

Abstract
Variation in methotrexate (MTX) efficacy represents a significant barrier to early and effective disease control in the treatment of autoimmune arthritis. We hypothesize that the utilization of metabolomic techniques will allow for an improved understanding of the biochemical basis for the pharmacological activity of MTX, and can promote the identification and evaluation of novel molecular biomarkers of MTX response. In this work, erythroblastoid cells were exposed to MTX at the physiologic concentration of 1,000 nM and analyzed using three metabolomic platforms to give a broad spectrum of cellular metabolites. MTX pharmacological activity, defined as cellular growth inhibition, was associated with an altered cellular metabolomic profile based on the analysis of 724 identified metabolites. By discriminant analysis, MTX treatment was associated with increases in ketoisovaleric acid, fructose, galactose, and 2‐deoxycytidine, and corresponding reductions in 2‐deoxyuridine, phosphatidylinositol 32:0, orotic acid, and inosine monophosphate. Inclusion of data from analysis of folate metabolism in combination with chemometric and metabolic network analysis demonstrated that MTX treatment is associated with dysregulated folate metabolism and nucleotide biosynthesis, which is in line with its known mechanism of action. However, MTX treatment was also associated with alterations in a diversity of metabolites, including intermediates of amino acid, carbohydrate, and lipid metabolism. Collectively, these findings support a robust metabolic response following exposure to physiologic concentrations of MTX. They also identify various metabolic intermediates that are associated with the pharmacological activity of MTX, and are, therefore, potential molecular biomarker candidates in future preclinical and clinical studies of MTX efficacy in autoimmune arthritis.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The need remains to identify clinical biomarkers of response to methotrexate (MTX) in the treatment of autoimmune arthritis. Although pharmacometabolomics represents a potentially powerful tool for biomarker identification, it has not been systematically applied to identify molecular markers of MTX efficacy.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ In this work, a systematic pharmacometabolomic approach is applied to an established in vitro model of MTX response to identify potential molecular markers of MTX response and key metabolic pathways impacted by the drug.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study identifies a number of key metabolites associated with the pharmacological activity of methotrexate, in vitro, including metabolites indicative of dysregulated folate and nucleotide metabolism, as well as intermediates of amino acid, carbohydrate, and lipid metabolism.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This study establishes the basis for a pharmacometabolomic‐based approach to identify molecular markers of MTX response in the treatment of autoimmune arthritis for translation into animal models and patient populations.
In chronic autoimmune arthritis, including juvenile idiopathic arthritis and rheumatoid arthritis (RA), rapid control of disease activity to prevent irreversible joint damage and promote improved long‐term outcomes continues to be a major therapeutic goal.1, 2, 3 However, in current practice, response to drug therapy is characterized by a highly variable and unpredictable course, which commonly necessitates multiple iterations of therapy modification to identify an effective treatment.4, 5 In the management of autoimmune arthritis, methotrexate (MTX) continues to be the cornerstone of initial and maintenance therapy, however, its onset of action is prolonged and approximately one in three patients fail to adequately respond to initial treatment.6, 7, 8, 9, 10, 11 To date, efforts to identify biomarkers to guide drug therapy have been unsuccessful, therapy remains effectively a trial‐and‐error process, and the mechanism of action of MTX remains incompletely understood.5, 12, 13 This approach to therapy wastes precious time and jeopardizes the overarching goal of early disease control. Therefore, a need exists to apply state‐of‐the‐art techniques, such as metabolomics, to identify biomarkers to guide clinicians in the early selection and optimization of drug therapy and further enhance the understanding of this extensively utilized drug.
Previous efforts to identify biomarkers of MTX response in autoimmune arthritis have primarily focused on the targeted analyses of MTX and its metabolites, or specific analytes hypothesized to be foundational to its impact on inflammation and immunity.14 A major limitation to these studies has been the biased investigative approach that largely focuses on a handful of metabolites or metabolic pathways, primarily revolving around the folate pathway and adenosine formation.15, 16, 17, 18 In fact, despite over 70 years of research on MTX, the biochemical basis through which MTX mediates its effects in autoimmune arthritis remains controversial and, therefore, has been a potential obstacle in the search for biochemical markers of efficacy.19 As an ‐omics approach, metabolomics offers a unique opportunity to apply a largely unbiased and untargeted approach to investigate potential biochemical markers of MTX response and to unravel the underlying biochemical basis of MTX efficacy.20, 21
Metabolomics references the study of the metabolome, which is the complete set of low‐molecular weight chemicals that can be found within a biological system.22 As a result, a patient's metabolome is the product of both endogenous and exogenous metabolism, thus various internal and external factors can impact a patient's metabolomic profile. In the case of RA, a number of metabolomic studies have been undertaken and have identified various metabolic pathways associated with disease activity, including nucleotide biosynthesis, glycolysis, amino acid metabolism, and the urea cycle.23, 24 Similar studies have evaluated differences in the metabolome between patients with RA based on their response to disease‐modifying therapy, and have similarly shown that changes in the metabolome are associated with drug efficacy.25, 26, 27, 28, 29 Despite these efforts to understand the metabolome in autoimmune arthritis, no concerted effort has been undertaken to define the relationship between the effect of drug therapy on the metabolome and pharmacological response to MTX as an approach to biomarker identification, which is the focus of the current work.
In this study, an untargeted global metabolomics approach is undertaken to evaluate the effect of exposure to physiologic concentrations of MTX on the metabolomic profile in an erythroblastoid cell line that has been previously utilized as a model for MTX disposition and pharmacological response.18, 30 The resulting metabolomic data are evaluated using multivariate statistical approaches along with chemometric and network enrichment analyses to interpret the impact of MTX on the cellular metabolome and to identify putative metabolomic markers of MTX response for interrogation in future studies of MTX response in autoimmune arthritis.
METHODS
Cell culture
The K562 human erythroblastoid cell line (catalog no. GM05372) was acquired from Coriell Cell Repositories (Camden, NJ) and maintained in Roswell Park Memorial Institute 1640 medium (catalog no. 61870‐127; ThermoFisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (catalog no. S11150; Atlanta Biologicals, Lawrenceville, GA) along with 100 units/mL penicillin and 1,000 units/mL streptomycin (catalog no. 15140122; ThermoFisher Scientific). Cells were maintained under normal growth conditions in a humidified, temperature and CO2‐controlled incubator at 37°C and 5% CO2. Cells were kept at density between 2.0 × 105 and 1.0 × 106 cells/mL to maintain logarithmic growth conditions to promote maximum proliferative activity over the experimental period. All experiments were conducted within six passages following removal from cryopreservation.
MTX treatment
Cell counts were conducted daily prior to, and on, the day of experimentation to ensure cells remained in a logarithmic growth phase. On the day of experimentation, K562 cells were seeded at a density of 4.0 × 105 cells/mL and exposed to 1,000 nM MTX or D‐phosphate‐buffered saline (i.e., vehicle) for 24 hours under normal growth conditions. At the end of the experiment, the cell density was measured and the volume of cells in media determined to contain 15 × 106 cells was removed and centrifuged at 1,000 g for 5 minutes. The cells were re‐suspended in 10 mL of ice‐cold D‐ phosphate‐buffered saline, centrifuged at 1,000 g for 5 minutes, and following the removal of the supernatant, the resulting cellular pellet was flash frozen in liquid nitrogen and stored in the vapor phase of a liquid nitrogen cryopreservation chamber prior to submission for metabolomics analysis. For both MTX treatment and control conditions, experiments were conducted using three replicate experiments using independently maintained cell culture lines on different days with three to four replicates per group resulting in a total sample size of 10 per group for the final analysis.
Anti‐proliferative activity
Cellular proliferation over the experimental period was determined based on the initial seeding density of 4.0 × 105 cells/mL for each sample and the measured cell density following the experimentation period. Cell densities were determined using a Sceptor 2.0 Cell Counter (MilliporeSigma, Burlington, MA). Fold‐change in cell density was calculated as the ratio of the density at the completion of the experiment relative to the seeding density, and the antiproliferative activity of MTX was determined based on comparison of fold‐change in cellular densities between the MTX‐treated and control groups.
Metabolomics analysis
Cryopreserved cellular pellets from MTX and control cells were submitted to the National Institutes of Health (NIH) West Coast Metabolomics Center at the University of California, Davis (Davis, CA) for untargeted analysis utilizing three independent standardized analytical methods for analysis to measure intermediates of primary metabolism, biogenic amines, and lipids.31, 32 Sample preparation was conducted at the West Coast Metabolomics Center using a biphasic liquid‐liquid extraction standard operating procedure developed for untargeted metabolomic analysis in plasma or serum samples across the three analytical platforms (File S1 ). Intermediates of primary metabolism were measured by automated liner exchange‐cold injection system gas chromatography time‐of‐flight mass spectrometry (File S2 ). Lipids were measured by charged surface hybrid chromatography electrospray ionization quadropole time‐of‐flight mass spectrometry (File S3 ). Biogenic amines were measured by hydrophilic interaction liquid chromatography electrospray ionization quadropole time‐of‐flight mass spectrometry (File S4 ). Detected peaks were identified based on retention times and mass spectra from MassBank of North America and reported as relative peak height intensities.33 Peak height intensity tables received following curation by the NIH West Coast Metabolomics Center were submitted to Metabolomics Workbench (https://www.metabolomicsworkbench.org/) under Project ID 1756 and are provided as supplemental files (Files S5–S7 ). Raw peak intensity data from each analytical platform underwent a normalization procedure.32 The normalization ratio was calculated as the ratio of the sum of peak heights for all identified metabolites for each sample to the total average metabolite total ion chromatogram for all samples. Peak heights for each metabolite were divided by the normalization ratio to arrive at the normalized peak height intensity. Metabolites measured in more than one analytical platform were combined by mean normalization to give equal weighting to each platform and averaged. The resulting normalized peak height intensities for known compounds (File S8 ) were uploaded into MetaboAnalyst 3.0 and further normalized by logarithmic transformation and Pareto scaling.34 The resulting data were analyzed for fold‐change and nonparametric unpaired analysis and visualized using volcano plots to identify initial metabolites of interest. The data were assessed by principle component analysis (PCA) and partial least squares discriminant analysis. A variable importance in projection was generated to identify and rank metabolites based on their ability to discriminate between the two treatment groups.
Enrichment analysis
The metabolites along with corresponding fold‐change and false discovery rate (FDR) adjusted P values were extracted and subjected to chemical and metabolic network enrichment analysis. Enrichment analysis based on chemical similarity was conducted using the open source software for Chemical Similarity Enrichment Analysis for Metabolomics (ChemRICH) that is independent of biochemical pathway assignments, but rather utilizes Tanimoto substructure similarity coefficients and medical subject headings ontology to generate nonoverlapping clusters of metabolites into distinct chemical classes.35 Metabolite predicted octanol:water partition coefficient (xlogP) values were collected using the PubChem chemical information database (https://pubchem.ncbi.nlm.nih.gov/). Additional chemometric and biochemical network characterization and visualization was conducted using MetaMapp to generate a network map based on chemical similarity utilizing the Kyoto Encyclopedia of Genes and Genomes metabolic network database and Tanimoto substructure similarity coefficients.36 The resulting mapping data were uploaded into Cytoscape 3.7.1 for visualization.
Statistical analysis
Fold‐change in cellular densities of K562 cells was determined by Wilcoxon rank‐sum analysis using JMP software version 11 (SAS Institute, Cary, NC). Analysis of differences in individual metabolites between groups was conducted using MetaboAnalyst 3.0 and included nonparametric univariate analysis and evaluated both statistical significance and fold‐change. An FDR of 0.05 was used to control for multiple testing. All metabolites achieving an FDR adjusted P value < 0.05 were considered significant and used in the chemometric and metabolic network enrichment analyses. Statistical testing within the chemometric analysis using the ChemRICH platform was determined by Kolmogorv–Smirnov testing and an FDR adjusted P value of < 0.05 was considered significant.
RESULTS
Antiproliferative activity of MTX is associated with an altered metabolic phenotype
Despite uncertainty about the mechanism of action of MTX in the treatment of autoimmune arthritis, MTX displays a quantifiable pharmacological response in the form of antiproliferative activity. In this study, K562 cells under normal growth conditions were either left untreated (i.e., controls) or treated with MTX at a concentration of 1,000 nM for 24 hours. The concentration of 1,000 nM was chosen based on MTX peak concentrations in the treatment of RA, and to allow analysis utilizing a complimentary data set including targeted metabolic profiling of folates in K562 cells treated with MTX.18, 37 Under these conditions, K562 cells are efficient at the uptake and polyglutamation of MTX, which is believed to be important in its pharmacological activity.30 Under normal growth conditions, control cells were found to more than double over the 24‐hour incubation period, whereas MTX‐treated cells failed to show any appreciable replication as represented by a median (interquartile range (IQR)) fold‐change in cell densities of 2.4 (IQR 2.0−2.5) and 1.1 (IQR 1.0−1.1), respectively (P = 0.0002; Figure 1).
Figure 1.
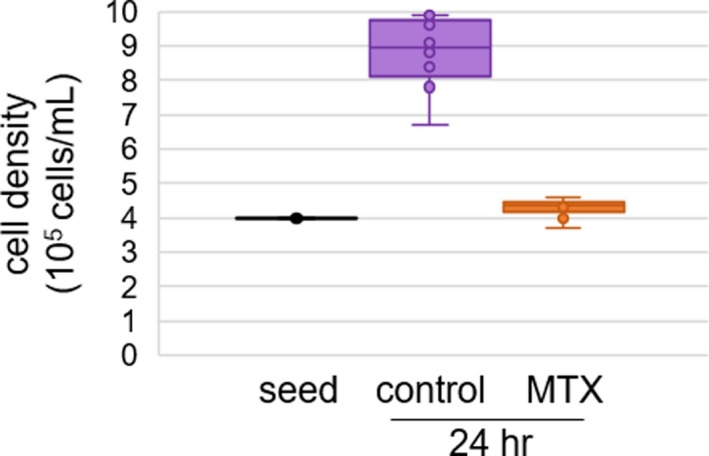
Pharmacological activity of methotrexate (MTX) in K562 cells. Pharmacological response was measured based on the antiproliferative effect of MTX following a 24‐hour exposure of K562 cells to MTX. Cell density data were collected from 10 samples per group over three independent experimental evaluations. Data points and representative box and whisker plots are shown based on the seeding density (i.e., seed) and density measurements after 24 hours of exposure to either vehicle control or 1,000 nM MTX. Cell densities were compared between groups using Wilcoxon rank‐sum testing and the resulting P values are provided.
A total of 15 million cells were collected from each sample with corresponding quantified antiproliferative response data and submitted for untargeted metabolomics analysis using three separate analytical platforms targeting the detection of markers of primary metabolism, complex lipids, and biogenic amines. Peak intensity data for known metabolites from each analytical platform underwent a normalization protocol and the data were combined and imported into MetaboAnalyst 3.0 for analysis. The analysis included 724 different known metabolites across the three analytical platforms. PCA was chosen for the initial visualization of the metabolomics data set (Figure 2 a). As an unsupervised and unbiased multivariate analytical technique, PCA allows for dimensional reduction of the data while preserving the variation of the high dimensional data set and allowing for visualization of the data distribution across the sample set. The control cells and MTX treated cells seem to be well separated by PCA. However, control cells formed a tighter cluster than MTX‐treated cells and were indicative of greater metabolic variation following MTX treatment. Therefore, based on the observed variance in the metabolomics data set, treatment with MTX results in significant alterations in the metabolomic profile of K562 cells.
Figure 2.
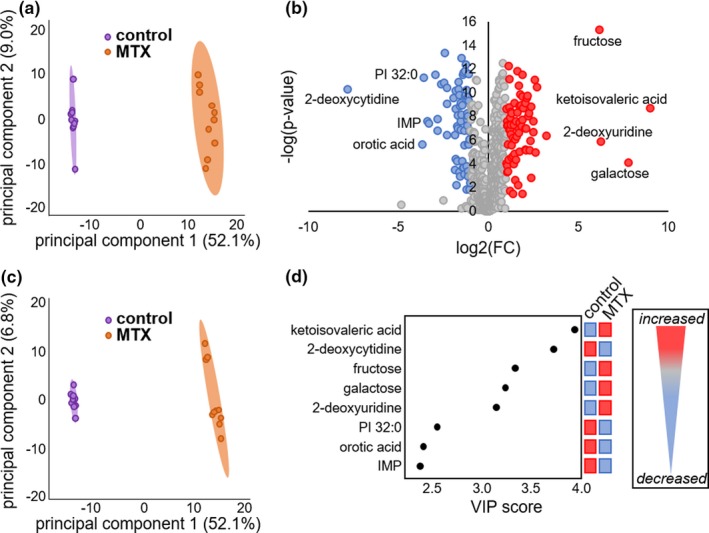
Identification of key metabolites associated with cellular response to methotrexate (MTX) by univariate and multivariate analysis of 724 identified metabolites. Metabolomics data were analyzed using MetaboAnalyst 3.0 and the resulting (a) principle component analysis scores plot, (b) volcano plot, (c) partial least squares discriminant analysis scores plot, and (d) variable importance in projection (VIP) score plot are presented. In the volcano plot, red‐colored metabolites represent those metabolites found to be increased in the MTX treated group, whereas the blue‐colored metabolites represent those metabolites that were found to be decreased and statistically significant metabolites (false discovery rate adjusted P value < 0.05) with a > 10‐fold change are labeled on the volcano plot. The top discriminating metabolites are presented in the VIP score plot and are the same metabolites identified in the volcano plot. FC, fold change; IMP, inosine monophosphate; PI, phosphatidylinositol.
Identification of metabolomic markers associated with the antiproliferative activity of MTX
Comparison of relative metabolite abundance between control and MTX‐treated cells was determined by unpaired univariate analysis. Calculated P values were adjusted based on an FDR of 0.05 and filtered by fold‐change analysis with metabolites displaying a greater than twofold change deemed significant. A total of 144 metabolites were found to be significantly different in the MTX‐treated group by this analysis; represented by an increase in 76 metabolites and a decrease in 68 metabolites. An additional step to identify the most significantly impacted metabolites included the selection of molecules displaying at least a 10‐fold change resulting in eight highly impacted metabolites (Figure 2 b). For illustrative purposes the metabolites reaching the 10‐fold change threshold are annotated on the resulting volcano plot, and include decreases in 2‐deoxycytidine, orotic acid, phosphatidylinositol (PI) 32:0, and inosine monophosphate (IMP), and increases in ketoisovaleric acid, galactose, fructose, and 2‐deoxyuridine.
The data set was interrogated by partial least squares discriminant analysis as a supervised multivariate approach to identify major discriminatory metabolites (Figure 2 c). The resulting scores plot demonstrated a good separation of metabolomic profiles for control and MTX treated cells and demonstrated good reliability by leave‐one‐out cross validation analysis (R 2 = 0.996, Q 2 = 0.991). Similar to PCA, control cells formed a tighter cluster than MTX treated cells. Generation of a variable importance in projection score plot demonstrated that the same eight metabolites identified in the univariate analysis were identified as the major discriminatory metabolites between the control and MTX‐treated cells (Figure 2 d).
Normalized log transformed peak intensity data for the eight metabolites identified by univariate and multivariate analysis are provided (Figure 3). Treatment with MTX resulted in increases in ketoisovaleric acid, fructose, galactose, and 2‐deoxyuridine that are represented by 496‐fold, 73‐fold, 324‐fold, and 76‐fold increases, respectively. In contrast, MTX treatment resulted in dramatic decreases in 2‐deoxycytidine, PI 32:0, orotic acid, and IMP that are represented by 248‐fold, 13‐fold, 13‐fold, and 10‐fold reductions, respectively.
Figure 3.
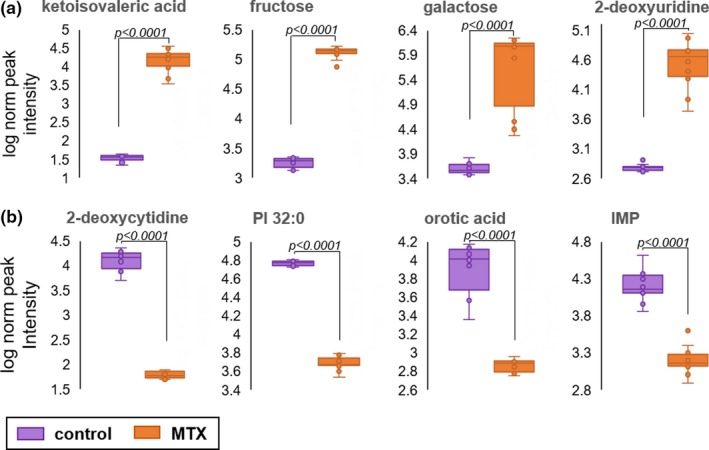
Relative abundance of metabolomic markers of methotrexate (MTX) response. The normalized peak ion intensity plots for the eight metabolites identified by univariate and multivariate analyses are presented. (a) Metabolites found to be increased in MTX‐treated cells includes ketoisovaleric acid, fructose, galactose, and 2‐deoxyuridine. (b) Metabolites found to be decreased in MTX‐treated cells includes 2‐deoxycytidine, phosphatidylinositol (PI) 32:0, orotic acid, and inosine monophosphate (IMP). Normalized peak ion intensity data were collected from 10 samples per group over three independent experimental evaluations. Data points and representative box and whisker plots are shown based on metabolomics analysis after 24 hours of exposure to either vehicle control or 1,000 nM MTX. Normalized peak ion intensities were compared between groups using unpaired nonparametric analysis with unequal variances. The resulting false discovery rate adjusted P values are provided.
Enrichment analysis supports folate and nucleotide metabolism as key pathways associated with the pharmacological activity of MTX
Following statistical analysis for individual analytes measured by the untargeted metabolomic analysis, chemometric and metabolic pathway analysis techniques were utilized to detect specific chemical classes and metabolic pathways associated with the observed pharmacological response to MTX. In addition to the metabolomic data collected in this study, targeted analysis of folates and select markers of purine and pyrimidine biosynthesis that were previously collected were included in the analysis.18, 30 Initial chemometric analysis was conducted using lipophilicity (i.e., xlogP) as a defining chemical descriptor to stratify the individual metabolites and plotted as a function of fold‐change with node size representing the negative logarithmic transformation of the FDR adjusted P value for each metabolite (Figure 4 a). Only metabolites with a greater than twofold change and an FDR adjusted P value of < 0.05 were included in the plot. The eight metabolites identified by univariate and multivariate analysis and an additional five metabolites of greater than 10‐fold change from the previous targeted analysis are labeled on the plot. Specifically, the previous study found reductions in 5‐methyl‐tetrahydrofolate (5m‐THF), formyl‐THF, and methylene‐THF of 67‐fold, 16‐fold, and 14‐fold; as well as 75‐fold increase in folic acid. Interestingly, except for PI 32:0, all of the significantly altered metabolites with a > 10‐fold change are relatively hydrophilic molecules with xlogP values < 1.0, and suggests MTX has a greater impact on intermediates of primary metabolism and biogenic amines than on lipids.
Figure 4.
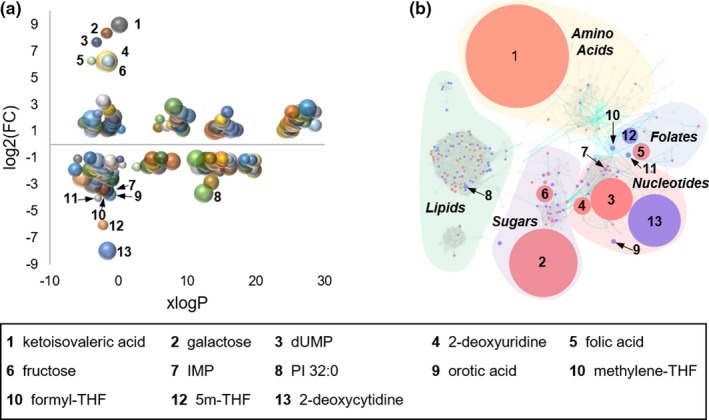
Chemometric and metabolic network mapping of metabolomic and targeted metabolic profiling data. (a) Metabolomic data combined with data from targeted analysis of folates and select intermediates of nucleotide biosynthesis was assessed based on lipophilicity, fold‐change (FC), and false discovery rate adjusted P value; with node size directly proportional to the inverse log of the P value for each metabolite. (b) A chemometric and metabolic network map was built using MetaMapp and visualized using Cytoscape. Red denotes metabolites found to significantly increase with methotrexate (MTX) treatment and blue denotes metabolites found to significantly decrease in cells treated with MTX. Node size is directly proportional to measured FC. Blue lines between nodes represent Kyoto Encyclopedia of Genes and Genomes reaction pairs, whereas gray lines represent pairing based on chemical similarity. Metabolites found to be significantly altered with a greater than 10‐fold change are labeled by number with the corresponding metabolite name provided in the key.
Integration of biochemical pathway and chemical similarity mapping for the metabolomics data set was conducted using MetaMapp and visualized using Cytoscape 3.7.1 (Figure 4 b). Node size is directly proportional to the observed median fold‐change and nodes are color‐coded based on whether the analyte was increased (i.e., red) or decreased (i.e., blue) when K562 cells were treated with MTX. Only analytes with an FDR adjusted P value < 0.05 and a > 2‐fold change were colored‐coded and the 13 significantly altered metabolites with a > 10‐fold change are labeled. The resulting network map structure is based on the Kyoto Encyclopedia of Genes and Genomes reactant pair database as well as Tanimoto chemical and National Institute of Standards and Technology mass spectral similarity scores. The resulting node clusters were further manually classified based on the chemical composition and associated metabolic pathways for the metabolites within the clusters.
The cluster of metabolites representing folate metabolism demonstrates that MTX has a profound impact on folate metabolism (Figure 5 a). Most notably, MTX treatment results in the accumulation of folic acid and a corresponding depletion of 5m‐THF, methylene‐THF, and formyl‐THF that is consistent with inhibition of dihydrofolate reductase (Figure 5 a).38 Evaluation of nucleotide biosynthesis depicts changes in pyrimidine biosynthesis that is of greater magnitude than the observed antifolate effects of MTX (Figure 5 b). In particular, MTX caused a dramatic accumulation of intracellular deoxyuridine monophosphate and 2‐deoxyuridine with a corresponding depletion of 2‐deoxycytidine and orotic acid. Additionally, a significant depletion of pyrimidines, including uridine diphosphate N‐acetylglucosamine, uridine, and 5‐methyluridine was observed despite an unanticipated increase in cellular thymidine monophosphate levels. Purines were also found to be markedly depleted with significant reductions in IMP, adenosine monophosphate, S‐adenosyl‐homocysteine, inosine, hypoxanthine, guanosine, and guanine. However, an increase in 8‐oxo‐2′‐deoxyadenosine was observed and may represent an increase in oxidative damage of DNA in MTX‐treated cells.39
Figure 5.
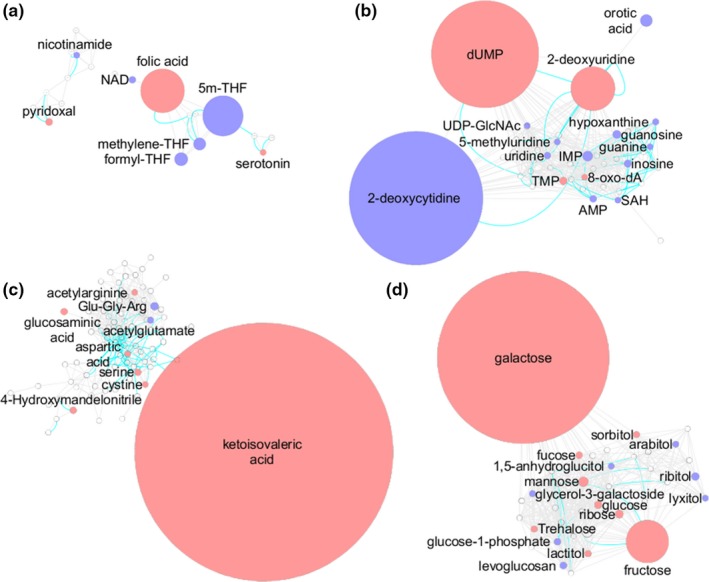
Key metabolic pathways associated with pharmacological activity of methotrexate (MTX) in K562 cells. The metabolic network built in Cytoscape was divided into clusters using the community cluster tool within Cytoscape. The resulting clusters represent (a) folate‐related metabolites, (b) nucleotides, (c) amino acids, and (d) carbohydrates. Red denotes metabolites found to significantly increase with MTX treatment and blue denotes metabolites found to significantly decrease with MTX treatment. Node size is directly proportional to measured fold‐change. Blue lines between nodes represent Kyoto Encyclopedia of Genes and Genomes reaction pairs, whereas gray lines represent pairing based on chemical similarity. 5m‐THF, 5‐methyl‐tetrahydrofolate; dUMP, deoxyuridine monophosphate; IMP, inosine monophosphate; SAH, S‐adenosyl‐homocysteine; THF, tetrahydrofolate; TMP, thymidine monophosphate.
Evaluation of the cluster of metabolites surrounding ketoisovaleric acid mainly represented intermediates of amino acid metabolism, and despite the high level of ketoisovaleric acid accumulation following MTX treatment, only minimal corresponding changes in amino acid metabolism were observed (Figure 5 c). However, in general, MTX treatment was associated with increased amino acid levels. Similarly, evaluation of metabolites related to fructose and galactose demonstrated a similar general increase in intermediates of carbohydrate metabolism with MTX treatment (Figure 5 d). Lipids and intermediates of the tricarboxylic acids (TCA) cycle, although less dramatic, also demonstrated sensitivity to MTX exposure suggesting inhibition of the TCA cycle and a shift from saturated to unsaturated fatty acid (FA) containing lipids (Figure S1). Enrichment analysis using ChemRICH to categorize the metabolites into chemical classes based on Tanimoto substructure similarity coefficients and medical subject headings was used to further describe the effect of MTX on various classes of metabolites (Figure 6). In the resulting plot, the fraction directional change for each class of metabolites is compared based on the median xlogP for the classes of metabolites that are found to be significantly different in the MTX‐treated group based on an FDR adjusted P value < 0.05. In this analysis, fraction directional change for each group of metabolites was calculated based on the net directional change in metabolite levels divided by the total number of metabolites measured in each group, for example, 88 metabolites classified as unsaturated phosphatidylcholines were measured with 50 found to be significantly increased and 15 found to be significantly reduced (i.e., net directional change = 50–15 = 35), therefore, fraction directional change was calculated as 0.43 (i.e., 35/88 = 0.43) and indicated a net increase in unsaturated phosphatidylcholines following MTX treatment. Node size is directly proportional to the negative log transformed FDR adjusted P value for each class of metabolites. From these data, it is evident that metabolites representing a diversity of chemical classes are significantly different in MTX‐treated cells, but based on fraction directional change the response to MTX is associated with generalized depletion of some sets of metabolites and accumulation of others. Most notably, in addition to the effect of MTX on folates, nucleotides, amino acids, and tricarboxylic acids, treatment with MTX was found to be associated with altered lipid homeostasis represented by reduced levels of cholesterol esters, diglycerides, phosphatidylinositols, lysophospholipids, and saturated triglycerides, phosphatidylcholines, and lysophosphatidylcholines, and corresponding increases in sphingomyelins, ceramides, galactosylceramides, plasmalogens, phosphatidylethanolamines, and unsaturated FAs, triglycerides, lysophosphatidylcholines, and phosphatidylcholines.
Figure 6.
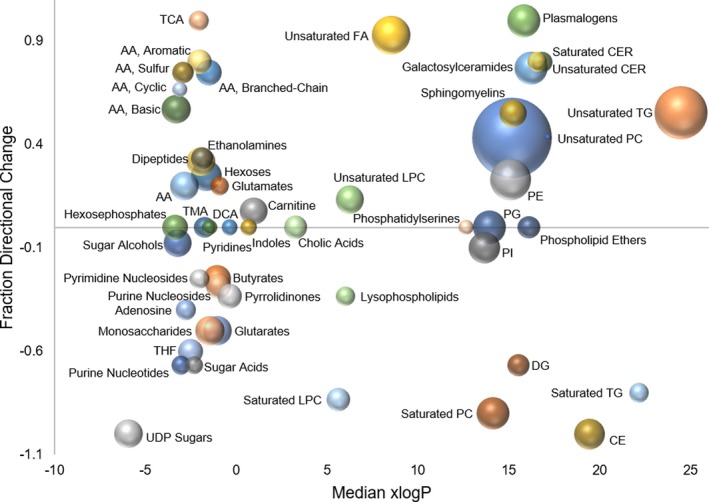
Association of methotrexate (MTX) pharmacological activity with changes by chemical group classification using chemical similarity enrichment analysis. Metabolomics data were analyzed using ChemRICH open source software to produce nonoverlapping chemical group classifications that was able to map 671 of the identified metabolites to 62 nonoverlapping chemical classes, of which 50 were found to be statistically significantly different between the MTX treated and control group (false discovery rate adjusted P value < 0.05). For each group of chemicals the fraction directional change is plotted as a function of the median xlogP and node size is directly proportional to the negative logarithm of the P value.
DISCUSSION
The findings from this analysis can be partially mapped onto the known biochemical pathways targeted by MTX (Figure 7).30, 40 In this illustration, the red “X” indicates known enzymatic targets of MTX, font color denotes the effect of MTX on the metabolite under the treatment conditions used in this study, and the enlarged font size represents the metabolites with a > 10‐fold change under the experimental conditions. Red denotes metabolites that were observed to increase, blue denotes metabolites that were observed to decrease, and black denotes metabolites that were not significantly altered (i.e., FDR adjusted P value > 0.05). Based on the known inhibitory activity of MTX against dihydrofolate reductase, as well as several folate‐dependent enzymes, it is clear that MTX exposure results in dysregulation of the intracellular folate pool. However, based on our experimental data, it seems that various downstream intermediates of nucleotide biosynthesis may represent more sensitive markers of MTX pharmacological response. In particular, several markers of pyrimidine and purine biosynthesis, including deoxyuridine monophosphate, 2‐deoxycytidine, 2‐deoxyuridine, and IMP may represent highly sensitive markers of MTX activity. These findings are consistent with previous studies that have found MTX treatment is associated with systemic depletion of circulating folates and corresponding dysregulation of intermediates of nucleotide metabolism.18, 28, 29, 41
Figure 7.
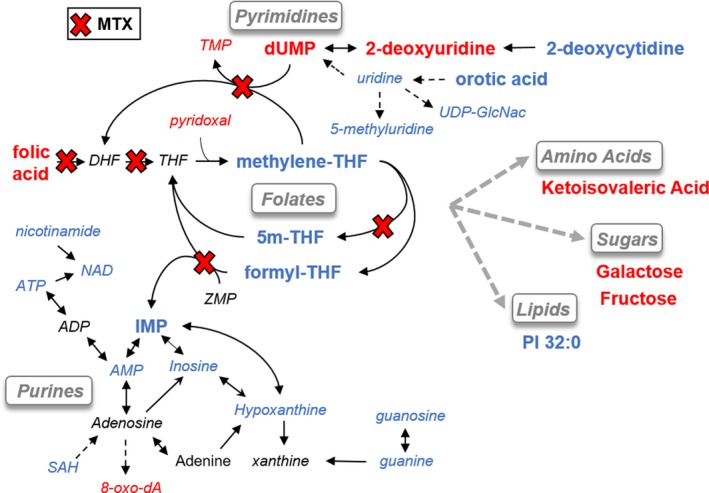
Mapping of biochemical data onto known pharmacologic targets of methotrexate (MTX). An illustration of the major metabolic pathways targeted by MTX is depicted and the findings of this study are shown based on the color and size of the labeled metabolites. Specifically, metabolites with large font represent metabolites with a > 10‐fold change following treatment with MTX, red font color indicates a decrease in the metabolite, blue font color represents an increase in the metabolite, and black font color represents a nonstatistically significant change in the measured metabolite. Known enzymatic targets of MTX and its metabolites are marked by a red “X.” 5m‐THF, 5‐methyl‐tetrahydrofolate; ATP, adenosine triphosphate; dUMP, deoxyuridine monophosphate; IMP, inosine monophosphate; PI, phosphatidylinositol; SAH, S‐adenosyl‐homocysteine; THF, tetrahydrofolate; TMP, thymidine monophosphate; UDP‐GlcNAc, uridine diphosphate N‐acetylglucosamine.
Additional metabolites outside of folate and nucleotide metabolism displayed a high sensitivity to MTX. These metabolites included a metabolic intermediate of valine metabolism (i.e., ketoisovaleric acid) and several sugars, including galactose and fructose. However, these intermediates are not directly linked to the known pharmacologic targets of MTX and may represent downstream metabolic changes related to the static growth state of cells treated with MTX. This interpretation would be supported by the observation that MTX treatment was also associated with the cellular accumulation of amino acids and TCA cycle intermediates, which would be suggestive of a generalized decrease in cellular metabolic activity. These findings are in agreement with previous studies that have demonstrated that, in addition to dysregulation in folates and nucleotide metabolism, MTX response in RA is also associated with downregulation of glycolysis and the TCA cycle, in combination with reductions in amino acid and lipid metabolism.28, 29
Analysis of the metabolomic data set using chemical similarity enrichment analysis revealed changes in several classes of biochemicals, most notably changes in the cellular lipid composition following treatment with MTX that suggest an effect of MTX on cellular lipid homeostasis in favor of less cholesterol esters, less saturated FA‐containing lipids, and an increase in unsaturated FA‐containing lipids. Together, such changes in lipid composition, if observed in vivo, may be expected to yield beneficial cardiovascular effects.42 Although MTX therapy in the treatment of autoimmune arthritis is associated with a decrease in cardiovascular risk, the recently completed cardiovascular inflammation reduction trial (CIRT) failed to demonstrate a cardiovascular benefit of MTX in a non‐RA population at risk of cardiovascular events.43, 44 Therefore, although these cellular data support some potentially beneficial effects on lipid metabolism, these effects need to be evaluated in ongoing animal and human studies to determine whether this effect on lipids is also observed in vivo and translated into cardiovascular benefit.
A major limitation of this study is the investigation of a single cell line without the analysis of the time‐dependence and dose‐dependence of the observed metabolic changes. Additionally, the K562 cell line is a transformed cancerous cell line and may not be representative of the metabolic effects of MTX at the site of action in untransformed human tissues in patients with autoimmune arthritis. As a descriptive metabolomics study, an additional weakness of this study is the lack of mechanistic studies to define how MTX impacts the various metabolic pathways identified and how these pathways are related.
Together, these data support a robust and diverse cellular metabolic response to MTX that encompasses various metabolites across a variety of biochemical classes. However, the fact that four of the eight metabolites identified in this metabolomic study represent intermediates of de novo purine and pyrimidine biosynthesis further highlights the importance of these pathways, both in the pharmacological activity of MTX and as potentially important pathways for the identification of metabolic biomarkers of MTX response. Further, exposure to MTX is observed to be associated with alteration in the TCA cycle, amino acid, and carbohydrate metabolism, and a shift in the cellular lipid profile in favor of reduced cellular cholesterol and saturated FA content in favor of unsaturated FA. The relevance of these findings depends on establishing whether the same effect occurs in vivo.
Funding
This work was supported by the University of Kansas and a Clinical and Translational Science Award (CTSA) grant from NCATS awarded to the University of Kansas for Frontiers: University of Kansas Clinical and Translational Science Institute (#KL2TR002367) in addition to support through the Kansas Institute for Precision Medicine funded by a Centers of Biomedical Research Excellence (COBRE) grant awarded by NIGMS (#P20GM130423). The article processing charges related to the publication of this article were partially supported by The University of Kansas (KU) One University Open Access Author Fund sponsored jointly by the KU Provost, KU Vice Chancellor for Research & Graduate Studies, and KUMC Vice Chancellor for Research and managed jointly by the Libraries at the Medical Center and KU ‐ Lawrence.
Conflict of interest
All authors declared no competing interests for this work.
Author contributions
R.S.F. and M.L.B. wrote the manuscript. R.S.F. and M.L.B. designed the research. R.S.F. and R.K.S. performed the research. R.S.F. analyzed the data.
Supporting information
Figure S1. Additional metabolic pathways associated with pharmacological activity of MTX in K562 cells.
File S1. Sample preparation standard operating procedure for metabolomics analysis.
File S2. Data dictionary for primary metabolism by ALEX‐CIS GCTOF MS.
File S3. Data dictionary for lipidomics by CSH‐ESI QTOF MS/MS.
File S4. Data dictionary for biogenic amines by HILIC‐ESI QTOF MS/MS.
File S5. Peak intensity tables for primary metabolism analysis.
File S6. Peak intensity tables for lipidomics analysis.
File S7. Peak intensity tables for biogenic amine analysis.
File S8. Normalized peak intensity table for combined metabolomics data.
Acknowledgments
The authors would like to acknowledge Kelly Paglia and members of the Fiehn Laboratory at the NIH West Coast Metabolomics Center for their help in the design and conduct of this metabolomics analysis.
Data availability statement
These data have been submitted to the NIH Common Fund's Metabolomics Data Repository and Coordinating Center (supported by NIH grant, U01‐DK097430) website, the Metabolomics Workbench, http://www.metabolomicsworkbench.org, where it has been assigned Project ID 1756.
References
- 1. Anderson, J.J. , Wells, G. , Verhoeven, A.C. & Felson, D.T. Factors predicting response to treatment in rheumatoid arthritis: the importance of disease duration. Arthritis Rheum. 43, 22–29 (2000). [DOI] [PubMed] [Google Scholar]
- 2. Lard, L.R. et al Early versus delayed treatment in patients with recent‐onset rheumatoid arthritis: comparison of two cohorts who received different treatment strategies. Am. J. Med. 111, 446–451 (2001). [DOI] [PubMed] [Google Scholar]
- 3. Albers, H.M. et al Time to treatment as an important factor for the response to methotrexate in juvenile idiopathic arthritis. Arthritis Rheum. 61, 46–51 (2009). [DOI] [PubMed] [Google Scholar]
- 4. Sizova, L. Approaches to the treatment of early rheumatoid arthritis with disease‐modifying antirheumatic drugs. Br. J. Clin. Pharmacol. 66, 173–178 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Funk, R.S. & Becker, M.L. Disease modifying anti‐rheumatic drugs in juvenile idiopathic arthritis: striving for individualized therapy. Expert Rev. Precision Med. Drug Dev. 1, 53–68 (2016). [Google Scholar]
- 6. Giannini, E.H. et al Methotrexate in resistant juvenile rheumatoid arthritis. Results of the U.S.A.‐U.S.S.R. double‐blind, placebo‐controlled trial. The Pediatric Rheumatology Collaborative Study Group and The Cooperative Children's Study Group. N. Engl. J. Med. 326, 1043–1049 (1992). [DOI] [PubMed] [Google Scholar]
- 7. Lambert, C.M. et al Dose escalation of parenteral methotrexate in active rheumatoid arthritis that has been unresponsive to conventional doses of methotrexate: a randomized, controlled trial. Arthritis Rheum. 50, 364–371 (2004). [DOI] [PubMed] [Google Scholar]
- 8. Ramanan, A.V. , Whitworth, P. & Baildam, E.M. Use of methotrexate in juvenile idiopathic arthritis. Arch. Dis. Child. 88, 197–200 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ruperto, N. et al A randomized trial of parenteral methotrexate comparing an intermediate dose with a higher dose in children with juvenile idiopathic arthritis who failed to respond to standard doses of methotrexate. Arthritis Rheum. 50, 2191–2201 (2004). [DOI] [PubMed] [Google Scholar]
- 10. Shinde, C.G. , Venkatesh, M.P. , Kumar, T.M. & Shivakumar, H.G. Methotrexate: a gold standard for treatment of rheumatoid arthritis. J. Pain Palliat. Care Pharmacother. 28, 351–358 (2014). [DOI] [PubMed] [Google Scholar]
- 11. Bathon, J.M. et al A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N. Engl. J. Med. 343, 1586–1593 (2000). [DOI] [PubMed] [Google Scholar]
- 12. Abud‐Mendoza, C. , Martinez‐Martinez, M.U. , Monsivais‐Urenda, A. & Gonzalez‐Amaro, R. Laboratory biomarkers for guiding therapy with methotrexate in rheumatoid arthritis. Curr. Pharm. Des. 21, 202–211 (2015). [DOI] [PubMed] [Google Scholar]
- 13. Chan, E.S. & Cronstein, B.N. Methotrexate–how does it really work? Nat. Rev. Rheumatol. 6, 175–178 (2010). [DOI] [PubMed] [Google Scholar]
- 14. Brown, P.M. , Pratt, A.G. & Isaacs, J.D. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat. Rev. Rheumatol. 12, 731–742 (2016). [DOI] [PubMed] [Google Scholar]
- 15. Calasan, M.B. et al Methotrexate polyglutamates in erythrocytes are associated with lower disease activity in juvenile idiopathic arthritis patients. Ann. Rheum. Dis. 74, 402–407 (2015). [DOI] [PubMed] [Google Scholar]
- 16. de Rotte, M.C. et al Association of low baseline levels of erythrocyte folate with treatment nonresponse at three months in rheumatoid arthritis patients receiving methotrexate. Arthritis Rheum. 65, 2803–2813 (2013). [DOI] [PubMed] [Google Scholar]
- 17. de Rotte, M.C. et al Methotrexate polyglutamates in erythrocytes are associated with lower disease activity in patients with rheumatoid arthritis. Ann. Rheum. Dis. 74, 408–414 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Funk, R.S. , van Haandel, L. , Leeder, J.S. & Becker, M.L. Folate depletion and increased glutamation in juvenile idiopathic arthritis patients treated with methotrexate. Arthritis Rheumatol. 66, 3476–3485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Friedman, B. & Cronstein, B. Methotrexate mechanism in treatment of rheumatoid arthritis. Joint Bone Spine. 86, 301–307 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beger, R.D. et al Metabolomics enables precision medicine: “A White Paper, Community Perspective”. Metabolomics 12, 149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaddurah‐Daouk, R. , Weinshilboum, R. & Pharmacometabolomics Research Network . Metabolomic signatures for drug response phenotypes: pharmacometabolomics enables precision medicine. Clin. Pharmacol. Ther. 98, 71–75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fiehn, O. Metabolomics–the link between genotypes and phenotypes. Plant Mol. Biol. 48, 155–171 (2002). [PubMed] [Google Scholar]
- 23. Gupta, L. , Ahmed, S. , Jain, A. & Misra, R. Emerging role of metabolomics in rheumatology. Int. J. Rheum. Dis. 21, 1468–1477 (2018). [DOI] [PubMed] [Google Scholar]
- 24. Guma, M. , Tiziani, S. & Firestein, G.S. Metabolomics in rheumatic diseases: desperately seeking biomarkers. Nat. Rev. Rheumatol. 12, 269–281 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Priori, R. et al (1)H‐NMR‐based metabolomic study for identifying serum profiles associated with the response to etanercept in patients with rheumatoid arthritis. PLoS One 10, e0138537 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cuppen, B.V. et al Exploring the inflammatory metabolomic profile to predict response to TNF‐alpha inhibitors in rheumatoid arthritis. PLoS One 11, e0163087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sweeney, S.R. et al Metabolomic profiling predicts outcome of rituximab therapy in rheumatoid arthritis. RMD Open. 2, e000289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang, Z. et al (1)H NMR‐based metabolomic analysis for identifying serum biomarkers to evaluate methotrexate treatment in patients with early rheumatoid arthritis. Exp. Ther. Med. 4, 165–171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang, M. et al Treatment of rheumatoid arthritis using combination of methotrexate and tripterygium glycosides tablets—a quantitative plasma pharmacochemical and pseudotargeted metabolomic approach. Front. Pharmacol. 9, 1051 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Funk, R.S. , van Haandel, L. , Becker, M.L. & Leeder, J.S. Low‐dose methotrexate results in the selective accumulation of aminoimidazole carboxamide ribotide in an erythroblastoid cell line. J. Pharmacol. Exp. Ther. 347, 154–163 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cajka, T. & Fiehn, O. Toward merging untargeted and targeted methods in mass spectrometry‐based metabolomics and lipidomics. Anal. Chem. 88, 524–545 (2016). [DOI] [PubMed] [Google Scholar]
- 32. Fiehn, O. Metabolomics by gas chromatography‐mass spectrometry: combined targeted and untargeted profiling. Curr. Protoc. Mol. Biol. 114, 30.4.1–30.4.32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blaženović, I. , Kind, T. , Ji, J. & Fiehn, O. Software tools and approaches for compound identification of LC‐MS/MS data in metabolomics. Metabolites 8, pii: E31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xia, J. & Wishart, D.S. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinform. 55, 14.10.11–14.10.91 (2016). [DOI] [PubMed] [Google Scholar]
- 35. Barupal, D.K. & Fiehn, O. Chemical Similarity Enrichment Analysis (ChemRICH) as alternative to biochemical pathway mapping for metabolomic datasets. Sci. Rep. 7, 14567 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barupal, D.K. et al MetaMapp: mapping and visualizing metabolomic data by integrating information from biochemical pathways and chemical and mass spectral similarity. BMC Bioinform. 13, 99 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoekstra, M. et al Bioavailability of higher dose methotrexate comparing oral and subcutaneous administration in patients with rheumatoid arthritis. J. Rheumatol. 31, 645–648 (2004). [PubMed] [Google Scholar]
- 38. White, J.C. & Goldman, I.D. Mechanism of action of methotrexate. IV. Free intracellular methotrexate required to suppress dihydrofolate reduction to tetrahydrofolate by Ehrlich ascites tumor cells in vitro. Mol. Pharmacol. 12, 711–719 (1976). [PubMed] [Google Scholar]
- 39. Singh, R. et al Simultaneous determination of 8‐oxo‐2′‐deoxyguanosine and 8‐oxo‐2′‐deoxyadenosine in DNA using online column‐switching liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass. Spectrom. 23, 151–160 (2009). [DOI] [PubMed] [Google Scholar]
- 40. Becker, M.L. & Funk, R.S. Reverse translation in advancing pharmacotherapy in pediatric rheumatology: a logical approach in rare diseases with limited resources. Clin. Transl. Sci. 11, 106–108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singh, R.K. et al Methotrexate disposition, anti‐folate activity and efficacy in the collagen‐induced arthritis mouse model. Eur. J. Pharmacol. 853, 264–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ference, B.A. , Graham, I. , Tokgozoglu, L. & Catapano, A.L. Impact of lipids on cardiovascular health: JACC health promotion series. J. Am. Coll. Cardiol. 72, 1141–1156 (2018). [DOI] [PubMed] [Google Scholar]
- 43. Everett, B.M. et al Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am. Heart J. 166, 199–207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Micha, R. et al Systematic review and meta‐analysis of methotrexate use and risk of cardiovascular disease. Am. J. Cardiol. 108, 1362–1370 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Additional metabolic pathways associated with pharmacological activity of MTX in K562 cells.
File S1. Sample preparation standard operating procedure for metabolomics analysis.
File S2. Data dictionary for primary metabolism by ALEX‐CIS GCTOF MS.
File S3. Data dictionary for lipidomics by CSH‐ESI QTOF MS/MS.
File S4. Data dictionary for biogenic amines by HILIC‐ESI QTOF MS/MS.
File S5. Peak intensity tables for primary metabolism analysis.
File S6. Peak intensity tables for lipidomics analysis.
File S7. Peak intensity tables for biogenic amine analysis.
File S8. Normalized peak intensity table for combined metabolomics data.
Data Availability Statement
These data have been submitted to the NIH Common Fund's Metabolomics Data Repository and Coordinating Center (supported by NIH grant, U01‐DK097430) website, the Metabolomics Workbench, http://www.metabolomicsworkbench.org, where it has been assigned Project ID 1756.