

Abstract
MicroRNAs (miRNAs) play an essential role in the self-renewal of breast cancer stem cells (BCCs). Our study aimed to clarify the role of proto-oncogene c-Jun-regulated miR-5188 in breast cancer progression and its association with Timeless-mediated cancer stemness. In the present study, we showed that miR-5188 exerted an oncogenic effect by inducing breast cancer stemness, proliferation, metastasis, and chemoresistance in vitro and in vivo. The mechanistic analysis demonstrated that miR-5188 directly targeted FOXO1, which interacted with β-catenin in the cytoplasm, facilitated β-catenin degradation, and impaired the nuclear accumulation of β-catenin, thus stimulating the activation of known Wnt targets, epithelial-mesenchymal transition (EMT) markers, and key regulators of cancer stemness. Moreover, miR-5188 potentiated Wnt/β-catenin/c-Jun signaling to promote breast cancer progression. Interestingly, c-Jun enhanced miR-5188 transcription to form a positive regulatory loop, and Timeless interacted with Sp1/c-Jun to induce miR-5188 expression by promoting c-Jun-mediated transcription, which further activated miR-5188-FOXO1/β-catenin-c-Jun loop and facilitated breast cancer progression. Importantly, miR-5188 was upregulated in breast cancer and was positively correlated with poor patient prognosis. This study identifies miR-5188 as a novel oncomiR and provides a new theoretical basis for the clinical use of miR-5188 antagonists in the treatment of breast cancer.
Keywords: miR-5188, timeless, FOXO1, β-catenin, ubiquitination, stemness, cancer stem cells, breast cancer
Graphical Abstract
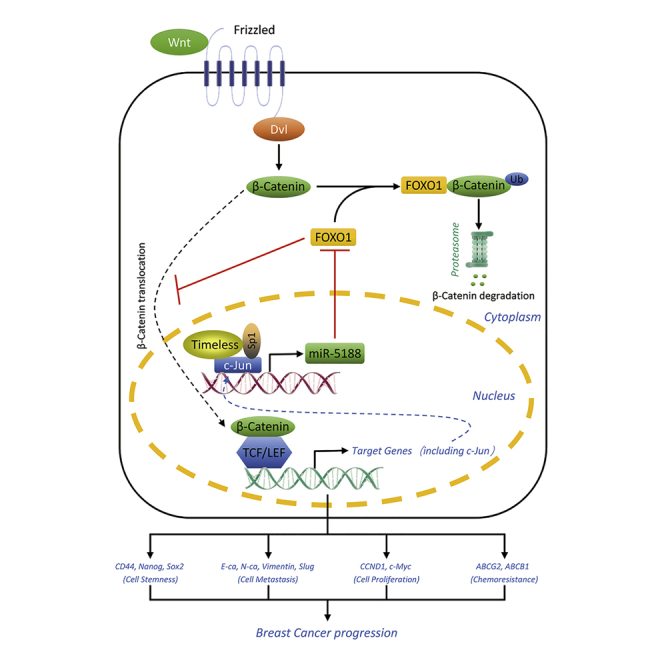
Molecular targeting therapy has become the most prevalent strategy for cancer treatment. miR-5188 is a significant inducer of cancer stemness that promotes breast cancer pathogenesis. miR-5188 may be a novel factor for the diagnosis and prognosis of breast cancer and may serve as a useful therapeutic target for breast cancer.
Introduction
Breast cancer is currently the second most common cancer in women worldwide and poses a serious health threat.1 Cancer stem cells (CSCs) are responsible for cancer recurrence, metastasis, and therapy resistance and are associated with poor patients’ prognosis in breast cancer.2, 3 Therefore, investigating the underlying mechanisms of CSC regulation is essential for the development of alternative breast cancer treatments.
c-Jun, a well-known oncogene, plays vital role in malignant transformation of different cancers, highly activated in breast cancer. Previous studies have indicated that JUN expression levels were positively correlated with breast cancer lung metastasis (BCLM)-associated gene expression and lung metastases in breast cancer patients.4 Furthermore, activation of c-Jun could regulate breast cancer tumorigenesis by promoting the CSC phenotype.5 However, the downstream molecules involved c-Jun signaling in breast cancer remains unclear.
MicroRNAs (miRNAs) are critical components of the noncoding RNA family and involved in multiple cellular functions.6 miRNAs are known to regulate initiation and progression of cancers, including breast cancer.7, 8, 9, 10 High miR-34a expression inhibited the tumor-initiating properties and proliferation of long-term maintenance breast CSCs derived from either clinical specimens or cell lines.11 Another study showed that VEGFA promoted breast CSC self-renewal and metastasis via downregulating SOX2-mediated expression of miR-452, which appeared to be a novel metastatic suppressor directly targeting Slug.12 Based on the above evidence, we concluded that miRNA may play a vital role in breast cancer stemness. Hence, we searched miRNAs regulated by the proto-oncogene c-Jun based on the chromosome immunoprecipitation sequencing (ChIP-seq) data in the Cistrome data browser (http://cistrome.org/db/) and identified miR-5188 as a potential miRNA among the top putative targets with a promoter region harboring c-Jun binding sites. However, the biological role of miR-5188 in the regulation of breast cancer stemness has never been reported.
Wnt/β-catenin signaling is required for the maintenance of breast cancer cell stemness.13 Moreover, Timeless was an oncogenic driver that induced breast cancer stemness,14 and was shown to promote epithelial-mesenchymal transition (EMT) and chemoresistance through Wnt/β-catenin signaling.15 However, whether Timeless exerts its functions by regulating breast cancer stemness via a specific miRNA is not fully understood.
Here, we identified a novel miR-5188-FOXO1/β-catenin-c-Jun feedback circuit, which was modulated by the Timeless/Sp1/c-Jun complex, and promoted breast cancer stemness, metastasis, proliferation, and chemoresistance. Data obtained from breast cancer cell lines, xenografts, and clinical samples consistently provided detailed information on the mechanisms by which miR-5188 exerted promotive effects on breast cancer progression through the Wnt/β-catenin/c-Jun signaling. Thus, targeting miR-5188 could be a practical approach for breast cancer treatments.
Results
miR-5188 Is Elevated in Breast Cancer and Predicts a Poor Prognosis in Breast Cancer Patients
c-Jun is well characterized as a proto-oncogene. To explore the roles of miRNAs in the development of breast cancer, we systematically searched c-Jun-regulated miRNAs in the Cistrome data browser database (http://cistrome.org/db/). Four published articles with the highest quality ChIP-seq data were enrolled in this study.16, 17, 18, 19 miR-5188 was screened as a potential miRNA among the top putative targets transcripted by c-Jun in breast cancer (Figure 1A). Moreover, bioinformatics analysis of breast invasive carcinoma (BRCA) data from the cancer genome atlas (TCGA) showed that compared with paracarcinoma tissues, miR-5188 levels were upregulated in breast cancer tissues (p = 0.0042) (Figure 1B). Intriguingly, miR-5188 expression was positively correlated with Oct4 and Nanog expression (Figure 1C). Furthermore, gene set enrichment analysis (GSEA) based on the TCGA BRCA dataset showed that miR-5188 negatively participated in regulating FOXO signaling and was positively associated with cell cycle regulation, DNA replication, transcription, cancer metastasis, and improved breast cancer outcomes (Figure 1D).
Figure 1.

The Bioinformatics Analysis of c-Jun-Regulated miR-5188 Based on the ChIP-Seq and TCGA Database
(A) A Venn diagram analysis was screened for c-Jun-regulated downstream effectors among the top putative targets. (B) Comparison of miR-5188 expression between breast cancer and para-carcinoma tissues. (C) The relationships between Oct4, Nanog, and miR-5188 expression (Spearman’s rank correlation test). (D) GSEA of miR-5188 shows significant enrichment of the gene set involved in the regulation of FOXO signaling, the cell cycle, DNA replication, transcription, cancer metastasis, and improved outcome of breast cancer patients. (E and F) Comparison of miR-5188 expression between breast cancer tissues and para-carcinoma tissues (E) and between tissues from patients with and without recurrence (F). Scale bars, 10 μm. (G) Overall survival analysis of breast cancer patients based on miR-5188 expression (log-rank test). (H) The analysis of relapse-free survival of breast cancer patients based on miR-5188 expression (log-rank test). The lines indicate median values, and the whiskers indicate minimum and maximum values (B), Wilcoxon rank sum test. ChIP-seq, chromatin immunoprecipitation sequencing; GSEA, gene set enrichment analysis; TCGA, the cancer genome atlas.
Based on these TCGA data mining results, we then detected miR-5188 levels by in situ hybridization in 140 breast cancer and 77 paracarcinoma tissues. The results from the clinical samples demonstrated that miR-5188 was upregulated in breast cancer tissues compared with paracarcinoma tissues (p = 0.002) (Figure 1E; Table S1). Interestingly, miR-5188 expression was higher in samples from patients with recurrent disease than that in samples from recurrence-free patients (p = 0.035) (Figure 1F; Table 1). Consistently, quantitative real-time PCR results revealed that miR-5188 was highly expressed in six breast cancer cell lines (MCF-7, SKBR-3, T47D, MDA-MB-468, MDA-MB-231, and MDA-MB-453) compared with the noncancerous immortalized breast tissue cell line, MCF-10A (Figure S1A). The relationships between miR-5188 expression and clinicopathological characteristics are summarized in Table 1. High miR-5188 expression was positively correlated with tumor recurrence (p = 0.035), but not correlated with other clinicopathological features.
Table 1.
Correlations between miR-5188 Expression and the Clinicopathological Features of Breast Cancer Patients
| miR-5188 Expression |
||||
|---|---|---|---|---|
| Characteristics | n | Low | High | p Value |
| Age (Years) | ||||
| ≤Median | 71 | 32 (45.1%) | 39 (54.9%) | 0.074 |
| >Median | 69 | 21 (30.4%) | 48 (69.6%) | |
| Gender | ||||
| Male | 0 | 0 (0.0%) | 0 (0.0%) | |
| Female | 140 | 53 (37.9%) | 87 (62.1%) | |
| AJCC Stage | ||||
| I–II | 94 | 38 (40.4%) | 56 (59.6%) | 0.370 |
| III–IV | 46 | 15 (32.6%) | 31 (67.4%) | |
| T Classification | ||||
| T1–T2 | 138 | 53 (38.4%) | 85 (61.6%) | 0.266 |
| T3–T4 | 2 | 0 (0.0%) | 2 (100.0%) | |
| N Classification | ||||
| N0 | 75 | 32 (42.7%) | 43 (57.3%) | 0.208 |
| N1–N3 | 65 | 21 (32.3%) | 44 (67.7%) | |
| Distant Metastasis | ||||
| No | 140 | 53 (37.9%) | 87 (62.1%) | |
| Yes | 0 | 0 (0.0%) | 0 (0.0%) | |
| Recurrence | ||||
| No | 102 | 44 (43.1%) | 58 (56.9%) | 0.035 |
| Yes | 38 | 9 (23.7%) | 29 (76.3%) | |
| Vascular Invasion | ||||
| No | 124 | 45 (36.3%) | 79 (63.7%) | 0.287 |
| Yes | 16 | 8 (50.0%) | 8 (50.0%) | |
| Histology Grade | ||||
| I–II | 98 | 39 (39.8%) | 59 (60.2%) | 0.470 |
| III–IV | 42 | 14 (33.3%) | 28 (66.7%) | |
| Tumor Site | ||||
| Left | 72 | 24 (33.3%) | 48 (66.7%) | 0.256 |
| Right | 68 | 29 (42.6%) | 39 (57.4%) | |
Breast cancer patients with high miR-5188 expression conferred a poorer overall survival and relapse-free survival (log rank test, p = 0.025 and p = 0.020, respectively) (Figures 1G and 1H). Finally, univariate and multivariate analyses suggested that miR-5188 expression was an independent prognostic factor for the overall survival of breast cancer patients (hazard ratio [HR] = 0.439, 95% CI 0.205–1.002, p = 0.051) (Table 2). The obtained data suggested that c-Jun-regulated miR-5188 may serve as an oncomiR in breast cancer.
Table 2.
Univariate and Multivariate Survival Analysis of Clinicopathological Variables of Breast Cancer Patients
| Overall Survival |
||||||
|---|---|---|---|---|---|---|
| Univariate Analysis |
Multivariate Analysis |
|||||
| Characteristics | HR | 95% CI | p Value | HR | 95% CI | p Value |
| miR-5188 expression | 0.419 | (0.195–0.897) | 0.025 | 0.439 | (0.205–1.002) | 0.051 |
| Low versus high | ||||||
| Age (years) | 0.513 | (0.247–1.067) | 0.074 | 0.449 | (0.191–1.059) | 0.067 |
| ≤Median versus >median | ||||||
| AJCC stage | 0.266 | (0.121–0.587) | 0.001 | 1.912 | (0.456–8.019) | 0.376 |
| I–II versus III–IV | ||||||
| T classification | 2.763 | (0.115–6.540) | 0.531 | |||
| T1–T2 versus T3–T4 | ||||||
| N classification | 0.368 | (0.177–0.767) | 0.008 | 0.435 | (0.109–1.742) | 0.240 |
| N0 versus N1–N3 | ||||||
| Recurrence | 0.039 | (0.016–0.096) | <0.001 | 0.090 | (0.032–0.252) | <0.001 |
| No versus yes | ||||||
| Vascular invasion | 0.569 | (0.182–1.782) | 0.333 | |||
| No versus yes | ||||||
| Histology grade | 0.411 | (0.181–0.932) | 0.033 | 0.537 | (0.253–1.140) | 0.106 |
| I–II versus III–IV | ||||||
| Tumor site | 1.155 | (0.557–2.394) | 0.698 | |||
| Left versus right | ||||||
miR-5188 Promotes Breast Cancer Stemness, Metastasis, Proliferation, and Chemoresistance In Vitro and In Vivo
To identify the role of miR-5188 in breast cancer progression, we introduced lentivirus particles, inhibitor, or mimics of miR-5188 into breast cancer cells according to the miR-5188 levels (Figure S1B). Results showed that stable overexpression of miR-5188 in MCF-7 and MDA-MB-468 cells increased the spheroid-forming capability, the proportion of side-population, and the percent of cells that stained with CD44 and CD133 (stemness marker) (Figures 2A–2C). Similarly, immunofluorescence analysis showed that miR-5188 increased the fluorescence intensity of stemness markers, CD44 and CD133, in breast cancer cells (Figure 2D). Moreover, the migration and invasion abilities of breast cancer cells were enhanced after transfections of miR-5188 (Figures 2E and 2F). miR-5188 overexpression stimulated cell proliferation as determined by the colony formation and 5-ethynyl-2'-deoxyuridine (EdU) incorporation assays (Figures 2G and 2H). Since cancer stemness is associated with chemoresistance, we performed 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assays. As shown in Figure 2I, the overexpression of miR-5188 led to chemoresistance to paclitaxel and epirubicin, in a dose- and time-dependent manner (Figure 2I). Conversely, miR-5188 downregulation in T47D and MDA-MB-231 cells treated with miR-5188 inhibitor induced the opposite results (Figures S1C–S1G).
Figure 2.

miR-5188 Promotes Breast Cancer Stemness, Metastasis, Proliferation, and Chemoresistance
(A) Mammosphere formation analysis during three serial passages (scale bars, 40 μm), (B and C) flow cytometry, (D) immunofluorescence analyses (scale bars, 10 μm), (E) wound-healing assays, (F) transwell assays (scale bars, 100 μm), (G) colony-formation assays, and (H) EdU incorporation assays (scale bars, 100 μm) of miR-5188-overexpressed MCF-7 and MDA-MB-468 cells and their control cells. (I) Drug sensitivity tests of MCF-7 and MDA-MB-468 cells treated with paclitaxel and epirubicin in a dose-dependent and time-dependent manner. *p < 0.05; **p < 0.01; ***p < 0.001.
Mouse models were adopted to evaluate the in vivo oncogenic efficacy of miR-5188. Stable overexpression of miR-5188 in MCF-7 cells (miR-5188 group) enhanced tumor formation ability in female nude mice compared with the lentivirus of negative control (NC) group (Figure 3A). Compared to the NC group, more metastatic nodules were detected in the pulmonary metastasis mouse model mice in miR-5188 group as shown in our fluorescent and histopathologic assays (Figure 3B). In addition, the miR-5188 group exhibited accelerated tumor growth and displayed higher Ki67 and PCNA expression compared with the NC group (Figure 3C). However, knockdown of miR-5188 (sh-miR-5188 group) in MDA-MB-231 cells induced the opposite results (Figures 3A–3C).
Figure 3.

miR-5188 Enhances Breast Cancer Stemness, Metastasis, Proliferation, and Chemoresistance In Vivo
(A) The subcutaneous xenograft mouse model was adopted to evaluate the effect of miR-5188 on the tumor-initiating frequency (n = 6). (B) The pulmonary metastasis model was chosen to assess the effect of miR-5188 on cancer metastasis (scale bar, 100 μm) (n = 5, Student’s t test). (C) The subcutaneous xenograft mouse model was used to evaluate the effect of miR-5188 on cancer proliferation (n = 5, general linear model). Xenograft tumors were directly stained with H&E (scale bar, 100 μm) and were subjected to immunohistochemical stains for Ki67 and PCNA expression (scale bars, 40 μm) (n = 5). (D) Survival analysis shows the cumulative overall survival time of the mice in miR-5188 group, paclitaxel/epirubicin-treated group, miR-5188 + paclitaxel/epirubicin group, miR-5188 antagomir group, antagomir + paclitaxel/epirubicin group, antagomir + si-FOXO1 group, and their controls (n = 10, log-rank test). *p < 0.05; **p < 0.01.
Furthermore, Kaplan-Meier analysis was adopted to calculate the survival times of the mice. The paclitaxel/epirubicin-treated group showed prolonged survival times compared with the untreated control group, with a shortened survival time in the miR-5188 group. Intriguingly, combining miR-5188 antagomir and chemo-therapeutic treatment further delayed breast cancer progression. However, no significant differences were detected between the miR-5188 + paclitaxel/epirubicin group and the control group (Figure 3D).
Collectively, these results demonstrated that miR-5188 promoted breast cancer stemness, metastasis, proliferation, and chemoresistance in breast cancer.
miR-5188 Enhances Breast Cancer Stemness, Metastasis, Proliferation, and Chemoresistance via Wnt/β-Catenin Signal Pathway
To determine the mechanisms by which miR-5188 promote breast cancer progression, we performed western blot to screen activation status of signaling pathways involved in miR-5188 regulation. The data revealed that miR-5188 upregulation increased the expression of known Wnt targets, including β-catenin, N-cadherin (N-ca), vimentin, Slug, Nanog, Sox2, Oct4, ABCB1, ABCG2, CCND1, c-Jun, and c-Myc, while it decreased E-cadherin (E-ca) expression (Figure 4A). In addition, we found knockdown of β-catenin or miR-5188 inhibitor abolished the expression pattern induced by miR-5188 upregulation (Figure 4A). Consistently, functional assays indicated that knockdown of β-catenin or miR-5188 inhibitor abrogated the promotive effects of miR-5188 on breast cancer stemness, metastasis, proliferation, and chemoresistance (Figures S2A–S2D). These results suggested that miR-5188 might exert its oncogenic role through Wnt/β-catenin signaling pathway in breast cancer.
Figure 4.

miR-5188 Directly Targets FOXO1 to Augment β-Catenin-Mediated Breast Cancer Stemness, Metastasis, Proliferation, and Chemoresistance
(A) Western blot analysis of stemness, metastasis, proliferation, chemoresistance, and Wnt/β-catenin signaling-associated protein expression in miR-5188-overexpressed breast cancer cells, miR-5188-overexpressed breast cancer cells with β-catenin or miR-5188 knockdown, and their control cells. (B) Bioinformatics analysis identified the binding sequence of miR-5188 within the FOXO1 3′ UTR. (C and D) Western blot (C) and quantitative real-time PCR (D) analysis of FOXO1 expression in miR-5188-overexpressed breast cancer cells, miR-5188-silenced breast cancer cells, and their control cells. (E) Immunohistochemistry analysis of FOXO1 expression in xenograft tumors derived from MCF-7 cells after stable miR-5188 overexpression, MDA-MB-231 cells after stable miR-5188 knockdown, and their controls (scale bar, 10 μm) (n = 5). (F) Luciferase reporter assays were conducted to validate the interaction between miR-5188 and the FOXO1 3′ UTR. (G) RIP was conducted to validate the interaction between the AGO2-bound miR-5188 and FOXO1 mRNA. RIP, RNA immunoprecipitation assay. ***p < 0.001.
FOXO1 Serves as a Direct Target Gene of miR-5188 in Breast Cancer
Since the function of miRNAs is dependent on the regulation of target genes, the potential target of miR-5188 including FOXO1 were selected by four prediction databases (TargetScan, RNAhybird, Microt4, and miRMap). In addition, results from GSEA based on the TCGA BRCA dataset showed that miR-5188 can negatively regulate FOXO signaling (Figure 1D). Hence, FOXO1 was chosen for further investigation.
Although no significant difference in FOXO1 mRNA levels was detected after transfection of miR-5188 mimic or inhibitor by quantitative real-time PCR (Figure 4D), the western blot results showed that miR-5188 downregulation increased p-FOXO1 and FOXO1 protein levels, and miR-5188 upregulation presented the opposite effects in breast cancer cells (Figure 4C). In addition, immunohistochemistry analysis showed FOXO1 expression was decreased in MCF-7 xenografts after transfection with miR-5188, while knockdown of miR-5188 with transfection of sh-miR-5188 increased FOXO1 expression in MDA-MB-231 xenografts (Figure 4E). These results indirectly suggest that FOXO1 may be one of the target genes of miR-5188.
To determine whether FOXO1 is a direct target of miR-5188, we performed Luciferase reporter assays. We constructed the wild-type (WT) 3′ UTR and the mutant (mt) 3′ UTR fragments according to the predicted binding sites (Figure 4B). These vectors and miR-5188 mimics or miR-5188 inhibitor were cotransfected into MCF-7 cells. As shown in Figure 4H, the luciferase assay showed that miR-5188 mimics and miR-5188 inhibitor have no apparent effects on FOXO1-mt 3′ UTR, while the luciferase activity of the reporter plasmid inserted with FOXO1-WT 3′ UTR was significantly decreased in miR-5188 mimics group and increased in miR-5188 inhibitor group. Furthermore, RNA immunoprecipitation verified the combination of the AGO2-bound miR-5188 and FOXO1 mRNA (Figure 4G). All together, these findings confirmed that FOXO1 is a direct target gene of miR-5188, and the binding site was the designated mutation site.
Restoring FOXO1 Expression Reverses Breast Cancer Stemness, Metastasis, Proliferation, and Chemoresistance Induced by miR-5188 via Wnt/β-Catenin Signaling in Breast Cancer
To demonstrate that FOXO1 suppression mediated the role of miR-5188, the effects of FOXO1 on miR-5188-modulated signals were evaluated in breast cancer cells, and the results showed that restoring FOXO1 abolished miR-5188 stimulation of β-catenin, N-ca, Vimentin, Slug, CD44, Nanog, Sox2, Oct4, ABCG2, ABCB1, CCND1, c-Jun, and c-Myc and miR-5188 suppression of E-ca (Figure 5A). Additionally, TOP/FOP luciferase reporter assays confirmed that miR-5188 and FOXO1 modulated Wnt signaling in breast cancer cells (Figure S3A). Consistently, FOXO1 inhibited the promotive effects of miR-5188 on stemness, metastasis, proliferation, and chemoresistance in breast cancer cells (Figures S3B–S3E). Intriguingly, the FOXO1 small interfering RNA (siRNA) shortened the survival time of the mice treated with miR-5188 antagomir in vivo (Figure 3D).
Figure 5.

miR-5188 Augments Breast Cancer Stemness, Metastasis, Proliferation, and Chemoresistance through FOXO1-Mediated β-Catenin Ubiquitination and Translocation
(A) Western blot analysis of stemness, metastasis, proliferation, chemoresistance, and Wnt/β-catenin signaling-associated proteins expression in FOXO1-overexpressed MCF-7 cells, miR-5188-overexpressed MCF-7 cells, miR-5188-overexpressed MCF-7 cells with FOXO1 overexpression, FOXO1-silenced MDA-MB-231 cells, miR-5188-silenced MDA-MB-231 cells, miR-5188-silenced MDA-MB-231 cells with FOXO1 knockdown, and their control cells. (B) Endogenous coimmunoprecipitation analysis validated the interaction between FOXO1 and β-catenin in MCF-7 cells. (C) Immunofluorescence costaining of FOXO1 and β-catenin was performed in MCF-7 cells. The fluorescence intensities along the dark arrow crossing the cytoplasm were calculated to show the colocalization of FOXO1 and β-catenin. (D) Western blot and quantification analysis of the effects of miR-5188 and FOXO1 on β-catenin stability in MCF-7 and MDA-MB-231 cells treated with cycloheximide at different time points. (E) Immunofluorescence costaining of FOXO1 and β-catenin was performed to detect their expression and subcellular localization in MCF-7 and MDA-MB-231 cells (scale bars, 10 μm). (F) Nucleic and cytoplasmic proteins were extracted for β-catenin detection by western blot. (G) Coimmunoprecipitation analysis of the effects of miR-5188 and FOXO1 on the interaction between β-catenin and ubiquitin in MCF-7 cells treated with MG132. (H) Western blot analysis of total β-catenin and active β-catenin expression in MCF-7 and MDA-MB-231 cells treated with FOXO1 plasmid, FOXO1 siRNA, or lithium (LiCl). (I) Immunohistochemistry analysis of Sox2, Oct4, Nanog, E-ca, N-ca, and Vimentin expression in xenograft tumors derived from MCF-7 cells after stable miR-5188 overexpression, MDA-MB-231 cells after stable miR-5188 knockdown, and their controls (scale bars, 40 μm) (n = 5). *p < 0.05; **p < 0.01; ***p < 0.001.
It has been reported that FOXO1 interacted with β-catenin to exert its functions in several diseases.20, 21, 22 In breast cancer cells, we observed the interaction between FOXO1 and β-catenin (Figure 5B), and the cytoplasmic colocalization of FOXO1 and β-catenin (Figure 5C). Since FOXO1 repressed β-catenin expression (Figure 5A), we explored the mechanisms of this regulation. Intriguingly, FOXO1 caused ubiquitin-mediated degradation of β-catenin, impaired the nuclear accumulation of β-catenin, and alleviated the inhibitory effects of miR-5188 on β-catenin degradation and the promotive effects of miR-5188 on nuclear β-catenin enrichment (Figures 5D–5G). Considering that β-catenin dephosphorylation facilitated its stabilization and accumulation in the nucleus,23 we proposed that the effect of FOXO1 on β-catenin ubiquitination depends on β-catenin phosphorylation. Intriguingly, we demonstrated that the facilitation of β-catenin ubiquitination by FOXO1 was independent of β-catenin phosphorylation (Figure 5H).
Furthermore, miR-5188-overexpressed MCF-7 cells exhibited higher expression of Sox2, Oct4, Nanog, N-ca, vimentin, Ki67, and PCNA and lower expression of E-ca compared with the control (NC) cells in the xenografts, and the knockdown of miR-5188 (sh-miR-5188) in MDA-MB-231 cells presented the opposite expression patterns (Figure 5I).
Together, these results suggest that miR-5188 activates Wnt/β-catenin signaling to promote breast cancer progression by directly targeting FOXO1.
c-Jun Induces miR-5188 Transcription Expression to Cooperatively Drive β-Catenin Signaling
The JASPAR (http://jaspar.genereg.net), UCSC (http://genome.ucsc.edu/), and Cistrome data browser (http://cistrome.org/db/) databases predicted that the miR-5188 promoter region contains three putative c-Jun-binding sites (site A from −1,424 to −1,412, site B from −761 to −755, site C from −353 to −345) (Figure 6A). In a further study, we showed that knockdown of c-Jun downregulated pre-miR-5188 and mature miR-5188 expression in breast cancer cells (Figure 6B), implying that c-Jun was an upstream regulator of miR-5188. Subsequently, ChIP assays indicated that endogenous c-Jun bound to the miR-5188 promoter region (Figure 6C), and electrophoretic mobility shift assay demonstrated that the three predicted c-Jun-binding sites in the miR-5188 promoter region were functional (Figure 6D). Besides, elevated luciferase activity of the WT miR-5188 promoter was observed in MCF-7 and MDA-MB-231 cells, and the effects could be eliminated in cells treated with pGL4.1 containing mutant miR-5188 promoter (Figure 6E). These data suggested that c-Jun enhanced miR-5188 transcription by functionally binding to the miR-5188 promoter in breast cancer.
Figure 6.

c-Jun Transcriptionally Promotes miR-5188 Expression to Form a Positive Regulatory Loop
(A) ChIP-seq binding peaks were searched using the Cistrome data browser, and bioinformatics analysis were performed to identify binding sites of c-Jun within the miR-5188 promoter. (B) Quantitative real-time PCR analysis of pre-miR-5188 and mature miR-5188 expression in c-Jun-depleted MCF-7 and MDA-MB-231 cells and their control cells. (C) Chromatin immunoprecipitation analysis verified c-Jun binding to the miR-5188 promoter. (D) The protein-DNA interaction between c-Jun and miR-5188 promoter was determined using the electrophoretic mobility shift assay. (E) Luciferase reporter assays were performed to confirm c-Jun binding to the miR-5188 promoter. (F) Immunohistochemistry analysis of c-Jun expression in xenograft tumors derived from MCF-7 cells after stable miR-5188 overexpression, MDA-MB-231 cells after stable miR-5188 knockdown, and their controls (scale bars, 40 μm) (n = 5). (G) Western blot analysis of stemness, metastasis, proliferation, chemoresistance, and Wnt/β-catenin signaling-associated protein expression in c-Jun-silenced MDA-MB-231 and MCF-7 cells, miR-5188-overexpressed MDA-MB-231 and MCF-7 cells, c-Jun-silenced MDA-MB-231 and MCF-7 cells with miR-5188 overexpression, and their control cells. (H) Chromatin immunoprecipitation analysis of c-Jun binding to miR-5188 promoter in FOXO1-overexpressed MDA-MB-231 cells, FOXO1-overexpressed MDA-MB-231 cells with β-catenin overexpression, FOXO1-silenced MCF-7 cells, FOXO1-silenced MCF-7 cells with β-catenin knockdown, and their control cells. ChIP-seq, chromatin immunoprecipitation sequencing. *p < 0.05; **p < 0.01; ***p < 0.001.
Immunohistochemistry of miR-5188-overexpressed MCF-7 xenografts showed elevations of c-Jun expression and the sh-miR-5188 MDA-MB-231 xenografts induced decreased c-Jun expression (Figure 6F). In addition, we found that c-Jun silence by si-c-Jun increased FOXO1 levels and decreased expression of β-catenin and known Wnt targets (Figure 6G). The functional assays showed that the silence of c-Jun impaired breast cancer stemness, metastasis, proliferation, and chemoresistance (Figure S4). Moreover, knockdown of c-Jun on breast cancer progression were reversed by overexpression of miR-5188 (miR-5188 mimics) in breast cancer cells (Figure S4). Finally, ChIP assays suggested that β-catenin alleviated the inhibitory effects of FOXO1 on c-Jun binding to the miR-5188 promoter region in breast cancer cells (Figure 6H). Collectively, these results demonstrated that c-Jun and miR-5188 formed a feedback loop to drive Wnt/β-catenin signaling and facilitated breast cancer progression.
Timeless Interacts with the Sp1/c-Jun Complex to Potentiate the Positive miR-5188-FOXO1/β-Catenin-c-Jun Regulatory Loop to Modulate Breast Cancer Stemness, Metastasis, Proliferation, and Chemoresistance
Previous study from our lab demonstrated that Timeless contributed to cancer stemness, metastasis, and tumor growth,14 which was consistent with another study that showed that Timeless promoted chemoresistance and the EMT via Wnt/β-catenin signaling.15 We questioned the specific mechanisms of Timeless-enhanced Wnt/β-catenin signaling and the relationship between Timeless and β-catenin/c-Jun-mediated miR-5188 expression. Timeless silence decreased pre-miR-5188 and mature miR-5188 expression, while c-Jun overexpression abolished the inhibitory effects, implying that Timeless regulated c-Jun-mediated miR-5188 expression at the transcription level (Figure S5A). The Biogrid database (https://thebiogrid.org/) suggested interaction between the Timeless and Sp1 transcription factor, which could combine with c-Jun to induce c-Jun-mediated transcription in breast cancer.24 We then questioned whether Timeless could form a complex with Sp1 and c-Jun to enhance miR-5188 expression. Exogenous and endogenous coimmunoprecipitation studies demonstrated that Timeless interacted with Sp1 and c-Jun in MCF-7 and MDA-MB-231 cells (Figure S5B). The nuclear colocalization of Timeless and Sp1, and Timeless and c-Jun, were observed in breast cancer cells (Figure S5C). However, Silence of Sp1 by si-Sp1 blocked Timeless and c-Jun interaction and impaired the stimulative effect of Timeless on c-Jun-mediated miR-5188 expression (Figures S5D and S5E). In subsequent investigations, we confirmed that miR-5188 mediated the stimulative effects of Timeless on breast cancer stemness, metastasis, proliferation, chemoresistance, and β-catenin/c-Jun signaling (Figure S6).
Finally, TOP/FOP luciferase reporter assays confirmed that Timeless enhanced miR-5188-mediated Wnt/β-catenin signaling (Figure S5F), and ChIP assays suggested that Timeless stimulated c-Jun-mediated miR-5188 expression through β-catenin signaling (Figure S5G). Overall, these data suggest that Timeless interacts with Sp1/c-Jun to facilitate the activation of the miR5188-FOXO1/β-catenin-c-Jun regulatory loop, thus activating Wnt/β-catenin signaling and promoting breast cancer development.
Relationships between miR-5188, FOXO1, β-Catenin, and c-Jun Expression and Clinicopathological Features of Breast Cancer Patients
Based on mRNA sequencing (mRNA-seq) and miRNA-seq data from TCGA, we found that Timeless expression was upregulated compared with paracarcinoma tissues, while FOXO1 expression was downregulated in breast cancer tissues (p < 0.001 and p < 0.001, respectively) (Figures S7A and S7B). Intriguingly, miR-5188 expression was positively correlated with Timeless expression (Figure S7C).
In situ hybridization and immunohistochemistry were then performed on 140 breast cancer and 77 paracarcinoma tissues. Results indicated a decrease in FOXO1, and an increase in β-catenin and c-Jun expression levels in breast cancer compared with para carcinoma tissues (Figure S7D). Moreover, miR-5188 expression was negatively correlated with FOXO1 expression (Kappa = −0.244, p = 0.001) and positively correlated with c-Jun expression (Kappa = 0.180, p = 0.033) (Figure S7E; Table S1). miR-5188 expression tended to be positively correlated with total β-catenin expression (Kappa = 0.152, p = 0.055) (Table S1). FOXO1 expression was negatively correlated with β-catenin expression (Kappa = −0.410, p < 0.001) and tended to be negatively correlated with β-catenin nuclear translocation (Kappa = −0.139, p = 0.057) (Table S1).
Discussion
CSCs properties are regarded as the main prognostic predictors of breast cancer. In this study, we identified that miR-5188 augmented Wnt/β-catenin/c-Jun signaling and its downstream signals, including the known Wnt targets, EMT markers, and key regulators of cancer stemness, thus facilitating the CSCs properties, metastasis, proliferation, and chemoresistance of breast cancer cells. miR-5188 knockdown delayed breast cancer progression in vitro and in vivo. Furthermore, miR-5188 was upregulated in breast cancer, and high miR-5188 expression conferred a poor prognosis for breast cancer patients.
FOXO1 is a transcription factor and member of the FOXO subfamily of the forkhead/winged-helix family. In mammals, FOXO1 has a wide range of biological functions. Previous reports have described FOXO1 as a tumor suppressor that suppressed tumor stemness, metastasis, proliferation, and chemoresistance in several cancers,25, 26, 27, 28, 29 including breast cancer.30, 31, 32 Several studies have shown the interaction of FOXO1 and β-catenin, and the regulatory function of FOXO1 in the Wnt/β-catenin signaling,21, 33, 34 However, this mechanism remains controversial. Here, we demonstrated that miR-5188 directly targeted FOXO1 to augment β-catenin/c-Jun signaling and its downstream signals. Previous study showed that FOXO1 could downregulate β-catenin expression, but FOXO1 did not affect nuclear β-catenin intensity.35 In the present study, we not only detected that FOXO1 suppressed β-catenin expression, but also found that FOXO1 coupled with β-catenin in the cytoplasm can prevent nuclear β-catenin accumulation, thus downregulating Wnt and c-Jun signaling activity, especially downstream key regulators of cancer stemness, metastasis, proliferation, and chemoresistance. The mechanistic study further clarified that FOXO1 interacted with β-catenin to facilitate ubiquitin-mediated proteolysis of β-catenin, revealing the mechanism by which FOXO1 elicited inhibitory effects on β-catenin expression and nuclear accumulation.
The proto-oncogene c-Jun serves as a crucial transcriptional regulator contributing to breast cancer development36, 37 and functions as an essential transcription factor modulating many protein coding genes, as well as the expression of miRNA in regulating breast cancer stemness,5, 38 metastasis,4, 39 proliferation,40 and chemoresistance.41 Interestingly, miR-5188 was identified as a potential oncomiR with a promoter region containing multiple c-Jun binding sites based on ChIP-seq data. Subsequently, our study confirmed that c-Jun transcriptionally promoted miR-5188 expression, suggesting that miR-5188 directly targeted FOXO1 and cooperated with the FOXO1/β-catenin complex and c-Jun to form a positive feedback loop. Intriguingly, miR-5188 induced its expression via the miR-5188-FOXO1/β-catenin-c-Jun loop to facilitate the activation of Wnt/β-catenin/c-Jun signaling, augmenting breast cancer progression.
The oncogenic functions of Timeless have been reported in different types of cancer.15, 42, 43, 44 Based on our previous study, which demonstrated that Timeless contributed to breast cancer progression,14 in the present study, we further demonstrated that Timeless interacted with Sp1 and c-Jun to form a complex, potentiating c-Jun-mediated miR-5188 transcription and promoting cancer stemness, metastasis, proliferation, and chemoresistance. Considering that miR-5188 was transcriptionally enhanced by the Timeless/Sp1/c-Jun complex and FOXO1 was confirmed as a direct miR-5188 target, we proposed that Timeless might promote miR-5188 to inhibit FOXO1 expression and thus activate Wnt/β-catenin/c-Jun signaling. As expected, Timeless suppressed FOXO1 expression and facilitated the activation of Wnt/β-catenin/c-Jun signaling through the miR-5188-FOXO1/β-catenin-c-Jun feedback loop.
Bioinformatics and immunohistochemical and in situ hybridization assays supported that miR-5188 served as an oncomiR promoting breast cancer progression and functioned as a prognostic factor for the overall survival of breast cancer patients. Furthermore, GSEA confirmed that miR-5188 was implicated in the regulation of FOXO signaling, cell cycle, DNA replication, transcription, and cancer metastasis. Importantly, high miR-5188 expression was associated with improved outcome of breast cancer patients. Correlation analysis of miR-5188, Oct4, Nanog, and Timeless expression based on the TCGA database, and the association of miR-5188, FOXO1, β-catenin, and c-Jun expression in clinical specimens further verified the results obtained from breast cancer cells. In summary, these findings suggest that miR-5188 cooperates with Timeless, FOXO1, β-catenin, and c-Jun to exert a crucial role in breast cancer progression.
Taken together, our present work elucidates a novel positive regulatory loop that involves miR-5188, FOXO1, β-catenin, and c-Jun in the regulation of breast cancer progression. Moreover, our study revealed a novel mechanism for FOXO1, where it could interact with β-catenin to facilitate ubiquitin-mediated β-catenin degradation, thus downregulating β-catenin expression and nuclear accumulation. Finally, the activation of the miR-5188-FOXO1/β-catenin-c-Jun feedback circuit is facilitated by the Timeless/Sp1/c-Jun complex and collaboratively enhances breast cancer progression by activating Wnt/β-catenin/c-Jun signaling. This study aids in unravelling the oncogenic roles of specific miRNAs and proteins in network crosstalk and highlights the biological basis for the clinical use of miR-5188 as a diagnostic and prognostic factor and as a potential target for breast cancer treatments.
Materials and Methods
Cell Cultures
Primary normal breast epithelial cell line was cultured in keratinocyte serum-free medium (Gibco, USA) supplemented with epithelial growth factor and bovine pituitary extract. Breast cancer cell lines including MCF-7, SKBR-3, T47D, MDA-MB468, MDA-MB231, and MDA-MB453 were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China) and the Cancer Research Institute of Southern Medical University (Guangzhou, China) and grown in DMEM supplemented with 10% fetal bovine serum (FBS) (Gibco, USA).
Cell Transfections
Lentiviral particles carrying the hsa-miR-5188 precursor (miR-5188) or miR-5188 shRNA (sh-miR-5188) were designed and constructed by GeneChem (Shanghai, China) (Table S2). The breast cancer cells were transfected with miR-5188 or sh-miR-5188, and transfection efficiencies were evaluated by miR-5188 expression, which was measured by quantitative real-time PCR. Plasmids were purchased from Vigene Biosciences (Shandong, China). siRNAs, mimics, and inhibitors were designed and synthesized by Guangzhou RiboBio (Guangzhou, China) (Table S2). Before transfection, exponentially growing cells were seeded in a cell culture plate or dish (NEST Biotech, China). Plasmids, siRNAs, mimics, and inhibitors were then transfected into cells using Lipofectamine TM 2000 (Invitrogen Biotechnology, Shanghai, China) according to the manufacturer’s protocol. Cells were collected 48–72 h after transfection for further experimentation.
Mammosphere Formation Analysis
Mammosphere formation analysis was performed according to a previous method.8 In brief, cells (5,000/well) were plated on 6-well ultra-low-attachment plates (Corning, NY, USA) and grown in serum-free DMEM/F12 (Gibco) supplemented with fibroblast growth factor (FGF) (20 ng/mL), epidermal growth factor (EGF) (20 ng/mL), and B27 (2%). Fresh medium was supplemented every 2 days. The mammospheres were photographed and counted on day 14. For secondary mammosphere formation, the primary mammospheres were dissociated into single cells and cultured in 6-well ultra-low-attachment plates for another 14 days. The tumor spheres were analyzed after continuous passaging for three generations. All experiments were repeated at least three times.
Flow Cytometry Analysis
For CD44highCD133high cells analysis, 1 × 106 cells were resuspended in 200 μL PBS containing fluorescein isothiocyanate (FITC)-conjugated anti-CD44 (555478, 1:100, BD Biosciences, USA) and allophycocyanin (APC)-conjugated anti-CD133 antibodies (566596, 1:100, BD Biosciences, USA). After incubation with the antibodies for 30 min on ice, the cells were washed with PBS and then subjected to flow cytometric analysis. All assays were independently performed in triplicate.
Side Population Analysis
Cells were resuspended at 1 × 106 cells per mL in DMEM containing 2% FBS and incubated with 5 μg/mL Hoechst 33342 (Sigma-Aldrich, MO, USA) for 90 min at 37°C with gentle inversion every 10 min. Samples were treated with 50 μmol/L verapamil (Sigma-Aldrich, MO, USA) to block the efflux of dye and served as a NC. Cells were washed with ice-cold PBS and then analyzed using flow cytometry. All experiments were repeated at least three times.
Immunofluorescence
Cells were seeded on coverslips in 24-well plates and cultured overnight and then fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 at room temperature. Cells were incubated with antibodies for 30–45 min at 37°C. Coverslips were mounted onto slides with a mounting solution containing 0.2 mg/mL DAPI and sealed with nail polish. Slides were stored in a dark box and observed under a fluorescent microscope. All assays were independently performed three times.
Migration and Invasion Assay
The transwell (BD Biosciences, NJ, USA) assay was used to test cell migration and invasion abilities. Cells were suspended in 100 μL DMEM without serum and seeded into the top chamber of the transwells coated with Matrigel (BD Biosciences, NJ, USA) or left uncoated, and the bottom chambers were filled with 500 μL DMEM supplemented with 10% FBS. The migrated cells were stained with crystal violet and then photographed and quantified by counting the cell numbers in five random fields. All assays were independently performed in triplicate.
Wound Healing Assay
Cells were seeded and grew in 6-well plates until a confluent monolayer was reached, and scratches (wounding) were created using a pipette tip. Progression of migration was photographed at initiation and 24, 48, and 72 h after wounding. All experiments were repeated at least three times.
MTT Assay
Cell proliferation and drug susceptibility tests were determined with the MTT assay. Cells were seeded in 96-well plates at a density of 1,000 cells/well. After incubation, each well was added using MTT (5 mg/mL) (Sigma-Aldrich, MO, USA), and incubated for 4 h. At the end of incubation, supernatants were removed, and dimethyl sulfoxide (Sigma-Aldrich, MO, USA) was added to each well. The absorbance value (OD) of each well was measured at 490 nm. The calculated rates were then used for curve fitting and half maximal inhibitory concentration (IC50) calculations. All assays were independently performed three times.
Colony-Formation Assay and EdU Incorporation Assays
For the colony-formation assay, cells were plated in 6-well plates at a density of 200 cells per well. The medium was refreshed after a 24 h incubation. After culturing for 14 days, colonies were stained with a hematoxylin solution after fixation with methanol for 15 min. The number of colonies containing ≥50 cells was counted under a microscope. For the EdU incorporation assay, EdU incorporation was performed using the Apollo567 in vitro imaging kit (RiboBio, Guangzhou, China) according to the manufacturer’s protocol. In brief, cells were incubated with 10 μM EdU for 2 h before fixation with 4% paraformaldehyde, permeabilization with 0.3% Triton X-100, and staining with Apollo fluorescent dyes. Cell nuclei were stained with 5 μg/mL DAPI for 10 min. The number of EdU-positive cells was counted under a microscope in five random fields. All assays were independently performed in triplicate.
Animal Studies
Animal experimental protocols were approved by The Institutional Animal Ethical Committee, Experimental Animal Center of Southern Medical University, China. The subcutaneous xenograft mouse model was adopted to evaluate tumor growth, in which 5 × 106 cells were injected into mammary fat pads of the 4- to 5-week-old female BALB/c-nu mice (N = 5 per group). Mice were sacrificed 20 or 30 days after cell inoculation, and tumors were excised, weighed, and processed for further experimentation. Tumor size was determined using measurements of the shortest diameter (A) and the longest diameter (B) with a caliper. The volume was calculated using the formula V = (A2B)/2.
The pulmonary metastasis model was adopted for the in vivo metastasis assay. A total of 1 × 106 cells were intravenously injected into the tail vein of mice (N = 5 per group). The optical and pathological images were visualized to monitor primary tumor growth and metastatic lesion formation.
The subcutaneous xenograft mouse model was also established to determine the tumor formation ability. A series of 5 × 105, 2 × 105, 1 × 105, and 5 × 104 cells were injected into mice (N = 6 per group), and the tumor-initiating frequency was calculated. Additionally, the subcutaneous xenograft mouse model was used to assess tumor chemoresistance. The 4- to 5-week-old female BALB/c-nu mice (N = 10 per group) were subcutaneously inoculated in the flanks with 5 × 106 cells and then intraperitoneally treated with paclitaxel at 10 mg/kg body weight for a total of 4 doses at 7, 10, 13, and 16 days after cell inoculation.45 Epirubicin was given at 2 mg/kg, two times weekly for 4 weeks.46 Two weeks after cell inoculation, 10 nmol siRNA in 0.1 mL saline buffer was locally injected into the tumor mass once every 3 days for 2 weeks. Survival curves were analyzed using Kaplan-Meier analysis.
Quantitative Real-Time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen, USA), and cDNA was synthesized using Hiscript reverse transcriptase (Vazyme Biotech, USA) according to manufacturer’s instructions. To measure the mRNA expression of FOXO1, total RNA was reversely transcribed using primeScript RT reagent kit (Takala, Dalian, China). Quantitative real-time PCR was performed using AceQ qPCR SYBR green master mix (Vazyme Biotech, USA) on the CFX-96 real-time PCR detection system (Bio-Rad, USA) according to the manufacturer’s protocol. For detection of miRNA, real-time PCR was performed using One Step PrimeScript miRNA cDNA synthesis kit and SYBR premix Ex Taq II kit. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or U6 small nuclear RNA (snRNA) was used as an endogenous normalization control. The primers are listed in Table S3. The cycle threshold (Ct) value was used for quantification using the 2−ΔΔCt method.47
Western Blot Analysis
Western blot analysis was performed as previous described.14 The primary antibodies were shown as follows: FOXO1, β-catenin, c-Jun, c-Myc, CCND1, CD44, Slug, Sox2, Oct4, Nanog, E-ca, N-ca, vimentin, ABCG2, ABCB1, GAPDH, and β-actin. Dilutions and sources of antibodies are shown in Table S4. Images were captured using a ChemiDoc XRS+ molecular imager (Bio-Rad, Hercules, CA, USA).
Coimmunoprecipitation (CoIP)
CoIP was carried out using a Pierce coimmunoprecipitation kit (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. In brief, total proteins were extracted from cells, and the concentration was quantified. A total of 5 mg protein was incubated with 10 μg specific antibodies or immunoglobulin G (IgG) overnight at 4°C. After elution, the recovered proteins were subjected to western blot analysis. IgG was used as a NC.
Luciferase Reporter Assay
FOXO1 was predicted to be a direct target miR-5188 using Bioinformatics software. A fragment of FOXO1 3′ UTR (WT 3′ UTR) was amplified. Site-directed mutagenesis of the miR-5188 binding site was generated using the GeneTailor site-directed mutagenesis system (Invitrogen, Guangzhou, China). The WT 3′ UTR or mutant (mt) 3′ UTR were cloned into psiCHECK-2 vectors to prepare for luciferase reporter assays. The vector was cotransfected with miR-5188 mimics/inhibitor or their control sequence into cells, and the luciferase activity was measured 48 h after transfection using the dual-luciferase reporter assay system (Promega Corporation, Madison, WI, USA).
RNA Immunoprecipitation (RIP) Assay
According to the manufacturer’s protocol, RNA immunoprecipitation assays were conducted using a RIP assay kit (Millipore, Billerica, MA, USA). Protein-RNA complexes were isolated, and anti-Ago2 or IgG was added to the reaction system for immunoprecipitation. After RNA purification, the immunoprecipitated RNA was detected by quantitative real-time PCR and/or PCR. IgG served as a NC.
TOP/FOP Luciferase Reporter Assay
Transcriptional activity assays were performed using the luciferase assay system according to the manufacturer’s instructions. In brief, cells were cotransfected with TOPflash or FOPflash with pRL (Millipore, Billerica, MA, USA) using Lipofectamine 2000. Twenty-four hours after transfection, cells were lysed, and luciferase activity was measured using the dual-luciferase reporter assay system (Promega, Madison, WI, USA) on a luminometer (BioTek Instruments, Winooski, VT, USA). The transcriptional activity of each sample was presented as the ratio of firefly to Renilla luciferase.
ChIP Assay
According to the manufacturer’s instructions, the ChIP assay was performed using a ChIP assay kit (Thermo Scientific, Waltham, MA, USA). Chromatin was crosslinked, isolated, and digested with micrococcal nuclease to obtain DNA fragments. Anti-c-Jun or IgG was added to the reaction system for immunoprecipitation. After elution and purification, PCR and qPCR were used to measure DNA fragments enrichment. IgG served as a NC.
Electrophoretic Mobility Shift Assay (EMSA)
Binding activity on the promoter region of miR-5188 (c-Jun-A, c-Jun-B, and c-Jun-C) was detected by the EMSA Kit (BersinBio, Guangzhou, China) according to the manufacturer’s protocol. EMSA was performed in a reaction mixture containing nuclear extracts and biotin-labeled probes. Competition or super-shift assays were performed by adding a 100-fold excess of cold competitors (unlabeled WT or mutant probes) (Table S5) or polyclonal rabbit anti-c-Jun (Cell Signaling Technology) to the reaction mixture. After electrophoresis and incubation, signals were recorded and analyzed. EMSA analysis was performed at Biosense Bioscience (Guangzhou, China).
Cell Fractionation Assay
The cell fractionation assay was performed using NE-PER@ nuclear and cytoplasmic extraction kit (Thermo Scientific Pierce, UK). According to the manufacturer’s instruction, cells were incubated with ice-cold CER I for 10 min at 4°C, and then the CER II extraction reagent was added to the reaction mixture, and the lysate was centrifuged at 16,000 × g for 5 min. The supernatant (cytoplasmic extract) was stored on ice. The pellet was resuspended in nuclear extraction reagent (NER) and incubated for 40 min on ice. The suspension was then centrifuged at 16,000 × g for 10 min, and the supernatant (nuclear extract) was transferred to a fresh microcentrifuge tube and stored on ice. The proteins were further analyzed by western blot.
CHX Chase Assay
Cells were transfected and then incubated with 50 μg/mL cycloheximide (CHX) at different time points. Subsequently, cells were harvested and quantitated using a bicinchoninic acid (BCA) protein assay kit and further analyzed using western blot analysis.
In Situ Hybridization (ISH)
Paraffin sections (4 μm) of tissue samples were deparaffinized in xylene and rehydrated in graded alcohols to distilled water and then incubated with proteinase K at room temperature for 30 min. The sections were rinsed, fixed, and prehybridized for 2 h. Hybridization was performed with miR-5188 digoxygenin (DIG)-labeled probes designed and synthesized by BersinBio (Guangzhou, China). The sections were washed and incubated with anti-DIG-horseradish peroxidase (HRP) Fab fragments for 1 h at room temperature. Signals were detected with the addition of 3,3'-diaminobenzidine (DAB) substrate (Maixin Biotech, China). Staining intensities were scored for statistical analysis.
Immunohistochemistry (IHC)
Paraffin sections (4 μm) from tissue samples were deparaffinized and rehydrated, and then antigen retrieval was performed in citrate buffer for 3 min. Endogenous peroxidase activity and nonspecific antigens were blocked with 3% H2O2 and goat serum which was followed by incubation with antibodies (Table S4) overnight at 4°C. After washing, the sections were incubated with HRP-conjugated secondary antibody and detected using the DAB substrate (Maixin Biotech, Fuzhou, China). Staining intensities were scored for statistical analysis.
Bioinformatics Analysis
The latest mRNA-seq and miRNA-seq data of breast cancer were extracted and used to perform differential expression analysis, correlation analysis, and survival analysis. The breast cancer expression profiles were analyzed based on the TCGA database. For statistical analysis, patients with mRNA or miRNA expression values greater than the median value were added to the high expression group, while the rest of the patients were classified into the low expression group.
Statistical Analysis
All the data were analyzed by SPSS 22.0 (SPSS, Chicago, IL, USA). The data are expressed as the means ± SD from at least three independent experiments. Statistical significance was determined using the Student’s two-tailed t test for two groups, one-way ANOVA for multiple groups, and a parametric generalized linear model with random effects for the MTT assay. The correlations between genes expression were conducted using Spearman rank correlation test. Kaplan-Meier survival curves were conducted for survival analysis. All statistical tests were two-sided, and a p value of < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, and ***p < 0.001.
Author Contributions
Z.W. and W.F. conceived and designed this study. Y.Z., X.L., J.B., Z.L., Y.C., Y.Q., H.M., and Y.T. performed the experiments. Y.Z. and X.L. conducted the data analyses. All authors read manuscript drafts, contributed edits, and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study was funded by the Natural Science Foundation of China (NSFC) (no. 81372769).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.08.015.
Contributor Information
Weiyi Fang, Email: fangweiyi1975@163.com.
Ziqing Wu, Email: hailian@fimmu.com.
Supplemental Information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Plaks V., Kong N., Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16:225–238. doi: 10.1016/j.stem.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawson D.A., Bhakta N.R., Kessenbrock K., Prummel K.D., Yu Y., Takai K., Zhou A., Eyob H., Balakrishnan S., Wang C.Y. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526:131–135. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shao W., Li S., Li L., Lin K., Liu X., Wang H., Wang H., Wang D. Chemical genomics reveals inhibition of breast cancer lung metastasis by Ponatinib via c-Jun. Protein Cell. 2019;10:161–177. doi: 10.1007/s13238-018-0533-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie X., Kaoud T.S., Edupuganti R., Zhang T., Kogawa T., Zhao Y., Chauhan G.B., Giannoukos D.N., Qi Y., Tripathy D. c-Jun N-terminal kinase promotes stem cell phenotype in triple-negative breast cancer through upregulation of Notch1 via activation of c-Jun. Oncogene. 2017;36:2599–2608. doi: 10.1038/onc.2016.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gebert L.F.R., MacRae I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019;20:21–37. doi: 10.1038/s41580-018-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang G., Gormley M., Qiao J., Zhao Q., Wang M., Di Sante G., Deng S., Dong L., Pestell T., Ju X. Cyclin D1-mediated microRNA expression signature predicts breast cancer outcome. Theranostics. 2018;8:2251–2263. doi: 10.7150/thno.23877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu F., Yao H., Zhu P., Zhang X., Pan Q., Gong C., Huang Y., Hu X., Su F., Lieberman J., Song E. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 9.Yin H., Xiong G., Guo S., Xu C., Xu R., Guo P., Shu D. Delivery of Anti-miRNA for Triple-Negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 2019;27:1252–1261. doi: 10.1016/j.ymthe.2019.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sang Y., Chen B., Song X., Li Y., Liang Y., Han D., Zhang N., Zhang H., Liu Y., Chen T. circRNA_0025202 Regulates Tamoxifen Sensitivity and Tumor Progression via Regulating the miR-182-5p/FOXO3a Axis in Breast Cancer. Mol. Ther. 2019;27:1638–1652. doi: 10.1016/j.ymthe.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin X., Chen W., Wei F., Zhou B.P., Hung M.C., Xie X. Nanoparticle Delivery of miR-34a Eradicates Long-term-cultured Breast Cancer Stem Cells via Targeting C22ORF28 Directly. Theranostics. 2017;7:4805–4824. doi: 10.7150/thno.20771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim M., Jang K., Miller P., Picon-Ruiz M., Yeasky T.M., El-Ashry D., Slingerland J.M. VEGFA links self-renewal and metastasis by inducing Sox2 to repress miR-452, driving Slug. Oncogene. 2017;36:5199–5211. doi: 10.1038/onc.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X., Jung Y.S., Jun S., Lee S., Wang W., Schneider A., Sun Oh Y., Lin S.H., Park B.J., Chen J. PAF-Wnt signaling-induced cell plasticity is required for maintenance of breast cancer cell stemness. Nat. Commun. 2016;7:10633. doi: 10.1038/ncomms10633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chi L., Zou Y., Qin L., Ma W., Hao Y., Tang Y., Luo R., Wu Z. TIMELESS contributes to the progression of breast cancer through activation of MYC. Breast Cancer Res. 2017;19:53. doi: 10.1186/s13058-017-0838-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S.L., Lin H.X., Lin C.Y., Sun X.Q., Ye L.P., Qiu F., Wen W., Hua X., Wu X.Q., Li J. TIMELESS confers cisplatin resistance in nasopharyngeal carcinoma by activating the Wnt/β-catenin signaling pathway and promoting the epithelial mesenchymal transition. Cancer Lett. 2017;402:117–130. doi: 10.1016/j.canlet.2017.05.022. [DOI] [PubMed] [Google Scholar]
- 16.Zhao C., Qiao Y., Jonsson P., Wang J., Xu L., Rouhi P., Sinha I., Cao Y., Williams C., Dahlman-Wright K. Genome-wide profiling of AP-1-regulated transcription provides insights into the invasiveness of triple-negative breast cancer. Cancer Res. 2014;74:3983–3994. doi: 10.1158/0008-5472.CAN-13-3396. [DOI] [PubMed] [Google Scholar]
- 17.Qiao Y., He H., Jonsson P., Sinha I., Zhao C., Dahlman-Wright K. AP-1 is a key regulator of proinflammatory cytokine TNFα-mediated triple-negative breast cancer progression. J. Biol. Chem. 2016;291:18309. doi: 10.1074/jbc.A115.702571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Z., Lan X., Wu D., Sunkel B., Ye Z., Huang J., Liu Z., Clinton S.K., Jin V.X., Wang Q. Ligand-dependent genomic function of glucocorticoid receptor in triple-negative breast cancer. Nat. Commun. 2015;6:8323. doi: 10.1038/ncomms9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zanconato F., Forcato M., Battilana G., Azzolin L., Quaranta E., Bodega B., Rosato A., Bicciato S., Cordenonsi M., Piccolo S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015;17:1218–1227. doi: 10.1038/ncb3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Essers M.A., de Vries-Smits L.M., Barker N., Polderman P.E., Burgering B.M., Korswagen H.C. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- 21.Qiao X., Rao P., Zhang Y., Liu L., Pang M., Wang H., Hu M., Tian X., Zhang J., Zhao Y. Redirecting TGF-β Signaling through the β-Catenin/Foxo Complex Prevents Kidney Fibrosis. J. Am. Soc. Nephrol. 2018;29:557–570. doi: 10.1681/ASN.2016121362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada K., Naito A.T., Higo T., Nakagawa A., Shibamoto M., Sakai T., Hashimoto A., Kuramoto Y., Sumida T., Nomura S. Wnt/β-Catenin Signaling Contributes to Skeletal Myopathy in Heart Failure via Direct Interaction With Forkhead Box O. Circ Heart Fail. 2015;8:799–808. doi: 10.1161/CIRCHEARTFAILURE.114.001958. [DOI] [PubMed] [Google Scholar]
- 23.Niehrs C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012;13:767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- 24.Wu Y., Zhang X., Zehner Z.E. c-Jun and the dominant-negative mutant, TAM67, induce vimentin gene expression by interacting with the activator Sp1. Oncogene. 2003;22:8891–8901. doi: 10.1038/sj.onc.1206898. [DOI] [PubMed] [Google Scholar]
- 25.Zhao M., Luo R., Liu Y., Gao L., Fu Z., Fu Q., Luo X., Chen Y., Deng X., Liang Z. miR-3188 regulates nasopharyngeal carcinoma proliferation and chemosensitivity through a FOXO1-modulated positive feedback loop with mTOR-p-PI3K/AKT-c-JUN. Nat. Commun. 2016;7:11309. doi: 10.1038/ncomms11309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi F., Li T., Liu Z., Qu K., Shi C., Li Y., Qin Q., Cheng L., Jin X., Yu T. FOXO1: Another avenue for treating digestive malignancy? Semin. Cancer Biol. 2018;50:124–131. doi: 10.1016/j.semcancer.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H., Pan Y., Zheng L., Choe C., Lindgren B., Jensen E.D., Westendorf J.J., Cheng L., Huang H. FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res. 2011;71:3257–3267. doi: 10.1158/0008-5472.CAN-10-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y., Hanifi-Moghaddam P., Hanekamp E.E., Kloosterboer H.J., Franken P., Veldscholte J., van Doorn H.C., Ewing P.C., Kim J.J., Grootegoed J.A. Progesterone inhibition of Wnt/beta-catenin signaling in normal endometrium and endometrial cancer. Clin. Cancer Res. 2009;15:5784–5793. doi: 10.1158/1078-0432.CCR-09-0814. [DOI] [PubMed] [Google Scholar]
- 29.Wu L., Li H., Jia C.Y., Cheng W., Yu M., Peng M., Zhu Y., Zhao Q., Dong Y.W., Shao K. MicroRNA-223 regulates FOXO1 expression and cell proliferation. FEBS Lett. 2012;586:1038–1043. doi: 10.1016/j.febslet.2012.02.050. [DOI] [PubMed] [Google Scholar]
- 30.Chakrabarty A., Bhola N.E., Sutton C., Ghosh R., Kuba M.G., Dave B., Chang J.C., Arteaga C.L. Trastuzumab-resistant cells rely on a HER2-PI3K-FoxO-survivin axis and are sensitive to PI3K inhibitors. Cancer Res. 2013;73:1190–1200. doi: 10.1158/0008-5472.CAN-12-2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gargini R., Cerliani J.P., Escoll M., Antón I.M., Wandosell F. Cancer stem cell-like phenotype and survival are coordinately regulated by Akt/FoxO/Bim pathway. Stem Cells. 2015;33:646–660. doi: 10.1002/stem.1904. [DOI] [PubMed] [Google Scholar]
- 32.Zeng Z., Lin H., Zhao X., Liu G., Wang X., Xu R., Chen K., Li J., Song L. Overexpression of GOLPH3 promotes proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor. Clin. Cancer Res. 2012;18:4059–4069. doi: 10.1158/1078-0432.CCR-11-3156. [DOI] [PubMed] [Google Scholar]
- 33.Hoogeboom D., Essers M.A., Polderman P.E., Voets E., Smits L.M., Burgering B.M. Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J. Biol. Chem. 2008;283:9224–9230. doi: 10.1074/jbc.M706638200. [DOI] [PubMed] [Google Scholar]
- 34.Zhu J., Wang P., Yu Z., Lai W., Cao Y., Huang P., Xu Q., Yu M., Xu J., Huang Z., Zeng B. Advanced glycosylation end product promotes forkhead box O1 and inhibits Wnt pathway to suppress capacities of epidermal stem cells. Am. J. Transl. Res. 2016;8:5569–5579. [PMC free article] [PubMed] [Google Scholar]
- 35.Almeida M., Han L., Martin-Millan M., O’Brien C.A., Manolagas S.C. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J. Biol. Chem. 2007;282:27298–27305. doi: 10.1074/jbc.M702811200. [DOI] [PubMed] [Google Scholar]
- 36.Smith L.M., Wise S.C., Hendricks D.T., Sabichi A.L., Bos T., Reddy P., Brown P.H., Birrer M.J. cJun overexpression in MCF-7 breast cancer cells produces a tumorigenic, invasive and hormone resistant phenotype. Oncogene. 1999;18:6063–6070. doi: 10.1038/sj.onc.1202989. [DOI] [PubMed] [Google Scholar]
- 37.Shao J., Teng Y., Padia R., Hong S., Noh H., Xie X., Mumm J.S., Dong Z., Ding H.F., Cowell J. COP1 and GSK3β cooperate to promote c-Jun degradation and inhibit breast cancer cell tumorigenesis. Neoplasia. 2013;15:1075–1085. doi: 10.1593/neo.13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balko J.M., Schwarz L.J., Bhola N.E., Kurupi R., Owens P., Miller T.W., Gómez H., Cook R.S., Arteaga C.L. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res. 2013;73:6346–6358. doi: 10.1158/0008-5472.CAN-13-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song K.H., Park M.S., Nandu T.S., Gadad S., Kim S.C., Kim M.Y. GALNT14 promotes lung-specific breast cancer metastasis by modulating self-renewal and interaction with the lung microenvironment. Nat. Commun. 2016;7:13796. doi: 10.1038/ncomms13796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barbetti V., Morandi A., Tusa I., Digiacomo G., Riverso M., Marzi I., Cipolleschi M.G., Bessi S., Giannini A., Di Leo A. Chromatin-associated CSF-1R binds to the promoter of proliferation-related genes in breast cancer cells. Oncogene. 2014;33:4359–4364. doi: 10.1038/onc.2013.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen L., Bourguignon L.Y. Hyaluronan-CD44 interaction promotes c-Jun signaling and miRNA21 expression leading to Bcl-2 expression and chemoresistance in breast cancer cells. Mol. Cancer. 2014;13:52. doi: 10.1186/1476-4598-13-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang X., Wood P.A., Hrushesky W.J. Mammalian TIMELESS is required for ATM-dependent CHK2 activation and G2/M checkpoint control. J. Biol. Chem. 2010;285:3030–3034. doi: 10.1074/jbc.M109.050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magne Nde C.B., Casas Gimeno G., Docanto M., Knower K.C., Young M.J., Buehn J., Sayed E., Clyne C.D. Timeless Is a Novel Estrogen Receptor Co-activator Involved in Multiple Signaling Pathways in MCF-7 Cells. J. Mol. Biol. 2018;430:1531–1543. doi: 10.1016/j.jmb.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida K., Sato M., Hase T., Elshazley M., Yamashita R., Usami N., Taniguchi T., Yokoi K., Nakamura S., Kondo M. TIMELESS is overexpressed in lung cancer and its expression correlates with poor patient survival. Cancer Sci. 2013;104:171–177. doi: 10.1111/cas.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blanchard Z., Paul B.T., Craft B., ElShamy W.M. BRCA1-IRIS inactivation overcomes paclitaxel resistance in triple negative breast cancers. Breast Cancer Res. 2015;17:5. doi: 10.1186/s13058-014-0512-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yip C., Foidart P., Somja J., Truong A., Lienard M., Feyereisen E., Schroeder H., Gofflot S., Donneau A.F., Collignon J. MT4-MMP and EGFR expression levels are key biomarkers for breast cancer patient response to chemotherapy and erlotinib. Br. J. Cancer. 2017;116:742–751. doi: 10.1038/bjc.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu N., Liu B., Lian C., Doycheva D.M., Fu Z., Liu Y., Zhou J., He Z., Yang Z., Huang Q. Long noncoding RNA AC003092.1 promotes temozolomide chemosensitivity through miR-195/TFPI-2 signaling modulation in glioblastoma. Cell Death Dis. 2018;9:1139. doi: 10.1038/s41419-018-1183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.