

Summary
Pancreatic endocrine cell development into differentiated α- and β-cells is highly regulated and involves multiple transcription factors. However, the mechanisms behind the determination of α- and β-cell masses remains unclear. We previously identified Foxo1 CoRepressor (FCoR), which inhibits Foxo1 by acetylation. Here we demonstrate that Fcor-knockout mice (FcorKO) exhibit significantly increased α-cell mass, expression of the master α-cell regulatory transcription factor Aristaless-related homeobox (Arx), which can be normalized by β-cell-specific FCoR overexpression (FcorKO-βFcor), and exhibit β-to-α-cell conversion. Compared with FcorKO, β-cell-specific Foxo1 knockout in the FcorKO (DKO) led to decreased Arx expression and α-cell mass. Foxo1 binding to Arx promoter led to DNA methyltransferase 3a (Dnmt3a) dissociation, Arx promoter hypomethylation, and increased Arx expression. In contrast, FCoR suppressed Arx through Foxo1 inhibition and Dnmt3a recruitment to Arx promoter and increased Arx promoter methylation. Our findings suggest that the FCoR-Foxo1 axis regulates pancreatic α-cell mass by suppressing Arx expression.
Subject Areas: Molecular Biology, Endocrinology
Graphical Abstract
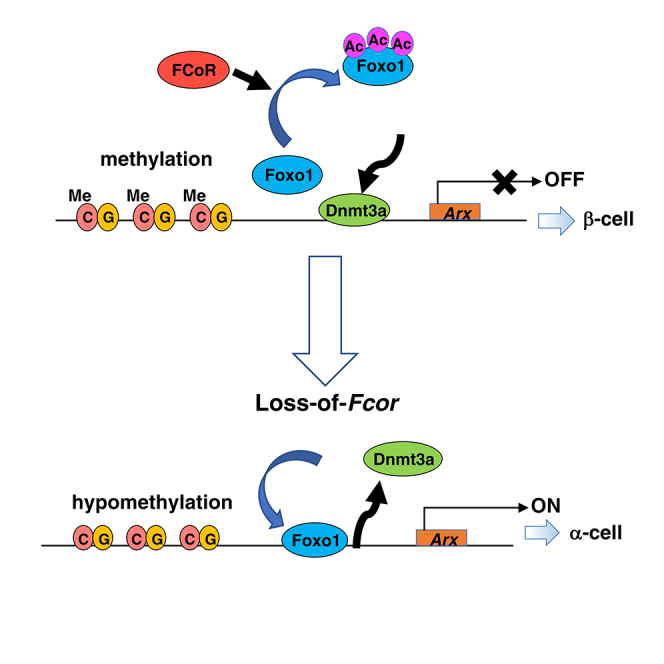
Highlights
-
•
FCoR increases DNA methylation of Arx promoter and decreases Arx expression
-
•
Loss-of-Fcor increases conversion from β- to α-cells
-
•
FCoR acetylates and inhibits Foxo1 in pancreatic β-cells
-
•
Inactivation of Foxo1 facilitates recruitment of Dnmt3a to Arx promoter
Molecular Biology; Endocrinology
Introduction
Type 2 diabetes results from insulin resistance in peripheral insulin responsive tissues, along with relatively deficient insulin secretion from pancreatic β-cells because of β-cell dysfunction and/or decreased mass. Increased glucagon secretion from α-cells is also found in type 2 diabetes (Dunning and Gerich, 2007, Menge et al., 2011). This hyperglucagonemia may result from decreased insulin-induced inhibition of α-cells or from a relative increase in α-cell mass (Dor and Glaser, 2013). Although apoptosis and proliferation regulate β-cell mass, little is known about the regulation of α-cell mass. “Intra-islet plasticity” has been suggested to be involved in the mechanism for regulating β-cell and other endocrine cell masses (Ziv et al., 2013). Glucagon-expressing α-cells or δ-cells can be converted to β-like cells by near-total genetic ablation of β-cells, forced Pax4 expression, or loss of Arx (Thorel et al., 2010) (Collombat et al., 2009) (Courtney et al., 2013). On the other hand, deletion of Dnmt1, Pdx1, or Foxo1 converts β-cells to α-cells or other endocrine cells (Dhawan et al., 2011) (Gao et al., 2014) (Talchai et al., 2012b). However, the precise molecular mechanism of conversions among endocrine cell types remains unknown.
Foxo1 may be a key molecule that determines endocrine cell fate at conversion. The loss of Foxo1 in neurogenin (Ngn)3-positive enteroendocrine progenitors or human fetal pancreatic explants gives rise to insulin-positive cells (Talchai et al., 2012a) (Bouchi et al., 2014). Furthermore, Foxo1 ablation in pancreatic and endocrine progenitors leads to expanded β-cell mass, whereas Foxo1 ablation in terminally differentiated β-cells does not have this effect (Talchai and Accili, 2015). These findings indicate that Foxo1 inhibits β-cell differentiation in endocrine progenitor cells. Foxo1 is phosphorylated and inhibited by insulin/IGF-1 through PI3-kinase/Akt and is translocated into the nucleus and activated by oxidative stress (Accili and Arden, 2004). Within the nucleus, Foxo1 is deacetylated and activated by Sirt1 or Hdacs (Accili and Arden, 2004) (Wang et al., 2011) (Mihaylova et al., 2011) (Banks et al., 2011). We recently identified a Foxo1 co-repressor (Foxo1 CoRepressor; FCoR) in adipocytes, which acetylates and inhibits Foxo1 activity (Nakae et al., 2012).
Here, we demonstrate that FcorKO exhibits significantly increased α-cell mass and expression of the master α-cell regulatory transcription factor Arx, which can be normalized by β-cell-specific FCoR overexpression, and that FCoR suppresses Arx expression through increased methylation of the Arx promoter region by inhibition on Foxo1 activity. In contrast, β-cell-specific Foxo1 knockout in the FcorKO led to decreased Arx expression and α-cell mass through loss of Foxo1-induced Arx expression. These findings indicate that the FCoR-Foxo1 axis regulates pancreatic α-cell mass via Foxo1 inhibition and suggest new avenues for the development of diabetes therapies.
Results
FCoR Is Expressed in Pancreatic Islets
FCoR is a Foxo1-binding protein that was originally identified in mouse adipose tissue. Null FcorKO exhibits glucose intolerance and insulin resistance (Figures S1A and S1B) (Nakae et al., 2012). Although insulin-resistant mice generally show increased insulin secretion as a result of compensated β-cell hypertrophy, we found that FcorKO exhibited decreased glucose-stimulated insulin secretion compared with control (Figure 1A). Additionally, compared with control, FcorKO showed significantly increased glucagon secretion at 15 min after glucose stimulation and in response to L-arginine (Figures 1B and 1C).
Figure 1.

FCoR Is Expressed in Pancreatic Islets
(A) Insulin secretion during an intraperitoneal glucose tolerance test (IPGTT) in control (open circle, n = 7) and FcorKO (closed circle, n = 10). Data represent means ± SEM. *p < 0.05 by two-way ANOVA with Fisher's test.
(B and C) Glucagon secretion during the IPGTT (control, n = 11; FcorKO, n = 15) (B), and L-arginine tolerance test (ATT) (control, n = 5; FcorKO, n = 9) (C). Data represent means ± SEM. *p < 0.05 and **p < 0.005 by two-way ANOVA with Fisher's test.
(D and E) Insulin (D) and glucagon (E) secretion in response to glucose or L-arginine tolerance tests in islets isolated from WT (white bar, n = 6) and FcorKO (black bar, n = 6). Data represent means ± SEM. *p < 0.05 by one-way ANOVA.
(F) Immunofluorescence of pancreatic islets for FCoR and glucagon or FCoR and insulin in islets from the 24-week-old C57Bl6J mouse. Scale bar, 20 μm. DAPI, 4′,6-diamidino-2-phenylindole.
(G and H) Immunofluorescence of pancreatic islets for FCoR and glucagon from mice fed a normal chow and a high-fat diet (G), and from Leprm+/m+ and Leprdb/db (H). Scale bar, 20 μm.
(I and J) Representative images of pancreatic islets for FCoR and insulin (I) or FCoR and glucagon (J) from embryos at embryonic days 14.5 (E14.5), 16.5 (E16.5), and 18.5 (E18.5), and postnatal day 1 (P1). Scale bar, 20 μm.
(K) The percentage of embryonic cells stained with FCoR among glucagon- or insulin-positive cells at embryonic day 14.5 (percentage of FCoR-stained cells/glucagon-positive cells versus FCoR-stained cells/insulin-positive cells 100 ± 4.76: 63.6 ± 12.0, n = 3; 42 glucagon-positive cells, 193 insulin-positive cells counted, consistent for all comparisons), embryonic day 16.5 (34.8 ± 24.3: 84.8 ± 15.2, n = 3; 40 glucagon-positive cells, 36 insulin-positive cells), embryonic day 18.5 (34 ± 11.5: 100 ± 0, n = 3; 71 glucagon-positive cells, 86 insulin-positive cells counted), and P1 (31.3 ± 3.86: 100 ± 0, n = 3; 136 glucagon-positive cells, 78 insulin-positive cells counted). Data represent means ± SEM. *p < 0.05 by one-way ANOVA.
We next isolated islets and examined their insulin and glucagon secretion in response to glucose or L-arginine. Compared with control islets, islets from FcorKO exhibited significantly lower insulin secretion upon stimulation with glucose, whereas insulin secretion in response to L-arginine was similar in islets from FcorKO and control (Figure 1D), suggesting decreased glucose sensing and/or normal insulin secretion after depolarization. However, real-time PCR revealed that expression levels of Slc2a2 and Gck in isolated islets of FcorKO were similar to those of control (Figure S1C) and the insulin content of FcorKO islets was similar to that of control islets (Figure S1D). Therefore, as far as we investigated, we could not reveal the mechanism by which islets from FcorKO have the insensitivity to glucose. Additionally, glucagon secretion after stimulation with glucose was significantly greater in FcorKO islets compared with control islets (Figure 1E). These data suggested that the dysfunction of insulin and glucagon secretion in FcorKO was pancreatic endocrine cell-intrinsic.
In pancreatic tissue from C57Bl6J mice, we performed immunofluorescence analyses for FCoR and glucagon or insulin, which revealed FCoR expression in both α- and β-cells, although FCoR expression in postnatal α-cells was limited (Figure 1F). FCoR moves from cytosol to nucleus through phosphorylation by protein kinase A (PKA) and binds to and acetylates Foxo1 both in cytosol and nucleus (Nakae et al., 2012). On the other hand, the acetylation status of Foxo1 has been known to change intracellular and/or intranuclear localization. Furthermore, deacetylation of Foxo1 by Sirtuins overrides the phosphorylation-dependent nuclear exclusion of Foxo1 caused by growth factors and causes nuclear translocation of Foxo1, leading to activation of Foxo1 (Frescas et al., 2005). Acetylation of Foxo1 by FCoR in cytosol may also inhibit Foxo1 owing to cytosolic retention. Therefore, FCoRs may be active both in cytosol and nucleus. Additionally, to investigate whether FCoR expression in islets was modulated at insulin-resistant state, we examined FCoR protein expression in islets of mice fed a high-fat diet (HFD) or of Leprdb/db. An HFD led to decreased FCoR expression in the islets of C57Bl6J mice (Figure 1G) and FCoR expression was also decreased in Leprdb/db (Figure 1H). These data suggested that FCoR may play some physiological roles in islets.
Furthermore, immunofluorescence analyses in mouse embryos demonstrated co-localization of FCoR in both insulin- and glucagon-positive cells starting at embryonic day 14.5 (Figures 1I–1K). Although FCoR expression was increased in insulin-positive cells after embryonic day 16.5 and was in almost all insulin-positive cells after embryonic day 18.5, FCoR expression was decreased in glucagon-positive cells after embryonic day 14.5 (Figures 1I–1K). At embryonic day 14.5, FCoR was expressed in most of the glucagon-positive cells, which later decreased. In contrast, after embryonic day 18.5, all insulin-positive cells expressed FCoR (Figure 1K). FCoR was not co-localized within neurogenin 3 (Ngn3)-positive cells during these embryonic stages (data not shown). Foxo1 was expressed mainly in glucagon-positive cells until embryonic day 17.5. Furthermore, after that, Foxo1 was expressed in insulin-positive cells rather than in glucagon-positive cells (Figures S1E–S1H). These data suggest that FCoR expression occurs just after the development of pancreatic endocrine progenitors (Puri and Hebrok, 2010) and may be related to the development of pancreatic endocrine cell differentiation and that FCoR and Foxo1 may be co-localized in glucagon-positive cells before embryonic day 15.5.
Fcor Ablation Increases Arx Expression and α-Cell Mass
To investigate the effects of Fcor deletion on pancreatic islets, we examined α-cell and β-cell masses. Immunofluorescence analyses revealed significantly increased α-cell mass in FcorKO compared with control, with no significant difference in β-cell mass (Figures 2A–2C). The sizes did not differ between control and FcorKO islets (data not shown). The architecture of islets from FcorKO was normal, which indicated peripheral localization of α-cells (Figure 2A). In contrast, transgenic mice overexpressing FCoR specifically in β-cells in a FcorKO genetic background (FcorKO-βFcor, developed using RIP-Cre transgenic mice; Figures S2A–S2D) showed an α-cell mass similar to that of controls (Figures 2A–2C). Furthermore, to investigate the effects of FCoR on α-cell mass, we generated transgenic mice overexpressing α-cell-specific FCoR (αFcor) (Figures S2B, S3A, and S3B). Immunofluorescence demonstrated significantly decreased α-cell mass in αFcor compared with controls, but β-cell mass in αFcor was similar to that of control (Figure S3C). These findings indicated that FCoR plays an important role in regulating α-cell mass.
Figure 2.

FCoR Regulates Arx Expression and α-Cell Mass
(A) Immunofluorescence of insulin and glucagon in islets from control, FcorKO, and FcorKO-βFcor at age 24 weeks. Scale bar, 50 μm.
(B and C) Quantification of α-cell (B) and β-cell masses (C) in control (n = 7 mice; 230 islets measured), FcorKO (n = 5 mice; 220 islets measured), and FcorKO-βFcor (n = 3 mice; 80 islets measured). Four sections ∼200 μm apart were covered systematically by accumulating images from non-overlapping fields for whole pancreas sections. Data represent means ± SEM. *p < 0.05 by one-way ANOVA.
(D) Expression levels of α-cell marker genes in islets isolated from control (white bar) (n = 4), FcorKO (black bar) (n = 4), and FcorKO-βFcor (gray bar) (n = 4) at the age of 20–24 weeks. Data represent means ± SEM. *p < 0.05 by one-way ANOVA.
(E and F) Representative images of Arx and glucagon immunostaining of islets from control and FcorKO at age 20 weeks (E), and the percentages of Arx-positive cells among glucagon-positive cells (F). We have calculated Arx-positive and glucagon-positive cells in 3–5 sections from each 5 mice. Data represent means ± SEM. *p < 0.05 by one-way ANOVA. Scale bar, 50 μm.
(G and H) Representative image of immunofluorescence for Arx and insulin in fetal pancreas on embryonic day 15.5 (G), and the ratio of numbers of nuclear Arx-positive cells and of insulin-positive cells per field; control, 0.75 ± 0.20, n = 5 mice, 90 Arx-positive cells and 166 insulin-positive cells counted; FcorKO, 2.20 ± 0.21; n = 5 mice, 261 Arx-positive cells and 114 insulin-positive cells counted (H). Data represent means ± SEM. *p < 0.05 by one-way ANOVA. Scale bar, 20 μm.
(I) The numbers of insulin-positive cells in each 2 section from fetal pancreases of control (n = 5) and FcorKO (n = 5) at embryonic day 15.5. Data represent means ± SEM.
(J) Representative immunofluorescence images of insulin and glucagon staining in fetal pancreas of control and FcorKO at embryonic day 17.5. Scale bar, 200 μm.
(K and L) The α-cell and β-cell numbers of control and FcorKO at embryonic day 17.5. Representative image (K) and β- to α-cell ratio are (L): control, 1 ± 0.24, n = 3, 3 sections for each mouse was measured; FcorKO, 0.59 ± 0.06, n = 3 mice, 3–4 sections for each mouse was measured. Data represent means ± SEM. *p < 0.05 by one-way ANOVA. Scale bar, 50 μm.
(M) Expression levels of β-cell marker genes in islets isolated from control (white bar) (n = 4), FcorKO (black bar) (n = 4), and FcorKO-βFcor (gray bar) (n = 4) at the age of 20–24 weeks. Data represent means ± SEM. *p < 0.05 by one-way ANOVA.
To investigate the mechanism behind the increased α-cell mass in FcorKO, we isolated islets from control, FcorKO, and FcorKO-βFcor and measured the expression levels of α-cell-specific genes. Real-time PCR revealed significantly increased expression of α-cell-specific genes (Gcg, Arx, and Mafb) in FcorKO compared with control (Figure 2D). In contrast, Gcg, Arx, and Mafb expressions in islets isolated from FcorKO-βFcor were similar to control levels (Figure 2D). Furthermore, Arx expression was significantly decreased in αFcor (Figure S3D). Moreover, immunofluorescence analyses of pancreas from 20- to 24-week-old adult mice revealed that the nuclear Arx protein was less detectable in α-cells from control mice, whereas nuclear Arx expression was present in most α-cells in FcorKO (Figures 2E and 2F). These data suggested that FCoR suppressed Arx expression in islets and that Fcor knockout led to increased Arx expression in islets.
To investigate whether the increased α-cell mass observed in FcorKO resulted from increased Arx expression during the embryonic stage, we performed immunofluorescence analysis with anti-Arx antibody. During pancreatic development, the master regulator of pancreatic islet differentiation and regeneration, Ngn3, was expressed in endocrine precursor cells starting at embryonic day 9.5 (Rukstalis and Habener, 2009). Subsequently, the Ngn3-positive cells that were also Arx positive differentiated into α-cells (Collombat et al., 2006). Immunofluorescence analysis revealed significantly more Arx-positive cells in FcorKO compared with controls on embryonic day 15.5 (Figures 2G and 2H). At embryonic day 15.5, there were no significant differences in numbers of insulin-positive cells between control and FcorKO (Figure 2I). Therefore, we counted the numbers of Arx-positive cells corrected by the numbers of insulin-positive cells counted. From embryonic day 16.5 to birth, endocrine cell lineages organize into clusters to form the islets (Habener et al., 2005, Rukstalis and Habener, 2009). Immunofluorescence of the fetal pancreas at embryonic day 17.5 revealed significantly more glucagon-positive cells in FcorKO compared with control (Figures 2J–2L). These results suggested that FCoR regulated Arx expression from the embryonic stage and controlled α-cell mass.
During development of pancreatic endocrine cells, a complex network of transcription factors, including Arx and Pax4, drives endocrine precursor cells toward the different endocrine cell fate. These two transcription factors mutually inhibit the other's transcription and display antagonistic activities for proper endocrine cell lineage allocation (Collombat et al., 2005). Indeed, expression levels of the β-cell-specific gene Pax4 in islets isolated from FcorKO were significantly decreased compared with control and FcorKO-βFcor (Figure 2M). However, mature β-cell-specific marker genes (Pdx1, Mafa, Neurod, and Unc3) expression levels in islets from FcorKO were significantly increased compared with control and FcorKO-βFcor cells (Figure 2M).
FCoR Regulates Arx Expression in Cell Lines
We next investigated the direct effects of FCoR on Arx expression by infecting the α-cell-line αTC1 cells with adenoviruses encoding LacZ or FLAG-tagged FCoR and examining the cellular expression of Arx. Real-time PCR analysis revealed that Fcor expression level in αTC1 cells was significantly lower than in the β-cell line MIN6 (Figure S4A). FCoR overexpression in αTC1 cells significantly and dose-dependently decreased Arx expression (Figure S4B). In contrast, expression levels of the β-cell-specific marker genes, Pax4, Mafa, and Ins1, were significantly increased in FCoR-infected cells compared with LacZ-infected cells, although expression levels of the α-cell-specific marker genes Mafb and Gcg were unaffected (Figure S4C). Furthermore, we examined the effects of FCoR knockdown on Arx expression in MIN6 cells. Arx expression was significantly increased in MIN6 cells infected with adenovirus encoding short hairpin RNA (shRNA) targeting FCoR (which showed 85% knockdown of endogenous FCoR), compared with that in MIN6 cells infected with adenovirus encoding SCR-shRNA (Figure S4D). In contrast, Pax4 and Mafa expression levels were significantly decreased in FCoR-knockdown cells compared with those in SCR shRNA-infected cells. Expression levels of α-cell-specific marker genes Mafb and Gcg tended to be increased in FCoR-knockdown MIN6 cells but not statistically significantly (Figure S4E). These data indicated that FCoR suppresses Arx and increases Pax4 expression.
Fcor Ablation Causes β-to-α-Cell Conversion
To examine whether the increased α-cell mass in FcorKO resulted from increased conversion from β-cells to α-cells, we performed a double immunostaining of EGFP and glucagon using control or FcorKO with the transgene RIP-Cre: EGFP (Figure S5). The confocal microscopy analyses demonstrated that EGFP was stained exclusively in the β-cell area in control mice. One EGFP-positive cell (0.17%) was detected among 574 glucagon-positive cells of 38 islets from three control mice. In contrast, FcorKO exhibited cells co-stained with EGFP and glucagon, indicating conversion from β-cells to α-cells. A total of 90 EGFP-positive cells (2.47%) were detected among 3,644 glucagon-positive cells of 195 islets from five FcorKO mice (Figures 3A and 3B). Moreover, a 3D reconstruction of z stack images of pancreas from a 5-μm section also identified EGFP- and glucagon-double-positive cells (Figure 3C). Furthermore, double immunostaining of insulin and glucagon revealed a significantly increased number of double-positive cells in islet of FcorKO compared with control (Figures 3D and 3E). The presence of these cells indicates conversion from β-cells to α-cells and bihormonal and immature cells.
Figure 3.

Fcor Ablation Causes β-to-α-Cell Conversion
(A) β-Cell lineage tracing of control (CAG-CAT-EGFP:RIP-Cre) and FcorKO (FcorKO:CAG-CAT-EGFP: RIP-Cre) at age 24 weeks. The sections were examined with a confocal microscope (Olympus Fluoview FV3000). White arrows indicate EGFP- and glucagon-double-positive cells. Scale bar, 20 μm.
(B) The percentage of EGFP-positive cells among glucagon-positive cells in islets from control and FcorKO. In control mice, one EGFP-positive cell (0.17%) was detected among 574 glucagon-positive cells of 38 islets from three control mice. In contrast, 90 EGFP-positive cells (2.47%) were detected among 3,644 glucagon-positive cells of 195 islets from five FcorKO.
(C) Z sectioning and 3D reconstruction of a 5-μm section of pancreas from FcorKO:CAG-CAT-EGFP: RIP-Cre shows a glucagon- and EGFP-double-positive cell.
(D and E) Representative image of immunofluorescence for insulin and glucagon in pancreas at the age of 24 weeks (D), and the percentage of numbers of insulin-positive cells among glucagon-positive cells (E); control, 2 sections from each mouse (n = 3), 0 insulin-positive cells among 46 islets counted; FcorKO, 2 sections from each mouse (n = 3), 12 insulin-positive cells among 77 islets counted. Data represent means ± SEM. *p < 0.05 by one-way ANOVA. Scale bar, 50 μm.
To explore the alternative mechanism that α-cells may undergo expansion, we performed Ki67-staining in glucagon-positive cells from control and FcorKO mice at the ages of e15.5, e17.5, and adulthood. However, we detected no recognizable differences in glucagon-positive cells between control and FcorKO (Figure S6). These data indicate that α-cell expansion in FcorKO does not result from increased proliferation of α-cells. Furthermore, to investigate whether β-cells proliferate more to compensate the loss of β-cells due to β-to-α-cell conversion, we performed double staining of insulin and Ki67 of islets using the same samples. However, there were also no significant differences in insulin-positive cells (data not shown). These indicate that there is no increased proliferation of β-cell in FcorKO mice. Moreover, no Ngn3-positive cells were present in islets isolated from adult FcorKO mice, excluding the possibility that the converted cells were not attributable to dedifferentiated cells (data not shown). These results suggested that the conversion from β- to α-cells contributed to increased α-cell mass in FcorKO.
FCoR Is a Regulator of Foxo1 Activity in Islets
FCoR enhances the acetylation of Foxo1 and Foxo3, thus inhibiting their transcriptional activity (Nakae et al., 2012). Indeed, compared with control, islets isolated from FcorKO showed significantly greater deacetylation of transduced ADA-Foxo1, which was a constitutively nuclear mutant Foxo1 (Nakae et al., 2001), and stronger binding to Sirt1 (Figure 4A). Furthermore, immunofluorescence analyses revealed nuclear localization of endogenous Foxo1 in islets from FcorKO, in both α-cells and non-α-cells (Figure 4B), suggesting that Foxo1 may have been active. In addition, western blotting showed that endogenous Foxo1 protein expression level was significantly decreased in islets isolated from FcorKO compared with controls (Figure 4C), consistent with previous findings demonstrating that deacetylated Foxo1 is quickly ubiquitinated and degraded (Kitamura et al., 2005). These data indicate that FCoR is one of the regulators of Foxo1 acetylation and intracellular localization in islets.
Figure 4.

FCoR-Foxo1 Axis Regulates Arx Expression and α-Cell Mass
(A) Transduced Foxo1 deacetylation in islets isolated from FcorKO. Islets isolated from control and FcorKO were transduced with adenoviruses encoding HA-tagged ADA-Foxo1 and harvested at 36 h after transduction. Cell lysates were immunoprecipitated with anti-HA and blotted with the indicated antibodies. The left panel indicates representative western blotting, and the right panel indicates the quantitative analysis of acetylated HA-tagged ADA Foxo1 in islets. The intensity of each band was measured using NIH ImageJ, and the intensity of band of acetylated HA-tagged ADA Foxo1 was corrected by total HA-tagged ADA Foxo1 and calculated as the percentage of control. Data represent means ± SEM from three independent experiments. *p < 0.05 by one-way ANOVA.
(B) Representative immunofluorescence of nuclear localization of endogenous Foxo1 and glucagon-positive cells in islets from 16-week-old control and FcorKO. Scale bar, 20 μm. Insets represent endogenous nuclear Foxo1 and glucagon-negative (left panel) or glucagon-positive cells (right panel). Arrows indicate endogenous nuclear Foxo1-positive cells. Scale bar, 20 μm.
(C) Endogenous Foxo1 protein expression level was decreased in islets isolated from FcorKO compared with control. The top panel indicates representative western blotting, and the bottom panel indicates the quantitative analysis of endogenous Foxo1 in islets. The intensity of each band was measured using NIH ImageJ, and the intensity of band of endogenous Foxo1 was corrected by tubulin and calculated as the percentage of control. Data represent means ± SEM from three independent experiments. *p < 0.05 by one-way ANOVA.
(D) Representative double immunofluorescence of islets from 24-week-old controls and from FcorKO, DKO, and βFoxo1KO using anti-glucagon and anti-FCoR, anti-Foxo1, or anti-insulin antibodies. Scale bar, 50 μm.
(E) Arx expression level in islets isolated from 20- to 24-week-old controls (n = 4) and from β Foxo1KO (n = 4), FcorKO (n = 4), and DKO (n = 4). Data represent means ± SEM from three independent experiments. *p < 0.05 and **p < 0.005 by one-way ANOVA.
(F and G) Quantification of α-cell (F) and β-cell masses (G) in 20- to 24-week-old control (n = 3 mice; 70 islets measured), βFoxo1KO (n = 3; 61 islets measured), FcorKO (n = 4 mice; 102 islets measured), and DKO (n = 4; 98 islets measured). Data represent means ± SEM. *p < 0.05 and **p < 0.005 by one-way ANOVA.
If Foxo1 activation in islets mediates the phenotypes of FcorKO, including Arx expression and α-cell mass, then additional knockout of Foxo1 specifically in β-cells could rescue the FcorKO phenotype. To examine this hypothesis, we generated double-knockout mice, β-cell-specific Foxo1KO (βFoxo1KO) in the background of FcorKO (DKO) (Figure 4D). Real-time PCR of islets isolated from each genotype revealed significantly decreased Foxo1 expression levels in βFoxo1KO and DKO mice compared with control and FcorKO. Foxo3 and Foxo4 expression levels were similar in islets isolated from each genotype, except for increased Foxo3 expression in FcorKO (Figure S7A).
Real-time PCR demonstrated that Arx expression level in islets isolated from DKO was significantly decreased compared with FcorKO and similar to controls. Furthermore, Arx expression level in islets isolated from βFoxo1KO was significantly lower than from any other genotyped islets (Figure 4E). Furthermore, immunofluorescence analyses also revealed that Arx staining in α-cells of FcorKO was more prominent than in control and DKOs. In contrast, Arx staining in α-cells from βFoxo1KO was difficult to be detected (Figures S7B and S7C). These data indicate that increased expression of Arx in islets from FcorKO results from Foxo1 activation in islets and that Foxo1 may have an important role in the regulation of Arx expression. Furthermore, immunofluorescence analyses revealed that the α-cell mass in DKO was significantly decreased compared with that in FcorKO, although it was still significantly increased compared with controls (Figure 4F). The groups did not significantly differ in β-cell mass or number of islets (Figure 4G). These data indicate that increased Arx expression and α-cell mass with the FcorKO genotypes results from Foxo1 activation.
Arx Is a Target Gene of Foxo1
As described above, loss of Foxo1 in β-cells significantly decreased Arx expression compared with control, FcorKO, and DKO, although it did not affect the architecture of islets (Figures 4E–4G). These data led us to speculate that Foxo1 itself may affect endogenous Arx expression. To investigate the influence of Foxo1 on endogenous Arx expression, we infected MIN6 cells with adenovirus encoding constitutively nuclear HA-ADA Foxo1 (Nakae et al., 2001). Real-time PCR demonstrated that Arx expression was significantly increased compared with LacZ-infected control cells (Figure 5A). These data indicated that Foxo1 induced Arx expression. The murine Arx gene contains a conserved forkhead responsive element (FRE) in its promoter region (Figures S8A and S8B). ChIP assay results confirmed that constitutively nuclear Foxo1 mutant (HA-ADA) bound to the Arx promoter region, including FRE, in MIN6 cells (Figure 5B).
Figure 5.

Arx Is a Target Gene of Foxo1
(A) Foxo1 induces Arx expression in MIN6 cells. MIN6 cells were transduced with adenoviruses encoding LacZ (white bar) or HA-ADA-Foxo1 (gray bar) and harvested at 48 h after transduction. Data represent means ± SEM from three independent experiments. *p < 0.05 and **p < 0.005 by one-way ANOVA.
(B) ChIP assay of MIN6 cells transduced with adenoviruses encoding LacZ (white bar) or HA-ADA-Foxo1 (gray bar) and harvested at 48 h after transduction. Samples were subjected to immunoprecipitation with anti-HA, followed by PCR amplification of the UR2 region. Data represent means ± SEM from three independent experiments. *p < 0.05 by one-way ANOVA.
(C) Effect of Foxo1 on Arx promoter activity. Data were obtained from 10 experiments and are shown as mean ± SEM of the fold-change from mock vector-transfected activity. Data represent means ± SEM from three independent experiments. **p < 0.005 by one-way ANOVA.
(D) EMSA of Foxo1 binding to DNA. The DNA probe was a DNA fragment of 31 base pairs covering the consensus Foxo1-binding site (−1552/−1547 nt) of the mouse Arx promoter (lanes 1–6). A mutant DNA with an altered Foxo1-binding motif was used as a control (lanes 7–9). The position of the gel-retarded complex is indicated as A and that of the super-shifted complex is indicated as B.
(E) Oligonucleotide probes corresponding to the Foxo1-binding site of the Arx promoter were incubated with nuclear extracts in the absence or presence of increasing amounts of unlabeled wild-type (lanes 1–3) or mutant oligonucleotide (lanes 4–6). The position of the gel-retarded complex is indicated as A.
To assess whether Foxo1 directly induced Arx expression, we performed the Arx promoter assay in MIN6 cells using Arx promoter regions of various lengths. We found that FLAG-tagged constitutively nuclear mutant Foxo1 (FLAG-3AFoxo1) (Nakae et al., 2006) significantly increased the luciferase activity in the FRE-containing Arx promoter (Figure 5C) but did not do so in the truncated Arx promoter lacking FRE or in the Arx promoter with mutated FRE (Figure 5C). Moreover, site-directed mutagenesis of FRE in the Arx promoter completely abolished Foxo1-induced luciferase activity (Figure 5C).
We next conducted an electrophoretic mobility shift assay (EMSA) to further examine the ability of this putative FRE to bind Foxo1. A cMyc-tagged Foxo1 caused significant retardation of the FRE DNA (Figure 5D, lane 4), and inclusion of the anti-cMyc antibody resulted in a supershifted DNA band (Figure 5D, lane 5). As a control, we performed the same EMSA using a mutant DNA containing five base substitutions within the FRE motif. Alteration of this consensus FRE motif abrogated its ability to bind Foxo1 (Figure 5D, lane 7). We incubated nuclear extracts from cells expressing cMyc-tagged Foxo1 with a probe encoding the 31-base pair FRE DNA sequence, which yielded a slower complex that was competed out by excess cold probe but not mutant probe (Figure 5E). These data indicated that Foxo1 directly induced Arx and that Arx was a target gene of Foxo1.
FCoR Induces DNA Methylation of the Arx Promoter Region
Epigenetic regulation in the Arx promoter region controls Arx expression. The Arx promoter region is methylated, and its expression is suppressed in β-cells, whereas in α-cells, this promoter is hypomethylated and Arx is expressed. One well-studied methylation locus is the CpG-rich UR2 region located upstream of the transcription start site, from −2,103 to around −1,992 nt (Papizan et al., 2011). Bisulfite sequencing analysis of the Arx upstream promoter region revealed that a vast majority of CpG dinucleotides in the UR2 region were methylated in MIN6 cells, but hypomethylated in αTC1 cells (Figure S9A), consistent with previous findings (Dhawan et al., 2011). Intrinsic FCoR expression level was low in αTC1 cells and high in MIN6 cells (Figure S4A). Therefore, to investigate the effects of FCoR on Arx methylation, we overexpressed FCoR in αTC1 cells (through infection with adenovirus encoding FLAG-FCoR) and we knocked down Fcor in MIN6 cells (through infection with adenovirus encoding FCoR-targeting shRNA). Bisulfite sequencing of the UR2 region revealed that methylation in the UR2 locus was significantly increased in FCoR-overexpressing αTC1 cells (Figure 6A) and was significantly decreased in FCoR-knockdown MIN6 cells (Figure 6B). Methylation of the CpG-rich UR2 region enables binding of methyl CpG binding protein 2 (MeCP2) to this area (Dhawan et al., 2011). We next conducted a chromatin immunoprecipitation (ChIP) assay in αTC1 cells infected with adenovirus encoding FLAG-FCoR or LacZ. The results revealed that MeCP2 bound to the UR2 regulatory region of the Arx promoter, whereas we observed no binding in αTC1 cells infected with adenovirus encoding LacZ (Figure S9B).
Figure 6.

FCoR Regulates DNA Methylation of the Arx Promoter
(A) Bisulfite-sequencing analysis of the UR2 region of the Arx promoter in αTC1 cells transduced with adenoviruses encoding LacZ (n = 45) or FLAG-FCoR (n = 50). Quantification of the bisulfite-sequencing data is shown as %DNA methylation.
(B) Bisulfite-sequencing analysis of the UR2 region of the Arx promoter in MIN6 cells transduced with adenoviruses encoding shRNA of scramble sequence (SCR) (n = 45) or FCoR knockdown sequence (n = 50). Quantification of the bisulfite-sequencing data is shown as %DNA methylation. Data represent means ± SEM. *p < 0.05 by one-way ANOVA.
(C and D) Bisulfite-sequencing analysis of the UR2 region of the Arx promoter in islets from control (n = 12, 263 colonies sequenced), FcorKO (n = 12, 274 colonies sequenced) and FcorKO-βFcor (n = 10, 192 colonies sequenced) (C), and quantification of the bisulfite-sequencing data is shown as %DNA methylation (D). Data represent means ± SEM. *p < 0.05 by one-way ANOVA.
DNA methylation requires conducive histone modifications at target genes (Martin and Zhang, 2007). For this reason, we explored histone modification associated with Arx in αTC1 cells overexpressing FCoR. A ChIP assay using anti-H3K4 trimethylation (H3K4me3) antibody, which is implicated in activation of transcription (Kouzarides, 2007), in αTC1 cells overexpressing FCoR showed significantly low levels of H3K4me3 at the UR2 region of Arx promoter compared with control αTC1 cells. This result indicates that FCoR overexpression induces a repressed chromatin state of the UR2 region of Arx promoter (Figure S9C). Furthermore, a ChIP assay using anti-H3K9 trimethylation (H3K9me3) showed significantly low levels of this histone modification at the Arx locus in Fcor-knockdown MIN6 cells compared with SCR-MIN6 cells (Figure S9D). Because H3K9me3 is implicated in the silencing of euchromatic genes as well as in forming silent heterochromatin, which is transcriptionally inert (Berger, 2007), a low level of H3K9me3 in the Arx UR2 region of Fcor-knockdown MIN6 cells indicates an open chromatin state and active transcription of Arx. These data suggest that FCoR can lead to increased DNA methylation and silent chromatin structure of the UR2 region of Arx promoter.
To confirm the effects of FCoR on methylation of the UR2 region of the Arx promoter in vivo, we performed bisulfite sequencing analysis using islets isolated from control, FcorKO, and FcorKO-βFcor. Although bisulfite sequencing data using whole islets may be less informative, the size of islets and β-cell mass of FcorKO were similar to that of control. Therefore, we thought that whole islets bisulfite sequencing data could indicate endogenous status of DNA methylation of Arx promoter in β-cells. Compared with islets from control and FcorKO-βFcor, islets from FcorKO showed significantly fewer methylated sites (Figures 6C and 6D). Higher DNA methylation in islets isolated from FcorKO-βFcor may be the result of excessive expression level of FCoR compared with endogenous FCoR expression level (Figure S2B). These data indicated that FCoR induced methylation of the UR2 region of the Arx promoter.
Foxo1 Inhibits Methylation of the Arx Promoter
As described above, DNA methylation of Arx promoter largely affects Arx gene expression level. Therefore, to investigate whether Foxo1 influences DNA methylation of the Arx promoter or not, we performed bisulfite sequencing to examine the methylation status of Arx promoters from islets isolated from control, FcorKO, DKO, and βFoxo1KO. The results revealed significantly increased methylation of the UR2 region of Arx in islets from βFoxo1KO compared with control, FcorKO, and DKO. Methylation of the Arx promoter in islets from DKO was similar to that of controls and was significantly increased compared with FcorKO (Figures 7A and 7B). These data indicated that Foxo1 decreased Arx promoter methylation and FCoR inhibits Foxo1-induced Arx hypomethylation.
Figure 7.

FCoR-Foxo1-regulated Dnmt3a Recruitment to the Arx Promoter
(A) Bisulfite-sequencing analysis of the UR2 region of the Arx promoter in islets isolated from 20- to 24-week-old control (n = 15, 263 colonies sequenced), βFoxo1 (n = 10, 157 colonies sequenced), FcorKO (n = 15, 274 colonies sequenced), and DKO (n = 10, 158 colonies sequenced).
(B) Quantification of the bisulfite-sequencing data is shown as %DNA methylation. Data represent means ± SEM. *p < 0.05 and **p < 0.005 by one-way ANOVA.
(C) ChIP assay of MIN6 cells transduced with adenoviruses encoding LacZ (white bar) or HA-ADA-Foxo1 (gray bar) and harvested at 48 h after transduction. Samples were subjected to immunoprecipitation with anti-DNMT1or anti-DNMT3A, followed by PCR amplification of the UR2 region. Data represent means ± SEM from three independent experiments. *p < 0.05 and **p < 0.005 by one-way ANOVA.
(D) ChIP assay of αTC1 cells transduced with adenoviruses encoding LacZ (white bar) or FLAG-FCoR (black bar) and harvested at 48 h after transduction. Samples were subjected to immunoprecipitation with anti-DNMT1or anti-DNMT3A, followed by PCR amplification of the UR2 region. Data represent means ± SEM from three independent experiments. *p < 0.05 and **p < 0.005 by one-way ANOVA.
Finally, to examine the molecular mechanism through which Foxo1 decreased methylation of the UR2 region of the Arx promoter, we performed a ChIP assay using MIN6 cells transduced with adenoviruses encoding LacZ or HA-ADAFoxo1. Our results showed significantly decreased Dnmt3a binding to the UR2 region in MIN6 cells transduced with adenovirus encoding HA-ADAFoxo1 compared with LacZ (Figure 7C). In contrast, the ChIP assay revealed that FCoR overexpression in αTC1 cells led to increased recruitment of Dnmt3a (but not Dnmt1) to the UR2 region of the Arx promoter (Figure 7D). These data indicated that Foxo1 bound to and released Dnmt3a from the UR2 region of the Arx promoter and that FCoR increased Dnmt3a recruitment to the UR2 region of the Arx promoter, leading to its increased DNA methylation.
Discussion
Our present results confirmed that FCoR plays important roles in the regulation of pancreatic α-cell mass. FCoR expression starts around embryonic day 14.5 in both insulin- and glucagon-positive cells. However, FCoR expression declines in glucagon-positive cells, so that by embryonic day 18.5, it is restricted to insulin-positive cells. This differential pattern promotes increased Arx expression in glucagon-positive cells and decreased Arx expression in insulin-positive cells, resulting in determination of terminally mature endocrine cells.
Of note, FCoR and Foxo1 are expressed in both insulin- and glucagon-positive cells during differentiation. We could not co-stain cells with FCoR and Foxo1 because antibodies for both FCoR and Foxo1 that we used are anti-rabbit antibodies. However, as the developmental phase proceeds, Foxo1 expression becomes restricted in insulin-positive cells, as does FCoR. Interestingly, Foxo1 was strongly co-stained with glucagon-positive cells at embryonic day 15.5, the early phase of endocrine progenitor cells. In contrast, FCoR expression in glucagon-positive cells after embryonic day 14.5 was declined. These data are consistent with the previous report of Foxo1 expression in embryonic glucagon-positive cells (Kitamura et al., 2009) and that Foxo1 may be activated due to loss of FCoR in α-cell-committed cells and facilitate α-cell differentiation at early phase of endocrine progenitor cells. FCoR loss results in decreased acetylation and increased activation of Foxo1, namely, increased nuclear Foxo1 because FCoR is a co-repressor and main acetyltransferase for Foxo1 in islets. From the present study, the most important significance of nuclear Foxo1 from Fcor loss in pancreatic endocrine cells was the induction of Arx expression. Especially, in fetal pancreatic endocrine cells, until embryonic day 14.5, FCoR was expressed in both α- and β-cells. Therefore, FCoR can inactivate Foxo1, and Arx expression is suppressed in pancreatic endocrine precursors. However, after embryonic day 14.5, FCoR expression in α-cells gradually declined, leading to Foxo1 activation and Arx induction, resulting in fate determination similar to α-cells.
Indeed, loss of Fcor causes Foxo1 activation and increased Arx expression in both differentiating pancreatic α-cells and insulin-positive β-cells, increasing both the α-cell generation and the conversion of β- to α-cells, ultimately raising the proportion of α-cell mass. However, the architecture of islets from the FcorKO genotype is almost normal, indicating the peripheral localization of α-cells. Several cases of β- to α-cell conversion have demonstrated that α-cells are localized in the core of the islets (Lu et al., 2018). Furthermore, the ratio of EGFP-positive cells among glucagon-positive cells was relatively low (2.47%). This low ratio of β-to-α-cell conversion might result from the period of activation of rat insulin promoter in RIP-Cre mice. It has been reported that rat insulin promoter was active in terminally differentiated β-cells (Talchai and Accili, 2015). In contrast, Fcor deletion is constitutive and occurs before defining a particular cell fate of pancreatic endocrine cells. Therefore, we could detect β-to-α-cell conversion only in adult β-cells. These findings indicate that conversion from β-cells to α-cells in the FcorKO genotype might occur in the earlier stages during differentiation of pancreatic endocrine cells and that the architecture of islets therefore should be normal. However, it is also possible that the increase in α-cell mass is due to unrestrained growth of those cells in the earlier stages during differentiation of pancreatic endocrine cells because increased Foxo1 activity due to loss-of-Fcor in embryonic α-cells may lead to α-cell hyperplasia (Kitamura et al., 2009), although we could not detect increased proliferation of α-cells in FcorKO at embryonic day 15.5.
The use of RIP-Cre to trigger FCoR overexpression specifically in β-cells of FcorKO (FcorKO-βFcor) normalized the proportion of α-cell mass attributable to the inhibition of Arx expression in cells that would otherwise have been converted from β- to α-cells. In contrast, the use of RIP-Cre to specifically delete Foxo1 in terminally differentiated β-cells does not alter the proportion of α-cells (Talchai and Accili, 2015) because decreased Arx expression due to loss-of-Foxo1 from terminally differentiated β-cells does not influence the β-cell fate. On the other hand, in αFcor, FCoR overexpression in glucagon-positive cells significantly decreased the α-cell mass compared with controls. This effect may become manifest just before the establishment of differentiated pancreatic endocrine cells, because endogenous glucagon-positive cells are reportedly detectable at embryonic day 9.5, before detection of the first insulin-positive cells at embryonic day 10.5–11 (Herrera et al., 2002). These findings suggest that FCoR overexpression using Glucagon-Cre leads to a decrease in the α-cell lineage itself and thus to a significantly decreased α-cell mass compared with controls. Of interest, DKO mice exhibited significantly greater α-cell mass compared with controls, although Arx expression level was normalized. Because FcorKO represents null knockout mice, in these animals, deletion of Fcor has occurred not only in the β-cell lineage but also in the α-cell lineage, resulting in increased α-cell lineage during fetal development of pancreatic endocrine cells. Therefore, even if deletion of Foxo1 specifically in Fcor-deleted β-cells prevents β-to-α-cell conversion, it does not affect α-cell lineage increases. These data indicate that FCoR regulates Arx expression through Foxo1. However, we cannot exclude the possibility that other factors mediate the function of FCoR in islets. Further investigation should be needed.
During the development of pancreatic endocrine cells, next to Ngn3 induction, a complex network of transcription factors, including Arx and Pax4, differentially promotes the specific endocrine fates. These two transcription factors, Arx and Pax4, have mutually antagonistic effects. The amount of Pax4 expression is up-regulated in Arx mutant mice, whereas the Arx expression is increased in Pax4-deficient pancreas (Collombat et al., 2003). This is consistent with the present study. Pax4 expression in FcorKO islets was significantly decreased compared with control islets. Furthermore, overexpression of FCoR in αTC1 cells significantly increased Pax4 expression but decreased Arx expression. In contrast, knockdown of Fcor in MIN6 cells significantly increased Arx expression but decreased Pax4 expression. However, interestingly, FcorKO islets exhibit significantly increased expression of mature β-cell markers, including Pdx1, Mafa, Neurod, and Ucn3, even if Pax4 expression is decreased. These findings in FcorKO islets are not consistent with decreased Mafa expression in Fcor-knockdowned-MIN6 cells. We speculate that decreased Mafa expression in Fcor-knockdowned-MIN6 cells is an acute effect of Fcor knockdown but that Fcor deletion in FcorKO is constitutive and has chronic effects on gene expression in islets, leading to compensatory induction of mature β-cell markers. Indeed, Mafa expression is regulated by several transcription factors, including Foxa2, Nkx2.2, and Pdx1, and glucose levels (Vanderford, 2011). Therefore, chronic effects resulting from Fcor deletion may affect expression levels of mature β-cell markers because null FcorKO exhibits glucose intolerance due to insulin resistance in adipose tissues (Nakae et al., 2012). Furthermore, these findings might indicate that the effect on glucose-stimulated insulin secretion in FcorKO may come from an α-cell-centric phenotype and these changes in β-cell-maturity-markers may be compensatory, resulting in the generation of dysfunctional α-cells, or the generation of dysfunctional β-cells, leading to decreased insulin secretion. Indeed, bihormonal or immature cells were increased significantly in islets of FcorKO compared with control. However, further investigation is needed to elucidate the precise molecular mechanism by which loss-of-Fcor induces the generation of dysfunctional β-cells. Furthermore, as already reported, activation of Foxo1 in pancreatic β-cells leads to protection of β-cells against oxidative stress through formation of a complex with the promyelocytic leukemia protein Pml and Sirt1 to activate expression of Mafa and Neurod (Kitamura et al., 2005) (Kobayashi et al., 2012). This finding is consistent with our present data, which indicate increased expression levels of Mafa and Neurod in islets isolated from FcorKO.
The molecular mechanism by which DNA methylation regulates Arx expression has been reported. In one proposed model, a repressor complex that includes Nkx2.2, Grg3, and Dnmt3a preferentially recruits HDAC1 to the Arx promoter to silence Arx expression specifically in the pancreatic β-cells, preventing β-to-α-cell conversion (Dhawan et al., 2011, Papizan et al., 2011). Of interest, the FRE in the Arx promoter is close to the consensus Nkx2.2-binding site (Figure S8A). The binding of Foxo1 to the FRE may dissociate the repressor complex from the Arx promoter, leading to Arx promoter hypomethylation and induction of Arx expression. In contrast, inhibition of Foxo1 by FCoR may make the repressor complex assemble on the Arx promoter, leading to hypermethylation as shown in the present study. Furthermore, our present findings confirmed that the binding of Foxo1 to the Arx promoter led to Dnmt3a dissociation from the Arx promoter. However, we performed bisulfite sequencing analysis using whole islets from each genotype. Therefore, these data may be affected by differences in the α-/β-cell ratios. But, because β-cell mass of each genotype is similar and the absolute value of α-cell mass is relatively low, the differences of α-/β-cell ratios may slightly affect the results of bisulfite sequencing data.
In the present study, the findings of significantly decreased acetylation of transduced Foxo1 in FcorKO islets are notable. Deacetylated Foxo1 can be imported to the nucleus, where it can bind to the FREs of its target genes, and should be active transcriptionally (Matsuzaki et al., 2005) (Qiang et al., 2010). The main acetyltransferases of Foxo1 are reported to be CBP, p300, and PCAF (Brunet et al., 2004) (Motta et al., 2004) (Yoshimochi et al., 2010). However, little is known about the regulation of Foxo1 acetylation in islets, although the mechanism by which Foxo1 in islets is deacetylated is well described (Kitamura et al., 2005). The present data indicate that FCoR is one of the regulators of Foxo1 acetylation in islets and works upstream of Foxo1.
In conclusion, here we provide evidence that the FCoR-Foxo1 axis is an important determinant in regulating the identities of pancreatic α-cell. Elucidating the molecular mechanisms underlying the regulation of α- and β-cell masses will be an important step toward developing strategies to cure diabetes. Our present findings indicate that FCoR offers a promising target molecule for the development of therapies for type 2 diabetes.
Limitations of the Study
In the present study, we demonstrated that FCoR increased DNA methylation of Arx promoter using the whole islets. However, to clarify DNA methylation of Arx promoter in β-cells, we should perform bisulfite sequencing analysis using the sorted β-cells. Furthermore, ChIP analysis of Dnmt3a should be performed using isolated islets. These experiments will reveal the conversion of β- to α-cells more precisely.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Jun-ichi Miyazaki (Osaka University, Osaka, Japan) for providing the CAG-CAT-EGFP mice and pBS II SK (+)-CAG-CAT-EGFP vector, Dr. Ken-ichirou Morohashi and Dr. Kanako Miyabayashi (Kyushu University, Fukuoka, Japan) for providing anti-Arx antibody, Dr. Naoki Mochizuki (National Cerebral and Cardiovascular Center, Osaka, Japan) for providing anti-EGFP antibody, and Dr. Ronald A DePinho (University of Texas MD Anderson Cancer Center, Houston, USA) and Dr. Domenico Accili (Columbia University, New York, USA) for providing the Foxo1flox/flox mice. This work was supported by Scientific Research on Innovative Areas, a MEXT Grant-in-Aid Project "Crosstalk between transcriptional control and energy pathways, mediated by hub metabolites" grant numbers 26116724 and JSPS KAKENHI 26670509 to J.N., and by a grant from Nippon Boehringer Ingelheim Co., Ltd. to H.I.
Author Contributions
J.N. designed the experiments. N.K., M.K., O.K., and J.N. performed the experiments investigating physiological and molecular phenotypes. T.K. prepared the embryonic pancreas samples. N.K. and J.N. wrote the manuscript. H.I. provided detailed comments regarding the manuscript.
Declaration of Interests
The authors declare that they have no competing interests.
Published: January 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.100798.
Supplemental Information
References
- Accili D., Arden K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- Banks A.S., Kim-Muller J.Y., Mastracci T.L., Kofler N.M., Qiang L., Haeusler R.A., Jurczak M.J., Laznik D., Heinrich G., Samuel V.T. Dissociation of the glucose and lipid regulatory functions of FoxO1 by targeted knockin of acetylation-defective alleles in mice. Cell Metab. 2011;14:587–597. doi: 10.1016/j.cmet.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S.L. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Bouchi R., Foo K.S., Hua H., Tsuchiya K., Ohmura Y., Sandoval P.R., Ratner L.E., Egli D., Leibel R.L., Accili D. FOXO1 inhibition yields functional insulin-producing cells in human gut organoid cultures. Nat. Commun. 2014;5:4242. doi: 10.1038/ncomms5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Sweeney L.B., Sturgill J.F., Chua K.F., Greer P.L., Lin Y., Tran H., Ross S.E., Mostoslavsky R., Cohen H.Y. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Collombat P., Hecksher-Sorensen J., Broccoli V., Krull J., Ponte I., Mundiger T., Smith J., Gruss P., Serup P., Mansouri A. The simultaneous loss of Arx and Pax4 genes promotes a somatostatin-producing cell fate specification at the expense of the alpha- and beta-cell lineages in the mouse endocrine pancreas. Development. 2005;132:2969–2980. doi: 10.1242/dev.01870. [DOI] [PubMed] [Google Scholar]
- Collombat P., Hecksher-Sorensen J., Serup P., Mansouri A. Specifying pancreatic endocrine cell fates. Mech. Dev. 2006;123:501–512. doi: 10.1016/j.mod.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Collombat P., Mansouri A., Hecksher-Sorensen J., Serup P., Krull J., Gradwohl G., Gruss P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17:2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P., Xu X., Ravassard P., Sosa-Pineda B., Dussaud S., Billestrup N., Madsen O.D., Serup P., Heimberg H., Mansouri A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell. 2009;138:449–462. doi: 10.1016/j.cell.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney M., Gjernes E., Druelle N., Ravaud C., Vieira A., Ben-Othman N., Pfeifer A., Avolio F., Leuckx G., Lacas-Gervais S. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet. 2013;9:e1003934. doi: 10.1371/journal.pgen.1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan S., Georgia S., Tschen S.I., Fan G., Bhushan A. Pancreatic beta cell identity is maintained by DNA methylation-mediated repression of Arx. Dev. Cell. 2011;20:419–429. doi: 10.1016/j.devcel.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y., Glaser B. Beta-cell dedifferentiation and type 2 diabetes. N. Engl. J. Med. 2013;368:572–573. doi: 10.1056/NEJMcibr1214034. [DOI] [PubMed] [Google Scholar]
- Dunning B.E., Gerich J.E. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr. Rev. 2007;28:253–283. doi: 10.1210/er.2006-0026. [DOI] [PubMed] [Google Scholar]
- Frescas D., Valenti L., Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- Gao T., McKenna B., Li C., Reichert M., Nguyen J., Singh T., Yang C., Pannikar A., Doliba N., Zhang T. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab. 2014;19:259–271. doi: 10.1016/j.cmet.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habener J.F., Kemp D.M., Thomas M.K. Minireview: transcriptional regulation in pancreatic development. Endocrinology. 2005;146:1025–1034. doi: 10.1210/en.2004-1576. [DOI] [PubMed] [Google Scholar]
- Herrera P.L., Nepote V., Delacour A. Pancreatic cell lineage analyses in mice. Endocrine. 2002;19:267–278. doi: 10.1385/ENDO:19:3:267. [DOI] [PubMed] [Google Scholar]
- Kitamura T., Kitamura Y.I., Kobayashi M., Kikuchi O., Sasaki T., Depinho R.A., Accili D. Regulation of pancreatic juxtaductal endocrine cell formation by FoxO1. Mol. Cell. Biol. 2009;29:4417–4430. doi: 10.1128/MCB.01622-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y.I., Kitamura T., Kruse J.P., Raum J.C., Stein R., Gu W., Accili D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Kobayashi M., Kikuchi O., Sasaki T., Kim H.J., Yokota-Hashimoto H., Lee Y.S., Amano K., Kitazumi T., Susanti V.Y., Kitamura Y.I. FoxO1 as a double-edged sword in the pancreas: analysis of pancreas- and beta-cell-specific FoxO1 knockout mice. Am. J. Physiol. Endocrinol. Metab. 2012;302:E603–E613. doi: 10.1152/ajpendo.00469.2011. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lu T.T., Heyne S., Dror E., Casas E., Leonhardt L., Boenke T., Yang C.H., Sagar L., Arrigoni L., Dalgaard K. The Polycomb-dependent epigenome controls beta cell dysfunction, dedifferentiation, and diabetes. Cell Metab. 2018;27:1294–1308.e7. doi: 10.1016/j.cmet.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C., Zhang Y. Mechanisms of epigenetic inheritance. Curr. Opin. Cell Biol. 2007;19:266–272. doi: 10.1016/j.ceb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H., Daitoku H., Hatta M., Aoyama H., Yoshimochi K., Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. U S A. 2005;102:11278–11283. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menge B.A., Gruber L., Jorgensen S.M., Deacon C.F., Schmidt W.E., Veldhuis J.D., Holst J.J., Meier J.J. Loss of inverse relationship between pulsatile insulin and glucagon secretion in patients with type 2 diabetes. Diabetes. 2011;60:2160–2168. doi: 10.2337/db11-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova M.M., Vasquez D.S., Ravnskjaer K., Denechaud P.D., Yu R.T., Alvarez J.G., Downes M., Evans R.M., Montminy M., Shaw R.J. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–621. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta M.C., Divecha N., Lemieux M., Kamel C., Chen D., Gu W., Bultsma Y., McBurney M., Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- Nakae J., Cao Y., Daitoku H., Fukamizu A., Ogawa W., Yano Y., Hayashi Y. The LXXLL motif of murine forkhead transcription factor FoxO1 mediates Sirt1-dependent transcriptional activity. J. Clin. Invest. 2006;116:2473–2483. doi: 10.1172/JCI25518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae J., Cao Y., Hakuno F., Takemori H., Kawano Y., Sekioka R., Abe T., Kiyonari H., Tanaka T., Sakai J. Novel repressor regulates insulin sensitivity through interaction with Foxo1. EMBO J. 2012;31:2275–2295. doi: 10.1038/emboj.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae J., Kitamura T., Silver D.L., Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Invest. 2001;108:1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papizan J.B., Singer R.A., Tschen S.I., Dhawan S., Friel J.M., Hipkens S.B., Magnuson M.A., Bhushan A., Sussel L. Nkx2.2 repressor complex regulates islet beta-cell specification and prevents beta-to-alpha-cell reprogramming. Genes Dev. 2011;25:2291–2305. doi: 10.1101/gad.173039.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri S., Hebrok M. Cellular plasticity within the pancreas–lessons learned from development. Dev. Cell. 2010;18:342–356. doi: 10.1016/j.devcel.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang L., Banks A.S., Accili D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J. Biol. Chem. 2010;285:27396–27401. doi: 10.1074/jbc.M110.140228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rukstalis J.M., Habener J.F. Neurogenin3: a master regulator of pancreatic islet differentiation and regeneration. Islets. 2009;1:177–184. doi: 10.4161/isl.1.3.9877. [DOI] [PubMed] [Google Scholar]
- Talchai C., Xuan S., Kitamura T., DePinho R.A., Accili D. Generation of functional insulin-producing cells in the gut by Foxo1 ablation. Nat. Genet. 2012;44:406–412. doi: 10.1038/ng.2215. S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai C., Xuan S., Lin H.V., Sussel L., Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai S.C., Accili D. Legacy effect of Foxo1 in pancreatic endocrine progenitors on adult beta-cell mass and function. Diabetes. 2015;64:2868–2879. doi: 10.2337/db14-1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorel F., Nepote V., Avril I., Kohno K., Desgraz R., Chera S., Herrera P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464:1149–1154. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderford N.L. Regulation of beta-cell-specific and glucose-dependent MafA expression. Islets. 2011;3:35–37. doi: 10.4161/isl.3.1.14032. [DOI] [PubMed] [Google Scholar]
- Wang B., Moya N., Niessen S., Hoover H., Mihaylova M.M., Shaw R.J., Yates J.R., 3rd, Fischer W.H., Thomas J.B., Montminy M. A hormone-dependent module regulating energy balance. Cell. 2011;145:596–606. doi: 10.1016/j.cell.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimochi K., Daitoku H., Fukamizu A. PCAF represses transactivation function of FOXO1 in an acetyltransferase-independent manner. J. Recept. Signal Transduct. Res. 2010;30:43–49. doi: 10.3109/10799890903517947. [DOI] [PubMed] [Google Scholar]
- Ziv O., Glaser B., Dor Y. The plastic pancreas. Dev. Cell. 2013;26:3–7. doi: 10.1016/j.devcel.2013.06.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.