

Summary
Recurrent respiratory papillomatosis (RRP) is characterized by benign exophytic lesions of the respiratory tract caused by the human papillomavirus (HPV), in particular low‐risk HPV6 and HPV11. Aggressiveness varies greatly among patients. Surgical excision is the current standard of care for RRP, with adjuvant therapy used when surgery cannot control disease recurrence. Numerous adjuvant therapies have been used to control RRP with some success, but none are curative. Current literature supports a polarization of the adaptive immune response to a T helper type 2 (Th2)‐like or T regulatory phenotype, driven by a complex interplay between innate immunity, adaptive immunity and HPV6/11 proteins. Additionally, certain immunogenetic polymorphisms can predispose individuals to an HPV6/11‐tolerant microenvironment. As a result, immunomodulatory efforts are being made to restore the host immune system to a more balanced T cell phenotype and clear viral infection. Literature has shown exciting evidence for the role of HPV vaccination with Gardasil or Gardasil‐9 as both primary prevention, by decreasing incidence through childhood vaccinations, and secondary prevention, by treating active RRP disease. Multi‐institution randomized clinical trials are needed to better assess their efficacy as treatment for active disease. Interestingly, a DNA vaccine has recently shown in‐vitro success in generating a more robust CD8+ T cell response. Furthermore, clinical trials for programmed death 1 (PD‐1) inhibitors are under investigation for RRP management. Molecular insights into RRP, in particular the interplay between RRP and the immune system, are needed to advance our understanding of this disease and may lead to the identification of immunomodulatory agents to better manage RRP.
Keywords: killer immunoglobulin‐like receptors (KIR), Th1/Th2 cells, tumor immunology, vaccination, viral
Current literature supports a polarization of the adaptive immune response to a Th2‐like or T‐regulatory phenotype, driven by a complex interplay between innate immunity, adaptive immunity, and HPV6/11 proteins in recurrent respiratory papillomatosis. Additionally, certain immunogenetic polymorphisms can predispose individuals to an HPV6/11‐tolerant microenvironment. As a result, immunomodulatory efforts are being made to restore the host immune system to a more balanced T cell phenotype and clear HPV infection.
Introduction
Recurrent respiratory papillomatosis (RRP) is a rare, benign, chronic disease that involves exophytic lesions of the mucosa of the respiratory tract 1. During the past two decades, incidence and prevalence have remained relatively consistent across the globe at less than 0·01%, with minor variability between adult and juvenile subpopulations 2, 3, 4, 5. It is most commonly caused by low‐risk human papillomavirus (HPV) types 6 and 11 6. The clinical course of RRP shows variability, with some patients experiencing spontaneous remission for periods of time and others showing aggressive recurrent growth 2. RRP is associated with significant morbidity secondary to airway obstruction and voice change and, on infrequent occasions, mortality or malignant transformation 7, 8. To date, there is no cure for the disease, but the treatment is directed toward symptomatic control and reduction of disease burden 9. The current standard of care for RRP is surgical excision, primarily with microdebriders and lasers, and adjuvant therapies as needed. Patients often require multiple surgeries during their lifetime, sometimes several each year, and less frequently can require tracheotomy or adjuvant drug administration, making RRP an expensive disease to treat 10. Clinical complications can arise from frequent surgical procedures, papillomatous lower airway involvement and therapeutic resistance 11.
Literature supports that juvenile‐onset RRP is transmitted vertically during pregnancy or is acquired at birth from an HPV‐infected mother 12. In contrast, scarce literature is available regarding the acquisition mechanism of adult‐onset RRP. One study showed that risk of adult‐onset RRP is associated with number of sexual partners, although not all studies corroborate this finding 9, 13. Of note, juvenile‐onset RRP has a more aggressive clinical course and a greater potential for airway compromise 14.
A 2017 study by the National Center for Health Statistics (NCHS) of the Centers for Disease Control and Prevention (CDC) examined the prevalence of oral HPV infection in more than 9000 participants aged 18–69 years in the United States 15. They estimated that 14·7% of the study population has detectable HPV DNA in the oral cavity. When separated by genotype, approximately 1% of the population has oral cavity infected with HPV6 or HPV11, yet only a small fraction of those develop RRP 16. Current hypotheses suggest that multiple genes predispose individuals to tolerate HPV6/11 infection. Genetic analyses of RRP patients have revealed an increased frequency of certain human leukocyte antigen (HLA) alleles, as well as a decreased frequency of certain innate immune cell receptors that may predispose individuals to RRP development and be correlated with disease severity 17, 18, 19. When HPV infects the epithelium of susceptible individuals, HPV oncoproteins, predominately E6, can interfere with innate immunity and skew the adaptive immune response to a type 2 T helper cell (Th2)‐like or T regulatory (Treg) cell phenotype. This skewed adaptive immune response, in combination with an altered innate immune response, produces an HPV‐tolerant cytokine microenvironment 20, 21, 22, 23. The aim of this review is to characterize the current understanding of RRP etiology, focusing on immune system tolerance of HPV6/11 in the setting of RRP as well as the current and future immunomodulatory approaches being investigated to manage this disease.
HPV and RRP
HPV structure and infection
The HPV genome is encapsulated in an icosahedral protein structure 55 nm in diameter. Its double‐stranded, circular DNA encodes for eight gene proteins 24. There are six ‘early’ genes (E1, E2, E4, E5, E6 and E7) responsible for cell cycle disruption and two ‘late’ genes (L1 and L2) that encode capsid proteins. E1 and E2 control viral gene replication and expression, while E4 releases the virus from its host cell. E5, E6 and E7 are responsible for tumor formation and malignancy 24.
HPVs are known for causing benign and malignant anogenital lesions as well as cutaneous warts; however, they can also infect the epithelium of the head and neck, including ocular conjunctiva, ear canal, nasal sinuses, oral cavity, tonsils, pharynx and larynx. When low‐risk HPV6/11 infection leads to papilloma formation in the larynx, it is called RRP 25. Adult‐onset RRP is most probably associated with orogenital spread of HPV; the leading risk factor for juvenile‐onset RRP is a maternal history of genital papilloma and vertical transmission from mother to child during childbirth 26, 27. HPV infects basal epithelial cells in the larynx and usually occurs via membrane disruption 28, 29. The mechanism of infection in the basal epithelium of the vocal folds is relatively unknown, but it is hypothesized that microtrauma from vocalization may be a mode of entry 30, 31. In general, after infection, HPV DNA can integrate into the host genome, or it can exist in the episomal form, with its circular double‐stranded DNA intact. In RRP, HPV DNA is maintained in the episomal state within the nucleus 32. Entry of the HPV genome into the basal cell nucleus requires mitosis, which then allows cell proteins to interact with the HPV genome and activate viral protein transcription. Viral oncoproteins E6 and E7 target the tumor suppressors p53 and pRB, respectively, induce cell division and promote differentiation of HPV‐infected basal cells into upper epithelial layers 33, 34. In‐situ hybridization shows HPV6/11 RNA expression to be strongest in the suprabasal layer of the epithelium 35. There is some evidence that the laryngeal squamo–columnar epithelial junction is more vulnerable to HPV infection than squamous epithelia due to the absence of the protective, non‐dividing outer squamous layers 25. Tracheal papilloma involvement may be aided by metaplastic squamous changes of the trachea permitting HPV infection; however, HPV has also been shown to directly infect tracheal ciliated columnar epithelium 36. Additionally, there is evidence that co‐infection with human immunodeficiency virus (HIV) proteins tat and gp120 may disrupt epithelial tight junctions, facilitating HPV entry and potentiating HPV‐associated lesions 37. Concurrent or co‐infection with herpes simplex virus‐1, cytomegalovirus or Epstein–Barr virus has also been shown in both juvenile and adult RRP patients and may be predictive of an aggressive clinical course 1, 38, 39.
The reason for recurrence of RRP is still not well understood. Some hypothesize that it is due to repeated reinfections with novel variants of HPV, while others point to evidence of persistent infection from latent HPV in nearby ‘normal’ epithelial tissue 40. Kocjan et al. reported that the HPV genome of 67 of 70 (95·7%) patients showed complete genetic identity between either the highly variable regions (LCR, E5a) of the genetic code or between entire HPV genomes from the initial HPV isolate and a second isolate obtained 1–22 years later 40. This observation indicates that the HPV genome is genetically stable and does not change during the course of the disease, suggesting that disease recurrence may be caused by reactivation of latent HPV in adjacent normal epithelia 40. Moreover, it has been suggested that adjacent tissue may provide a reservoir for the latent virus and wait for trigger signals. For example, the healing process after surgical removal of papillomas may promote the reactivation and replication of latent viruses in the surrounding tissue 25, 41, 42. Another study showed that exposure to acid or gastric enzymes may cause epithelial irritation, producing an inflammatory response and contributing to the spread or recurrence of RRP 43. Therefore, controlling gastroesophageal reflux may assist in the management of RRP 44, 45, 46, 47.
Pathogenesis in larynx
HPV6/11‐infected laryngeal papilloma cells exhibit an altered cellular biology. E6 and E7 are the most important HPV proteins in pathogenesis and interfere with tumor suppressors p53 and pRb, respectively, as well as several other proteins involved with cell cycle regulation, proliferation and apoptosis 48. Furthermore, laryngeal papilloma cells over‐express epithelial growth factor receptor (EGFR) and contain multiple altered EGFR‐dependent signaling pathways, including elevated phosphatidylinositol 3‐kinase (PI3K) activity 49. Cyclo‐oxygenase‐2 (COX2), a normal mediator of inflammation, is also over‐expressed in papilloma cells and has been shown to inhibit epithelial differentiation and apoptosis. Wu et al. showed that COX‐2 over‐expression is mediated by EGFR‐dependent Rac1 over‐expression, a Rho family GTPase that controls cell cycle progression, apoptosis, gene expression, cell motility and tumor invasiveness 50, 51. Additionally, in‐vitro inhibition of COX‐2 in papilloma cells with the selective COX‐2 inhibitor celecoxib showed reduced cell growth and increased apoptotic activity 50. Contrary to these data, a randomized double‐blind controlled study was recently completed that assessed the safety and efficacy of celecoxib in both pediatric and adult RRP patients (NCT 00571701). The primary objective of this trial was to determine the efficacy of celecoxib in comparison to conventional endoscopy and surgical treatment. The data showed that celecoxib did not affect the mean percentage change in papilloma growth rate during treatment compared to baseline (P = 0·57). It should be noted that the study celecoxib dose was reduced by regulators to a dose lower than that typically administered because of concerns for cardiac side effects.
High‐ versus low‐risk HPVs
HPV subtypes are stratified into low‐ and high‐risk groups based on their oncogenic potential; however, this does not mean that low‐risk HPV subtypes cannot also cause cancer. While both low‐ and high‐risk HPVs infect and replicate in the same cellular environments and overcome the same cellular defenses, it is interesting that their respective pathologies and cellular targets differ markedly. Much less is known about the function of low‐risk HPV proteins than those of high‐risk HPV types, and RRP is almost exclusively caused by low‐risk HPV6 and HPV11 52. Research has shown that the E7 protein from HPV6 and HPV11 has a lower binding affinity to pRb than the E7 protein from high‐risk HPV types, which may be one reason that HPV6 and HPV11 are less efficient at malignant transformation 48, 53. In addition, E7 from high‐risk strains induce proteasome‐mediated degradation of pRb, freeing E2F transcription factor to stimulate cells into S phase, and only high‐risk E6 can degrade p53 in an E6AP‐dependent manner, compromising host cell ability to complete cell cycle checkpoints 54. E6 from both high‐ and low‐risk HPV strains can bind to p300/CBP to prevent acetylation of p53, but high‐risk E6 has a higher binding affinity to p300/CBP than low‐risk E6 55, 56, 57.
HPV 6 versus 11
Between the two low‐risk types, a more aggressive clinical course of RRP is associated with HPV11 lesions 58. This includes diagnosis at a younger age, need for more surgeries per year and decreased remission rate. Gerein et al. evaluated the use of interferon (IFN)‐α in the treatment of JO‐ and AO‐RRP caused by both HPV6 and HPV11 59. For HPV6‐positive patients, 16 of 17 (94%) had remission after treatment, and 11 of 17 (65%) maintained remission during a follow‐up of a mean 172 months. In the HPV11‐positive patients, only four of 14 (29%) showed remission after treatment, and two of 14 (14%) maintained remission. Therefore, HPV6‐positive RRP was more responsive to IFN‐α therapy and was less likely recur during the follow‐up period than HPV11‐positive RRP. The severity score for RRP aggressiveness was also significantly higher in HPV11‐positive than in HPV6‐positive patients 59. There is limited literature on the oncogenic differences between HPV6 and HPV11. First, HPV11 E6 and E7 could have more ‘oncogenic’ targets than HPV6 or be more efficient at interacting with host cell proteins. Vambutas et al. proposed that HPV11 E7 inhibits ATP‐dependent peptide transport due to interaction with TAP‐1, the protein that transports peptides into the endoplasmic reticulum for presentation by major histocompatibility complex (MHC) class I 60. Furthermore, it has been hypothesized that HPV11 E7 interacts more strongly with TAP‐1 than HPV6 E7, leading to decreased immune responsiveness and the more aggressive disease course observed in HPV11 RRPs 61. Secondly, subtle life cycle differences may make HPV11 more oncogenic than HPV6. Peh et al. showed that the life cycles of low‐risk HPV11 and high‐risk HPV16 were organized similarly. Both their sequence of viral gene expression and pattern of viral genome amplification in the epithelium were closely related, suggesting a closer association between HPV11 and HPV16 than previously thought 62.
RRP and malignancy
Approximately 2% of patients with RRP develop a carcinoma in the region of the papilloma 52. In the above‐mentioned Gerein et al. study evaluating IFN‐α in the treatment of juvenile and adult RRP there were no cases of malignancy in HPV6‐positive patients, but five of 14 (36%) of the HPV11‐positive patients developed malignancy in the area of the papilloma at some point during the 20‐year follow‐up. This study provides further evidence that HPV11 leads to a more severe clinical course and a higher rate of malignant transformation 59. It should be noted that this study included only patients with RRP disease that was aggressive enough to require adjuvant therapy, so a higher malignancy rate would be expected in this study versus reality. A case–series of three children with HPV11‐induced RRP who developed bronchogenic squamous cell carcinoma provided evidence that HPV11‐induced RRP may undergo malignant transformation 58, 63, 64, 65. In rare instances, RRP can be aggressive enough to cause lung involvement, and many of these cases result in lung cancer. Interestingly, HPV genome analysis in these patients reveal substantial mutations; specifically, amplification of regions that promote increased expression of HPV E6 and/or E7. This suggests that even low‐risk HPVs can cause malignant transformation under the right circumstances 66. In addition, two separate cohort studies found that a history of tobacco use was not a risk factor for dysplasia or malignancy in RRP 7, 67.
There is also evidence that low‐risk HPVs can modify cellular processes such as that seen in some malignancies. Chemokine (C‐X‐C motif) ligand (CXCL)1, CXCL6, CXCL8 and vascular endothelial growth factor (VEGF)‐A are differentially expressed in papillomas and associated with malignancy, operating as growth and angiogenic factors 68. Microarray analysis revealed that CXCL1, CXCL6 and VEGF‐A were significantly correlated with severe RRP, providing evidence that angiogenesis is integral to disease pathology 69. They also identified reduced expression of certain tumor suppressors; namely, insulin‐like growth factor‐binding protein (IGFbp5), four‐and‐a‐half LIM domains 1 (FHL1) and SPARC‐like 1 (mast9, hevin) (SPARCL1), and over‐expression of several proto‐oncogenes, including IGFbp3, placental growth factor (PGF), and parathyroid hormone‐like hormone (PTHLH) 69. Three members of the S100 family, which have both oncogenic and tumor‐suppressive functions, were also differentially expressed in papillomas 69.
Immunology
Studies have shown that RRP patients can mount an initial, measurable serum antibody response to both HPV6 and HPV11, indicating that there is viral immune recognition in these patients 70, 71, 72. Thus, immune dysfunction in RRP is suggestive of HPV6/11 tolerance, not a lack of viral recognition 20. Interestingly, RRP patients have demonstrated normal immunological responses to other pathogens without chronic disease manifestations, indicating a site‐specific immune tolerance of HPV6/11 that does not necessarily apply to other mucosal sites 20, 73. Therefore, it is hypothesized that RRP is a multi‐gene disease that polarizes innate and adaptive immunity to tolerate chronic and local HPV6/11 infection (Fig. 1).
Figure 1.
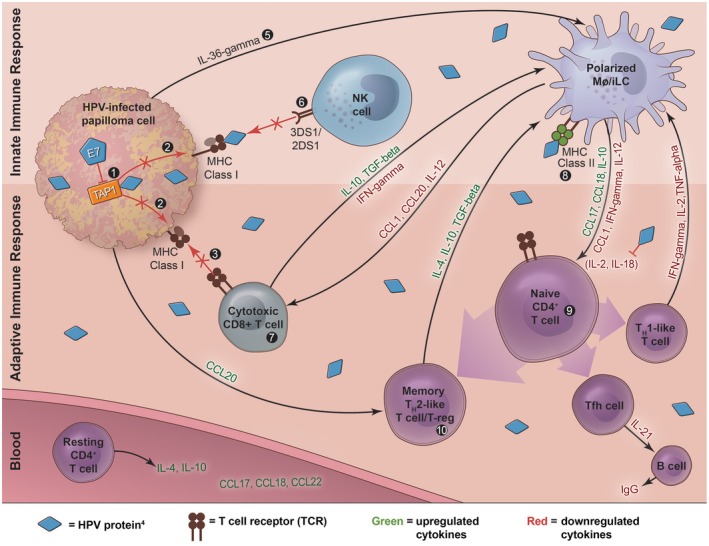
- TAP‐1 polymorphisms in ATPase domain
- MHC Class I expression is downregulated for an unknown reason, so there is less antigen presentation for NK cell‐ and CD8+ cytotoxic T cell‐mediated killing
- Less HPV‐specific cytotoxic T cell killing due to MHC Class I downregulation
- MHC Class II complexes present E2 and E6 peptides; however, E6 is the dominant inducer of the Th2‐like phenotype in RRP
- iLCs have decreased response to the proinflammatory IL‐36‐γ
- KIR genes 3DS1 and 2DS1 are protective against severe disease
- Papillomas are enriched for CD8+CD28– T cells that expressed the Th2‐like cytokines IL‐10 and TGF‐beta
- MHC Class II alleles: DRB1*0102, DRB1*0301, DQB1*0201, DQB1*0202 skew to Th2‐like phenotype; DQB1*0602 may be protective
- Naïve CD4+ T cells are induced predominately to a Th2‐like or T‐reg phenotype either directly by HPV proteins or indirectly by iLCs, which then overproduce Th2‐like cytokines
- CD4+CD25+CD127lowFOXp3+ T‐reg cells
Innate and adaptive immunity
Recent studies have shown that HPV may be a part of the commensal microflora of primary epithelial cells, and approximately 1% of the population harbors HPV6/11 DNA in the larynx 15. However, a very small percentage of HPV‐exposed individuals develop RRP 16, 74. This suggests that altered immune function may contribute to the development of RRP. However, it is unclear why some individuals do not mount a sufficient anti‐HPV response to the initial HPV6‐ and HPV11‐infected keratinocytes 75. One study showed that the number of CD4+ T helper cells, CD8+ cytotoxic T cells, B cells and natural killer (NK) cells was similar between healthy individuals and RRP patients 76. That same group later reported that RRP patients have increased numbers of a specific subset of circulating CD4+ T cells that express Th2‐like cytokines; therefore, the adaptive immune response in patients with RRP becomes polarized towards a Th2‐like or T regulatory (Treg) phenotype 20. Antigen‐presenting cells (APCs), including polarized macrophages and immature Langerhans cells (iLCs) and naive T cell‐derived cells, namely Th2‐like and Treg cells, drive this in response to HPV proteins 20. HPV E6 protein is the dominant inducer of immune dysregulation in RRP 21. In fact, there is evidence that E6 can directly inhibit IL‐2 and IL‐18 expression, two cytokines that contribute to maintaining a balanced adaptive immune response and an HPV‐specific cytotoxic T cell function 21, 77. Polarization towards a Th2‐like phenotype up‐regulates the Th2 effector cytokine IL‐10. IL‐10 suppresses Th1 cells that aid in clearing viral infection via production of cytokines such as interferon (IFN)‐γ, IL‐2 and tumor necrosis factor (TNF)‐α 20, 78. In support of this observation, several studies have shown that levels of the Th2‐like chemokines CCL17, CCL18 and CCL22 were found to be high in the serum of patients with RRP 22, 79. Over‐expression of these cytokines is correlated with RRP disease severity, and may provide biomarkers for predicting disease prognosis 23. Another study suggested that RRP patients have more circulating CD4+ T cells that constitutively express the Th2‐like cytokines IL‐4 and IL‐10 20, 80. Treg cells [CD4+CD25+CD127lowforkhead box protein 3 (FoxP3+)] were also enriched in papilloma samples compared to autologous peripheral blood mononuclear cell (PBMC) samples, indicating that Treg cells may contribute to immune dysfunction 81. Furthermore, the HPV‐specific effector CD8+ T cell population and its killing function is altered in RRP, possibly to a more immature or Th1 suppressor function. One study found that papillomas were enriched for CD8+ CD28– T cells that expressed the Th2‐like cytokines IL‐10 and TGF‐β and were correlated with disease severity 82. Because LCs are required for microbial‐specific CD8+ T cell development, Devoti et al. hypothesized that deficient LC production of IL‐12 and CCL1 could explain the loss of CD8+CD28– T cell response to HPV‐infected cells and reduced IFN‐γ expression 83. Immunoglobulin (Ig)G levels were also found to be extremely low in JO‐RRP patients compared to healthy controls, and B cell maturation and antibody production were limited in these patients, due possibly to reduced IL‐21 secretion by T follicular helper (Tfh) cells 84.
Innate immunity has also been shown to be altered in the RRP microenvironment. In fact, dysfunctional activation of adaptive immunity by macrophages and dendritic cells (Langerhans cells) in the papilloma microenvironment may perpetuate the polarization to a Th2‐like phenotype, suppressing HPV clearance. Memory Th2‐like T cells expressing IL‐4, IL‐10 and TGF‐β alternatively activate macrophages to express the Th2‐like chemokines CCL17 and CCL18, which polarize naive CD4+ T cells to become memory Th2‐like T cells and Tregs 19. This is because HPV6/11 E6 has been shown to reduce the expression in papilloma of IFN‐γ, a Th1‐like cytokine which normally balances macrophage activation 21. Immature dendritic cells also contribute to a Th2‐like T cell response via production of CCL18 and low baseline expression of CCL1 83. IL‐12 and IL‐18, constitutively expressed by dendritic cells to support Th1‐like T cell maturation and IFN‐γ expression, were also deficient in severe RRP cases 21. Another study found that iLCs in RRP patients have a decreased response to the proinflammatory cytokine IL‐36‐γ, further contributing to an ineffective adaptive immune response to HPV 83. Additionally, polymorphic killer‐cell immunoglobulin‐like receptors (KIRs) on NK cells may aid in the early clearance of HPV6/11 infection, and their absence may be positively correlated with RRP disease severity 17, 18. Furthermore, one group used microarray analysis to identify genes that may contribute to RRP etiology by measuring changes in the transcriptional profiles between papilloma tissue and autologous laryngeal epithelium 69. They found elevated expression of IL‐1F9, which was strongly correlated with severe RRP and may alter innate signaling to stimulate a Th2‐like response 69, 85. Their data also corroborate that the papilloma microenvironment contains an expression profile consistent with an altered innate immune response and defective Th1‐like response. They found that mRNA expression was decreased for the Th1‐like chemokines CCL19 and CCL21 and elevated for the Th2‐like chemokine CCL20 23. Interestingly, a different study by the same group found that CCL20 was only differentially over‐expressed in the basal cell layer. They hypothesized that CCL20 expression in these cells may form a barrier, selectively admitting Th2‐like T cells and restricting access to more superficial epithelial layers, where HPV expression is greatest 23.
Recent research is exploring the role of immune inhibitory receptors in HPV tolerance and RRP development. Programmed death ligand 1 (PD‐L1), a negative immune regulator found on APCs, was shown to be up‐regulated in both papilloma and infiltrating immune cells 86, 87. Furthermore, T cell immunoglobulin and ITIM domain (TIGIT), another immune inhibitory receptor, has been found on Treg cells in papilloma tissue (personal communication, 10 October 2019) 88. These findings may show how direct down‐regulation of the immune system allows for persistent HPV infection and the development of RRP.
Immunogenetics
There is evidence that multiple genes may predispose individuals to the development of RRP from HPV infection and explain varying disease severity among the RRP population. RRP patients have an increased frequency of HLA class II alleles DRB1*0102, DRB1*0301, DQB1*0201 and DQB1*0202, associated with reduced IFN‐γ expression. Alternatively, DQB1*0602 may be protective in RRP patients 19. Single nucleotide polymorphisms in the TAP‐1 gene have also been correlated with disease severity, and this severity may be mitigated by the proximity of the TAP‐1 gene to HLA class II genes 61. In addition, KIRs control early NK cell response against viral infection and tumor transformation. Bonagura et al. showed that the absence of select KIR genes 3DS1 and 3DS2 may be predictive of disease severity, and reactivating them in 3DS1‐ and 3DS2‐deficient patients may decrease RRP aggressiveness 17, 18. These findings may explain why certain patients are predisposed to defective clearance of HPV‐infected keratinocytes.
Collectively, these findings provide initial insight toward the intricate interaction between the immune system and low‐risk HPV. Additional research is critically needed to expand our understanding of immune system dysfunction in RRP, enabling the rational development of immunomodulatory approaches to better manage RRP patients.
Immunomodulatory therapies
Surgery is the primary treatment modality to manage RRP, and adjuvant therapy is required when surgery cannot control the disease. Early attempts at immunological therapies were directed at the use of IFN‐α, with some promising results. However, systemic side effects of IFN‐α have limited its use, and adjuvant therapies have moved toward intralesional bevacizumab, a monoclonal humanized antibody against the angiogenic factor VEGF‐A, and cidofovir, a cytosine nucleotide that blocks viral DNA polymerase. However, these are still not curative, so immunomodulatory efforts are being made to restore host immune function and clear HPV infection.
HPV vaccine
Vaccination has the most exciting potential to manage RRP. The quadrivalent vaccine Gardasil, with activity against HPV subtypes 6, 11, 16 and 18, and the new nonavalent vaccine Gardasil‐9, with activity against HPV subtypes 6, 11, 16, 18, 31, 33, 45, 52 and 58, were developed from the HPV L1 viral capsid protein as a primary prevention measure to prevent healthy patients from initial HPV infection or infected patients from reinfection 43. Its efficacy in reducing RRP incidence through herd immunity was demonstrated in Australia in 2007, when an extensive Gardasil vaccination program was initiated for females aged 12–26 years and a school‐based program that provided Gardasil to boys and girls aged 12–13 years. A prospective study of the Australian juvenile RRP population showed a decline in incidence rate from 0·16 per 100 000 in 2012 to 0·022 per 100 000 in 2016 [95% confidence interval (CI), P < 0·001]. Furthermore, male genital warts declined, suggesting that herd vaccination effects may be decreasing horizontal transmission 89. These data suggest that juvenile‐onset RRP may be a vaccine‐preventable disease. Currently, the Centers for Disease Control’s (CDC) Advisory Committee on Immunization Practices (AICP) recommends the use of Gardasil‐9 for girls aged 13–26 and boys aged 13–21, but some health insurance policies may even cover HPV vaccination for older adults 90.
A systematic review of HPV vaccination as secondary prevention for active RRP disease found that nine of 12 (75%) studies reported decreased disease recurrence, decreased disease burden or increased intersurgical interval after adjuvant treatment with HPV vaccine 91. Tjon Pian Gi et al. reported, in a group of RRP patients, that antibody activity against their particular HPV type rose significantly when comparing pre‐ and post‐Gardasil vaccination serologies (P < 0·001) 92. Furthermore, anti‐HPV IgG titers were found in cord blood samples of Gardasil‐vaccinated pregnant women, demonstrating that anti‐HPV antibodies cross the placenta 93. These data suggest that Gardasil vaccination may be viable as both a prophylactic and therapeutic treatment regimen; however, multi‐center randomized controlled trials are needed to fully assess the efficacy of the HPV vaccination as a therapeutic vaccine in the RRP population.
One explanation for the therapeutic successes of L1 viral capsid vaccines, such as Gardasil and Gardasil‐9, in RRP may be related to HPV’s existence in the episomal state within the papilloma cell 32. It has been documented that the integration of HPV DNA into host cell DNA causes deletion of HPV late genes L1 and L2 94. This renders prophylactic vaccines generated with the L1 viral capsid protein ineffective at targeting these cells 95. However, because RRP cells harbor episomal HPV DNA, these late genes are expressed, promoting antigen recognition. This may be one reason why Gardasil and Gardasil‐9 demonstrate some efficacy as a secondary prevention measure in RRP. However, the limitations of protein‐based vaccines are based on their low immunogenicity. They are presented primarily via the MHC class II pathway and mildly via the MHC class I pathway, which generates a strong antibody response, but only a mild cytotoxic T cell response, respectively. This allows for effective antibody‐mediated neutralization and phagocytosis of HPV, but inefficient cytotoxic killing of infected cells. Furthermore, literature supports that certain MHC class II alleles predispose patients to generating a skewed Th2‐like response in RRP, making this route of immunogenicity less likely to be successful in clearing persistent infection.
Unlike preventative vaccines aimed at antibody generation, therapeutic vaccines may have greater therapeutic benefit by generating a more robust cell‐mediated immune response. DNA vaccines, comprised of a plasmid encoding HPV E6 and E7 proteins, generate a more sustained immune response than capsid protein vaccines through continuous expression of viral antigens. Dendritic cells are integral in the immunogenicity DNA vaccination, specifically in activating MHC class I‐mediated cytotoxic T cell killing. They may either present exogenous antigens generated from transfected cells or endogenous antigens from direct transfection of dendritic cells 96, 97. Ahn et al. investigated a novel calreticulin‐linked DNA vaccine encoding HPV11 E6 and E7 proteins in mice that were inoculated with HPV11 E6/E7 tumors. Not only did they find a robust CD8+ T cell response, they also showed a reduction in tumor growth and volume 98. As more research works to understand immune‐tolerant microenvironment of RRP, therapeutic vaccination may be an exciting development to restore host immune function and clear HPV infection.
PD‐1 inhibitors
PD‐1 on leukocytes binding to its ligands, PD‐L1 and PD‐L2, on APCs negatively regulates the immune system, and PD‐L1 has been shown to be highly expressed in HPV‐associated head and neck squamous cell carcinoma (HNSCC) 99, 100. Furthermore, PD‐L1 was upregulated in both papilloma and infiltrating immune cells; therefore, the PD‐1/PD‐L1 interaction may contribute to immunosuppression in RRP 86, 87. PD‐1 inhibitors, such as pembrolizumab and avelumab, block the interaction between PD‐1 and its ligands and have clinical efficacy in numerous advanced solid tumors, including HPV‐associated HNSCC 101. The activity against HPV‐associated HNSCC has prompted investigators to initiate a Phase II clinical trial to assess the efficacy of pembrolizumab in RRP (NCT 02632344). RRP patients will be administered 200 mg pembrolizumab as a 30‐min intravenous (i.v.) infusion every 3 weeks on day 1 of each cycle after all procedures and assessments have been completed. Another Phase II clinical trial will assess the efficacy of avelumab in RRP by treating patients with 10 mg/kg avelumab as an i.v. infusion every 2 weeks for up to six total doses (NCT 02859454).
Conclusion
HPV6/11 infection is present in an estimated 1% of the general population; however, only a small subset of infected individuals will develop RRP. There is some evidence that expression of certain HLA alleles may predispose people to develop RRP and may be linked to disease aggressiveness 19. Another study showed that the absence of certain NK cell receptors was positively correlated with RRP severity 18. These are mainly unexplored areas of research in RRP, and certainly a more comprehensive longitudinal study focusing on HPV infection and RRP development would provide more insight on genetic events that predispose an individual to RRP.
The literature suggests that HPV6/11 proteins dampen immune system surveillance of HPV6/11‐infected cells, but the mechanism is unclear. Evidence points toward the polarization of the adaptive immune system to a Th2‐like or Treg phenotype in RRP patients, which subsequently suppresses Th1 cell‐mediated clearance of the infection 20. One group found this polarization to be directed by E6 21. Another study proposed that E7 interferes with viral peptide presentation to adaptive immune cells via an interaction with TAP‐1 60. These preliminary findings suggest an interaction between the immune system and low‐risk HPV proteins. Evidence of a defect in the innate proinflammatory responses showed that NK cells failed to detect and/or lyse HLA class I‐deficient cells, and that macrophages and Langerhans cells failed to initiate a proper adaptive immune response 17, 83. These interactions between low‐risk HPV proteins, innate immune cells and adaptive immune cells perpetuate an immunosuppressive cycle, quelling anti‐HPV function. Further understanding the molecular interactions of low‐risk E6 and E7 proteins with the immune system is necessary to discover how HPV6/11‐infected cells evade immune surveillance. If immune system dysfunction is the major contributing factor to the recurrence of RRP, then immunomodulatory approaches can be tailored to manage RRP patients more effectively.
Currently, the standard of care for RRP patients is surgical excision, with use of intralesional adjuvant therapies as needed and reservation of systemic adjuvant therapies for the most severe cases. Current adjuvants are not curative, so immunomodulatory approaches are under investigation with the goal of clearing HPV infection and preventing recurrence. HPV vaccines, including the newly developed nonavalent Gardasil‐9, have shown benefit as both primary and secondary prevention, but additional work, in particular a multi‐center randomized clinical trial, is needed to evaluate the efficacy of HPV vaccines to manage active disease. Furthermore, there is increasing evidence that widespread HPV vaccination reduces HPV genital wart acquisition and, as a result, secondary laryngeal infections and RRP incidence. Additionally, DNA vaccines stimulate a more robust, cytotoxic CD8+ T cell response, and an in‐vitro study demonstrates reduction in both tumor growth and volume in an HPV11‐driven tumor 98. Due to evidence of PD‐L1 overexpression in both papilloma and infiltrating immune cells, two ongoing Phase II clinical trials have been initiated to assess the efficacy of the anti‐PD‐1 antibodies in RRP.
In conclusion, RRP is a recalcitrant disease, incurable with current treatment modalities. Adjuvant therapies are often utilized to control aggressive disease; however, these treatments are not curative and typically only increase the interval between surgical procedures. Research is accumulating to support the notion that the RRP microenvironment is immunocompromised and, thus, regaining Th1 cell function may be a durable approach to prevent persistent infection. A better understanding of the mechanisms used by HPV6/11 to escape immune surveillance may lead to the development of novel immunomodulatory approaches to manage RRP patients more effectively.
Disclosure
The authors certify that they have no affiliations with or involvement in any organization that has financial or non‐financial interest in the material discussed in this manuscript.
Acknowledgements
The illustration in Figure 1 was created by Anthony S. Baker, CMI, ©The Ohio State University.
References
- 1. Derkay CS, Wiatrak B. Recurrent respiratory papillomatosis: a review. Laryngoscope 2008; 118:1236–47. [DOI] [PubMed] [Google Scholar]
- 2. Larson DA, Derkay CS. Epidemiology of recurrent respiratory papillomatosis. APMIS 2010; 118:450–4. [DOI] [PubMed] [Google Scholar]
- 3. Armstrong LR, Preston EJ, Reichert M et al Incidence and prevalence of recurrent respiratory papillomatosis among children in Atlanta and Seattle. Clin Infect Dis 2000; 31:107–9. [DOI] [PubMed] [Google Scholar]
- 4. Armstrong LR, Derkay CS, Reeves WC. Initial results from the national registry for juvenile‐onset recurrent respiratory papillomatosis. RRP Task Force. Arch Otolaryngol Head Neck Surg 1999; 125:743–8. [DOI] [PubMed] [Google Scholar]
- 5. Derkay CS. Task force on recurrent respiratory papillomas. A preliminary report. Arch Otolaryngol Head Neck Surg 1995; 121:1386–91. [DOI] [PubMed] [Google Scholar]
- 6. Duggan MA, Lim M, Gill MJ, Inoue M. HPV DNA typing of adult‐onset respiratory papillomatosis. Laryngoscope 1990; 100:639–42. [DOI] [PubMed] [Google Scholar]
- 7. Blumin JH, Handler EB, Simpson CB, Osipov V, Merati AL. Dysplasia in adults with recurrent respiratory papillomatosis: incidence and risk factors. Ann Otol Rhinol Laryngol 2009; 118:481–5. [DOI] [PubMed] [Google Scholar]
- 8. Derkay CS, Malis DJ, Zalzal G, Wiatrak BJ, Kashima HK, Coltrera MD. A staging system for assessing severity of disease and response to therapy in recurrent respiratory papillomatosis. Laryngoscope 1998; 108:935–7. [DOI] [PubMed] [Google Scholar]
- 9. Ruiz R, Achlatis S, Verma A et al Risk factors for adult‐onset recurrent respiratory papillomatosis. Laryngoscope 2014; 124:2338–44. [DOI] [PubMed] [Google Scholar]
- 10. Chesson HW, Forhan SE, Gottlieb SL, Markowitz LE. The potential health and economic benefits of preventing recurrent respiratory papillomatosis through quadrivalent human papillomavirus vaccination. Vaccine 2008; 26:4513–8. [DOI] [PubMed] [Google Scholar]
- 11. Doyle DJ, Gianoli GJ, Espinola T, Miller RH. Recurrent respiratory papillomatosis: juvenile versus adult forms. Laryngoscope 1994; 104:523–7. [DOI] [PubMed] [Google Scholar]
- 12. Rodier C, Lapointe A, Coutlee F et al Juvenile respiratory papillomatosis: risk factors for severity. J Med Virol 2013; 85:1447–58. [DOI] [PubMed] [Google Scholar]
- 13. Kashima HK, Shah F, Lyles A et al A comparison of risk factors in juvenile‐onset and adult‐onset recurrent respiratory papillomatosis. Laryngoscope 1992; 102:9–13. [DOI] [PubMed] [Google Scholar]
- 14. Reeves WC, Ruparelia SS, Swanson KI, Derkay CS, Marcus A, Unger ER. National registry for juvenile‐onset recurrent respiratory papillomatosis. Arch Otolaryngol Head Neck Surg 2003; 129:976–82. [DOI] [PubMed] [Google Scholar]
- 15. Sonawane K, Suk R, Chiao EY et al Oral human papillomavirus infection: differences in prevalence between sexes and concordance with genital human papillomavirus infection, NHANES 2011 to 2014. Ann Intern Med 2017; 167:714–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abramson AL, Steinberg BM, Winkler B. Laryngeal papillomatosis: clinical, histopathologic and molecular studies. Laryngoscope 1987; 97:678–85. [DOI] [PubMed] [Google Scholar]
- 17. Bonagura VR, Du Z, Ashouri E et al Activating killer cell immunoglobulin‐like receptors 3DS1 and 2DS1 protect against developing the severe form of recurrent respiratory papillomatosis. Hum Immunol 2010; 71:212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonagura VR, Du Z, Luo L et al KIR3DS1, KIR2DS1, and KIR2DS5 protect against the development of severe recurrent respiratory papillomatosis (RRP) in HPV‐6/11‐infected patients. J Allergy Clin Immunol 2009; 123:S165–S165. [Google Scholar]
- 19. Bonagura VR, Vambutas A, DeVoti JA et al HLA alleles, IFN‐gamma responses to HPV‐11 E6, and disease severity in patients with recurrent respiratory papillomatosis. Hum Immunol 2004; 65:773–82. [DOI] [PubMed] [Google Scholar]
- 20. Bonagura VR, Hatam LJ, Rosenthal DW et al Recurrent respiratory papillomatosis: a complex defect in immune responsiveness to human papillomavirus‐6 and ‐11. APMIS 2010; 118:455–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DeVoti JA, Steinberg BM, Rosenthal DW et al Failure of gamma interferon but not interleukin‐10 expression in response to human papillomavirus type 11 E6 protein in respiratory papillomatosis. Clin Diagn Lab Immun 2004; 11:538–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rosenthal DW, DeVoti JA, Schmidtmayerova H, Steinberg BM, Bonagura VR. Human papillomavirus causes a TH2‐like chemokine predominance in recurrent respiratory papillomatotosis (RPR). J Allergy Clin Immunol 2008; 121:S15. [Google Scholar]
- 23. Rosenthal DW, DeVoti JA, Steinberg BM, Abramson AL, Bonagura VR. T(H)2‐like chemokine patterns correlate with disease severity in patients with recurrent respiratory papillomatosis. Mol Med 2012; 18:1338–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nebesio CL, Mirowski GW, Chuang TY. Human papillomavirus: clinical significance and malignant potential. Int J Dermatol 2001; 40:373–9. [DOI] [PubMed] [Google Scholar]
- 25. Chow LT, Broker TR, Steinberg BM. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS 2010; 118:422–49. [DOI] [PubMed] [Google Scholar]
- 26. Born H, Ruiz R, Verma A et al Concurrent oral human papilloma virus infection in patients with recurrent respiratory papillomatosis: a preliminary study. Laryngoscope 2014; 124:2785–90. [DOI] [PubMed] [Google Scholar]
- 27. Silverberg MJ, Thorsen P, Lindeberg H, Grant LA, Shah KV. Condyloma in pregnancy is strongly predictive of juvenile‐onset recurrent respiratory papillomatosis. Obstet Gynecol 2003; 101:645–52. [DOI] [PubMed] [Google Scholar]
- 28. Rautava J, Syrjanen S. Biology of human papillomavirus infections in head and neck carcinogenesis. Head Neck Pathol 2012; 6(Suppl 1):S3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aaltonen LM, Rihkanen H, Vaheri A. Human papillomavirus in larynx. Laryngoscope 2002; 112:700–7. [DOI] [PubMed] [Google Scholar]
- 30. Rousseau B, Suehiro A, Echemendia N, Sivasankar M. Raised intensity phonation compromises vocal fold epithelial barrier integrity. Laryngoscope 2011; 121:346–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sivasankar M, Erickson E, Rosenblatt M, Branski RC. Hypertonic challenge to porcine vocal folds: effects on epithelial barrier function. Otolaryngol Head Neck Surg 2010; 142:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Major T, Szarka K, Sziklai I, Gergely L, Czegledy J. The characteristics of human papillomavirus DNA in head and neck cancers and papillomas. J Clin Pathol 2005; 58:51–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aydin I, Weber S, Snijder B et al Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLOS Pathog 2014; 10:e1004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Calton CM, Bronnimann MP, Manson AR et al Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLOS Pathog 2017; 13:e1006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Steinberg BM, Gallagher T, Stoler M, Abramson AL. Persistence and expression of human papillomavirus during interferon therapy. Arch Otolaryngol Head Neck Surg 1988; 114:27–32. [DOI] [PubMed] [Google Scholar]
- 36. Abramson AL, Nouri M, Mullooly V, Fisch G, Steinberg BM. Latent human papillomavirus infection is comparable in the larynx and trachea. J Med Virol 2004; 72:473–7. [DOI] [PubMed] [Google Scholar]
- 37. Tugizov SM, Herrera R, Chin‐Hong P et al HIV‐associated disruption of mucosal epithelium facilitates paracellular penetration by human papillomavirus. Virology 2013; 446:378–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rimell FL, Shoemaker DL, Pou AM, Jordan JA, Post JC, Ehrlich GD. Pediatric respiratory papillomatosis: prognostic role of viral typing and cofactors. Laryngoscope 1997; 107:915–8. [DOI] [PubMed] [Google Scholar]
- 39. Pou AM, Rimell FL, Jordan JA et al Adult respiratory papillomatosis: human papillomavirus type and viral coinfections as predictors of prognosis. Ann Otol Rhinol Laryngol 1995; 104:758–62. [DOI] [PubMed] [Google Scholar]
- 40. Kocjan BJ, Gale N, Hocevar Boltezar I et al Identical human papillomavirus (HPV) genomic variants persist in recurrent respiratory papillomatosis for up to 22 years. J Infect Dis 2013; 207:583–7. [DOI] [PubMed] [Google Scholar]
- 41. Pignatari S, Smith EM, Gray SD, Shive C, Turek LP. Detection of human papillomavirus infection in diseased and nondiseased sites of the respiratory tract in recurrent respiratory papillomatosis patients by DNA hybridization. Ann Otol Rhinol Laryngol 1992; 101:408–12. [DOI] [PubMed] [Google Scholar]
- 42. Camilleri IG, Milner RH. Human papilloma virus proliferation in a healing burn. Burns 1996; 22:162–3. [DOI] [PubMed] [Google Scholar]
- 43. Carifi M, Napolitano D, Morandi M, Dall’Olio D. Recurrent respiratory papillomatosis: current and future perspectives. Ther Clin Risk Manag 2015; 11:731–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Borkowski G, Sommer P, Stark T, Sudhoff H, Luckhaupt H. Recurrent respiratory papillomatosis associated with gastroesophageal reflux disease in children. Eur Arch Oto‐Rhino‐Laryngol 1999; 256:370–2. [DOI] [PubMed] [Google Scholar]
- 45. Harcourt JP, Worley G, Leighton SE. Cimetidine treatment for recurrent respiratory papillomatosis. Int J Pediatr Otorhinolaryngol 1999; 51:109–13. [DOI] [PubMed] [Google Scholar]
- 46. Holland BW, Koufman JA, Postma GN, McGuirt WF Jr. Laryngopharyngeal reflux and laryngeal web formation in patients with pediatric recurrent respiratory papillomas. Laryngoscope 2002; 112:1926–9. [DOI] [PubMed] [Google Scholar]
- 47. McKenna M, Brodsky L. Extraesophageal acid reflux and recurrent respiratory papilloma in children. Int J Pediatr Otorhinolaryngol 2005; 69:597–605. [DOI] [PubMed] [Google Scholar]
- 48. Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev 2004; 68:362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johnston D, Hall H, DiLorenzo TP, Steinberg BM. Elevation of the epidermal growth factor receptor and dependent signaling in human papillomavirus‐infected laryngeal papillomas. Cancer Res 1999; 59:968–74. [PubMed] [Google Scholar]
- 50. Wu R, Abramson AL, Shikowitz MJ, Dannenberg AJ, Steinberg BM. Epidermal growth factor‐induced cyclooxygenase‐2 expression is mediated through phosphatidylinositol‐3 kinase, not mitogen‐activated protein/extracellular signal‐regulated kinase kinase, in recurrent respiratory papillomas. Clin Cancer Res 2005; 11:6155–61. [DOI] [PubMed] [Google Scholar]
- 51. Wu R, Coniglio SJ, Chan A, Symons MH, Steinberg BM. Up‐regulation of Rac1 by epidermal growth factor mediates COX‐2 expression in recurrent respiratory papillomas. Mol Med 2007; 13:143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hartley C, Hamilton J, Birzgalis AR, Farrington WT. Recurrent respiratory papillomatosis – the Manchester experience, 1974–1992. J Laryngol Otolo 1994; 108:226–9. [PubMed] [Google Scholar]
- 53. Heck DV, Yee CL, Howley PM, Münger K. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proc Natl Acad Sci 1992; 89:4442–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus‐16 induces degradation of retinoblastoma protein through the ubiquitin‐proteasome pathway. Cancer Res 1996; 56:4620–4. [PubMed] [Google Scholar]
- 55. Thomas MC, Chiang CM. E6 oncoprotein represses p53‐dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell 2005; 17:251–64. [DOI] [PubMed] [Google Scholar]
- 56. Patel D, Huang SM, Baglia LA, McCance DJ. The E6 protein of human papillomavirus type 16 binds to and inhibits co‐activation by CBP and p300. EMBO J 1999; 18:5061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zimmermann H, Degenkolbe R, Bernard HU, O'Connor MJ. The human papillomavirus type 16 E6 oncoprotein can down‐regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J Virol 1999; 73:6209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rabah R, Lancaster WD, Thomas R, Gregoire L. Human papillomavirus‐11‐associated recurrent respiratory papillomatosis is more aggressive than human papillomavirus‐6‐associated disease. Pediatr Dev Pathol 2001; 4:68–72. [DOI] [PubMed] [Google Scholar]
- 59. Gerein V, Rastorguev E, Gerein J, Jecker P, Pfister H. Use of interferon‐alpha in recurrent respiratory papillomatosis: 20‐year follow‐up. Ann Otol Rhinol Laryngol 2005; 114:463–71. [DOI] [PubMed] [Google Scholar]
- 60. Vambutas A, DeVoti J, Pinn W, Steinberg BM, Bonagura VR. Interaction of human papillomavirus type 11 E7 protein with TAP‐1 results in the reduction of ATP‐dependent peptide transport. Clin Immunol 2001; 101:94–9. [DOI] [PubMed] [Google Scholar]
- 61. Vambutas A, Bonagura VR, Reed EF et al Polymorphism of transporter associated with antigen presentation 1 as a potential determinant for severity of disease in recurrent respiratory papillomatosis caused by human papillomavirus types 6 and 11. J Infect Dis 2004; 189:871–9. [DOI] [PubMed] [Google Scholar]
- 62. Peh WL, Middleton K, Christensen N et al Life cycle heterogeneity in animal models of human papillomavirus‐associated disease. J Virol 2002; 76:10401–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rady PL, Schnadig VJ, Weiss RL, Hughes TK, Tyring SK. Malignant transformation of recurrent respiratory papillomatosis associated with integrated human papillomavirus type 11 DNA and mutation of p53. Laryngoscope 1998; 108:735–40. [DOI] [PubMed] [Google Scholar]
- 64. Moore CE, Wiatrak BJ, McClatchey KD et al High‐risk human papillomavirus types and squamous cell carcinoma in patients with respiratory papillomas. Otolaryngol Head Neck Surg 1999; 120:698–705. [DOI] [PubMed] [Google Scholar]
- 65. Cook JR, Hill DA, Humphrey PA, Pfeifer JD, El‐Mofty SK. Squamous cell carcinoma arising in recurrent respiratory papillomatosis with pulmonary involvement: emerging common pattern of clinical features and human papillomavirus serotype association. Mod Pathol 2000; 13:914–8. [DOI] [PubMed] [Google Scholar]
- 66. Pim D, Banks L. Interaction of viral oncoproteins with cellular target molecules: infection with high‐risk vs low‐risk human papillomaviruses. APMIS 2010; 118:471–93. [DOI] [PubMed] [Google Scholar]
- 67. Lee LA, Cheng AJ, Fang TJ et al High incidence of malignant transformation of laryngeal papilloma in Taiwan. Laryngoscope 2008; 118:50–5. [DOI] [PubMed] [Google Scholar]
- 68. Moriconi A, Cesta MC, Cervellera MN et al Design of noncompetitive interleukin‐8 inhibitors acting on CXCR1 and CXCR2. J Med Chem 2007; 50:3984–4002. [DOI] [PubMed] [Google Scholar]
- 69. DeVoti JA, Rosenthal DW, Wu R, Abramson AL, Steinberg BM, Bonagura VR. Immune dysregulation and tumor‐associated gene changes in recurrent respiratory papillomatosis: a paired microarray analysis. Mol Med 2008; 14:608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tachezy R, Hamsikova E, Valvoda J et al Antibody response to a synthetic peptide derived from the human papillomavirus type 6/11 L2 protein in recurrent respiratory papillomatosis: correlation between Southern blot hybridization, polymerase chain reaction, and serology. J Med Virol 1994; 42:52–9. [DOI] [PubMed] [Google Scholar]
- 71. Sameshima A, Fujiyoshi T, Pholampaisathit S et al Demonstration of antibodies against human papillomavirus type‐11 E6 and L2 proteins in patients with recurrent respiratory papillomatosis. Auris Nasus Larynx 1997; 24:185–91. [DOI] [PubMed] [Google Scholar]
- 72. Bonnez W, Kashima HK, Leventhal B et al Antibody response to human papillomavirus (HPV) type 11 in children with juvenile‐onset recurrent respiratory papillomatosis (RRP). Virology 1992; 188:384–7. [DOI] [PubMed] [Google Scholar]
- 73. Israr M, Rosenthal D, Frejo‐Navarro L, DeVoti J, Meyers C, Bonagura VR. Microarray analysis of human keratinocytes from different anatomic sites reveals site‐specific immune signaling and responses to human papillomavirus type 16 transfection. Mol Med 2018; 24:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Broker TR, Jin G, Croom‐Rivers A et al Viral latency–the papillomavirus model. Dev Biol (Basel) 2001; 106:443–451; discussion 452–443, 465–475. [PubMed] [Google Scholar]
- 75. Faria AM, Weiner HL. Oral tolerance. Immunol Rev 2005; 206:232–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bonagura VR, Siegal FP, Abramson AL et al Enriched HLA‐DQ3 phenotype and decreased class I major histocompatibility complex antigen expression in recurrent respiratory papillomatosis. Clin Diagn Lab Immunol 1994; 1:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cho YS, Kang JW, Cho M et al Down modulation of IL‐18 expression by human papillomavirus type 16 E6 oncogene via binding to IL‐18. FEBS Lett 2001; 501:139–45. [DOI] [PubMed] [Google Scholar]
- 78. Armstrong L, Jordan N, Millar A. Interleukin 10 (IL‐10) regulation of tumour necrosis factor alpha (TNF‐alpha) from human alveolar macrophages and peripheral blood monocytes. Thorax 1996; 51:143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rosenthal DW, Schmidtmayerova H, Steinberg BM et al Recurrent respiratory papillomatosis (RRP): Disease severity associates with enhanced T<sub>H</sub>2‐like dendritic cell chemokine (DC‐CK1) plasma expression. J Allergy Clin Immunol 2005; 115:S81. [Google Scholar]
- 80. Romagnani S. Th1 and Th2 in human diseases. Clin Immunol Immunopathol 1996; 80:225–35. [DOI] [PubMed] [Google Scholar]
- 81. Hatam LJ, Rosenthal DW, DeVoti JA et al CD4(+)Foxp3(+)CD127(+low) T‐Regulatory cells are increased in HPV infected papillomas in patients with recurrent respiratory papillomatosis (RRP). J Allergy Clin Immunol 2008; 121:S211. [Google Scholar]
- 82. Bonagura VR, Hatam L, DeVoti J, Zeng F, Steinberg BM. Recurrent respiratory papillomatosis: altered CD8(+) T‐cell subsets and T(H)1/T(H)2 cytokine imbalance. Clin Immunol 1999; 93:302–11. [DOI] [PubMed] [Google Scholar]
- 83. DeVoti J, Hatam L, Lucs A et al Decreased Langerhans cell responses to IL‐36gamma: altered innate immunity in patients with recurrent respiratory papillomatosis. Mol Med 2014; 20:372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wu X, Wang G, Chen X et al Impaired T cell‐dependent humoral immune response associated with juvenile‐onset recurrent respiratory papillomatosis progression. Sci Rep 2016; 6:36378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ramadas RA, Li X, Shubitowski DM, Samineni S, Wills‐Karp M, Ewart SL. IL‐1 Receptor antagonist as a positional candidate gene in a murine model of allergic asthma. Immunogenetics 2006; 58:851–5. [DOI] [PubMed] [Google Scholar]
- 86. Ahn J, Bishop JA, Roden RBS, Allen CT, Best SRA. The PD‐1 and PD‐L1 pathway in recurrent respiratory papillomatosis. Laryngoscope 2018; 128:E27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hatam LJ, DeVoti JA, Rosenthal DW et al Immune suppression in premalignant respiratory papillomas: enriched functional CD4(+)Foxp3(+) regulatory T cells and PD‐1/PD‐L1/L2 expression. Clin Cancer Res 2012; 18:1925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev 2017; 276:112–20. [DOI] [PubMed] [Google Scholar]
- 89. Novakovic D, Cheng ATL, Zurynski Y et al A prospective study of the incidence of juvenile‐onset recurrent respiratory papillomatosis after implementation of a national HPV vaccination program. J Infect Dis 2018; 217:208–12. [DOI] [PubMed] [Google Scholar]
- 90. Petrosky E, Bocchini JA, Hariri S et al Use of 9‐valent human papillomavirus (HPV) vaccine: updated HPV vaccination recommendations of the Advisory Committee on Immunization Practices. Morb Mort Wly Rep 2015; 64:300–4. [PMC free article] [PubMed] [Google Scholar]
- 91. Dion GR, Teng S, Boyd LR et al Adjuvant human papillomavirus vaccination for secondary prevention: a systematic review. JAMA Otolaryngol Head Neck Surg 2017; 143:614–22. [DOI] [PubMed] [Google Scholar]
- 92. Tjon Pian Gi RE, San Giorgi MR, Pawlita M et al Immunological response to quadrivalent HPV vaccine in treatment of recurrent respiratory papillomatosis. Eur Arch Oto‐Rhino‐Laryngol 2016; 273:3231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Matys K, Mallary S, Bautista O et al Mother–infant transfer of anti‐human papillomavirus (HPV) antibodies following vaccination with the quadrivalent HPV (type 6/11/16/18) virus‐like particle vaccine. Clin Vaccine Immunol 2012; 19:881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Klaes R, Woerner SM, Ridder R et al Detection of high‐risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res 1999; 59:6132–6. [PubMed] [Google Scholar]
- 95. Yang A, Jeang J, Cheng K et al Current state in the development of candidate therapeutic HPV vaccines. Expert Rev Vaccines 2016; 15:989–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chattergoon MA, Robinson TM, Boyer JD, Weiner DB. Specific immune induction following DNA‐based immunization through in vivo transfection and activation of macrophages/antigen‐presenting cells. J Immunol 1998; 160:5707–18. [PubMed] [Google Scholar]
- 97. Dupuis M, Denis‐Mize K, Woo C et al Distribution of DNA vaccines determines their immunogenicity after intramuscular injection in mice. J Immunol 2000; 165:2850–8. [DOI] [PubMed] [Google Scholar]
- 98. Ahn J, Peng S, Hung CF, Roden RBS, Wu TC, Best SR. Immunologic responses to a novel DNA vaccine targeting human papillomavirus‐11 E6E7. Laryngoscope 2017; 127:2713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Freeman GJ, Long AJ, Iwai Y et al Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lyford‐Pike S, Peng S, Young GD et al Evidence for a role of the PD‐1:PD‐L1 pathway in immune resistance of HPV‐associated head and neck squamous cell carcinoma. Cancer Res 2013; 73:1733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Brahmer JR, Drake CG, Wollner I et al Phase I study of single‐agent anti‐programmed death‐1 (MDX‐1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010; 28:3167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]