured, and only energy expenditure showed a significant but subtle increase in ABKO mice (fig. S5), which might also explain the accumulated body weight difference between the two genotypes over long-time HFD challenge.
Ablation of β-catenin in mature adipocytes reduces the proliferation of preadipocytes and iWAT mass in HFD feeding
It is well known that obesity comes with expansion of WATs, which generally contains two distinct processes, one in the form of hypertrophy (characterized by an enlargement in adipocyte size) and the other in the form of hyperplasia (characterized by new adipocyte generation from progenitor and preadipocyte cells) (13, 15, 28). We found that iWAT weight reduced significantly in ABKO mice, while the weights of eWAT and BAT were unaltered (Fig. 5, A to C). Thereafter, the adipocyte size of iWAT was carefully examined but appeared to be highly similar between ABKO and control mice (Fig. 5, D to F). Meanwhile, the expression of genes related to fatty acid synthesis and lipolysis was further examined and also showed no significant alterations either in iWAT, eWAT, or BAT between the two groups (fig. S6, A to F). The expression of adipogenesis-related genes was not different in adipose tissues either (fig. S6, G to I). These results strongly suggested that β-catenin regulated iWAT mass not through affecting hypertrophy. It is previously reported that iWAT expansion involves hyperplasia due to the notable increase in adipocyte progenitor proliferation in response to HFD (28). We hypothesized that the reduced iWAT mass in ABKO mice might result from decreased progenitor or preadipocyte proliferation and the consequent lower hyperplasia for newborn adipocytes. As shown in Fig. 5G, Pdgfrα+ preadipocytes, which reside in SVFs and contribute to new white adipocyte generation in response to HFD (8), were significantly decreased in ABKO mice (Fig. 5G). The percentage of proliferated preadipocytes, marked by BrdU+ Pdgfrα+ double labeling, was also decreased in ABKO mice (Fig. 5H), indicating that the blunted proliferation of preadipocytes was associated with the restricted mass expansion in iWAT of ABKO mice. Consistently, the progenitors marked by other recently identified markers, such as Cd142 and Icam1, were also significantly reduced in the iWAT of ABKO mice (fig. S6, J to Q) (10). There were no obvious changes in these progenitors in eWAT of ABKO mice (fig. S6, J to Q). These data together may suggest that ablation of β-catenin in mature adipocytes reduced hyperplastic adipose expansion through reducing the proliferation of preadipocytes.
Fig. 5. Ablation of β-catenin in mature adipocyte reduces preadipocyte proliferation and subcutaneous fat mass in long-term HFD feeding.
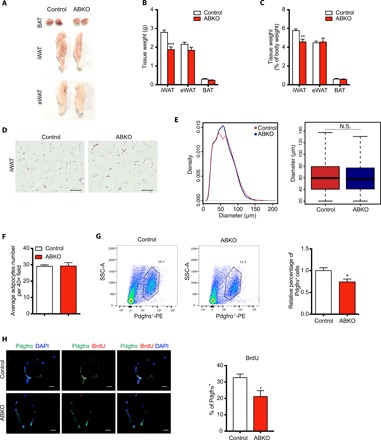
(A) Representative images of BAT, iWAT, and eWAT depots in 26-week HFD-fed control and ABKO mice. (B and C) Absolute tissue weight (B) and percentage of tissue weight to body weight (C) for iWAT, eWAT, and BAT depot in 26-week HFD-fed control and ABKO mice (n = 13 for each group). (D) Representative images of HE staining of iWAT in 26-week HFD-fed control and ABKO mice. Scale bars, 100 μm. (E) Left: The frequency distribution of adipocyte cell size in iWAT of 26-week HFD-fed control and ABKO mice. Right: Box plot of adipocyte diameter in two groups, Wilcoxon rank-sum test, boxes represent the interquartile range (IQR) between the first and third quartiles, and the line inside the box represents the median; whiskers represent the lowest or highest values within 1.5 times IQR from the first or third quartiles. (F) Cell counting per field in HE-stained sections under microscopy in iWAT of 26-week HFD-fed control and ABKO mice. (G) Left: Representative cell sorting of Pdgfrα+ cells from iWAT SVF of 26-week HFD-fed control and ABKO mice by FACS. Right: Quantification of the percentage of Pdgfrα+ cells to SVF cells in iWAT (n = 7 for each group). (H) Left: Representative images of Pdgfrα and BrdU staining in BrdU-incorporated iWAT sections of 26-week HFD-fed control and ABKO mice. Scale bars, 10 μm. Right: Quantification of the percentage of Pdgfrα+BrdU+ cells to Pdgfrα+ cells in iWATs (n = 4 for each group). ABKO, Adiponectin-cre; β-cateninflox/flox; HFD, high-fat diet; BAT, brown adipose tissue; iWAT, inguinal white adipose tissue; eWAT, epididymal white adipose tissue; SVF, stromal vascular fraction; SSC-A, side scatter area. Data are shown as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. N.S., not significant.
β-catenin regulates the transcription of serum amyloid A3 (Saa3) through the TCF binding site
As β-catenin was specifically deleted in mature adipocytes but not in preadipocytes of adipose tissues (Fig. 3E), we hypothesized that β-catenin deletion may regulate certain factors that can be secreted by mature adipocytes and, in turn, affect the preadipocyte proliferation. We then performed unbiased whole transcriptome profiling analysis on iWATs of the two genotypes. Among the altered genes (Fig. 6A), the secreted factors were further examined, and serum amyloid A3 (Saa3), which was an adipokine highly expressed in WAT (29) and validated by real-time polymerase chain reaction (PCR) (Fig. 6B), was included for further analysis, while the other factor Slit2 was excluded for its antiobese effects (30), which was opposite to the effects of β-catenin. We next examined the cell-autonomous regulation of Wnt/β-catenin on Saa3. β-catenin knockdown by lentivirus short hairpin RNA (shRNA) in differentiated adipocytes from 3T3-L1 preadipocytes significantly reduced the expression of Saa3 (Fig. 6C). Meanwhile, recombinant Wnt3a stimulation, which can robustly lead to downstream β-catenin activation, significantly up-regulated Saa3 expression (Fig. 6D), while PKF115, a small molecule as a canonical Wnt signaling inhibitor, decreased Saa3 expression (Fig. 6E) in mature adipocytes differentiated from SVFs. Wisp2 was examined as a positive control of canonical Wnt signaling target and activation (Fig. 6, C to E). We further demonstrated that β-catenin overexpression significantly increased Saa3 transcriptional activity (Fig. 6F). As β-catenin acts as a cofactor of the transcription factor Tcf4 (also named as Tcf7l2) to promote downstream targets, we further analyzed the promoter region of Saa3 gene, and six potential TCF binding sites, CTTTG(A/T)(A/T) (31), were indicated, of which deletion of TCF binding site (−84 to −90) largely abolished β-catenin–induced transcriptional activity, while deletion or mutation of other sites showed marginal alterations (fig. S7A). Chromatin immunoprecipitation (ChIP) assay further showed the interaction of the β-catenin–Tcf4 complex with Saa3 promoter containing the above binding site in white adipocytes, which were differentiated from SVFs for 8 days (Fig. 6, G and H). These data together demonstrated Saa3 as a new downstream target of the canonical Wnt/β-catenin pathway in mature adipocytes.
Fig. 6. β-catenin ablation in mature adipocyte inhibits preadipocyte proliferation partially mediated by repressing Saa3 and decreasing macrophage recruitment.
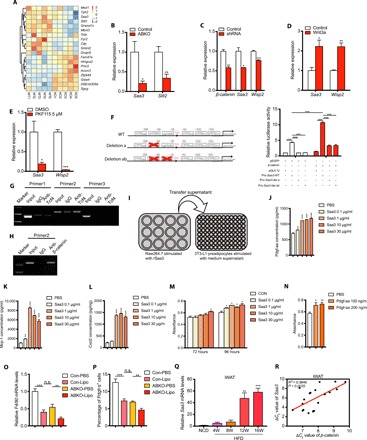
(A) Gene expression profiles of iWAT in 24-week HFD-fed control and ABKO mice. Heatmap of genes that are differentially expressed between two groups, with a fold change larger than 1.5 and the corresponding P values less than 0.05. (B) Validation of Saa3 expression in iWATs between the two groups by real-time polymerase chain reaction (PCR). (C) Saa3 expression detected in mature adipocytes differentiated from 3T3-L1 cells after β-catenin knockdown by shRNA. (D) Saa3 expression detected in mature adipocytes differentiated from 3T3-L1 cells after canonical Wnt signaling activation by Wnt3a (10 ng/ml) treatment for 16 hours. (E) Saa3 expression detected in mature adipocytes differentiated from SVF cells after canonical Wnt signaling inhibition by 5 μM PKF115 treatment for 48 hours. (F) Left: Schematic of wild-type Saa3 promoter and mutations deleting the indicated Tcf4 binding sites. Right: Luciferase reporter assay performed in the 3T3-L1 cell line 48 hours after transfection with the indicated plasmids. pRL-TK (expressing Renilla luciferase) was used as the normalized control. (G) Chromatin immunoprecipitation (ChIP) assay in white adipocytes differentiated from SVF immunoprecipitated with anti-Tcf4 antibody followed by PCR detection with the indicated primers for different parts of Saa3 promoter. Of note, products with primer 2 were designed to amplify the region containing Tcf4 binding sites, while primer 1 and primer 3 were not. IgG, immunoglobulin G. (H) ChIP assay in differentiated white adipocytes immunoprecipitated with anti–β-catenin antibody followed by PCR detection with the primer 2. (I) Schematic of experiment design in which Raw264.7 was stimulated with recombinant Saa3 for 6 hours, and the supernatant was then added to 3T3-L1 cells and refreshed every 24 hours. (J to L) Concentrations of Pdgf-aa (J), Mcp-1 (K), and Cxcl2 (L) in the conditioned medium of Raw264.7 cells treated with recombinant Saa3 at 0.1, 1, 10, and 30 μg/ml, respectively. (M) Viability of 3T3-L1 cells treated with the conditioned medium of Raw264.7 cells in (I). (N) Cell viability of 3T3-L1 cells treated with the recombinant Pdgf-aa protein with different concentrations for 120 hours. (O and P) The relative mRNA levels of F4/80 (O) and the percentage of Pdgfrα+ cells calculated (P) in the indicated groups (n = 6 to 8 for each group). (Q) Saa3 expression was induced in iWAT during long-term HFD feeding (n = 4 for each group). (R) Saa3 expression was associated with β-catenin expression in iWAT. ΔCt value of Saa3 and β-catenin were used to examine the association in the linear regression analysis. ABKO, Adiponectin-cre;β-cateninflox/flox; HFD, high-fat diet; Con-PBS, control mice injected with PBS; Con-Lipo, control mice injected with liposome; ABKO-PBS, ABKO mice injected with PBS; ABKO-Lipo, ABKO mice injected with liposome. Data are shown as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. n.s., not significant.
Saa3-activated macrophages promote preadipocyte proliferation
We have demonstrated that β-catenin deficiency in mature adipocyte largely decreased the expression of Saa3, while Saa3, similar to its homolog Saa1/2, can be secreted into the tissue matrix as a secreted adipokine (32). We next examined whether Saa3 could further function on preadipocytes. Unexpectedly, we did not observe the direct effects of recombinant Saa3 protein or Saa3 overexpression on the growth rate of SVF cells or 3T3-L1 preadipocytes (fig. S7, B and C). Previous studies have shown that Saa3 is also referred as a chemokine that played a role in macrophage recruitment in WATs (29), and ablation of Saa3 inhibits macrophage accumulation and then resists HFD-induced obesity (33). We further examined whether Saa3 functioned through recruiting macrophages to induce the secreted factors, which then promoted the proliferation of adipocyte progenitors. Saa3 stimulation significantly increased the mRNA expression of several inflammatory and secreted factors in macrophages (fig. S7D and Fig. 6, I to L), among which Pdgf-aa was significantly elevated in the conditioned medium in a dose-dependent manner, and other factors such as Mcp-1 and Cxcl2 were also significantly increased (Fig. 6, J to L, and fig. S7, E to G). The conditioned medium of Saa3-treated macrophages further increased the growth rate of preadipocytes (Fig. 6M), suggesting that Saa3 indirectly promoted preadipocyte proliferation through activating macrophages. This was also consistent with reduced macrophage infiltration and the improved inflammation reaction in iWAT of ABKO mice (Fig. 4, J to M). To further identify the potential secreted factors that contributed to the effects of Saa3/macrophages on the proliferation of preadipocytes, we next treated 3T3-L1 preadipocytes with different combination formula consisting of the recombinant proteins of these up-regulated factors. Of note, the combination containing Pdgf-aa or Pdgf-aa alone both significantly increased the growth rate of preadipocytes (Fig. 6N and fig. S7, H and I). These findings indicated that Pdgf-aa may be one of the contributing factors linking Saa3/macrophages and preadipocytes.
To further demonstrate that macrophages in iWAT may contribute to the maintenance of Pdgfrα+ cells in iWAT, clodronate liposomes were respectively injected into one side of iWAT pads of control and ABKO mice fed with HFD to clear macrophages, while an equal amount of control liposomes was injected into the other side. We found that macrophages marked with F4/80 in the clearance side were remarkably reduced in both control and ABKO mice (Fig. 6O). Moreover, the percentage of Pdgfrα+ preadipocytes was also decreased upon clodronate liposome treatment in the two groups (Fig. 6P), in parallel with the changes of macrophage markers. These results implied that the reduction in Pdgfrα+ preadipocyte percentage in iWAT of ABKO mice was most likely due to the lower number of macrophages. As such, Saa3 may mediate the effects of β-catenin ablation on adipose expansion by recruiting macrophages and their effects on the proliferation of preadipocytes. Furthermore, in agreement with the gradually increased β-catenin levels upon HFD, Saa3 was also robustly induced in iWAT in response to HFD (Fig. 6Q) and showed tightly positive correlation with β-catenin expression (Fig. 6R). These results together supported the findings that ablation of β-catenin in mature adipocytes decreased macrophages infiltration in WAT and resisted long-term HFD–induced adipose expansion.
DISCUSSION
Canonical Wnt/β-catenin signaling plays important biological roles in adipocyte differentiation and metabolism (21). However, the in vivo physiological roles of its key effector CTNNB1/β-catenin in human obesity and mature adipocyte function are still not clear. Here, we identify that rare gain-of-function mutations in CTNNB1 are associated with increased obesity risk in humans. A significant elevation of β-catenin expression is also detected in sWAT of obese subjects. By using mouse models, we further reveal the involvement of β-catenin in sWAT expansion, pointing to possible mechanisms of Wnt/β-catenin/Saa3-macrophage in the cross-talk between mature adipocytes and nonadipocytes in controlling adipose tissue expansion and obesity.
More and more genetic evidence has linked canonical Wnt signaling to human adiposity and metabolic disorders. Loss-of-function mutations in LRP5 and LRP6, which encode the coreceptors in the Wnt/Fzls signaling pathway, are associated with osteoporosis (34) and cardiovascular risk (35), respectively. In addition, gain-of-function mutations in LRP5 are associated with osteosclerosis (36) and altered fat distribution, enhanced lower-body sWAT accumulation, and reduced android fat content (17). GWAS also identify common variants in ZNRF3 and RSPO3, which encode the modulators of canonical Wnt signaling pathway, associated with WHR (19, 20). Our previous studies demonstrate that a low-frequency activating variant in LGR4, which encodes the receptor of canonical Wnt modulator RSPO1-4, is associated with increased risk of human obesity (18, 24). Here, we reported that the common variants in and around CTNNB1 were significantly associated with BMI, and further, we identified that rare germline missense mutations in CTNNB1 conferred a moderate risk of human obesity in a young, severely obese cohort. Of note, three of seven missense mutations showed a consistent gain of biological function, indicated by the enhanced transcriptional activities and nuclear accumulation. Somatic activating CTNNB1 mutations promote the development of several types of cancers, in particular colorectal cancers (37). As these germline gain-of-function CTNNB1 mutation obese carriers are much younger at present, they are suggested for intensive clinical follow-up for primary prevention of tumors later in life. In addition, by comparing with matched obese subjects without CTNNB1 mutations, we found that these functional CTNNB1 mutant carriers showed similar BMI but had reduced visceral fat mass and serum liver enzyme levels than obese controls, portraying the special features of these obese carriers. The humanized mice carrying these CTNNB1 mutations are needed to further illuminate the causal effects of these gain-of-function CTNNB1 mutations on fat distribution. However, we could not exclude the possibility that the CTNNB1 mutations, which could increase β-catenin activity in other tissues such as hypothalamus, may also contribute to obesity risk in other ways, for example by regulating appetite. In agreement with our reports of genetic CTNNB1 activation predisposing risk to adiposity, a line of clinical intervention evidence that Lithium salts, a first-line long-term bipolar drug that activates β-catenin by inhibiting GSK3β, also promote body weight gain in patients (38). These results suggested that β-catenin activation was associated with the development of human obesity. Future studies are warranted to validate these genetic findings in other ethnic and much larger-scale populations.
To further investigate the potential role of β-catenin in regulating obesity, we examined its expression in obese human and mice and consistently found that β-catenin expression was significantly increased in sWAT of obese subjects and HFD-induced obese mice, but less significant in vWAT. Moreover, β-catenin is more abundant in mature adipocytes than in preadipocytes. These results suggest that β-catenin may play important roles in mature adipocyte function contributing to more fat accumulation in sWAT rather than vWAT. Here, we used adiponectin-cre to specifically knock out β-catenin in mature adipocytes, which significantly reduced body weight and fat mass especially sWAT mass, in response to long-term HFD feeding, to further support the human findings. It should also be noted that activation or inactivation of Wnt pathway in different development stages may result in discordant phenotypes (39). A previous study reported that constitutive activation of β-catenin with the PPARγ-tTA;TRE-Cre switched the fate of adipose progenitors into a fibroblastic lineage in iWAT and showed lower visceral fat mass but increased single adipocyte hypertrophy in eWAT, while activation with aP2-cre showed no effects on adiposity and metabolism in chow diet (22). In our study, aP2-cre–induced β-catenin knockout mice produced haplomorphic mutant mice with impaired birth rate, and the mutant mice that survived showed developmental defects of iWAT and absolute absence of eWAT. According to a series of studies, PPARγ expresses in preadipocytes and mature adipocytes earlier than aP2, while adiponectin-cre functions much later and specifically expresses in mature adipocytes (40). These data together suggested that Wnt/β-catenin plays distinct physiological roles along different stages of fat tissue development, and β-catenin in mature adipocyte is crucial for promoting adiposity. It is also worth mentioning that Wnt/β-catenin activation in PPARγ-expressing cells promoted muscle glucose uptake through extrinsic signals (22), while we did not detect marked muscle phenotypes in adiponectin-cre–induced β-catenin knockout mice (data not shown), which may also reflect the various effects of Wnt/β-catenin in different stages of adipocytes.
In our study, β-catenin–specific ablation in mature adipocytes resulted in no differences in body weight and body composition compared with control mice under normal chow diet, which was consistent with the phenotype of β-catenin activation using aP2-cre in chow diet (22). Although many studies have demonstrated that activation of Wnt signaling in adipose precursors or preadipocytes could block the differentiation of adipocytes, none of them focused on the function of β-catenin in mature adipocytes under overnutrition, including mice with aP2-cre–induced β-catenin activation. We unexpectedly found that ABKO mice resisted long-term HFD–induced iWAT expansion and adiposity. These results may suggest that long-term overnutrition is one important triggering factor for the effects of Wnt/β-catenin on mature adipocytes. Detailed mechanism regarding the response of mature adipocyte Wnt/β-catenin to nutrition load needs to be clarified in future studies. Adipocyte hypertrophy and hyperplasia both contribute to HFD-induced obesity. We did not detect significant differences in adipocyte size between ABKO and control mice. However, Pdgfrα+ preadipocytes together with other subpopulations of preadipocytes were significantly decreased in ABKO mice, indicating that the reduced fat mass may result from the reduced number of adipose progenitors or preadipocytes contributing to new-turn adipocyte generation. This was consistent with the observation that iWAT expansion involves hyperplasia in response to HFD (28). There are also studies reporting that iWAT expansion originates from hypertrophy before 12 weeks of HFD feeding (13), while in our study, β-catenin ablation showed consistent weight loss effects later after 14 weeks of HFD feeding in three independent experiments and did not affect adipocyte hypertrophy in the early stage of HFD feeding, which might also explain the delayed effects of β-catenin ablation on obesity. To bridge the gap between β-catenin ablation in mature adipocytes and proliferated preadipocytes, we next performed an unbiased screening and found a secreted protein,Saa3, which was down-regulated in mature adipocytes of ABKO mice and was identified as a new target gene of canonical Wnt/β-catenin/TCF signaling. A previous study showed that Saa3 directly activates macrophages by the TLR4/MyoD88 pathway and enhances inflammatory factor release (41). Saa3 is also reported to increase during adipose tissues expansion and recruit monocytes for local inflammation in adipose tissues (29). Abundant evidence already demonstrates that macrophages have strong promoting effects on proliferation of preadipocytes via secreted growth factors (42). Our in vitro studies further found that the conditioned medium of Saa3-stimulated macrophages promoted the proliferation of preadipocytes, in which several secreted proteins were activated. Of note, Pdgf-aa was dose-dependently accumulated in the conditioned medium of macrophages and further promoted the growth rate of preadipocytes, pointing to its contribution to the cross-talk between Saa3/macrophage and preadipocytes. Other potential contributing factors still need to be identified in the future. Since clodronate liposomes could achieve incomplete clearance of macrophages in target tissues or organs, by this method, we found that when macrophages in iWAT of control mice decreased to the same level as those in ABKO mice, Pdgfrα+ preadipocytes in iWAT of control mice were also reduced to a similar level in ABKO mice. Future studies are warranted to better elucidate this by using a genetic or chemical approach to completely abolish the effects of macrophages. These findings may indicate a potential cross-talk among mature adipocytes, macrophages, and preadipocytes mediated by secreted adipokine and growth factors. Consistently, we also detected reduced inflammation factors in iWAT and circulation along with metabolic improvement, including improved glucose tolerance and enhanced insulin sensitivity, upon reduction in β-catenin and Saa3 in ABKO mice. Consistent to these phenotypes, Saa3−/− mice are also reported to gain less body weight and have reduced adipose inflammation in high-fat high-sucrose diet (33). Future studies using mature adipocyte–specific Saa3 knockout mice would be more helpful to evaluate the effects of Saa3 on macrophages and preadipocytes.
In summary, we identify that germline gain-of-function mutations in the CTNNB1 gene are associated with higher risk for human obesity. Further, using animal models, we demonstrate that β-catenin plays an essential role in mature adipocytes to regulate WAT expansion and obesity, and point to the possible cross-talk circuit from mature adipocytes to macrophages and further hyperplastic WAT expansion by β-catenin/Saa3 signaling. This study extends our understanding of the canonical Wnt/β-catenin signaling in human obesity and overnutrition-induced adipocyte hyperplasia following chronic mature adipocyte hypertrophy, providing the basis for developing drugs targeting CTNNB1/β-catenin to intervene in obesity.
MATERIALS AND METHODS
Human genetic study
For genetic evaluation of rare variants in the coding region of CTNNB1/β-catenin, we analyzed our in-home database consisting of the WES data of 1408 obese patients and the published database of 1455 ethnically matched control subjects (fig. S2A) (26). Obese patients were identified and recruited continuously from the specialized obesity outpatient clinic of Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. In brief, patients with body mass index (BMI) ≥ 30 kg/m2 were recruited and secondary or syndromatic obesity were excluded. Basic characteristic information was obtained from the Genetics of Obesity in Chinese Youngs study, which was previously established by Ruijin Hospital and registered in ClinicalTrials.gov (ClinicalTrials reg. no. NCT01084967, http://www.clinicaltrials.gov/) (18, 24, 43). By filtering on common variants with MAF > 5% and synonymous variants, we obtained a list of low-frequency or rare missense and indel variants in the CTNNB1 gene with MAF < 5% for further analysis. All the included variants were verified by Sanger sequencing (fig. S2B). The clinical procedures for anthropometric assessment, biochemical measurement, blood glucose, and lipid ascertainment were performed according to previously described protocols (18, 24, 43, 44). This study was approved by the Institutional Review Board of the Ruijin Hospital, Shanghai Jiao Tong University School of Medicine and was performed in accordance with the principle of the Helsinki Declaration II. A written informed consent was obtained from each participant.
Mice
APBKO mice and ABKO mice were generated using the Cre/Loxp system. In general, β-cateninflox/flox mice were mated with aP2-cre or adiponectin-cre mice, respectively. The F1 generation β-cateninflox/+;aP2-cre mice or β-cateninflox/+;adiponectin-cre mice was further intercrossed to generate β-cateninflox/flox;aP2-cre (APBKO) mice or β-cateninflox/flox;adiponectin-cre (ABKO) mice, respectively, and β-cateninflox/flox mice were used as control mice. Mice were maintained under 12-hour dark-light cycles with unrestricted access to food and water. A whole-body composition analyzer (EchoMRI) was used to evaluate fat mass and lean mass on awake animals. Mice were placed in metabolic cages (Columbus Instruments) to assess their energy expenditure, O2 consumption, CO2 production, and physical activity. Twenty-four–hour fecal caloric content was measured with a 6400 Automatic Isoperibol Calorimeter (Parr Instrument Company) according to the manufacturer’s instructions. In some experiments, mice were fed 60 kcal% HFD (Research Diet 12492i). For BrdU (bromodeoxyuridine) incorporation, BrdU (Sigma-Aldrich) was resolved in phosphate-buffered saline (PBS; 20 mg/ml) and diluted in drinking water at a final concentration of 0.4 ml/ml. Drinking water containing BrdU was given to HFD-fed male mice for 1 week and refreshed every other day. BrdU was detected by immunostaining as described below. For the clearance of macrophages located in iWAT, control and ABKO mice fed with HFD for 24 weeks were injected with clodronate liposomes (110 mg/kg; Clodronateliposomes.org) into one side of iWAT pad or equal amount of control liposomes into the other side of iWAT pad, and the injection was repeated once 3 days later. Mice were euthanized 6 days later after the first injection, and iWATs were fixed in 4% paraformaldehyde (PFA). Male mice were used in the experiments unless otherwise indicated. All procedures were approved by the Animal Care Committee of Shanghai Jiao Tong University School of Medicine.
IPGTT and ITT
Mice were fasted overnight for 16 hours and injected with d-glucose (2 g/kg for normal chow diet and 1 g/kg for HFD mice) intraperitoneally for the glucose tolerance test. HFD mice were fasted for 6 hours and injected with recombinant human insulin (1.5 U/kg, Eli Lilly) intraperitoneally for the insulin tolerance test.
Blood glucose and plasma insulin and biochemical measurements
Mouse blood glucose levels were measured with whole blood from the tail vein using a glucose meter (LifeScan). Plasma insulin, leptin, resistin, PAI-1 (Millipore), triglyceride, and total cholesterol (Ke Hua Biotechnology) levels were measured using commercial enzyme-linked immunosorbent assay kits according to the manufacturer’s instructions.
Hematoxylin and eosin staining and cell counting
Liver and adipose tissues were fixed in 4% PFA and embedded in paraffin. Sections of 5-μm thickness were stained with hematoxylin and eosin (HE) according to standard protocols. HE images from iWAT were used for morphometric analysis. Cell counting was achieved using the ImageJ software and AdipoCount software (http://www.csbio.sjtu.edu.cn/bioinf/AdipoCount/).
Immunofluorescence
Immunostaining was performed in paraffin sections according to the standard protocols. In brief, samples were deparaffinized and rehydrated, followed by antigen retrieval in 10 mM sodium citrate. Blocking and staining were performed in antibody diluent with background-reducing components (Dako). Sections were incubated with rat anti-BrdU (Abcam, #ab6326, 1:500) and rabbit anti-Pdgfrα (Cell Signaling Technology, #3174, 1:500) antibodies overnight at 4°C, followed by incubation with the corresponding secondary antibodies goat anti-rat Alexa Fluor 594 and goat anti-rabbit Alexa Fluor 488 (Thermo Fisher Scientific, both used at 1:500), respectively. For HeLa cells overexpressing human wild-type or mutant β-catenin plasmids, cells were seeded in eight-well glass slides (Millipore). After transfection, cells were fixed in 4% PFA at 37°C for 30 min and then washed sequentially with PBS, 0.2% PBST (Phosphate buffered saline with Tween-20), and PBS. Blocking and staining proceeded as described above. Slides were mounted with DAPI (4′,6-diamidino-2-phenylindole) Fluoromount-G mounting media (Southern Biotech) and imaged by fluorescence microscopy (Olympus). Cell counting was achieved using the ImageJ software.
RNA isolation and real-time PCR
Total RNA was extracted from cells or tissues using TRIzol reagent (Life technologies). One microgram of RNA was transcribed to complementary DNA with the Reverse Transcription System (Promega) and diluted with ddH2O 10 times. Real-time PCR was carried out on the LC480 system using SYBR Green Supermix (Takara). Data were normalized to 36B4 and analyzed using the ΔΔCt method.
Western blot
Proteins from iWAT or HEK293T cells were isolated with cold radioimmunoprecipitation assay buffer, followed by concentration quantification with the BCA Protein Assay kit (Thermo Fisher Scientific) and immunoblot with p-Akt Ser473 (Cell Signaling Technology, #4060, 1:1,000), total-Akt antibodies (Cell Signaling Technology, #9272, 1:1,000), and β-catenin (Cell Signaling Technology, #9562, 1:1,000). β-Actin (Santa Cruz Biotechnologies, #sc-47778, 1:1,000) or Hsp90 (Cell Signaling Technology, #4877, 1:1,000) was used as the internal control. The membranes were visualized using LAS 4000 (Life Science) according to the manufacturer’s instructions.
Cell culture
Murine preadipocyte 3T3-L1 cell line and murine macrophage RAW264.7 cell line were purchased from the American Type Culture Collection. 3T3-L1 preadipocytes were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Raw264.7 cells were cultured in RPMI 1640 supplemented with 10% FBS.
SVF isolation, cell sorting, and white adipocyte differentiation
SVFs were isolated as described previously (24). In brief, adipose tissue was minced and digested with collagenase type II (2 mg/ml; Sigma-Aldrich) in Hepes (Invitrogen) supplemented with 1% bovine serum albumin for 30 min at 37°C, followed by quenching with complete medium. Cell suspensions were centrifuged, washed, and filtered through a 40-μm strainer (BD Biosciences). For fluorescence-activated cell sorting (FACS) analysis, SVFs were resuspended in red blood cell lysis buffer (BD Biosciences) for 15 min at room temperature and then further incubated with anti-mouse CD140a-PE (BioLegend, #135906), anti-mouse CD140a-APC (BD Pharmingen, #562777), anti-mouse CD45-FITC (BD Pharmingen, #561874), anti-mouse CD45-APC/Cy7 (BD Pharmingen, #560694), anti-mouse CD31-PE (eBioscience, #12-0311-81), anti-mouse CD26-PerCP/Cyanine5.5 (eBioscience, #45-0261-82), anti-mouse CD54-BV510 (BD Pharmingen, #563628), anti-mouse CD142 (Sino Biological, #50413-R001), anti-mouse F4/80-PerCP-Cyanine5.5 (eBioscience, #45-4801-82), and anti-mouse CD11b-AF700 (BD Pharmingen, #557960) and subjected to FACS (BD Biosciences). SVFs were cultured in DMEM supplemented with 10% FBS and murine basic fibroblast growth factor (10 ng/ml; R&D Systems). White adipocyte differentiation was conducted in a cocktail medium containing insulin (5 μg/ml), 0.5 mM isobutylmethylxanthine (Sigma-Aldrich), and 1 μM dexamethasone (Sigma-Aldrich).
Plasmids and luciferase reporter assay
Plasmids encoding green fluorescent protein (GFP)–β-catenin and TOP-Flash used in this study were described previously (45). Saa3 promoter (−400 to 590 from transcriptional start site) was amplified from mouse genomic DNA and inserted into the pGL4-basic vector (Promega). Various mutations were further introduced using the QuikChange II site-directed mutagenesis kit (Stratagene). Plasmids encoding human wild-type or mutant CTNNB1/β-catenin gene were constructed using pcDNA3.1(+)-IRES:EGFP vector. All constructs were verified by DNA sequencing. For the luciferase reporter assay, 3T3-L1 preadipocytes were transfected with the indicated Saa3 promoter reporter constructs, pRL-TK (expressing Renilla luciferase), and pEGFP–β-catenin or vector for 48 hours, followed by luciferase activity measurement using a dual-luciferase reporter assay system (Promega).
ChIP assay
ChIP assay was performed to detect the interaction of Tcf4 or β-catenin with the Saa3 promoter using a commercial kit (Millipore, #17-371) according to the manufacturer’s instructions. In brief, SVFs were differentiated toward white adipocytes for 8 days, and then cell lysates were incubated with rabbit anti-Tcf4 antibody (Cell Signaling Technology, #2569, 1:400) or anti–β-catenin (Cell Signaling Technology, #9562, 1:400) or rabbit immunoglobulin G (IgG). The precipitated DNA fragments were further analyzed by PCR with the following mouse Saa3 primers: primer1, 5-CCTGTTCCCTAAGGCTCCAG-3 (forward) and 5-GTTAGGACTTTCTGAAGTCT-3 (reverse); primer2, 5-TTTCCAACCAGGATGGCGAA-3 (forward) and 5-CGAGCTCCTTCTGGGACC-3 (reverse); and primer3, 5-GGTCCCAGAAGGAGCTCGCA-3 (forward) and 5-AGCACACTACAAGTTCCATG-3 (reverse).
Cell proliferation
Raw264.7 cells were seeded in a 12-well plate and then treated with recombinant Saa3 at different doses for 6 hours. The medium was then replaced with DMEM containing 2.5% FBS for another 24 hours, and the conditioned medium of Raw264.7 cells was collected, mixed with fresh DMEM containing 7.5% FBS to acquire medium with 5% FBS, and then added to 3T3-L1 preadipocytes. We refreshed the medium every 24 hours. MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide] assay was further performed to measure cell viability and growth rate of 3T3-L1 preadipocytes at 72, 96, and 120 hours after treatment with the conditioned medium or the following recombinant proteins: mouse Mcp-1 (R&D, #479-JE-010), mouse Cxcl2 (R&D, #452-M2-010), mouse Cxcl10 (R&D, #466-CR-010), mouse Cxcl12 (R&D, #460-SD-010), mouse osteopontin (R&D, #441-OP-050), and human PDGF-AA (R&D, #221-AA-010). For some experiments, cell number was assessed by cell counting with a hemocytometer. The protein levels in the conditioned medium were measured with a commercial kit (R&D, #LXSAMSM) according to the manufacturer’s protocols.
Microarray
RNA used for microarray was extracted using TRIzol Reagent (Life Technologies) following the manufacturer’s instructions and checked for a RNA integrity number (RIN) number to inspect RNA integrity by an Agilent Bioanalyzer 2100 (Agilent technologies). Qualified total RNA was further purified by RNeasy micro kit (QIAGEN) and RNase-Free DNase Set (QIAGEN). Total RNA was amplified, labeled, and purified by using GeneChip 3’IVT Express kit (Affymetrix) according to the manufacturer’s instructions. The biotin-labeled cRNA was then hybridized and washed by using GeneChip Hybridization, Wash and Stain Kit (Affymetrix) in Hybridization Oven 645 (Affymetrix) and Fluidics Station 450 (Affymetrix) following the manufacturer’s instructions. Hybridized slides were scanned by GeneChip Scanner 3000 (Affymetrix) and Command Console Software 3.1 (Affymetrix) with default settings. Raw data were normalized by the MAS 5.0 algorithm, Gene Spring Software 11.0 (Agilent technologies). Differential genes shown in the heatmap were visualized by using R packed “Pheatmap” under two selection criteria including (i) a fold change larger than 1.5 and (ii) the corresponding P values less than 0.05.
Statistical analysis
Data were shown as mean ± SEM and analyzed by Student’s t test for comparison between the two groups. In human genetic study, for continuous variables, the Shapiro-Wilk test was performed for normal distribution examination. Differences in categorical variables such as the gene-based frequency distribution of CTNNB1 mutations were calculated using the Fisher’s exact test. General linear regression analysis was performed to examine the difference in clinical indicators between CTNNB1 mutation carriers and noncarriers with adjustment for age and sex, and logistic transformation was applied where appropriate. Energy expenditure, O2 consumption, and CO2 production were corrected for lean mass. Energy expenditure was also analyzed using analysis of covariance (ANCOVA) with body weight as a covariate according to previous report (46). A P value less than 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank K. Kristiansen from the University of Copenhagen, T. Hansen from the University of Southern Denmark, and M. Prentki from the Université de Montréal for their valuable comments and help during the draft preparation. Funding: This work was supported by grants from National Key Research and Development Program of China (2018YFC1313801 and 2018YFC1313802), the National Natural Science Foundation of China (81522011, 81822009, 81730023, 81570758, 81570757, 81621061, 81870585, 81670799, and 61725302), 973 Foundation (2015CB553600), Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (20161306 and 20171903), Shanghai Rising-Star Program (17QA1403300), and Shanghai Municipal Commission of Health and Family Planning (2017YQ002). Author contributions: J.W., R.L., and G.N. designed the experiments and supervised the study. M.C., P.L., Q.M., and N.C. carried out the animal and molecular experiments. B.C. performed the FACS analysis. H.S. developed the “AdipoCount” software. J.W., W.L., J.She., and Q.L. analyzed the cohort data. Y.C., S.Z., Y.S., L.S., J.Shi, J.H., W.G., Y.Z., and W.W. provided the cohort resources. M.C., P.L., R.L., and J.W. wrote the original draft. G.N. and W.W. reviewed and edited the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/2/eaax9605/DC1
Fig. S1. Common noncoding variants in and around CTNNB1 show significant association signals with BMI in GIANT UK Biobank study.
Fig. S2. Genetic mutations in the CTNNB1 gene in young obese and control subjects.
Fig. S3. Phenotypes of APBKO mice.
Fig. S4. Metabolic parameters in ABKO mice fed normal chow diet at young and old ages.
Fig. S5. Food intake, fecal calorie excretion, and energy expenditure in ABKO mice fed HFD.
Fig. S6. The expression changes of genes involved in adipogenesis and the percentage changes of progenitors in adipose tissues of ABKO mice fed HFD.
Fig. S7. Cell viability of Saa3-treated preadipocytes and the expression of secreted factors in Saa3-treated macrophages.
Table S1. The BMI association signals of common variants in and around CTNNB1 from GIANT UK Biobank GWAS.
Table S2. Rare missense variants in the CTNNB1 gene in young, severely obese cases and controls.
Table S3. The clinical parameters related to obesity in gain-of-function CTNNB1 carriers.
REFERENCES AND NOTES
- 1.Xu Y., Wang L., He J., Bi Y., Li M., Wang T., Wang L., Jiang Y., Dai M., Lu J., Xu M., Li Y., Hu N., Li J., Mi S., Chen C.-S., Li G., Mu Y., Zhao J., Kong L., Chen J., Lai S., Wang W., Zhao W., Ning G.; China Noncommunicable Disease Surveillance Group , Prevalence and control of diabetes in Chinese adults. JAMA 310, 948–959 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Heymsfield S. B., Wadden T. A., Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 376, 254–266 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Sun K., Kusminski C. M., Scherer P. E., Adipose tissue remodeling and obesity. J. Clin. Invest. 121, 2094–2101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jo J., Gavrilova O., Pack S., Jou W., Mullen S., Sumner A. E., Cushman S. W., Periwal V., Hypertrophy and/or hyperplasia: Dynamics of adipose tissue growth. PLOS Comput. Biol. 5, e1000324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xue H., Wang C., Li Y., Chen J., Yu L., Liu X., Li J., Cao J., Deng Y., Guo D., Yang X., Huang J., Gu D., Incidence of type 2 diabetes and number of events attributable to abdominal obesity in China: A cohort study. J. Diabetes 8, 190–198 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Boutens L., Stienstra R., Adipose tissue macrophages: Going off track during obesity. Diabetologia 59, 879–894 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodeheffer M. S., Birsoy K., Friedman J. M., Identification of white adipocyte progenitor cells in vivo. Cell 135, 240–249 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Lee Y.-H., Petkova A. P., Granneman J. G., Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 18, 355–367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berry R., Rodeheffer M. S., Characterization of the adipocyte cellular lineage in vivo. Nat. Cell Biol. 15, 302–308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merrick D., Sakers A., Irgebay Z., Okada C., Calvert C., Morley M. P., Percec I., Seale P., Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 364, eaav2501 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wernstedt Asterholm I., Tao C., Morley T. S., Wang Q. A., Delgado-Lopez F., Wang Z. V., Scherer P. E., Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 20, 103–118 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molgat A. S. D., Gagnon A., Sorisky A., Preadipocyte apoptosis is prevented by macrophage-conditioned medium in a PDGF-dependent manner. Am. J. Phys. Cell Phys. 296, C757–C765 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Wang Q. A., Tao C., Gupta R. K., Scherer P. E., Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 19, 1338–1344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spalding K. L., Arner E., Westermark P. O., Bernard S., Buchholz B. A., Bergmann O., Blomqvist L., Hoffstedt J., Näslund E., Britton T., Concha H., Hassan M., Rydén M., Frisén J., Arner P., Dynamics of fat cell turnover in humans. Nature 453, 783–787 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Jeffery E., Church C. D., Holtrup B., Colman L., Rodeheffer M. S., Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat. Cell Biol. 17, 376–385 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gesta S., Blüher M., Yamamoto Y., Norris A. W., Berndt J., Kralisch S., Boucher J., Lewis C., Kahn C. R., Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc. Natl. Acad. Sci. U.S.A. 103, 6676–6681 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loh N. Y., Neville M. J., Marinou K., Hardcastle S. A., Fielding B. A., Duncan E. L., Mc Carthy M. I., Tobias J. H., Gregson C. L., Karpe F., Christodoulides C., LRP5 regulates human body fat distribution by modulating adipose progenitor biology in a dose- and depot-specific fashion. Cell Metab. 21, 262–272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou Y., Ning T., Shi J., Chen M., Ding L., Huang Y., Kauderer S., Xu M., Cui B., Bi Y., Liu S., Hong J., Liu R., Ning G., Wang J., Association of a gain-of-function variant in LGR4 with central obesity. Obesity 25, 252–260 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Heid I. M., Jackson A. U., Randall J. C., Winkler T. W., Qi L., Steinthorsdottir V., Thorleifsson G., Zillikens M. C., Speliotes E. K., Mägi R., Workalemahu T., White C. C., Bouatia-Naji N., Harris T. B., Berndt S. I., Ingelsson E., Willer C. J., Weedon M. N., Luan J., Vedantam S., Esko T., Kilpeläinen T. O., Kutalik Z., Li S., Monda K. L., Dixon A. L., Holmes C. C., Kaplan L. M., Liang L., Min J. L., Moffatt M. F., Molony C., Nicholson G., Schadt E. E., Zondervan K. T., Feitosa M. F., Ferreira T., Allen H. L., Weyant R. J., Wheeler E., Wood A. R., MAGIC, Estrada K., Goddard M. E., Lettre G., Mangino M., Nyholt D. R., Purcell S., Smith A. V., Visscher P. M., Yang J., McCarroll S. A., Nemesh J., Voight B. F., Absher D., Amin N., Aspelund T., Coin L., Glazer N. L., Hayward C., Heard-Costa N. L., Hottenga J.-J., Johansson Å., Johnson T., Kaakinen M., Kapur K., Ketkar S., Knowles J. W., Kraft P., Kraja A. T., Lamina C., Leitzmann M. F., Knight B. M., Morris A. P., Ong K. K., Perry J. R. B., Peters M. J., Polasek O., Prokopenko I., Rayner N. W., Ripatti S., Rivadeneira F., Robertson N. R., Sanna S., Sovio U., Surakka I., Teumer A., van Wingerden S., Vitart V., Zhao J. H., Cavalcanti-Proença C., Chines P. S., Fisher E., Kulzer J. R., Lecoeur C., Narisu N., Sandholt C., Scott L. J., Silander K., Stark K., Tammesoo M.-L., Teslovich T. M., Timpson N. J., Watanabe R. M., Welch R., Chasman D. I., Cooper M. N., Jansson J.-O., Kettunen J., Lawrence R. W., Pellikka N., Perola M., Vandenput L., Alavere H., Almgren P., Atwood L. D., Bennett A. J., Biffar R., Bonnycastle L. L., Bornstein S. R., Buchanan T. A., Campbell H., Day I. N. M., Dei M., Dörr M., Elliott P., Erdos M. R., Eriksson J. G., Freimer N. B., Fu M., Gaget S., Geus E. J. C., Gjesing A. P., Grallert H., Gräßler J., Groves C. J., Guiducci C., Hartikainen A.-L., Hassanali N., Havulinna A. S., Herzig K.-H., Hicks A. A., Hui J., Igl W., Jousilahti P., Jula A., Kajantie E., Kinnunen L., Kolcic I., Koskinen S., Kovacs P., Kroemer H. K., Krzelj V., Kuusisto J., Kvaloy K., Laitinen J., Lantieri O., Lathrop G. M., Lokki M.-L., Luben R. N., Ludwig B., McArdle W. L., Carthy A. M., Morken M. A., Nelis M., Neville M. J., Paré G., Parker A. N., Peden J. F., Pichler I., Pietiläinen K. H., Platou C. G. P., Pouta A., Ridderstråle M., Samani N. J., Saramies J., Sinisalo J., Smit J. H., Strawbridge R. J., Stringham H. M., Swift A. J., Teder-Laving M., Thomson B., Usala G., van Meurs J. B. J., van Ommen G.-J., Vatin V., Volpato C. B., Wallaschofski H., Walters G. B., Widen E., Wild S. H., Willemsen G., Witte D. R., Zgaga L., Zitting P., Beilby J. P., James A. L., Kähönen M., Lehtimäki T., Nieminen M. S., Ohlsson C., Palmer L. J., Raitakari O., Ridker P. M., Stumvoll M., Tönjes A., Viikari J., Balkau B., Ben-Shlomo Y., Bergman R. N., Boeing H., Smith G. D., Ebrahim S., Froguel P., Hansen T., Hengstenberg C., Hveem K., Isomaa B., Jørgensen T., Karpe F., Khaw K.-T., Laakso M., Lawlor D. A., Marre M., Meitinger T., Metspalu A., Midthjell K., Pedersen O., Salomaa V., Schwarz P. E. H., Tuomi T., Tuomilehto J., Valle T. T., Wareham N. J., Arnold A. M., Beckmann J. S., Bergmann S., Boerwinkle E., Boomsma D. I., Caulfield M. J., Collins F. S., Eiriksdottir G., Gudnason V., Gyllensten U., Hamsten A., Hattersley A. T., Hofman A., Hu F. B., Illig T., Iribarren C., Jarvelin M.-R., Kao W. H. L., Kaprio J., Launer L. J., Munroe P. B., Oostra B., Penninx B. W., Pramstaller P. P., Psaty B. M., Quertermous T., Rissanen A., Rudan I., Shuldiner A. R., Soranzo N., Spector T. D., Syvanen A.-C., Uda M., Uitterlinden A., Völzke H., Vollenweider P., Wilson J. F., Witteman J. C., Wright A. F., Abecasis G. R., Boehnke M., Borecki I. B., Deloukas P., Frayling T. M., Groop L. C., Haritunians T., Hunter D. J., Kaplan R. C., North K. E., O’Connell J. R., Peltonen L., Schlessinger D., Strachan D. P., Hirschhorn J. N., Assimes T. L., Wichmann H.-E., Thorsteinsdottir U., van Duijn C. M., Stefansson K., Cupples L. A., Loos R. J. F., Barroso I., McCarthy M. I., Fox C. S., Mohlke K. L., Lindgren C. M., Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat. Genet. 42, 949–960 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shungin D., Winkler T. W., Croteau-Chonka D. C., Ferreira T., Locke A. E., Mägi R., Strawbridge R. J., Pers T. H., Fischer K., Justice A. E., Workalemahu T., Wu J. M. W., Buchkovich M. L., Heard-Costa N. L., Roman T. S., Drong A. W., Song C., Gustafsson S., Day F. R., Esko T., Fall T., Kutalik Z., Luan J., Randall J. C., Scherag A., Vedantam S., Wood A. R., Chen J., Fehrmann R., Karjalainen J., Kahali B., Liu C.-T., Schmidt E. M., Absher D., Amin N., Anderson D., Beekman M., Bragg-Gresham J. L., Buyske S., Demirkan A., Ehret G. B., Feitosa M. F., Goel A., Jackson A. U., Johnson T., Kleber M. E., Kristiansson K., Mangino M., Leach I. M., Medina-Gomez C., Palmer C. D., Pasko D., Pechlivanis S., Peters M. J., Prokopenko I., Stančáková A., Sung Y. J., Tanaka T., Teumer A., Van Vliet-Ostaptchouk J. V., Yengo L., Zhang W., Albrecht E., Ärnlöv J., Arscott G. M., Bandinelli S., Barrett A., Bellis C., Bennett A. J., Berne C., Blüher M., Böhringer S., Bonnet F., Böttcher Y., Bruinenberg M., Carba D. B., Caspersen I. H., Clarke R., Daw E. W., Deelen J., Deelman E., Delgado G., Doney A. S. F., Eklund N., Erdos M. R., Estrada K., Eury E., Friedrich N., Garcia M. E., Giedraitis V., Gigante B., Go A. S., Golay A., Grallert H., Grammer T. B., Gräßler J., Grewal J., Groves C. J., Haller T., Hallmans G., Hartman C. A., Hassinen M., Hayward C., Heikkilä K., Herzig K.-H., Helmer Q., Hillege H. L., Holmen O., Hunt S. C., Isaacs A., Ittermann T., James A. L., Johansson I., Juliusdottir T., Kalafati I.-P., Kinnunen L., Koenig W., Kooner I. K., Kratzer W., Lamina C., Leander K., Lee N. R., Lichtner P., Lind L., Lindström J., Lobbens S., Lorentzon M., Mach F., Magnusson P. K. E., Mahajan A., McArdle W. L., Menni C., Merger S., Mihailov E., Milani L., Mills R., Moayyeri A., Monda K. L., Mooijaart S. P., Mühleisen T. W., Mulas A., Müller G., Müller-Nurasyid M., Nagaraja R., Nalls M. A., Narisu N., Glorioso N., Nolte I. M., Olden M., Rayner N. W., Renstrom F., Ried J. S., Robertson N. R., Rose L. M., Sanna S., Scharnagl H., Scholtens S., Sennblad B., Seufferlein T., Sitlani C. M., Smith A. V., Stirrups K., Stringham H. M., Sundström J., Swertz M. A., Swift A. J., Syvänen A.-C., Tayo B. O., Thorand B., Thorleifsson G., Tomaschitz A., Troffa C., van Oort F. V. A., Verweij N., Vonk J. M., Waite L. L., Wennauer R., Wilsgaard T., Wojczynski M. K., Wong A., Zhang Q., Zhao J. H., Brennan E. P., Choi M., Eriksson P., Folkersen L., Franco-Cereceda A., Gharavi A. G., Hedman Å. K., Hivert M.-F., Huang J., Kanoni S., Karpe F., Keildson S., Kiryluk K., Liang L., Lifton R. P., Ma B., Mc Knight A. J., Pherson R. M., Metspalu A., Min J. L., Moffatt M. F., Montgomery G. W., Murabito J. M., Nicholson G., Nyholt D. R., Olsson C., Perry J. R. B., Reinmaa E., Salem R. M., Sandholm N., Schadt E. E., Scott R. A., Stolk L., Vallejo E. E., Westra H.-J., Zondervan K. T.; The ADIPOGen Consortium; The CARDIOGRAMplus CD Consortium; The CKDGen Consortium; The GEFOS Consortium; The GENIE Consortium; GLGC; ICBP; The International Endogene Consortium; The Life Lines Cohort Study; The MAGIC Investigators; The Mu THER Consortium; The PAGE Consortium; The Repro Gen Consortium; Amouyel P., Arveiler D., Bakker S. J. L., Beilby J., Bergman R. N., Blangero J., Brown M. J., Burnier M., Campbell H., Chakravarti A., Chines P. S., Claudi-Boehm S., Collins F. S., Crawford D. C., Danesh J., de Faire U., de Geus E. J. C., Dörr M., Erbel R., Eriksson J. G., Farrall M., Ferrannini E., Ferrières J., Forouhi N. G., Forrester T., Franco O. H., Gansevoort R. T., Gieger C., Gudnason V., Haiman C. A., Harris T. B., Hattersley A. T., Heliövaara M., Hicks A. A., Hingorani A. D., Hoffmann W., Hofman A., Homuth G., Humphries S. E., Hyppönen E., Illig T., Jarvelin M.-R., Johansen B., Jousilahti P., Jula A. M., Kaprio J., Kee F., Keinanen-Kiukaanniemi S. M., Kooner J. S., Kooperberg C., Kovacs P., Kraja A. T., Kumari M., Kuulasmaa K., Kuusisto J., Lakka T. A., Langenberg C., Marchand L. L., Lehtimäki T., Lyssenko V., Männistö S., Marette A., Matise T. C., McKenzie C. A., Knight B. M., Musk A. W., Möhlenkamp S., Morris A. D., Nelis M., Ohlsson C., Oldehinkel A. J., Ong K. K., Palmer L. J., Penninx B. W., Peters A., Pramstaller P. P., Raitakari O. T., Rankinen T., Rao D. C., Rice T. K., Ridker P. M., Ritchie M. D., Rudan I., Salomaa V., Samani N. J., Saramies J., Sarzynski M. A., Schwarz P. E. H., Shuldiner A. R., Staessen J. A., Steinthorsdottir V., Stolk R. P., Strauch K., Tönjes A., Tremblay A., Tremoli E., Vohl M.-C., Völker U., Vollenweider P., Wilson J. F., Witteman J. C., Adair L. S., Bochud M., Boehm B. O., Bornstein S. R., Bouchard C., Cauchi S., Caulfield M. J., Chambers J. C., Chasman D. I., Cooper R. S., Dedoussis G., Ferrucci L., Froguel P., Grabe H.-J., Hamsten A., Hui J., Hveem K., Jöckel K.-H., Kivimaki M., Kuh D., Laakso M., Liu Y., März W., Munroe P. B., Njølstad I., Oostra B. A., Palmer C. N. A., Pedersen N. L., Perola M., Pérusse L., Peters U., Power C., Quertermous T., Rauramaa R., Rivadeneira F., Saaristo T. E., Saleheen D., Sinisalo J., Slagboom P. E., Snieder H., Spector T. D., Thorsteinsdottir U., Stumvoll M., Tuomilehto J., Uitterlinden A. G., Uusitupa M., van der Harst P., Veronesi G., Walker M., Wareham N. J., Watkins H., Wichmann H.-E., Abecasis G. R., Assimes T. L., Berndt S. I., Boehnke M., Borecki I. B., Deloukas P., Franke L., Frayling T. M., Groop L. C., Hunter D. J., Kaplan R. C., O’Connell J. R., Qi L., Schlessinger D., Strachan D. P., Stefansson K., van Duijn C. M., Willer C. J., Visscher P. M., Yang J., Hirschhorn J. N., Zillikens M. C., Mc Carthy M. I., Speliotes E. K., North K. E., Fox C. S., Barroso I., Franks P. W., Ingelsson E., Heid I. M., Loos R. J. F., Cupples L. A., Morris A. P., Lindgren C. M., Mohlke K. L., New genetic loci link adipose and insulin biology to body fat distribution. Nature 518, 187–196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross S. E., Hemati N., Longo K. A., Bennett C. N., Lucas P. C., Erickson R. L., MacDougald O. A., Inhibition of adipogenesis by Wnt signaling. Science 289, 950–953 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Zeve D., Seo J., Suh J. M., Stenesen D., Tang W., Berglund E. D., Wan Y., Williams L. J., Lim A., Martinez M. J., McKay R. M., Millay D. P., Olson E. N., Graff J. M., Wnt signaling activation in adipose progenitors promotes insulin-independent muscle glucose uptake. Cell Metab. 15, 492–504 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christodoulides C., Scarda A., Granzotto M., Milan G., Dalla Nora E., Keogh J., de Pergola G., Stirling H., Pannacciulli N., Sethi J. K., Federspil G., Vidal-Puig A., Farooqi I. S., O’Rahilly S., Vettor R., WNT10B mutations in human obesity. Diabetologia 49, 678–684 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J., Liu R., Wang F., Hong J., Li X., Chen M., Ke Y., Zhang X., Ma Q., Wang R., Shi J., Cui B., Gu W., Zhang Y., Zhang Z., Wang W., Xia X., Liu M., Ning G., Ablation of LGR4 promotes energy expenditure by driving white-to-brown fat switch. Nat. Cell Biol. 15, 1455–1463 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Yengo L., Sidorenko J., Kemper K. E., Zheng Z., Wood A. R., Weedon M. N., Frayling T. M., Hirschhorn J., Yang J., Visscher P. M.; GIANT Consortium , Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang H., Jin X., Li Y., Jiang H., Tang X., Yang X., Cheng H., Qiu Y., Chen G., Mei J., Zhou F., Wu R., Zuo X., Zhang Y., Zheng X., Cai Q., Yin X., Quan C., Shao H., Cui Y., Tian F., Zhao X., Liu H., Xiao F., Xu F., Han J., Shi D., Zhang A., Zhou C., Li Q., Fan X., Lin L., Tian H., Wang Z., Fu H., Wang F., Yang B., Huang S., Liang B., Xie X., Ren Y., Gu Q., Wen G., Sun Y., Wu X., Dang L., Xia M., Shan J., Li T., Yang L., Zhang X., Li Y., He C., Xu A., Wei L., Zhao X., Gao X., Xu J., Zhang F., Zhang J., Li Y., Sun L., Liu J., Chen R., Yang S., Wang J., Zhang X., A large-scale screen for coding variants predisposing to psoriasis. Nat. Genet. 46, 45–50 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Jiang X., Cao Y., Li F., Su Y., Li Y., Peng Y., Cheng Y., Zhang C., Wang W., Ning G., Targeting β-catenin signaling for therapeutic intervention in MEN1-deficient pancreatic neuroendocrine tumours. Nat. Commun. 5, 5809 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joe A. W. B., Yi L., Even Y., Vogl A. W., Rossi F. M. V., Depot-specific differences in adipogenic progenitor abundance and proliferative response to high-fat diet. Stem Cells 27, 2563–2570 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Han C. Y., Subramanian S., Chan C. K., Omer M., Chiba T., Wight T. N., Chait A., Adipocyte-derived serum amyloid A3 and hyaluronan play a role in monocyte recruitment and adhesion. Diabetes 56, 2260–2273 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Svensson K. J., Long J. Z., Jedrychowski M. P., Cohen P., Lo J. C., Serag S., Kir S., Shinoda K., Tartaglia J. A., Rao R. R., Chédotal A., Kajimura S., Gygi S. P., Spiegelman B. M., A secreted slit2 fragment regulates adipose tissue thermogenesis and metabolic function. Cell Metab. 23, 454–466 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giese K., Amsterdam A., Grosschedl R., DNA-binding properties of the HMG domain of the lymphoid-specific transcriptional regulator LEF-1. Genes Dev. 5, 2567–2578 (1991). [DOI] [PubMed] [Google Scholar]
- 32.Fasshauer M., Klein J., Kralisch S., Klier M., Lossner U., Bluher M., Paschke R., Serum amyloid A3 expression is stimulated by dexamethasone and interleukin-6 in 3T3-L1 adipocytes. J. Endocrinol. 183, 561–567 (2004). [DOI] [PubMed] [Google Scholar]
- 33.den Hartigh L. J., Wang S., Goodspeed L., Ding Y., Averill M., Subramanian S., Wietecha T., O'Brien K. D., Chait A., Deletion of serum amyloid A3 improves high fat high sucrose diet-induced adipose tissue inflammation and hyperlipidemia in female mice. PLOS ONE 9, e108564 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ai M., Heeger S., Bartels C. F., Schelling D. K.; Osteoporosis-Pseudoglioma Collaborative Group , Clinical and molecular findings in osteoporosis-pseudoglioma syndrome. Am. J. Hum. Genet. 77, 741–753 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mani A., Radhakrishnan J., Wang H., Mani A., Mani M. A., Nelson-Williams C., Carew K. S., Mane S., Najmabadi H., Wu D., Lifton R. P., LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 315, 1278–1282 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Wesenbeeck L., Cleiren E., Gram J., Beals R. K., Bénichou O., Scopelliti D., Key L., Renton T., Bartels C., Gong Y., Warman M. L., de Vernejoul M.-C., Bollerslev J., Van Hul W., Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am. J. Hum. Genet. 72, 763–771 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morin P. J., Sparks A. B., Korinek V., Barker N., Clevers H., Vogelstein B., Kinzler K. W., Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 275, 1787–1790 (1997). [DOI] [PubMed] [Google Scholar]
- 38.McKnight R. F., Adida M., Budge K., Stockton S., Goodwin G. M., Geddes J. R., Lithium toxicity profile: A systematic review and meta-analysis. Lancet 379, 721–728 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Ho R., Papp B., Hoffman J. A., Merrill B. J., Plath K., Stage-specific regulation of reprogramming to induced pluripotent stem cells by Wnt signaling and T cell factor proteins. Cell Rep. 3, 2113–2126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z. V., Deng Y., Wang Q. A., Sun K., Scherer P. E., Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology 151, 2933–2939 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hiratsuka S., Watanabe A., Sakurai Y., Akashi-Takamura S., Ishibashi S., Miyake K., Shibuya M., Akira S., Aburatani H., Maru Y., The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 10, 1349–1355 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Couturier J., Patel S. G., Iyer D., Balasubramanyam A., Lewis D. E., Human monocytes accelerate proliferation and blunt differentiation of preadipocytes in association with suppression of C/EBPα mRNA. Obesity (Silver Spring) 20, 253–262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu R., Zou Y., Hong J., Cao M., Cui B., Zhang H., Chen M., Shi J., Ning T., Zhao S., Liu W., Xiong H., Wei C., Qiu Z., Gu W., Zhang Y., Li W., Miao L., Sun Y., Yang M., Wang R., Ma Q., Xu M., Xu Y., Wang T., Chan K.-h. K., Zuo X., Chen H., Qi L., Lai S., Duan S., Song B., Bi Y., Liu S., Wang W., Ning G., Wang J., Rare loss-of-function variants in NPC1 predispose to human obesity. Diabetes 66, 935–947 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Liu R., Hong J., Xu X., Feng Q., Zhang D., Gu Y., Shi J., Zhao S., Liu W., Wang X., Xia H., Liu Z., Cui B., Liang P., Xi L., Jin J., Ying X., Wang X., Zhao X., Li W., Jia H., Lan Z., Li F., Wang R., Sun Y., Yang M., Shen Y., Jie Z., Li J., Chen X., Zhong H., Xie H., Zhang Y., Gu W., Deng X., Shen B., Xu X., Yang H., Xu G., Bi Y., Lai S., Wang J., Qi L., Madsen L., Wang J., Ning G., Kristiansen K., Wang W., Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 23, 859–868 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Cao Y., Liu R., Jiang X., Lu J., Jiang J., Zhang C., Li X., Ning G., Nuclear-cytoplasmic shuttling of menin regulates nuclear translocation of β-catenin. Mol. Cell. Biol. 29, 5477–5487 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mina A. I., LeClair R. A., LeClair K. B., Cohen D. E., Lantier L., Banks A. S., CalR: A web-based analysis tool for indirect calorimetry experiments. Cell Metab. 28, 656–666.e1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaser R., Adusumalli S., Leng S. N., Sikic M., Ng P. C., SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., Kondrashov A. S., Sunyaev S. R., A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/2/eaax9605/DC1
Fig. S1. Common noncoding variants in and around CTNNB1 show significant association signals with BMI in GIANT UK Biobank study.
Fig. S2. Genetic mutations in the CTNNB1 gene in young obese and control subjects.
Fig. S3. Phenotypes of APBKO mice.
Fig. S4. Metabolic parameters in ABKO mice fed normal chow diet at young and old ages.
Fig. S5. Food intake, fecal calorie excretion, and energy expenditure in ABKO mice fed HFD.
Fig. S6. The expression changes of genes involved in adipogenesis and the percentage changes of progenitors in adipose tissues of ABKO mice fed HFD.
Fig. S7. Cell viability of Saa3-treated preadipocytes and the expression of secreted factors in Saa3-treated macrophages.
Table S1. The BMI association signals of common variants in and around CTNNB1 from GIANT UK Biobank GWAS.
Table S2. Rare missense variants in the CTNNB1 gene in young, severely obese cases and controls.
Table S3. The clinical parameters related to obesity in gain-of-function CTNNB1 carriers.