

Summary
The remarkable cellular and genetic heterogeneity of soft tissue sarcomas (STSs) limits the clinical benefit of targeted therapies. Here, we show that expression of the gluconeogenic isozyme fructose-1,6-bisphosphatase 2 (FBP2) is silenced in a broad spectrum of sarcoma subtypes, revealing an apparent common metabolic feature shared by diverse STSs. Enforced FBP2 expression inhibits sarcoma cell and tumor growth through two distinct mechanisms. First, cytosolic FBP2 antagonizes elevated glycolysis associated with the “Warburg effect”, thereby inhibiting sarcoma cell proliferation. Second, nuclear-localized FBP2 restrains mitochondrial biogenesis and respiration in a catalytic activity-independent manner by inhibiting the expression of nuclear respiratory factor and mitochondrial transcription factor A (TFAM). Specifically, nuclear FBP2 colocalizes with the c-Myc transcription factor at the TFAM locus and represses c-Myc-dependent TFAM expression. This unique dual function of FBP2 provides a rationale for its selective suppression in STSs, identifying a potential metabolic vulnerability of this malignancy and possible therapeutic target.
Keywords: fructose-1, 6-bisphosphatase 2; sarcoma progression; glycolysis; mitochondrial function; Myc
Graphical Abstract
eTOC blurb
Huangyang et al. demonstrate that the loss of fructose-1,6-bisphosphatase 2 (FBP2) is a common metabolic feature of soft tissue sarcomas (STSs). Restoration of FBP2 in STS cells suppresses sarcoma growth through two mechanisms, including inhibiting glycolysis and restraining mitochondrial biogenesis by inhibiting c-Myc-driven transcriptional activity.
Introduction
Soft tissue sarcomas (STSs) encompass a diverse group of mesenchymal tumors arising from connective tissues, such as muscle, fat and cartilage. Each year, approximately 13,000 new cases are diagnosed in the United States, and 5,000 patients succumb to this disease (Siegel et al., 2019). Collectively, STSs are classified into more than 70 subtypes based on pathological and clinical features, ranging from indolent to highly invasive and metastatic (Cancer Genome Atlas Research Network, 2017; Fletcher, 2014). Liposarcoma, undifferentiated pleomorphic sarcoma (UPS), and fibrosarcoma represent 40% of newly diagnosed sarcomas in adults (Lehnhardt et al., 2009). Although recent studies have integrated genome-scale analyses of the molecular mechanisms underlying sarcomagenesis and progression (Cancer Genome Atlas Research Network, 2017; Taylor et al., 2011), these cancers remain understudied due to their extensive heterogeneity. Current treatment options are limited to standard surgical resection, radiotherapy and chemotherapy (Mehren et al., 2018); however, response rates to cytotoxic chemotherapy are only 10–25% (Linch et al., 2014). Highly divergent genomic alterations and low response rates to traditional treatments necessitate development of effective therapies that exploit common features of sarcoma progression.
Various oncogenic signaling pathways and microenvironmental stresses converge to modify cellular metabolism, adapting it to limited nutrient and oxygen availability (Vander Heiden and DeBerardinis, 2017). A broad array of oncogenes and tumor suppressors that regulate metabolic pathways are mutated in sarcomas, such as PIK3CA (catalytic subunit of phosphatidylinositol 3-kinase), TP53, and NF1 (Barretina et al., 2010; Cancer Genome Atlas Research Network, 2017). In addition to effects imposed by genetic mutations, hypoxic (O2-deprived) tumor microenvironments characteristic of STS alter metabolism and are associated with worse prognosis (Brizel et al., 1996; Sadri and Zhang, 2013). While reprogrammed metabolic activities likely promote sarcoma growth and progression, they also create unique vulnerabilities and therefore new opportunities for therapeutic intervention.
Previously, labeled isotope infusion of individuals with sarcoma revealed elevated tissue glucose uptake and turnover, accompanied by decreased glucose oxidation (Shaw et al., 1988), suggesting abnormal glucose metabolism in these tumors. Glycolysis is counterbalanced by anabolic gluconeogenesis to maintain glucose homeostasis, and gluconeogenic enzymes play important roles in regulating tumor cell growth and behavior (Wang and Dong, 2019). Fructose-1,6-bisphosphatase (FBP) is a rate-limiting enzyme that catalyzes the irreversible hydrolysis of fructose-1,6-bisphosphate to fructose-6-phosphate and inorganic phosphate. Vertebrates possess two highly conserved FBP isozymes exhibiting 76.6% sequence identity: FBP1 is detected primarily in liver and kidney, whereas FBP2 expression is more ubiquitous although highest in skeletal muscle and other mesenchymal tissues. Recently, FBP1 loss has been found to contribute to the progression of multiple epithelial tumors, including clear cell renal cell carcinoma (ccRCC), breast cancer, hepatocellular carcinoma and pancreatic ductal adenocarcinoma (Dong et al., 2013; Hirata et al., 2016; Li et al., 2014; Zhu et al., 2015). Several mechanisms are implicated in downregulating FBP1, including transcription factor repression (Zhu et al., 2015), epigenetic silencing (Bigl et al., 2008; Chen et al., 2011; Yang et al., 2017) and proteasome degradation (Jin et al., 2017).
Restoration of FBP1 expression in breast cancer and ccRCC cells strongly antagonizes glycolysis through its catalytic activity (Dong et al., 2013; Li et al., 2014); however, FBP1 also regulates genes in ccRCC cells through an unanticipated nuclear function. Specifically, we demonstrated that FBP1 directly suppresses the transcriptional activity of hypoxia-inducible factors (HIFs) through an enzymatic activity-independent mechanism (Li et al., 2014). HIFs regulate several hundred genes, including those encoding the glycolytic enzymes GLUT1, HK2, PFK1 and LDHA, to facilitate cellular adaptation to hypoxia (Nakazawa et al., 2016b); thus, FBP1 loss further enhances HIF responses.
Whereas FBP1 has been studied in a variety of carcinomas, little is known about the role of FBP2 in mesenchymal cells or STS. We demonstrate here that FBP2 transcription is markedly silenced in the majority of STS subtypes, and that restoring FBP2 expression dramatically inhibits sarcoma cell proliferation in vitro and tumor growth in vivo, implicating FBP2 loss as an important general event during sarcoma progression. Isotope tracing and unbiased mass spectrometry analyses of liposarcoma, fibrosarcoma and UPS cells demonstrated that glycolysis and TCA cycle activity are inhibited by FBP2 restoration. Additionally, RNA-seq analysis indicates that mitochondrial function and oxidative phosphorylation (OXPHOS) gene signatures are significantly repressed by FBP2. Both FBP1 and FBP2 isozymes share a consensus nuclear localization sequence (NLS) (Gizak et al., 2009a; 2009b), and a nucleus-excluded FBP2 mutant has no effect on mitochondrial biogenesis and OXPHOS, indicating that nuclear FBP2 regulates mitochondrial function independent of its enzymatic activity. Finally, we determined that FBP2 co-localizes with c-Myc at the promoter region of TFAM, which encodes a master regulator of mitochondrial biogenesis, and inhibits c-Myc-mediated TFAM expression.
Results
FBP2 is Frequently Lost in STS
To assess their potential role in regulating sarcoma cell glucose metabolism, we measured the expression levels of three rate-limiting gluconeogenic enzymes, glucose-6-phosphatase catalytic subunit (G6PC), phosphoenolpyruvate carboxykinase 1 (PCK1), and fructose-1,6-bisphosphatase (FBP), using published microarray data from human STS (Barretina et al., 2010; Detwiller et al., 2005). As previously reported (Mizunuma and Tashima, 1990; Tejwani, 1983), FBP1 is the predominant isozyme in liver, kidney and small intestine (Figure S1A), whereas FBP2 expression predominates in muscle (Figure S1B). Further analysis revealed that FBP2 mRNA levels are significantly reduced in liposarcoma, fibrosarcoma, leiomyosarcoma, and UPS samples relative to either normal skeletal muscle (Figure 1A) or adipose tissue (Figure 1B). Moreover, mouse UPS tumors isolated from a previously described autochthonous “KP” (LSL-KrasG12D/+; Trp53fl/fl) model (Kirsch et al., 2007; Mito et al., 2009) exhibited decreased FBP2 immunostaining relative to adjacent skeletal muscle tissue (Figure 1C). Analysis of a representative human sarcoma tissue array showed undetectable FBP2 protein accumulation, compared to normal mesenchymal tissue, in almost 100% of sarcoma tumors examined (n = 70, Figure 1D; Figure S1C–D). FBP2 protein abundance was also decreased in a variety of human sarcoma and mouse UPS cell lines, relative to mesenchymal stem cell (MSC) and human skeletal muscle myoblast (HSMM) or mouse C2C12 myoblasts (Figure 1E). Even though FBP2 expression is widely repressed in STS, intertumoral heterogeneity still results in different levels of repression among sarcoma samples, and a liposarcoma data set (Gobble et al., 2011) revealed that lower FBP2 mRNA levels correlated with worse overall survival in liposarcoma patients (Figure 1F). We concluded that FBP2 loss is a common feature in STS subtypes and may contribute directly to tumorigenesis.
Figure 1. FBP2 is severely downregulated in a variety of sarcomas. Also see Figure S1 and S2.

(A and B) FBP2 mRNA expression from Oncomine analysis of the (A) Detwiller et al. and (B) Barretina et al. sarcoma patient samples data set (Barretina et al., 2010; Detwiller et al., 2005). Values are normalized to median-centered intensity and shown on a log2 scale. Abbreviations: dediff. lipo., dedifferentiated liposarcoma; MFH, malignant fibrous histiocytoma; MFH/Pleo., UPS; pleomorphic lipo., pleomorphic liposarcoma; RC lipo., round cell liposarcoma. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n.s., not significant.
(C) Representative immunohistochemical (IHC) FBP2 staining in mouse UPS tumor and surrounding muscle. Scale bars: 100 μm.
(D) Representative IHC FBP2 staining in human muscle and sarcoma samples from human sarcoma tissue array, including fibrous histiocytoma, liposarcoma, neurilemmoma, and leiomyosarcoma. Scale bars: 100 μm.
(E) Immunoblot analysis of FBP2 protein level in various human (left) and mouse (right) sarcoma cell lines. MSC, HSMM and C2C12 served as normal control. MSC, mesenchymal stem cell; HSMM, human skeletal muscle myoblast; (N), normoxia; (H) hypoxia; D-, differentiated; LMS, leiomyosarcoma; RD, rhabdomyosarcoma; FS, fibrosarcoma. GAPDH served as loading control.
(F) Kaplan–Meier curve of overall survival of liposarcoma patients from the Gobble et al. data set (Gobble et al., 2011), segregated into the top 33% FBP2 expression (High FBP2, n = 32), middle 33% FBP2 expression (Medium FBP2, n = 32), and bottom FBP2 expression (Low FBP2, n = 32). The p-value was calculated using a log-rank (Mantel–Cox) test.
In contrast to FBP2, G6PC and PCK1 mRNA levels were similar in liposarcoma, fibrosarcoma, leiomyosarcoma, and other sarcoma subtypes compared with normal skeletal muscle (Figure S2A–C), and liposarcoma cell lines exhibited PCK1 and G6PC protein abundance comparable to control MSCs (Figure S2D). FBP1 was undetectable in tumors and sarcoma cell lines of diverse histological subtypes or controls, including MSC and human skeletal muscle myoblast (HSMM) (Figure S2E–F).
FBP2 Re-Expression Suppresses Sarcoma Growth
To investigate its functional effects in different STS subtypes, we expressed FBP2 ectopically in human liposarcoma (LPS246, T1000, SW872), fibrosarcoma (HT1080), and mouse UPS (KP250) cell lines to levels observed in control HSMM or C2C12 cells, respectively (Figure S3A–B). FBP2 significantly inhibited cell proliferation under either low serum (1% serum, 25 mM glucose) or low glucose (10% serum, 5 mM glucose) conditions (Figures 2A, S3C), reminiscent of nutrient starved tumor microenvironments, as well as replete culture conditions (Figure S3D). In addition, FBP2 re-expression in SW872, HT1080 and KP250 cells dramatically impaired anchorage-independent growth in 3D soft agar colony assays (Figure 2B). We also exploited a doxycycline (dox)-inducible system to restore FBP2 expression in LPS246 cells (LPS246 TetO-FBP2) and identified dox concentrations that produced FBP2 protein in LPS246 TetO-FBP2 cells comparable to control HSMM cells (Figure S3E). To further evaluate the role of FBP2 in tumor growth and maintenance in vivo, highly immunodeficient NSG mice were injected subcutaneously with LPS246 TetO-FBP2 cells and tumors allowed to grow to 100 mm3. Subsequent dox-induced FBP2 expression decreased tumor growth (volume and mass) without affecting mouse body weight (Figure 2C–E, Figure S3F). The FBP2-expressing tumor cohorts exhibited decreased tumor cell proliferation as indicated by decreased phospho-histone H3 staining (Figure 2F). FBP2 expression was maintained during dox treatment as shown by western blot (Figure 2G) and immunohistochemistry (IHC) (Figure S3G). These results demonstrate that FBP2 restoration inhibits the in vitro proliferation and in vivo tumor growth of multiple STS cell types, including UPS, fibrosarcoma and liposarcoma.
Figure 2. FBP2 re-expression inhibits sarcoma cell proliferation and xenograft tumor growth. Also see Figure S3.

(A) Growth of LPS246, T1000, SW872, and KP250 cells in low serum medium (1% FBS), with or without ectopic FBP2 expression. n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
(B) Soft agar colony formation assay of SW872, HT1080, and KP250 cells with or without FBP2 expression. Representative pictures of colony size (left) and quantification of colony number (right). Error bars represent SD of three replicate wells for each condition. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
(C) Tumor volume of LPS246 TetO-FBP2 liposarcoma xenografts with or without dox-induced FBP2 expression (each group includes ten tumors from five mice). ****p < 0.0001.
(D and E) LPS246 xenograft tumor weight (D) and body weight (E) with or without dox treatment at time of euthanasia, measured in grams. ****p < 0.0001, n.s., not significant.
(F) Representative image (left) of immunofluorescent staining of phosphor-Histone 3 and quantifications (right) in xenograft tumors with or without dox treatment. Five fields per slide were quantified. Scale bars: 100 μm.
(G) FBP2 induction in xenograft tumors were validated by western blot. GAPDH served as loading control.
FBP2 Re-Expression Inhibits Glycolysis
Glucose metabolism is balanced by catabolic glycolysis/oxidative phosphorylation (OXPHOS) and anabolic gluconeogenesis/glycogen production. Since FBP2 is a rate-limiting enzyme in gluconeogenesis, FBP2 re-expression is likely to antagonize glycolysis and therefore reduce glucose uptake. We ectopically expressed FBP2 in five STS cell lines and examined glucose metabolism. FBP2 significantly decreased glucose uptake and lactate secretion without affecting glutamine uptake in LPS224, LPS246, T1000, HT1080, and KP250 cells cultured in 10 mM glucose (Figure 3A–C). To further assess glycolytic pathway, we investigated the metabolic fate of [1,2-13C]glucose, which produces glycolytic and TCA cycle intermediates containing two 13C atoms, as well as intermediates containing one 13C atom from the pentose phosphate pathway (PPP) (Figure 3D). Analysis of culture media confirmed reduced [1,2-13C]glucose uptake and M1- and M2-labeled 13C-lactate secretion in FBP2-expressing LPS246 cells (Figure 3E–F), indicating decreased conversion of glucose to lactate. [1,2-13C]glucose releases its first carbon in the form of CO2 to generate M1 species when catabolized through the oxidative portion of PPP (Lee et al., 1998); therefore, PPP flux can be determined by the ratio of M1- to M2-labeled lactate. Interestingly, we found that FBP2 re-expression reduced PPP flux (Figure 3G), which is important for the synthesis of ribonucleotides and NADPH. The same cell extracts were further analyzed by GC-MS to quantify M2 enrichment of glucose-derived TCA intermediates (citrate, α-ketoglutarate, fumarate, and malate), as well as TCA cycle products (aspartate and glutamate). We observed decreased M2 enrichment of four TCA intermediates (Figure 3H), as well as glutamate and aspartate (Figure 3I) in FBP2-expressing cells.
Figure 3. FBP2 restoration opposes glycolysis. Also see Figure S4.

(A–C) Glucose uptake (A), lactate secretion (B), and glutamine uptake (C) were assessed in LPS224, LPS246, T1000, HT1080, and KP250 cells with or without FBP2 re-expression by YSI bioanalyzer.
(D) Carbon fate map showing the isotopomer distribution of indicated metabolites derived from [1,2-13C]glucose. 13C atoms are depicted as filled circles. 13C atoms directly going through the glycolytic pathway are colored in red, while 13C atoms going through the PPP and recycled back to glycolysis are colored in blue.
(E and F) [1,2-13C]glucose consumption (E), M1 and M2 isotopomer distribution of lactate (F) measured from culture medium of LPS246 cells expressing vector control or FBP2.
(G) Calculated PPP flux (relative to vector control) in LPS246 cells with or without FBP2 expression based on the M1 to M2 13C-lactate ratio in cell extracts.
(H and I) M2 isotopomer distribution of indicated TCA metabolites (H) and amino acids (I) in LPS246 cells with or without FBP2 re-expression, labelled with [1,2-13C]glucose. M2 enrichment represents the mole per cent excess of M2 species above natural abundance. αKG, α-ketoglutarate.
Values represent mean ± SD of three experimental replicates. *p < 0.05, **p < 0.01, ***p < 0.001.
In addition, we employed an unbiased approach to determine relative abundance of intermediates from glycolysis, serine metabolism and the TCA cycle in KP250 and HT1080 cells. In both cell lines, ectopic FBP2 expression significantly reduced steady-state abundance of metabolites in glycolysis (glucose-6-phosphate, pyruvate, lactate) (Figure S4A), serine metabolism (serine and glycine) (Figure S4B) and the TCA cycle (citrate, α-ketoglutarate, fumarate, malate, oxaloacetate) (Figure S4C). In contrast to these decreased metabolic intermediates in FBP2-expressing cells, we observed enrichment of metabolites in other pathways (Figure S4D), indicating a global metabolic adaptation in FBP2-expressing cells. Together, these results indicate that FBP2 inhibits glycolysis, which further affects serine and TCA cycle metabolism.
Nuclear FBP2 Inhibits Mitochondrial Gene Expression
To investigate whether the growth inhibitory effect of FBP2 is dependent on its enzymatic activity, we generated a catalytically inactive FBP2 mutant by replacing a glycine residue at position 260 with arginine (FBP2G260R) as previously described (Åsberg et al., 2010) (Figure S5A). Upon wild-type FBP2 and FBP2G260R expression at comparable levels in LPS246 cells (Figure S5B), FBP2G260R inhibited cell growth, although to a lesser extent than wild-type FBP2 (Figure S5C), implying that FBP2 has catalytic activity-independent cellular functions.
Accumulating evidence demonstrates that multiple glycolytic enzymes exhibit nuclear localization and interact with transcription factors to regulate gene expression, in addition to their cytoplasmic roles (Huangyang and Simon, 2018). Interestingly, we observed strong nuclear staining of FBP2 within xenograft tumor sections (Figure S3G), suggesting that FBP2 possesses nuclear function. Specifically, immunofluorescence indicated overlap between endogenous FBP2 staining and DAPI staining in C2C12 cells (Figure 4A), and subcellular fractionation of HSMM and C2C12 cells further indicated that FBP2 co-purifies with both nuclear and cytosolic fractions (Figure 4B). A nuclear localization sequence (NLS) (203KKKGK207) was previously identified in FBP2 (Gizak et al., 2009a; 2009b). By replacing four lysine residues in the NLS with alanine, we generated a nucleus-excluded form of FBP2 without disrupting its catalytic activity (Figure 4C–D), and FBP24KA expression in LPS246 cells decreased cell proliferation albeit, not as dramatically as wild-type FBP2, suggesting that nuclear FBP2 contributes functionally to its growth inhibitory properties (Figure 4E–F).
Figure 4. Nuclear FBP2 inhibits mitochondrial gene expression. Also see Figure S5.

(A) Immunofluorescent staining of mouse myoblast C2C12 cells with FBP2 antibody. Asterisks indicate representative sites with nuclear FBP2. DAPI is a fluorescent nuclear dye.
(B) FBP2 protein levels detected in cytosolic and nuclear fractions of C2C12 and HSMM. HSP90, a cytosolic protein, and HDAC1, a nuclear protein, reflect the purity of respective subcellular fractionations.
(C) Western blot analysis of V5-tagged FBP2 or FBP24KA (4 lysines in nuclear localization sequence were substituted with alanine) in the cytosolic and nuclear fractions of transfected LPS246 cells.
(D) Enzymatic activity of FBP2 in 293T cells expressing control vector, wild-type FBP2 and FBP24KA.
(E) Protein levels of ectopically expressed FBP2 and FBP24KA mutant in LPS246 cells. GAPDH serves as a loading control.
(F) Growth of vector control, FBP2- or FBP24KA-expressing LPS246 cells in 1% serum medium.
(G) GSEA comparing vehicle-treated (n = 5) and dox-treated (n = 4) LPS246 TetO-FBP2 cells. The 50-gene “Hallmark signatures” set from MsigDB was queried. Top 10 gene sets downregulated in dox-treated groups are shown with the normalized enrichment score (NES). Relevant gene sets are highlighted with red.
(H) Differentially expressed genes were analyzed using ingenuity pathway analysis (IPA) software. Top 10 relevant and significant biological pathways were identified according to p-value from Fisher’s exact test.
(I) Heatmap showing the relative expression of OXPHOS genes and mitochondrial dysfunction pathway from IPA. Expression signals are depicted using pseudocoloring, in which expression for each gene is shown as high (red) or low (blue).
(J) qRT-PCR analysis of MT-ND1, MT-CYB, MT-CO1, and MT-ATP6 in LPS246 cells constitutively expressing TetO-FBP2 or Tet-FBP24KA, treated with vehicle or dox.
Error bars represent SD of three experimental replicates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s., not significant.
To gain further mechanistic insights into the function of nuclear FBP2, we compared the transcriptomes of vehicle (n = 5) or dox-treated (n = 4) LPS246 TetO-FBP2 cells by RNA-seq. Gene set enrichment analysis of FBP2-restored cells revealed reduced gene expression signatures for E2F targets, MYC targets, the G2M checkpoint, and OXPHOS (Figure 4G), as well as increased expression signatures related to protein secretion, UV responses, and heme metabolism (Figure S5D). Ingenuity Pathway Analysis (IPA) also identified OXPHOS and mitochondrial dysfunction as the primary differentially modulated pathways in FBP2-expressing cells, compared to controls (Figure 4H). These functional categories contain genes encoding essential subunits of mitochondrial complex I, III, IV and V of the electron transport chain and genes critical for mitochondrial function (UCP2 and RHOT2) (Figure 4I). We confirmed differential expression of several genes involved in OXPHOS (MT-ND1, MT-MYB, MT-CO1 and MT-ATP6) by qRT-PCR analysis and found that FBP2, but not FBP24KA, significantly decreased their expression (Figure 4J). Intriguingly, catalytically inactive FBP2G260R downregulated mitochondrial gene expression to a similar level as wild-type FBP2 (Figure S5E). Taken together, these data suggest that nuclear FBP2 is required for inhibiting OXPHOS in an enzymatic activity-independent manner.
Nuclear FBP2 Impairs Mitochondrial Biogenesis
To determine whether reduced transcription of OXPHOS genes in FBP2-expressing cells correlated with decreased mitochondrial biogenesis, we first measured mitochondrial DNA content by using real-time quantitative PCR. Mitochondrial to nuclear DNA ratios were dramatically decreased in FBP2-expressing LPS246 cells (Figure 5A), and flow cytometric analysis of cells stained with MitoTracker further revealed that FBP2 induction reduced mitochondrial mass (Figure 5B). Similarly, we observed a dramatic decrease in citrate synthase activity after restoring expression of FBP2 in LPS246 cells (Figure 5C). Decreased mitochondrial biogenesis in FBP2-expressing LPS246 cells was further confirmed by transmission electron microscopy, indicated by reduced number of mitochondria and swollen mitochondria with disorganized cristae (Figure 5D). Consistently, LPS246 xenograft sections exhibited reduced staining of Vdac (voltage-dependent anion channel) and Tomm20 (a constitutively expressed mitochondrial protein) in dox-treated tumors, supporting decreased mitochondrial mass caused by FBP2 expression (Figure 5E). As doxycyline has been reported to interfere with mammalian protein synthesis and disrupt mitochondrial proteostasis and function (Moullan et al., 2015), we confirmed that mtDNA content, MitoTracker staining, and citrate synthase activity were comparable in both vehicle and dox-treated LPS246 TetO-FBP24KA cells (Figure 5A–C). Collectively, these data demonstrate that nuclear FBP2 inhibits mitochondrial biogenesis in LPS246 cells.
Figure 5. Nuclear FBP2 impairs mitochondrial biogenesis.

(A) qPCR analysis of mitochondrial (MT-ND1) versus nuclear (β-globin) DNA content in indicated cells (three experimental replicates).
(B) LPS246 TetO-FBP2 cells and TetO-FBP24KA cells stained with MitoTracker Green FM probe. Flow cytometry plots (left) show the fluorescence intensity corresponding to mitochondrial mass. Histograms (right) show the quantification (three experimental replicates).
(C) Citrate synthase activity, served as a marker for mitochondrial content, was measured in indicated cells (three experimental replicates).
(D) Ultrastructural analysis of mitochondria in LPS246 Teto-FBP2 cells using transmission electron microscopy (TEM). Scale bars: 1 μm. Quantification of the number of mitochondria per cell in the imaged section (vehicle treated, n = 10 cells; dox treated, n = 10 cells).
(E) Immunohistochemical staining for expression of voltage-dependent anion channel (Vdac) and Tomm20 on sections from LPS246 TetO-FBP2 xenografts treated with vehicle or dox. Five fields per slide were quantified. Scale bars: 100 μm.
Data are represented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s., not significant.
FBP2 Suppresses Mitochondrial Respiration and the TCA Cycle
The effects of FBP2 expression on mitochondrial OXPHOS activity were investigated by culturing LPS246 cells in glucose-free medium containing 5 mM galactose, which cannot be fermented, requiring cells to rely on mitochondrial metabolism to generate sufficient ATP for survival (Rossignol et al., 2004). We observed increased apoptosis in dox-treated LPS246 TetO-FBP2 cells cultured in galactose medium (Figure 6A, S6A) compared to vehicle-treated cells, indicating that mitochondrial OXPHOS is critically impaired upon FBP2 induction. Furthermore, oxygen consumption rates (OCR) and ATP production were lower in FBP2-expressing LPS246 cells compared to controls (Figure 6B, 6D, S6B). In contrast, FBP24KA expression had no effect on OCR in LPS246 cells (Figure 6C, S6C). Loss of ATP-generating capacity stimulated glycolytic compensation in both LPS246 TetO-FBP2 and TetO-FBP24KA cells, as indicated by extracellular acidification rate (ECAR) (Figure S6D). Interestingly, ECAR was lower in both FBP2- and FBP24KA-expressing LPS246 cells than control cells (Figure S6D), indicating inhibition of glycolysis by each protein. These results are consistent with our previous conclusion that FBP2 suppresses glucose uptake and lactate secretion (Figure 2). In active mitochondria, a small percentage of electrons are prematurely leaked to O2 from complex I and/or complex III, resulting in the formation of reactive oxygen species (ROS) (Chatterjee et al., 2011). As expected, reduced OCRs observed in FBP2-expressing LPS246 cells correlated with a substantial decrease in ROS levels; whereas FBP24KA had no effect on ROS accumulation (Figure 6E).
Figure 6. FBP2 suppresses mitochondrial respiration. Also see Figure S6.

(A) LPS246 cells cultured in 5 mM glucose and 5 mM galactose medium. Apoptotic cells measured through Annexin V/PI staining followed by flow cytometry.
(B and C) Relative oxygen consumption rate normalized to protein abundance in LPS246 TetO-FBP2 cells (B) or LPS246 TetO-FBP24KA cells (C) treated with vehicle or dox. Data are presented as mean ± SD of three reading cycles of n = 9 wells pooled from three independent experiments.
(D) ATP production was measured in indicated cells.
(E) Fluorescence intensity of MitoSOX analyzed by flow cytometry. Flow cytometry plots (left) show the fluorescence intensity. Histograms (right) display their quantification (three experimental replicates).
(F) Carbon fate map showing the isotopomer distribution of indicated metabolites derived from [3-13C]pyruvate. 13C atoms are depicted as filled circles. 13C atoms directly going through the acetyl-CoA are colored in red, while 13C atoms going through oxaloacetate and combined with acety-CoA to generate citrate are colored in blue.
(G–J) M1 isotopomer distribution of indicated TCA metabolites (G) and amino acids (H), and M2 isotopomer distribution of indicated TCA metabolites (I) and amino acids (J) in LPS246 TetO-FBP2 cells with vehicle or dox treatment, labelled with [3-13C]pyruvate.
Error bars represent SD of three experimental replicates except in B and C. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s., not significant.
To assess the effects of FBP2 expression on TCA cycle metabolite levels, we performed isotope tracing experiments by labeling vehicle- or dox-treated LPS246 TetO-FBP2 cells with [3-13C]pyruvate. Labeled pyruvate is converted to acetyl-CoA by mitochondrial pyruvate dehydrogenase, which then combines with oxaloacetate to produce M1-labeled citrate. When acetyl-CoA is abundant, pyruvate decarboxylase can also generate M1-labeled oxaloacetate, which then incorporates M1-labeled acetyl-CoA to produce M2-labeled citrate (Figure 6F). We observed decreased M1 and M2 enrichment of three TCA intermediates (citrate, fumarate and malate), as well as glutamic acid and aspartic acid (Figure 6G–J) in FBP2-expressing cells. In contrast, ectopic FBP24KA expression had no effect on M1 and M2 enrichment of TCA intermediates and products compared with control cells (Figure S6E–H), further indicating that it is nuclear FBP2 that restrains mitochondrial function and metabolism.
FBP2 Transcriptionally Represses Mitochondrial Biogenesis
Mitochondrial biogenesis and function are controlled by a set of nuclear-encoded transcription factors, including PPAR-γ coactivators (PGC-1α), nuclear respiratory factor 1 (NRF1), and mitochondrial transcription factor A (TFAM) (Scarpulla et al., 2012). As nuclear FBP2 regulates mitochondrial function, we hypothesized that FBP2 dampens mitochondrial biogenesis through inhibiting one or more of these nuclear regulators. Quantitative RT-PCR analyses revealed significant downregulation of NRF1 and TFAM expression upon FBP2 restoration (Figure 7A). Ectopic expression of TFAM or NRF1 reversed the suppression of target genes associated with OXPHOS (MT-ND1, MT-CYB, MT-CO1 and MT-ATP) (Figure 7B–C, Figure S7A–B), and partially restored mitochondrial mass (Figure 7D, Figure S7C). Moreover, ectopic TFAM or NRF1 expression partially reversed cell apoptosis (Figure 7E, Figure S7D) and proliferation (Figure 7F, Figure S7E) in dox-treated LPS246 TetO-FBP2 cells.
Figure 7. FBP2 transcriptionally represses mitochondrial biogenesis. Also see Figure S7.

(A) qRT–PCR analysis of NRF1 and TFAM in vehicle treated or dox-treated LPS246 TetO-FBP2 cells.
(B) Immunoblot analysis for ectopic expression of vector control or TFAM in LPS246 TetO-FBP2 cells.
(C) qRT-PCR analysis of MT-ND1, MT-CYB, MT-CO1, and MT-ATP6 in indicated cells.
(D) LPS246 TetO-FBP2 cells with or without TFAM expression stained with MitoTracker Green FM probe. Fluorescence intensity corresponding to mitochondrial mass is shown in histograms.
(E) Indicated cells cultured in 5 mM glucose and 5 mM galactose medium, where apoptotic cells were measured through Annexin V/PI staining followed by flow cytometry.
(F) Growth of indicated cells in low serum medium (1% FBS).
(G) c-Myc reporter activity measured in LPS246 TetO-FBP2/FBP24KA cells transfected with Myc/Max luciferase reporter, in the presence of vehicle or dox. Transfection efficiencies were normalized to co-transfected pRenilla-luciferase.
(H) qRT-PCR analysis of c-Myc target genes (CCND2, eIF2A, NPM1, PSAT1) in indicated cells.
(I) Diagram of 2 kb upstream of exon 1 to exon 2 of human TFAM. Exons are represented by green boxes. The E box in amplicon 1 is illustrated. Horizontal bars labeled Neg, 1, 2 indicate the regions amplified for ChIP analysis.
(J) ChIP assays evaluating FBP2 association with chromatin at a c-Myc binding site in TFAM promoter (amplicon 1 and 2), or to a site negative for c-Myc binding (amplicon Neg). IgG, isotype-matched immunoglobulin G; V5, V5-tagged FBP2.
(K) ChIP-reChIP analysis examining the co-localization of c-Myc and FBP2 at amplicon 1 and 2 at TFAM promoters.
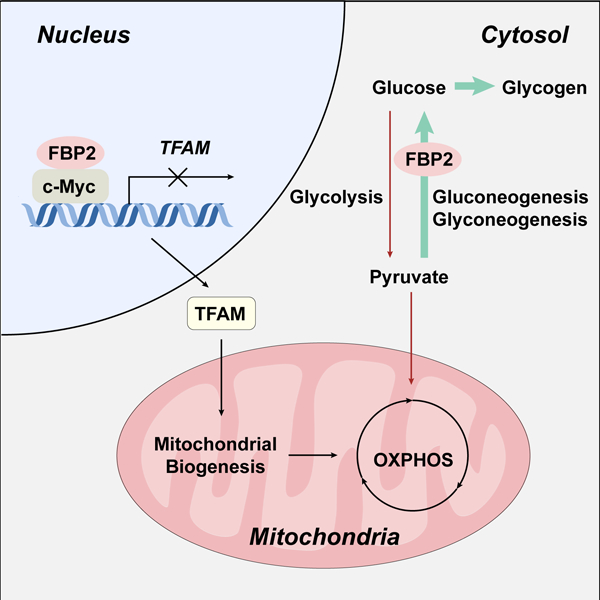
(L) Model depicting the metabolic status, transcription activity and mitochondrial change of sarcoma cells upon FBP2 re-expression.
Data are represented as mean ± SD (three experimental replicates) except in J and K, which indicate standard error of the mean (three technical replicates from a representative experiment). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s., not significant.
To elucidate the molecular mechanisms whereby FBP2 alters the expression of nuclear transcription factors associated with mitochondrial biogenesis, we examined the top differentially expressed pathways based on RNA-seq data and identified Myc target genes as one of the top downregulated gene sets (Figure 4G). Previous studies have demonstrated that c-Myc regulates mitochondrial biogenesis and gene expression (Lee et al., 2017; Scarpulla, 2008); for example, c-Myc induces NRF1 target genes by binding to a canonical NRF1 binding site, leading to the sensitization of cells to apoptosis (Morrish et al., 2003). In addition, c-Myc potentiates mitochondrial biogenesis through induction of TFAM and other nuclear-encoded mitochondrial genes (Li et al., 2005). To ascertain that FBP2 diminishes c-Myc transcriptional activity, we measured c-Myc luciferase-reporter intensity and found FBP2 decreased c-Myc functionality (Figure 7G). Transcript levels of c-Myc target genes (CCND2, eIF2A, NPM1, PSAT1) were also decreased in FBP2-expressing cells (Figure 7H). To determine whether FBP2 represses mitochondrial biogenesis in a predominantly c-Myc dependent manner, we assessed the expression of mitochondrial genes (MT-ND1, MT-CO1 and MT-ATP6) in the presence and absence of c-Myc using RNA interference. Interestingly, dox-induced FBP2 expression or repression of c-Myc expression by sic-Myc resulted in comparable repression of mitochondrial gene transcription (Figure S7F–G), and combined FBP2 expression and sic-Myc showed no additive effects on gene expression. MitoTracker staining indicated a slight but perceptible decrease in mitochondrial mass in cells with combined dox and sic-Myc treatment, relative to cells with either treatment alone (Figure S7H). We conclude that FBP2’s inhibition of mitochondrial biogenesis is largely, but not entirely dependent on the c-Myc pathway.
c-Myc has been reported to bind TFAM in the region of amplicons 1 and 2, approximately 900 bp upstream of the transcription start site (Figure 7I), thereby stimulating TFAM expression (Li et al., 2005). Remarkably, chromatin immunoprecipitation (ChIP) analyses indicated that FBP2 protein was enriched at amplicon 1, which contains a 5’-CACGTG-3’ c-Myc binding site, but not at amplicon 2, which contains a Myc/Max 5’-GCG-3’ half site (Figure 7J). As expected, nucleus-excluded FBP24KA was not detected at either amplicon (Figure 7J). Importantly, ChIP-reChIP analyses revealed co-localization of c-Myc and FBP2 proteins at TFAM promoter amplicon 1 (Figure 7K). We also demonstrated the association of epitope-tagged FBP2 or FBP24KA and endogenous c-Myc by co-immunoprecipitations in LPS246 cell lysates (Figure S7I), suggesting their physical interaction can occur in both the nucleus and cytosol (albeit at lower levels). Finally, GST pull-down assays of bacterially expressed proteins further revealed that FBP2 directly binds to c-Myc (Figure S7J). Collectively, these results indicate that FBP2 inhibits c-Myc transcriptional activation of TFAM, by direct physical association at its promoter.
Discussion
STSs are a complex set of mesenchymal malignancies, compromising more than 70 subtypes (Fletcher, 2014). Despite rapid advances in the molecular analysis of individual subtypes, the heterogeneous nature of sarcomas limits the efficacy of targeted therapies (Cancer Genome Atlas Research Network, 2017; Linch et al., 2014), emphasizing the need to identify therapeutic vulnerabilities common to multiple STS subtypes. Although a diverse spectrum of oncogenic mutations has been recently described for in STS (Barretina et al., 2010; Cancer Genome Atlas Research Network, 2017; Eisinger-Mathason et al., 2015; Frith et al., 2013; Nakazawa et al., 2016a), it is increasingly clear that all tumors must adapt cell metabolism to support growth. Whereas metabolic reprogramming in STS has long been observed (Shaw et al., 1988), how this influences sarcoma growth is unclear. In the current study, we demonstrate that the gluconeogenic enzyme FBP2 is uniformly depleted in multiple sarcoma subtypes. Restoring FBP2 expression in sarcoma cells dramatically inhibited tumor cell growth both in vitro and in vivo (Figure 2), suggesting that FBP2 has tumor suppressive activities in sarcomas. We further discovered that FBP2 exhibits two distinct tumor-suppressive functions that depend on its subcellular localization. These results identify a specific metabolic adaptation as a common feature of STS and could help inform therapeutic strategies.
Tumor cells are typified by enhanced glycolysis, as a means of generating ATP and providing building blocks for macromolecule biosynthesis, sometimes known as the “Warburg effect” (Vander Heiden and DeBerardinis, 2017; Warburg, 1956). Reprogrammed glucose metabolism is induced by multiple mechanisms, such as the deregulation of oncoproteins and tumor suppressors (Hay, 2016). Previous studies uncovered frequently mutated genes in STS, including TP53, NF1 and PIK3CA (Barretina et al., 2010; Frith et al., 2013), further confirmed by recent large-scale analyses of 206 adult soft tissue sarcomas representing 6 major subtypes using multi-platform molecular profiling (Cancer Genome Atlas Research Network, 2017). The p53 protein is involved in regulation of glucose metabolism by promoting OXPHOS and dampening glycolysis (Humpton and Vousden, 2016). Furthermore, PIK3CA mutations, among the most frequent somatic mutations found in STS along with gain of function mutations in c-KIT and PDGFRα, are implicated in activation of the AKT/mTOR pathway (Fruman et al., 2017; Yuan and Cantley, 2008), stimulating a shift towards aerobic glycolysis (Lien et al., 2016). We show here that FBP2 loss also underlies increased glycolytic activity observed in sarcomas, as FBP2 re-expression inhibits glucose uptake and lactate secretion in distinct sarcoma cell lines. We also observed decreased glycolytic intermediates for biosynthesis in FBP2-expressing cells, such as glucose-6-phosphate, serine and glycine, consistent with previous speculation that FBP2 participates in the regulation of glycolysis (Newsholme and Crabtree, 1970). Furthermore, FBP2 restoration reduces PPP activity needed for ribonucleotide production and reducing equivalents in the form of NADPH.
Increasing evidence shows that multiple essential glycolytic/gluconeogenic enzymes localize to the nucleus and influence gene transcriptional regulation (Boukouris et al., 2016; Huangyang and Simon, 2018). We suggest a model in which metabolic enzymes transduce signals from growth factors, nutrient and oxygen availability, and external stress to modulate gene expression. We reported previously that nuclear FBP1 functions as a transcriptional corepressor to inhibit HIF-1α and its downstream targets, including genes associated with glucose metabolism (GLUT1, LDHA, and PDK1), therefore decreasing glycolytic phenotypes and enhancing glutamine uptake in tumor cells (Li et al., 2014). FBP2 has been previously shown to reside in nuclei of myogenic progenitor cells, cardiomyocytes and smooth muscle cells (Gizak and Dzugaj, 2003; Gizak et al., 2005; 2006), although its nuclear function was unclear. We determined that FBP2 nuclear activity suppresses sarcoma cell growth by inhibiting mitochondrial biogenesis and OXPHOS in a catalytic activity-independent manner, at least in part by repressing the expression of critical transcription factors NRF1 and TFAM. Of note, nuclear FBP2 also plays a role in suppressing glycolysis, as nucleus-excluded FBP24KA exhibited less potent inhibition of glucose uptake and lactate secretion than wild-type FBP2, as indicated by [1,2-13C]glucose labeling experiments (e.g. 26% decrease in glucose consumption by FBP2 versus 14% by FBP24KA). We can account for this based on decreased expression of c-Myc target genes in FBP2-expressing cells from RNA-seq (Figure 4G). c-Myc regulates virtually all genes involved in glycolysis (Dang et al., 2006), including the essential glycolytic enzymes LDHA, PDK1 and ENO1, consistent with our observation that nuclear FBP2 also contributes to changes in glucose catabolism.
It’s noteworthy that even though FBP1 and FBP2 have different tissue distributions, they share many functional similarities due to 77% sequence homology. For example, enforced expression of FBP1 in sarcoma cells or FBP2 in renal cancer cells suppresses tumor cell proliferation (data not shown). In addition, we showed that c-Myc transcriptional activity can also be suppressed by FBP1 in sarcoma cells. Interestingly, in contrast to c-Myc, the effect of FBP1 and FBP2 on HIF signaling is tissue type-dependent. We consistently observed increased HRE luciferase activity upon FBP1/FBP2 expression in different sarcoma cell lines (data not shown). However, both proteins inhibit HIF transcriptional output in renal cancer cells. These results are in line with our previous findings that HIF2α is actually a tumor suppressor in soft tissue sarcoma, unlike kidney cancer (Nakazawa et al., 2016a) and increased HIF activity might also be involved in FBP2-mediated growth inhibition.
Recent isotope tracing analysis of human ccRCC confirmed enhanced glycolysis and suppressed glucose oxidation by the TCA cycle in these tumors (Courtney et al., 2018), likely due to HIF stabilization caused by frequent von Hippel-Lindau (VHL) mutations (Nickerson et al., 2008). However, sarcoma cells exhibited more active mitochondrial metabolism than ccRCC, as the incorporation rate of 13C from [1,2-13C]glucose into TCA cycle intermediates is faster (Figure 3H–I; see (Li et al., 2014)), suggesting that mitochondrial activity is critical for sarcoma progression. While earlier studies emphasized the importance of glycolysis, mitochondrial biosynthesis, bioenergetics, and signaling are also essential for tumorigenesis (Weinberg and Chandel, 2015). Intermediates from the TCA cycle, such as citrate, aspartate and glutamate, are precursors for macromolecule synthesis (lipid and nucleotides) to support biomass accumulation (Birsoy et al., 2015; Faubert et al., 2017; Hosios et al., 2016; Sellers et al., 2015). In addition, the mitochondrial ETC produces ROS via oxidative metabolism. Elevated ROS levels activate signaling pathways to promote cell proliferation and tumor progression (Schieber and Chandel, 2014). By inhibiting mitochondrial function, FBP2 reduces the abundance of TCA cycle intermediates, ATP, and ROS, therefore inhibiting cell growth.
A key observation is that FBP2 co-localizes with the c-Myc oncogene at the TFAM promoter, suggesting that FBP2 acts as a nuclear c-Myc transcriptional corepressor. c-Myc affects a large spectrum of genes involved in mitochondrial function (Dang et al., 2006) and directly activates TFAM expression. Given the multitude of biological processes c-Myc regulates, the precise mechanisms whereby FBP2 specifically targets c-Myc-induced mitochondrial biogenesis remain to be elucidated. Presumably, additional factors are required to regulate c-Myc driven targets associated with mitochondrial functions. Another nuclear-encoded transcription factor, NRF1 is key to mitochondrial biogenesis and also an upstream regulator of TFAM (Kelly and Scarpulla, 2004). Decreased NRF1 expression in FBP2-expressing cells further explains reduced TFAM activity in this context.
Interestingly, FBP2 has been reported to translocate from the nucleus to the cytosol during myoblast differentiation (Gizak et al., 2006), which corresponds to a shift from a highly glycolytic state to increased OXPHOS needed in fully developed muscle cells (Remels et al., 2010). We suggest that FBP2 is restricted to the nucleus in muscle progenitors to transcriptionally suppress mitochondrial biogenesis and OXPHOS, as undifferentiated cells frequently favor glycolytic metabolism. At the onset of differentiation, FBP2 exits the nucleus and functions as a gluconeogenic/glycogen producing enzyme to antagonize glycolysis in the cytosol. How FBP2 translocation is regulated and whether subcellular shuttling of FBP2 contributes to muscle differentiation are unclear, and remain part of our future studies. It will also be important to evaluate the role of FBP2 in adipogenic differentiation, another lineage derived from mesenchymal stem cells.
In summary, our findings demonstrate a role for FBP2 in suppressing sarcoma progression and further establish dual tumor-suppressive functions of FBP2, including promoting gluconeogenesis and inhibiting mitochondrial biogenesis by inhibiting c-Myc transcriptional activity (Figure 7L).
Limitations of Study
We have not delineated consequences of FBP2 loss during sarcoma initiation thus far; future studies will assess Fbp2 deletion in genetically engineered mouse models of STS, such as a “KP” UPS model (Kirsch et al., 2007; Nakazawa et al., 2016a). In addition, how FBP2 subcellular localization is dynamically controlled by extracellular stimuli remains unclear. It is critical to define how FBP2 shuttles from cytoplasm to nucleus, therefore shifting from predominantly gluconeogenic to nuclear activity in the process. Finally, it is important to identify additional factor(s) that specifically direct FBP2 to attenuate c-Myc-driven TFAM expression and other c-Myc transcriptional targets that may be affected.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, M. Celeste Simon (celeste2@pennmedicine.upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Xenograft tumor experiments were approved by the Animal Care and Use Committee at the University of Pennsylvania. Briefly, ten female NSG mice (the Jackson Laboratory, 6 weeks, 005557) were injected subcutaneously into both flanks with 2 million LPS246 cells stably expressing TetO-FBP2. Before injection, cells were resuspended in 100 μl PBS mixed with an equal volume of Matrigel (Corning, 356234). Once palpable tumors were established, tumor volume was measure with a digital caliper. When the average tumor size reached 100 mm3, ten mice were randomly separated into two groups: (1) Doxycycline diet (Bio-Serv, S3888) and (2) control diet (Bio-Serv, S4207). Upon completion of the experiment, the animals were sacrificed by CO2 inhalation and xenograft tumors were dissected for downstream analyses. Mice were housed in a controlled environment (12 h light/12 h dark cycle, humidity 30~70%, temperature 20~22°C), and had free access to water and rodent diet. All animal experiments were performed in accordance with the Guide for Care and Use of Laboratory Animals of the NIH. All animal studies were approved by Institutional Animal Care and Use Committee Office of Animal Welfare of University of Pennsylvania.
Cell Culture
LPS224, LPS246, T1000, SW872, HT1080, KP250 and 293T cells were tested to confirm they are mycoplasma negative, and cultured in DMEM (Life Technologies, 11965–084) containing 10% FBS (Gemini, 900–108). C2C12 myoblasts (ATCC, CRL-1772) were propagated in DMEM containing 20% FBS. To evaluate differentiation, myoblasts were grown to 80 to 90% confluence and switched to 2% horse serum (Life Technologies, 16050–122) in DMEM. For metabolic labelling assays, cells were maintained in glucose-free DMEM (Life Technologies, 11966–025) supplemented with 10% dialyzed FBS (Gemini, 100–108) and 10 mM [1, 2-13C]glucose (Sigma-Aldrich, 453188), or supplemented with 10% dialyzed FBS and 2 mM [3-13C]sodium pyruvate (Sigma-Aldrich, 490733).
METHOD DETAILS
Constructs and Viral Transduction
Lentivirus was produced by transfecting 2.5 × 106 HEK293 cells with the 10 μg indicated expression plasmid, 2.5 μg pRSV-Rev, 6.5 μg pMDL, and 3.5 μg pCMV-VSV-G plasmids (4th generation lentiviral system) using FuGENE 6 Transfection Kit (Promega, E2691). The virus was harvested 48 h after transfection by filtering the virus-containing medium through Amicon Ultra-15 Centrifugal Filter (Millipore, UFC901024). Virus infection was performed by incubating cells with medium containing indicated virus and 8 μg/ml polybrene (Sigma, 107689) for 24 h. Cells were allowed to recover in complete medium for 24 h and then selected with puromycin for 48 h. Surviving pools were subjected to indicated experiments.
Sequence verified cDNA constructs were obtained from the Mammalian Gene Collection (GE Dharmacon) and were subcloned into the pCDH-CMV-MCS-EF1-Puromycin mammalian expression vector (System Biosciences CD510B-1) and pLVX-TetOne-Puro (Clontech, 631847). FBP2 cDNA construct: MHS6278–211687897 (Accession, BC117477). The FBP24KA and FBP2G260R mutants were generated using Q5 Site-Directed Mutagenesis Kit (NEB, E0554). FBP24KA Forward primer: 5’- caggagcgatttacagcctgaatgag-3’, Reverse primer: 5’-ccgccgcaatcttgacatctttttccac-3’. FBP2G260R Forward primer: 5’-cctggtctatagaggaatcttcctgtaccc-3′, Reverse primer: 5′-gtgcggtgcacgtcagcc-3′.
Cell Growth Assays
Multiple cultures of LPS246, T449, T778, T1000, HT1080, and KP250 cells were plated in 60 mm plates at a density of 8 × 104 cells supplemented with either DMEM containing 1% FBS or glucose-free DMEM supplemented with 10% FBS and 5 mM D-(+)-Glucose (Sigma-Aldrich, G8270). Every other day, one set of cultures was collected and counted.
Anchorage-Independent Growth Assay
Sarcoma cells (SW872, HT1080, KP250) stably expressing FBP2 or vector control were plated in 6-well plates at a density of 6,000 cells per well in complete medium containing 0.3% agarose (low-melt 2-hydroxyethylagarose, Sigma-Aldrich, A4018), onto underlays composed of medium containing 0.6% agarose. Additional media was added to the cultures once per week, and after two weeks of growth the colonies were quantified.
Western Blot Analysis
Cells were harvested in lysis buffer (150 mM NaCl, 10 mM Tris pH 7.6, 0.1% SDS and 5 mM EDTA) containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, 78445). For western blots of xenograft tissue, approximately 10–20 mg of tissue was suspended in 500 μl lysis buffer and homogenized on ice using a Tissue-Tearor (Biospec, 985370). Samples were centrifuged at 12,000 rpm for 20 min at 4 °C. Protein lysates were resolved by Tris-Glycine SDS-PAGE and were transferred to nitrocellulose membranes (Bio-Rad, 162–0115, 0.45 μm pore size for all experiments). All membranes were incubated with the indicated primary antibodies overnight at 4 °C and were diluted in TBST (20 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween-20) supplemented with 5% bovine serum albumin (BSA, Sigma-Aldrich, A7906). Primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies followed by exposure to ECL reagents.
Quantitative RT-PCR
Total RNA was isolated from cells using the RNAeasy purification kit (Qiagen, 74104). cDNA was synthesized using a High Capacity RNA-to-cDNA kit (Applied Biosystems, 4368814). qRT-PCR was performed on a ViiA7 Real-Time PCR system from Applied Biosystems. Pre-designed Taqman primers were obtained from Life Technologies for the following genes: 18S (HS03928985_G1), MT-ND1 (HS02596873_S1), MT-MYB (HS02596867_S1), MT-CO1 (HS02596864_G1), and MT-ATP6 (HS02596862_G1).
Chromatin Immunoprecipitation (ChIP) and ChIP-reChIP Assay
ChIP and ChIP-reChIP were performed as described previously (Si et al., 2015). Briefly, cells were cross-linked using 1% formaldehyde at room temperature for 10 min. The cross-linking reaction was quenched by glycine. Cells were lysed in SDS buffer containing protease inhibitor cocktail and lysates were sonicated at 4 °C. The sheared chromatin was precleared, and then subjected to immunoprecipitation with 5 μg IgG (Cell Signaling Technology, 5415) and V5 (Thermo Fisher Scientific, R960–25) antibodies. After washing with low- and high-salt buffers, immunoprecipitated DNA fragments were eluted and purified with the QIAquick PCR Purification Kit and then analyzed by qRT-PCR. For ChIP-reChIP, the first ChIP was performed using c-Myc antibody (Santa Cruz Biotechnology, sc-40x), until the washing steps. The immunoprecipitated protein-DNA complexes were incubated in ChIP-reChIP elution buffer (10 mM Tris-HCl, pH 8.0, 2 mM EDTA, 2% SDS, 15 mM DTT, supplemented with protease inhibitor cocktail) for 30 min at 37 °C. T he isolated supernatant was diluted at least 20 times and subjected to the second ChIP using 5 μg IgG or V5 antibodies.
mtDNA Content
Total DNA was extracted from cell samples using Wizard Genomic DNA Purification Kit (Promega, A1120) according to the manufacturer’s instructions. To quantify mtDNA copy number, real-time PCR was performed using a on a ViiA7 Real-Time PCR system from Applied Biosystems against MT-ND1 (HS02596873_S1) as the standard for mtDNA. The β-globin (HBB) was used as the nuclear gene (nDNA) normalizer for calculation of the mtDNA/nDNA ratio, as previously described (Dickinson et al., 2013). A fragment of HBB gene was amplified using forward primer, 5’-caacttcatccacgttcacc-3’, and reverse primer, 5’-gaagagccaaggacaggtac-3’. The relative mtDNA content was calculated using the formula: mtDNA content = 1/2ΔCt, where ΔCt = CtmtDNA − CtHBB.
Subcellular Fractionation
Cytosolic and nuclear fractionation of indicated cells were performed using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific, 78833) by following the manufacturer’s protocol.
FBP2 Enzymatic Activity Assay
Vector control 293T cells, or 293T cells expressing FBP2, FBP24KA or FBP2G260R were harvested in lysis buffer used for western blot analysis. 20 μg lysates were added to 100 μl reaction buffer containing 50 mM Tris (pH 6.8), 10 mM MgCl2, and 100 μM D-fructose 1,6- bisphosphate trisodium salt (Sigma-Aldrich, F6803). The reaction was started by incubating solutions at 37 °C for 15 min and stopped by a deproteinization protocol using Deproteinizing Sample Preparation Kit (BioVision, K808). FBP2 catalytic activity was quantified according to the yield of fructose 6-phosphate (enzyme product), measured by Fructose-6-Phosphate Fluorometric Assay Kit (BioVision, K689–100).
Cell Apoptosis Assay
LPS246 TetO-FBP2 (PBS or Dox treated) and LPS246 TetO-FBP24KA (PBS or Dox treated) cells were seeded in 6-well plates at a density of 1.5 × 105 per well. These cells were incubated in 5 mM glucose medium (glucose-free DMEM, 10% FBS, 5 mM glucose) or 5 mM galactose medium (glucose-free DMEM, 10% FBS, 5 mM galactose). After 24 h, cells and supernatant were collected and centrifuged. The cell pellet was suspended with 110 μl binding buffer. Cells were then stained with the FITC-Annexin V Apoptosis Detection Kit (BD Biosciences, 556547) with Annexin V Alexa Fluor 488 and propidium iodide (PI) and incubated for 15 min at room temperature in the dark. After incubation, 400 μl of binding buffer was added and cells were analyzed by flow cytometry using the BD Accuri C6 instrument. Double-negative cells were determined viable.
MitoTracker and MitoSOX Staining
Cells were incubated with 5 μM MitoSOX reagent (Thermo Fisher Scientific, M36008) or 25–100 nM MitoTracker Green FM probe (Thermo Fisher Scientific, M7514) at 37 °C for 30 min. After staining was complete, the cells were gently washed three times with warm PBS. Detach cells from plates and ensure a single cell suspension. Analyze the samples by flow cytometry using BD Accuri C6.
ATP Measurements
ATP production was measured using the ATP Determination Kit (Thermo Fisher Scientific, A22066) according to the manufacturer’s protocol. Briefly, cells were homogenized in lysis buffer (1% Triton X-100, 0.1% SDS, 150 mM NaCl, 50 mM Tris-HCl pH 7.5) supplemented with protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, 78445). Data were determined by luminescence (Promega, Glomax 20/20 luminometer). Data were collected from multiple replicate wells for each experiment and were normalized to protein concentration.
Citrate Synthase Activity
Citrate synthase activity was measured on total cell extracts by Citrate Synthase Activity Assay Kit (Abcam, ab119692). In brief, 100 μl of cell lysates was added to the pre-coated microplate strips, sealed, and incubated at room temperature for 3 hours. The wells were aspirated and washed twice with wash buffer. Activity solution was added to the well and the plate was immediately read every 20 seconds for 30 min at a wavelength of 412 nm using a plate reader (BioTek, Synergy H1).
Immunohistochemistry
LPS246 tumor sections and tissue microarray slides (US Biomax, SO801b) were deparaffinized by baking slides at 60 °C for 30 min. The slides were rehydrated in series of ethanol solutions and endogenous peroxidase activities were quenched by 1% H2O2 in distilled water for 20 min. After three washes in TT buffer (500 mM NaCl, 10 mM Trizma, and 0.05% Tween-20), antigen retrieval was performed by boiling slides for 20 min in a citrate-based antigen unmasking solution (Vector labs, H3300). After cooling down to room temperature, slides were blocked in 2% normal goat serum and 4% BSA in TT buffer for 1 h. Next, tissue slides were incubated with various primary antibodies at 4 °C overnight. After three washes in TT buffer, biotinylated secondary antibody was added onto these slides for 1 h, following by 1 h treatment of the Vectastain Elite ABC reagents (Vector Labs, PK-6100). After three TT washes, the slides were processed with DAB peroxidase substrate kit (Vector Labs, SK-4100), and hematoxylin solutions for immunohistochemistry staining, dehydrated in a standard ethanol/xylenes series, and mounted in 75% v/v Permount (Fischer Scientific, SP15–500) in xylenes.
Tissue Immunofluorescence
LPS246 tumors were fixed in 4% paraformaldehyde (PFA)/PBS overnight at 4 °C. Fixed tissues were dehydrated in 30% sucrose solution and embedded in OCT for frozen sectioning. Tissue samples were submitted to the Molecular Pathology & Imaging Core (MPIC) at the University of Pennsylvania for processing and sectioning. Frozen sections were rinsed in cold PBS to remove OCT, and then incubated with 0.25% Triton-X-100 in PBS for 10 min at room temperature. The slides were incubated in a blocking buffer (4% BSA, 2% serum in PBS) for 1 h at room temperature. The slides were then incubated with diluted primary antibodies in blocking buffer overnight at 4 °C. After washing in PBS, the slides were further incubated with fluorescein-conjugated secondary antibodies diluted in blocking buffer for 1 hour at room temperature. The slides were finally mounted with ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific, P36962). Slides were examined and images were captured on a Leica DM5000B microscope.
Cell Immunofluorescence
Cells were plated in 2-well Chamber slides (Lab-Tek, 154461). After 24 h, the cells were washed three times with PBS and fixed for 20 min in 4% paraformaldehyde at room temperature. Cells were permeabilized in PBS containing 0.5% Triton X-100 and blocked in PBS containing 5% BSA (blocking buffer). Primary antibodies against FBP2 (Abcam, ab131253) and secondary antibodies (Thermo Fisher Scientific, Alexa Fluor 488) were diluted in blocking buffer. Slides were washed three times in PBS, rinsed in H2O, and mounted on slides using VectaShield mounting medium containing 4’6’-diamidino-2-phenylindole (DAPI) (Vector Labs, H-1200).
Metabolic Quantification
The rate of glucose uptake and lactate secretion was determined using Multiparameter Bioanalytical System (YSI Life Sciences, YSI 2950). Briefly, cells were seeded in 6 cm plates starting with 4 replicates. The next day, the medium was changed to 10 mM glucose medium (glucose-free and glutamine-free DMEM, 10% dialyzed FBS, 10 mM glucose, 2 mM glutamine). Cells in one replicate ware trypsinized and counted. The cell number was labeled as Q1 (starting cell number). 24 h later, culture medium was collected and read by YSI analyzer. Cell number was counted as Q2 (final cell cell number). Measurements were normalized to cell number area under the curve, as previously described (Gentile et al., 2013). Metabolite consumption was defined as v = V(xmedium control − xfinal)/A, where v is metabolite consumption/production, V is culture volume, x is metabolite concentration, and A is cell number area under the curve. A was calculated as N(T)d/ln2(1 − 2−T/d), where N(T) is the final cell count, d is doubling time, and T is time of experiment. Doubling time was calculated as d = (T)[log(2)/log(Q2/Q1)].
13C-metabolic Flux Analysis
Cells were seeded to 10 cm plates at a density of 1 million cells per plate. For [1,2-13C]glucose labeling, cells were incubated in 10 mM [1,2-13C]glucose medium (glucose-free DMEM, 10% FBS, 10 mM [1,2-13C]glucose) for 24 h. For [3-13C]pyruvate labeling, cells were incubated in 2 mM [3-13C]pyruvate medium (glucose-free DMEM, 10% FBS, 2 mM [3-13C]pyruvate) for 6 h. To measure the 13C enrichment in TCA cycle intermediates, [1,2-13C]glucose-labelled or [3-13C]pyruvate-labelled cells were extracted by 4% perchloric acid (PCA). Briefly, cells were washed twice with ice-cold PBS and then scraped in 4% PCA on dry ice, which was then freeze-thawed for three cycles. Cell extracts were neutralized using 5 M KOH and analyzed as previously described (Nissim et al., 2012). The neutralized PCA extract were subjected to an AG-1 column (100–200 mesh, 0.5 × 2.5 cm, Bio-rad) for enriching the organic acids, glutamate and aspartate, which were then converted to t-butyldimethylsilyl derivatives. The production of 13C-labeled metabolites was measured using GC-MS. Isotopic enrichment in [13C]glutamate isotopomers was monitored using ions at m/z 432, 433, 434, 435, 436 and 437 for M0, M1, M2, M3, M4 and M5 (containing 1 to 5 13C atoms above M0, the natural abundance), respectively. Isotopic enrichment in [13C]aspartate isotopomers was monitored using ions at m/z 418, 419, 420, 421 and 422 for M0, M1, M2, M3 and M4 (containing 1 to 4 13C atoms above M0, the natural abundance), respectively. Isotopic enrichment in [13C]lactate was determined using ions at m/z 261, 262, 263 and 264 for M0, M1, M2 and M3 (containing 1 to 3 13C atoms above natural abundance), respectively. Isotopic enrichment in [13C]pyruvate isotopomers was monitored using ions at m/z 259, 260, 261 and 262 for M0, M1, M2 and M3 (containing 1 to 3 13C atoms above M0, the natural abundance), respectively. Isotopic enrichment in [13C]malate isotopomers was monitored using ions at m/z 419, 420, 421, 422 and 423 for M0, M1, M2, M3 and M4 (containing 1 to 4 13C atoms above M0, the natural abundance), respectively, and 13C enrichment in [13C]citrate isotopomers was assayed using ions at m/z 459, 460, 461, 462, 463, 464 and 465 for M0, M1, M2, M3, M4, M5 and M6 (containing 1 to 6 13C atoms above M0, the natural abundance), respectively.
Quantification of the Pentose Phosphate Pathway (PPP)
Metabolic flux going through the PPP was quantified as previously described (Lee et al., 1998), using [1,2-13C]glucose as the labelling tracer. Briefly, direct glycolysis of [1,2-13C]glucose (without going through the PPP) produces M2-labelled lactate, while flux going through the oxidative portion of PPP removes the first carbon from glucose and releases it in the form of CO2. The resultant M1-labelled intermediates are recycled back to glycolysis to produce M1-labelled lactate through the non-oxidative potion of PPP. The ratio of M1 to M2-labelled lactate indicates the ratio of flux going through the PPP versus flux directly going through glycolysis. Therefore, PPP flux was calculated based on the following formula: PPP flux = glucose consumption × M1 lactate enrichment / (M1 lactate enrichment + M2 lactate enrichment). Calculated PPP flux was then normalized to vector control cells by setting the vector control flux equal to 1.
Seahorse XF Cell Mito Stress Analysis
The mitochondrial respiratory capacity was determined using XF Cell Mito Stress Test Kit (Agilent Technologies, 103015–100). Cells were seeded in the XF96 Cell Culture Microplate at a density of 2 × 104 cells per well. 6 replicates were set up for each of the following groups: (1) LPS246 TetO-FBP2 + Veh; (2) LPS246 TetO-FBP2 + Dox; (3) LPS246 TetO-FBP24KA + Veh; (4) LPS246 TetO-FBP24KA + Dox. Microplate was incubated for 24 h at 37 °C. Seahorse XF96 FluxPak sensor cartridge was hydrated in the utility plate filled with 200 μl of Seahorse Calibrant overnight in a non-CO2 incubator at 37 °C. Next day, cells were incubated with the base medium containing 2 mM L-glutamine, 1 mM sodium pyruvate, and 10 mM glucose for 1 hour prior to assay. The oxygen consumption rate (OCR) was measured by XFe96 extracellular flux analyzer with sequential injection of 1 μM oligomycin A, 1 μM FCCP, and 0.5 μM rotenone/antimycin A. After the experiment, cells lysates were harvested with 20 μl western blot lysis buffer and protein concentration was quantified using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225). OCR value was normalized to the protein concentration in each well.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were conducted using GraphPad Prism 7.0. All error bars are presented as mean ± s.d. unless otherwise specified. Data were reported as biological replicates except for ChIP and ChIP-reChIP experiments using technical replicates. In the xenograft experiments, 10 tumors per group formed in mice were subjected to analyze. To quantify mitochondrial number per cell by TEM, 10 cells each group were selected for counting. Five fields per slide were selected for the quantification of IHC staining. If not specified, three biological replicates were used for statistical analysis. Two-way ANOVA was performed for group analysis. When two groups were analyzed, P values were calculated based on two-tailed, unpaired Student’s t-tests with 95% confidence. No particular methods were used to determine whether the data met assumptions of the statistical approach. Detailed statistical parameters are presented in the legend for each figure. Significance was defined as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DATA AND SOFTWARE AVAILABILITY
Software
FlowJo (https://www.flowjo.com/) software was used to analyze FACS-based data. For quantification of IHC and IF images, ImageJ (https://imagej.nih.gov/ij/index.html) was used.
Data resources
The RNA-seq data is deposited in GEO and the accession number is GSE137755.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-FBP2 | Abcam | #ab131253 |
| Rabbit polyclonal anti-FBP2 for IHC | Sigma-Aldrich | #HPA012513 |
| Mouse polyclonal anti-FBP1 | Sigma-Aldrich | #SAB1405798 |
| Rabbit polyclonal anti-PCK1 | Abcam | #ab28455 |
| Rabbit polyclonal anti-G6PC | Abcam | #ab83690 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | #2118 |
| Mouse monoclonal anti-HDAC1 | Cell Signaling Technology | #5356 |
| Rabbit monoclonal anti-HSP90 | Cell Signaling Technology | #4877 |
| Rabbit polyclonal anti-Phospho-Histone H3 | Cell Signaling Technology | #9701 |
| Anti-rabbit IgG HRP-linked | Cell Signaling Technology | #7074 |
| Anti-mouse IgG HRP-linked | Cell Signaling Technology | #7076 |
| Mouse (G3A1) mAb IgG1 isotype control | Cell Signaling Technology | #5415 |
| Normal rabbit IgG | Cell Signaling Technology | #2729 |
| NRF1 (D9K6P) rabbit mAb | Cell Signaling Technology | #46743 |
| Rabbit monoclonal anit-VDAC1 | ABclonal | #A11242 |
| Rabbit polyclonal anti-TOMM20 | ABclonal | #A6774 |
| TFAM polyclonal antibody | ABclonal | #A13552 |
| Biotinylated goat anti-rabbit IgG | Vector Labs | #BA-1000 |
| Mouse monoclonal anti-V5 Tag | Thermo Fisher Scientific | #R960–25 |
| Goat anti-rabbit IgG (H+L) secondary antibody, Alexa Fluor 488 conjugated | Thermo Fisher Scientific | #A-11008 |
| c-Myc monoclonal antibody (9E10) | Thermo Fisher Scientific | #13–2500 |
| Biological Samples | ||
| Soft tissue malignant tumor tissue array | US Biomax | SO801b |
| Deposited Data | ||
| RNA-seq raw data | GEO | GSE137755 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Life Technologies | 11965–084 |
| Glucose-free DMEM | Life Technologies | 11966–025 |
| Glucose-free and glutamine-free DMEM | Life Technologies | A1443001 |
| L-Glutamine | Life Technologies | 25030–081 |
| Sodium pyruvate | Life Technologies | 11360–070 |
| Horse serum | Life Technologies | 16050–122 |
| Standard FBS | Gemini | 900–108 |
| Dialyzed FBS | Gemini | 100–108 |
| HSMM cell growth media (SkGM-2 BulletKit) | Lonza | CC-3245 |
| [1,2-13C]glucose | Sigma-Aldrich | 453188 |
| [3-13C]sodium pyruvate | Sigma-Aldrich | 490733 |
| D-(+)-Glucose | Sigma-Aldrich | G8270 |
| D-(+)-Galactose | Sigma-Aldrich | G0750 |
| D-fructose 1,6-bisphosphate trisodium salt hydrate | Sigma-Aldrich | F6803 |
| Perchloric acid, 70% | Sigma-Aldrich | 311421 |
| Polybrene | Sigma-Aldrich | 107689 |
| Doxycycline hydrochloride | Sigma-Aldrich | D3447 |
| 2-Hydroxyethyl agarose | Sigma-Aldrich | A4018 |
| Bovine serum albumin | Sigma-Aldrich | A7906 |
| MitoSOX | Thermo Fisher Scientific | M36008 |
| Hoechst | Thermo Fisher Scientific | 62249 |
| MitoTracker Green FM | Thermo Fisher Scientific | M7514 |
| Propidium Iodide | Thermo Fisher Scientific | P3566 |
| ProLong Diamond Antifade Mountant with DAPI | Thermo Fisher Scientific | P36962 |
| Halt Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | 78445 |
| FuGENE 6 Transfection Reagent | Promega | E2691 |
| Seahorse XFe96 FluxPaks | Agilent | 102601–100 |
| Seahorse XF Base Medium | Agilent | 102353–100 |
| Matrigel matrix | Corning | 356234 |
| Glutathione Sepharose 4B Resin | GE Healthcare | 17075601 |
| Doxycycline diet - Sterile | Bio-Serv | S3888 |
| Control diet - Sterile | Bio-Serv | S4207 |
| Critical Commercial Assays | ||
| Seahorse XF Cell Mito Stress Test Kit | Agilent | 103015–100 |
| Seahorse XF Glycolysis Stress Test Kit | Agilent | 103020–100 |
| Citrate Synthase Activity Assay Kit | Abcam | ab119692 |
| FITC Annexin V Apoptosis Detection Kit | BD Biosciences | 556547 |
| Deproteinizing Sample Preparation Kit | BioVision | K808 |
| Fructose-6-Phosphate Fluorometric Assay Kit | BioVision | K689–100 |
| Nuclear and Cytoplasmic Extraction Reagents | Thermo Fisher Scientific | 78833 |
| ATP Determination Kit | Thermo Fisher Scientific | A22066 |
| HisPur Ni-NTA Spin Purification Kit | Thermo Fisher Scientific | 88228 |
| Q5 Site-Directed Mutagenesis Kit | New England BioLabs | E0554 |
| Wizard Genomic DNA Purification Kit | Promega | A1120 |
| RNeasy Mini Kit | Qiagen | 74104 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | 4368814 |
| Experimental Models: Cell Lines | ||
| 293T | ATCC | CRL-3216 |
| HT1080 | ATCC | CCL-121 |
| C2C12 | ATCC | CRL-1772 |
| SW872 | ATCC | HTB-92 |
| LPS224 | Core Facilities, MD Anderson Cancer Center, Houston, TX | A gift from Dr. Dina Lev |
| LPS246 | Core Facilities, MD Anderson Cancer Center, Houston, TX | A gift from Dr. Dina Lev |
| T1000 | Established from a well-differentiated liposarcoma | CVCL_M809 |
| KP250 | Derived from UPS mouse tumor | |
| HSMM | Lonza | CC-2580 |
| Experimental Models: Organisms/Strains | ||
| NOD-scid IL2Rgammanull mice | The Jackson Laboratory | 005557 |
| Oligonucleotides | ||
| c-Myc siRNA | Santa Cruz Biotechnology | sc-29226 |
| ON-TARGETplus Non-targeting Pool | Dharmacon | D-001810-10-05 |
| Primers for qRT-PCR, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pCDH-CMV-MCS-EF1-Puromycin | System Biosciences | CD510B-1 |
| pLVX-TetOne-Puro | Clontech | 631847 |
| pLV-EF1a-IRES-Blast | Addgene | 85133 |
| pGEX | A gift from Dr. Xiaolu Yang | |
| pET-28a (+) | A gift from Dr. Xiaolu Yang | |
| FBP2 cDNA | Dharmacon | MHS6278–211687897 |
| TFAM cDNA | Dharmacon | MHS6278–211689190 |
| NRF1 cDNA | Dharmacon | MHS6278–202757860 |
| Software and Algorithms | ||
| GraphPad Prism 7.0 | GraphPad Software |
http://www.graphpad.com |
| ImageJ | NIH |
https://fiji.sc |
| FlowJo | FlowJo |
https://www.flowjo.com |
| Other | ||
| Seahorse XFe96 Analyzer | Agilent | |
| YSI 2950 | YSI | |
| Tissue Tearor | Biospec | 985370 |
Highlights
FBP2 is significantly underexpressed in a variety of STS subtypes
Ectopic FBP2 expression suppresses STS progression
Glycolysis and the pentose phosphate pathway are downregulated by FBP2 re-expression
Nuclear FBP2 impairs mitochondrial biogenesis by suppressing c-Myc activity
Context and Significance
Soft tissue sarcomas (STSs) are diverse cancers that arise from connective tissues. Although they have highly divergent genetic alterations, abnormal glucose metabolism is a universal feature of these lesions, though the mechanism to explain this hallmark has been unclear. Here, Celeste Simon and her colleagues show that fructose-1,6-bisphosphatase 2 (FBP2) is lost in many STS subtypes, and FBP2 re-expression dramatically inhibits sarcoma growth. Furthermore, the team define two independent tumor suppressive functions for FBP2 depending on its subcellular localization: cytosolic FBP2 inhibits glucose catabolism through its catalytic activity, while nuclear FBP2 represses the expression of a key factor in the creation and function of mitochondria. Both these aspects of FBP2 starve the cancer of energy, providing a rationale for why loss of FBP2 is so often seen in STSs and why its re-expression is a therapeutic option.
Acknowledgements
We thank members of Simon lab for helpful discussion and insightful comments on the manuscript. We also thank John Tobias for help with processing the RNA-seq data, and O. Horyn and Ilanna Nissim for performing the isotopomer enrichment analysis and metabolite measurements in the Metabolomics Core Facility, Children’s Hospital of Philadelphia. Finally, Michael S. Nakazawa and T.S. Karin Eisinger-Mathason provided technical assistance. This work was supported by National Cancer Institute (NCI) Grant CA158301 and Guangdong Innovative and Entrepreneurial Research Team Program (2016ZT06S638).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Åsberg C, Hjalmarson O, Alm J, Martinsson T, Waldenström J, and Hellerud C (2010). Fructose 1,6-bisphosphatase deficiency: enzyme and mutation analysis performed on calcitriol-stimulated monocytes with a note on long-term prognosis. J Inherit Metab Dis 33, 113–121. [DOI] [PubMed] [Google Scholar]
- Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, Shah K, Socci ND, Weir BA, Ho A, et al. (2010). Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet 42, 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigl M, Jandrig B, Horn L-C, and Eschrich K (2008). Aberrant methylation of human L- and M-fructose 1,6-bisphosphatase genes in cancer. Biochemical and Biophysical Research Communications 377, 720–724. [DOI] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, and Sabatini DM (2015). An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukouris AE, Zervopoulos SD, and Michelakis ED (2016). Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends in Biochemical Sciences 41, 712–730. [DOI] [PubMed] [Google Scholar]
- Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR, and Dewhirst MW (1996). Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 56, 941–943. [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2017). Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 171, 950–965.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Dasgupta S, and Sidransky D (2011). Mitochondrial Subversion in Cancer. Cancer Prev Res 4, 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Zhang J, Li N, Qian Z, Zhu M, Li Q, Zheng J, Wang X, and Shi G (2011). Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PLoS ONE 6, e25564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KD, Bezwada D, Mashimo T, Pichumani K, Vemireddy V, Funk AM, Wimberly J, McNeil SS, Kapur P, Lotan Y, et al. (2018). Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metabolism 28, 793–800.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, and Li F (2006). The c-Myc target gene network. Semin. Cancer Biol. 16, 253–264. [DOI] [PubMed] [Google Scholar]
- Detwiller KY, Fernando NT, Segal NH, Ryeom SW, D’Amore PA, and Yoon SS (2005). Analysis of hypoxia-related gene expression in sarcomas and effect of hypoxia on RNA interference of vascular endothelial cell growth factor A. Cancer Res. 65, 5881–5889. [DOI] [PubMed] [Google Scholar]
- Dickinson A, Yeung KY, Donoghue J, Baker MJ, Kelly RD, McKenzie M, Johns TG, and John JCS (2013). The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell Death Differ 20, 1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Yuan T, Wu Y, Wang Y, Fan TWM, Miriyala S, Lin Y, Yao J, Shi J, Kang T, et al. (2013). Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 23, 316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisinger-Mathason TSK, Mucaj V, Biju KM, Nakazawa MS, Gohil M, Cash TP, Yoon SS, Skuli N, Park KM, Gerecht S, et al. (2015). Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc. Natl. Acad. Sci. U.S.a. 112, E3402–E3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher CDM (2014). The evolving classification of soft tissue tumours - an update based on the new 2013 WHO classification. Histopathology 64, 2–11. [DOI] [PubMed] [Google Scholar]
- Frith AE, Hirbe AC, and Van Tine BA (2013). Novel pathways and molecular targets for the treatment of sarcoma. Curr Oncol Rep 15, 378–385. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, and Abraham RT (2017). The PI3K Pathway in Human Disease. Cell 170, 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile TL, Lu C, Lodato PM, Tse S, Olejniczak SH, Witze ES, Thompson CB, and Wellen KE (2013). DNMT1 Is Regulated by ATP-Citrate Lyase and Maintains Methylation Patterns during Adipocyte Differentiation. Mol. Cell. Biol 33, 3864–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizak A, and Dzugaj A (2003). FBPase is in the nuclei of cardiomyocytes. FEBS Letters 539, 51–55. [DOI] [PubMed] [Google Scholar]
- Gizak A, Rakus D, and Dzugaj A (2005). Nuclear Localization of Fructose 1,6-bisphosphatase in Smooth Muscle Cells. J Mol Hist 36, 243–248. [DOI] [PubMed] [Google Scholar]
- Gizak A, Wrobel E, Moraczewski J, and Dzugaj A (2006). Changes in subcellular localization of fructose 1,6-bisphosphatase during differentiation of isolated muscle satellite cells. FEBS Letters 580, 4042–4046. [DOI] [PubMed] [Google Scholar]
- Gizak A, Maciaszczyk-Dziubinska E, Maciaszczyk-Dziubinska E, Jurowicz M, Jurowicz M, and Rakus D (2009a). Muscle FBPase is targeted to nucleus by its 203KKKGK 207sequence. Proteins 77, 262–267. [DOI] [PubMed] [Google Scholar]
- Gizak A, Zarzycki M, and Rakus D (2009b). Nuclear targeting of FBPase in HL-1 cells is controlled by beta-1 adrenergic receptor-activated Gs protein signaling cascade. Biochimica Et Biophysica Acta 1793, 871–877. [DOI] [PubMed] [Google Scholar]
- Gobble RM, Qin L-X, Brill ER, Angeles CV, Ugras S, O’Connor RB, Moraco NH, Decarolis PL, Antonescu C, and Singer S (2011). Expression profiling of liposarcoma yields a multigene predictor of patient outcome and identifies genes that contribute to liposarcomagenesis. Cancer Res. 71, 2697–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N (2016). Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer 16, 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata H, Sugimachi K, Komatsu H, Ueda M, Masuda T, Uchi R, Sakimura S, Nambara S, Saito T, Shinden Y, et al. (2016). Decreased Expression of Fructose-1,6-bisphosphatase Associates with Glucose Metabolism and Tumor Progression in Hepatocellular Carcinoma. Cancer Res. 76, 3265–3276. [DOI] [PubMed] [Google Scholar]
- Hosios AM, Hecht VC, Danai LV, Johnson MO, Rathmell JC, Steinhauser ML, Manalis SR, and Vander Heiden MG (2016). Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 36, 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangyang P, and Simon MC (2018). Hidden features: exploring the non-canonical functions of metabolic enzymes. Dis Model Mech 11, dmm033365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humpton TJ, and Vousden KH (2016). Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb Perspect Med 6, a026146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Pan Y, Wang L, Zhang L, Ravichandran R, Potts PR, Jiang J, Wu H, and Huang H (2017). MAGE-TRIM28 complex promotes the Warburg effect and hepatocellular carcinoma progression by targeting FBP1 for degradation. Oncogenesis 6, e312–e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DP, and Scarpulla RC (2004). Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes & Development 18, 357–368. [DOI] [PubMed] [Google Scholar]
- Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, Nielsen GP, Quade BJ, Chaber CJ, Schultz CP, et al. (2007). A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat. Med 13, 992–997. [DOI] [PubMed] [Google Scholar]
- Lee K-M, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, Nixon MJ, Estrada MV, Sánchez V, Sanders ME, et al. (2017). MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metabolism 26, 633–647.e637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WN, Boros LG, Puigjaner J, Bassilian S, Lim S, and Cascante M (1998). Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2–13C2]glucose. Am. J. Physiol 274, E843–E851. [DOI] [PubMed] [Google Scholar]
- Lehnhardt M, Daigeler A, Homann HH, Schwaiberger V, Goertz O, Kuhnen C, and Steinau HU (2009). MFH revisited: outcome after surgical treatment of undifferentiated pleomorphic or not otherwise specified (NOS) sarcomas of the extremities -- an analysis of 140 patients. Langenbecks Arch Surg 394, 313–320. [DOI] [PubMed] [Google Scholar]
- Li B, Li B, Qiu B, Qiu B, Lee DSM, Lee DSM, Walton ZE, Walton ZE, Ochocki JD, Ochocki JD, et al. (2014). Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, Kim J-W, Yustein JT, Lee LA, and Dang CV (2005). Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol 25, 6225–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien EC, Lyssiotis CA, and Cantley LC (2016). Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. Recent Results Cancer Res. 207, 39–72. [DOI] [PubMed] [Google Scholar]
- Linch M, Miah AB, Thway K, Judson IR, and Benson C (2014). Systemic treatment of soft-tissue sarcoma-gold standard and novel therapies. Nat Rev Clin Oncol 11, 187–202. [DOI] [PubMed] [Google Scholar]
- Mehren, von M, Randall RL, Benjamin RS, Boles S, Bui MM, Ganjoo KN, George S, Gonzalez RJ, Heslin MJ, Kane JM, et al. (2018). Soft Tissue Sarcoma, Version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 16, 536–563. [DOI] [PubMed] [Google Scholar]
- Mito JK, Riedel RF, Dodd L, Lahat G, Lazar AJ, Dodd RD, Stangenberg L, Eward WC, Hornicek FJ, Yoon SS, et al. (2009). Cross species genomic analysis identifies a mouse model as undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma. PLoS ONE 4, e8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizunuma H, and Tashima Y (1990). Survey of fructose 1, 6-bisphosphatase isoenzyme in rat organs and ontogenic expression of the enzyme in rat fetus. Int. J. Biochem 22, 883–887. [DOI] [PubMed] [Google Scholar]
- Morrish F, Giedt C, and Hockenbery D (2003). c-MYC apoptotic function is mediated by NRF-1 target genes. Genes & Development 17, 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B, et al. (2015). Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep 10, 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa MS, Eisinger-Mathason TSK, Sadri N, Ochocki JD, Gade TPF, Amin RK, and Simon MC (2016a). Epigenetic re-expression of HIF-2α suppresses soft tissue sarcoma growth. Nat Commun 7, 10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa MS, Keith B, and Simon MC (2016b). Oxygen availability and metabolic adaptations. Nat Rev Cancer 16, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsholme EA, and Crabtree B (1970). The role of fructose-1,6-diphosphatase in the regulation of glycolysis in skeletal muscle. FEBS Letters 7, 195–198. [DOI] [PubMed] [Google Scholar]
- Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, et al. (2008). Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin. Cancer Res. 14, 4726–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissim I, Horyn O, Nissim I, Daikhin Y, Wehrli SL, Yudkoff M, and Matschinsky FM (2012). Effects of a glucokinase activator on hepatic intermediary metabolism: study with 13C-isotopomer-based metabolomics. Biochem. J 444, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remels AHV, Langen RCJ, Schrauwen P, Schaart G, Schols AMWJ, and Gosker HR (2010). Regulation of mitochondrial biogenesis during myogenesis. Mol. Cell. Endocrinol 315, 113–120. [DOI] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, and Capaldi RA (2004). Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 64, 985–993. [DOI] [PubMed] [Google Scholar]
- Sadri N, and Zhang PJ (2013). Hypoxia-inducible factors: mediators of cancer progression; prognostic and therapeutic targets in soft tissue sarcomas. Cancers 5, 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev 88, 611–638. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC, Vega RB, and Kelly DP (2012). Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab 23, 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber M, and Chandel NS (2014). ROS function in redox signaling and oxidative stress. Curr. Biol 24, R453–R462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers K, Fox MP, Bousamra M, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, et al. (2015). Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Invest 125, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JHF, Humberstone DM, and Wolfe RR (1988). Energy and Protein Metabolism in Sarcoma Patients. Annals of Surgery 207, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si W, Huang W, Zheng Y, Yang Y, Liu X, Shan L, Zhou X, Wang Y, Su D, Gao J, et al. (2015). Dysfunction of the Reciprocal Feedback Loop between GATA3- and ZEB2-Nucleated Repression Programs Contributes to Breast Cancer Metastasis. Cancer Cell 27, 822–836. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2019). Cancer statistics, 2019. CA Cancer J Clin 97, 3133. [DOI] [PubMed] [Google Scholar]
- Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, and Ladanyi M (2011). Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer 11, 541–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejwani GA (1983). Regulation of fructose-bisphosphatase activity. Adv. Enzymol. Relat. Areas Mol. Biol 54, 121–194. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, and Dong C (2019). Gluconeogenesis in Cancer: Function and Regulation of PEPCK, FBPase, and G6Pase. Trends Cancer 5, 30–45. [DOI] [PubMed] [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- Weinberg SE, and Chandel NS (2015). Targeting mitochondria metabolism for cancer therapy. 11, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Jin X, Yan Y, Shao Y, Pan Y, Roberts LR, Zhang J, Huang H, and Jiang J (2017). Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep 7, 43864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, and Cantley LC (2008). PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Shi M, Chen H, Gu J, Zhang J, Shen B, Deng X, Xie J, Zhan X, and Peng C (2015). NPM1 activates metabolic changes by inhibiting FBP1 while promoting the tumorigenicity of pancreatic cancer cells. Oncotarget 6, 21443–21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Software
FlowJo (https://www.flowjo.com/) software was used to analyze FACS-based data. For quantification of IHC and IF images, ImageJ (https://imagej.nih.gov/ij/index.html) was used.
Data resources
The RNA-seq data is deposited in GEO and the accession number is GSE137755.