

Abstract
The goal of this study was to compare dysphagia phenotypes in low and high copy number (LCN and HCN) transgenic superoxide dismutase 1 (SOD1) mouse models of ALS to accelerate the discovery of novel and effective treatments for dysphagia and early amyotrophic lateral sclerosis (ALS) diagnosis. Clinicopathological features of dysphagia were characterized in individual transgenic mice and age-matched controls utilizing videofluoroscopy in conjunction with postmortem assays of the tongue and hypoglossal nucleus. Quantitative PCR accurately differentiated HCN-SOD1 and LCN-SOD1 mice and nontransgenic controls. All HCN-SOD1 mice developed stereotypical paralysis in both hindlimbs. In contrast, LCN-SOD1 mice displayed wide variability in fore-and hindlimb involvement. Lick rate, swallow rate, inter-swallow interval, and pharyngeal transit time were significantly altered in both HCN-SOD1 and LCN-SOD1 mice compared to controls. Tongue weight, tongue dorsum surface area, total tongue length, and caudal tongue length were significantly reduced only in the LCN-SOD1 mice compared to age-matched controls. LCN-SOD1 mice with lower body weights had smaller/lighter weight tongues, and those with forelimb paralysis and slower lick rates died at a younger age. LCN-SOD1 mice had a 32% loss of hypoglossal neurons, which differed significantly when compared to age-matched control mice. These novel findings for LCN-SOD1 mice are congruent with reported dysphagia and associated tongue atrophy and hypoglossal nucleus pathology in human ALS patients, thus highlighting the translational potential of this mouse model in ALS research.
Keywords: Deglutition, Deglutition disorders, Dysphagia, Amyotrophic lateral sclerosis (ALS), Superoxide dismutase 1 (SOD1) transgenic mouse, Videofluoroscopy
Introduction
Dysphagia is considered one of the most serious clinical symptoms of amyotrophic lateral sclerosis (ALS) because it results in malnutrition and aspiration pneumonia [1–6], both of them being independent risk factors for early mortality [7, 8]. Moreover, dysphagia in ALS contributes to major depression [7, 9–12]. Predominant treatments include diet modification (e.g., avoiding specific food/liquid consistencies), postural changes (e.g., tucking the chin when swallowing), and eventually tube feedings once swallowing becomes unsafe and/or nonfunctional. However, these palliative treatments fail to target the underlying etiology and pathogenesis of dysphagia in ALS, which remain largely unexplored.
Dysphagia in ALS is predominantly attributed to tongue weakness as a result of progressive degeneration and death of motor neurons in the hypoglossal nucleus [13–16]. For bulbar-onset ALS patients, tongue weakness may be the first sign of the disease. However, tongue weakness is also common in spinal-onset ALS patients without perceptible swallowing (or speech) deficits [13]. This finding suggests hypoglossal neuron degeneration occurs early in ALS, regardless of disease-onset phenotype (spinal or bulbar) [16]. Moreover, impaired tongue strength has been identified as an independent prognostic indicator of earlier mortality in ALS patients [16, 17]. Beyond the tongue, the disease spreads with no known pattern to involve other muscles involved in swallowing, particularly muscles of the jaw, lips, pharynx, and larynx [18]. Given that dysphagia is highly associated with morbidity and mortality, elucidating the underlying pathological mechanisms that affect musculature and neuronal pathways involved in swallowing may provide novel therapeutic targets to significantly improve the life span and quality of life of ALS patients. Moreover, specifically investigating hypoglossal neurons and associated swallowing dysfunction may provide insight into the etiology and pathogenesis of ALS in general. However, this line of research requires good translational animal models, which have not yet been optimized for dysphagia therapeutic discovery in ALS.
Our study focused on two transgenic superoxide dismutase 1 (SOD1) mouse models of ALS: the widely used high copy number (HCN) SOD1-G93A model [B6SJL-Tg(SOD1*G93A)1Gur/J] [19, 20], and the rarely used low copy number (LCN) SOD1-G93A model [B6SJL-Tg(SOD1*G93A)dl1Gur/J] [19]. Here on, we refer to these models as HCN-SOD1 and LCN-SOD1. The HCN-SOD1 model, which expresses ~ 25 copies of the mutant transgene, develops progressive paralysis of the hindlimbs starting ~ 3 months of age, and dies by 4–5 months [19–22]. Our preliminary work with this model revealed a tongue motility deficit (i.e., significantly reduced lick rate during drinking) at weaning (postnatal day 21–23; P21–P23), which was the earliest time point tested [23]. This finding raises serious concerns regarding the appropriateness of the HCN-SOD1 model for translational ALS research, especially given that human ALS is an adult-onset rather than infantile disease [24–26].
In contrast, the LCN-SOD1 model expresses ~ 8 copies of the mutant transgene [25, 27], resulting in delayed clinical onset of hindlimb dysfunction (~ 6–7 months of age) [28] and a longer life span (7–10 months) [27, 29, 30]. Therefore, reducing the transgene copy number by approximately 30% (compared to HCN-SOD1 mice) significantly lengthens the preclinical (asymptomatic) phase as well as disease duration. Moreover, the preclinical window of LCN-SOD1 mice spans well into adulthood, making low copy number SOD1 models ideal for studying early-stage pathological mechanisms as well as testing therapies designed for the early disease stage [24, 25]. However, this model has rarely been studied, likely because it is only available through costly embryo cryorecovery services, compared to the large repository of live HCN-SOD1 mice that are readily available for immediate use in research. Here, we characterized the clinicopathological features of dysphagia in both models (HCN-SOD1 and LCN-SOD1) utilizing the clinical gold standard videofluoroscopic swallow study (VFSS) protocol that we previously adapted for use with mice [31, 32], in conjunction with postmortem assays of the tongue and hypoglossal nucleus.
Materials and Methods
Animals
All mice included in this IACUC-approved study were offspring from one of two separate SOD1 transgenic colonies (HCN and LCN), both maintained on a C57B6/SJL hybrid background, and both with a glycine-93-alanine (G93A) substitution [19, 20] that was randomly integrated into the genome on distal chromosome 12, band E [33]. Our HCN colony was started in 2010 by mating repository live transgenic males [B6SJL-Tg(SOD1*G93A)1Gur/J; stock #002726] [19, 20] with F1 hybrid females [B6SJLF1/J; stock #100012], both purchased from The Jackson Laboratory (Bar Harbor, ME). Our LCN colony was started in 2013 by mating cryorecovered transgenic males [B6SJL-Tg(SOD1*G93A)dl1Gur/J; stock #002300] [19, 30] with F1 hybrid females [B6SJLF1/J; stock #100012], also purchased from The Jackson Laboratory. The LCN-SOD1 strain arose at The Jackson Laboratory from a spontaneous meiotic intralocus recombination event during initial establishment of the HCN-SOD1 line, resulting in approximately 30 percent reduction in transgene copy number [30]. Once established at our facility, both colonies were continually maintained by breeding transgenic male colony offspring with F1 hybrid females [B6SJLF1/J; stock #100012], purchased from The Jackson Laboratory ~ 2–3 times per year. This strict backcross breeding scheme is in accordance with industry standard guidelines to minimize the possibility of genetic drift in transgenic lines [34]. Breeders from both colonies typically produced large litters (~ 10 pups) of an approximate 50:50 ratio of transgenic (hemizygous SOD1-G93A) and nontrans-genic (wild-type, WT) mice. At 19–21 days of age, offspring were weaned, ear punched for identification purposes, and tail snipped for genotyping (described below), followed by separation into cages for group housing of up to five mice per cage, based on colony (HCN versus LCN), sex, and litter.
A total of 97 offspring from 17 breeder pairs were randomly selected (based on genotype and sex) throughout 2014 and 2015 for allocation to the following four study groups: HCN-SOD1 (n = 24; 14 males and 10 females), HCN-control (n = 25; 15 males and 10 females), LCN-SOD1 (n = 25; 11 males and 14 females), and LCN-control (n = 23; 13 males and 10 females). Throughout the study, mice retained their original group housing assignment in microisolator (static) caging under standard vivarium conditions, including strict regulation of ambient temperature 20–26 °C, humidity 30–70%, and standard 12-h light cycle. Free access to food (Purina 5008 Mouse Diet) and water (filtered tap water; pH adjusted to 3.5) was provided in the home cage, except during experimental testing (described below). To minimize aggressive and/or over-barbering behaviors that are common in group-housed mice, cages were provided with fresh enrichment materials (e.g., hut, dental treats, wire trapeze, nestlet, etc.) weekly at cage change. Daily healthcare monitoring and routine surveillance for common rodent illnesses were provided by veterinary staff. All animal handling was performed within the guidelines of The Guide for the Care and Use of Laboratory Animals (8th ed., 2011) within our AAALAC-accredited academic institution.
Genotyping
Quantitative real-time polymerase chain reaction (qPCR) was performed on tail snips collected at weaning to determine genotype (transgenic versus nontransgenic) and confirm colony lineage (HCN versus LCN) for each mouse. qPCR was performed using a BIO-RAD CFX Connect Real-Time System and iTaq Universal SYBR Green Supermix (BIO-RAD CAT #172–5121). SOD1 forward (5′-GGG-AAG-CTG-TTG-TCC-CAA-G-3′) and reverse (5′-CAA-GGG-GAG-GTA-AAA-GAG-AGC-3′) primers were used to amplify the mutant human SOD1 gene. Apolipoprotein B (ApoB) was used as the internal control gene with forward (5′-CAC-GTG-GGC-TCC-AGC-ATT-3′) and reverse (5′-TCA-CCA-GTC-ATT-TCT-GCC-TTT-G-3′) primers. Cycle parameters were as follows: 5 min at 95 °C, 40 cycles of 5 s at 95 °C and 1 min at 60 °C, 5 s at 65 °C, and 5 s at 95 °C. An amplification plot was produced from each qPCR run, which shows the difference in threshold cycle between the transgene and the reference gene. The threshold cycle (Ct) number for each sample describes the point in which the fluorescence signal increases above the background during the exponential phase of the reaction [35, 36]. Distinct clustering of Ct values within each genotype (HCN-SOD1, LCN-SOD1, and controls) was expected; the mean Ct value was then used to estimate the reduction in transgene copy number for LCN-SOD1 mice compared to HCN-SOD1 mice. Upon completion of genotyping, researchers were blinded to the results to ensure unbiased data collection and analysis throughout the study.
Data Collection Time Point
The confounding effects of repeated X-ray exposure on the swallowing function of SOD1-G93A transgenic mice are unknown. Therefore, we limited VFSS to a single time point corresponding to an advanced stage of ALS, when mice had lost ~ 20% of their maximum body weight and developed limb paralysis but were still able to independently forage for food and water in the home cage. When this humane endpoint criterion was met, individual transgenic mice and age-matched controls were subjected to a single VFSS assay (described below), followed by postmortem assays of the tongue and hypoglossal nucleus (described below). The age (months) at which mice reached the study endpoint was recorded for subsequent survival analysis. For our calculations, 1 month equals 30 days.
Videofluoroscopic Swallow Study (VFSS)
VFSS was performed on advanced-stage HCN-SOD1 and LCN-SOD1 mice and age-matched controls using our previously established protocol and customized equipment [31, 32]. Briefly, following an overnight water restriction (12–14 h) to motivate participation, individual mice were subjected to ~ 2 min of low-dose radiation (~ 30 kV and ~ 0.2 mA at a source–image distance of 6–7 cm) using our miniaturized fluoroscope (The LabScope, Glenbrook Technologies, Randolph, NJ). During testing, mice were confined within a narrow, translucent/radiolucent chamber that facilitated voluntary drinking in the lateral plane. The test chamber was secured to a platform that permitted remote-controlled positioning of the mouse within the fluoroscope beam. A webcam (Logitech, HD Pro C920) was positioned above the chamber for real-time behavioral monitoring of the mouse. Swallowing was visualized via fluoroscopy while mice drank an oral contrast agent (Omnipaque, GE Healthcare, 350 mg iodine/mL), diluted to a 25% solution with deionized water and chocolate flavoring (viscosity = 5.41 centiPoise). The contrast agent was administered via syringe delivery system into a small bowl secured to the end of the test chamber closest to the X-ray source. The fluoroscope was manually activated (via foot pedal) only when the mouse was actively drinking from the bowl. The oropharyngeal stage of swallowing was visualized first, followed by remote-controlled repositioning of the chamber to visualize the esophageal stage of swallowing, as shown in Fig. 1. Approximately 30 s to 1 min of drinking was recorded for each stage of swallowing (i.e., oropharyngeal and esophageal). Fluoroscopic video recordings (1–2 min per mouse) were captured at 30 frames per second (fps) and stored as AVI files.
Fig. 1.

Videofluoroscopic swallow study (VFSS) assay. The oropharyngeal (a, b) and esophageal (c, d) stages of swallowing were assessed in the lateral plane while mice drank thin liquid contrast from a bowl (white asterisk). Jaw motion (open/closed cycles) and bolus flow (red arrows) were tracked frame-by-frame to quantitate several swallow metrics. UES upper esophageal sphincter
At a later time, videos were analyzed by two trained reviewers using video editing software (Pinnacle Studio, version 14; Pinnacle Systems, Inc., Mountain View, CA). Reviewer 1 identified 3–5 two-second episodes per mouse of uninterrupted drinking for each stage of swallowing, from which several previously validated VFSS metrics (Table 1) were quantified manually in frame-by-frame fashion [31, 32]. Each 2-s episode started with a swallow event, in which the bolus transited from the vallecular space (i.e., the stereotypical swallow trigger point in mice) [31, 32] to the esophagus. Reviewer 2 was blinded to values determined by Reviewer 1, with the exception of the start frame for each 2-s episode. Reviewer discrepancies were resolved by group consensus. Values for each VFSS metric were averaged for each mouse before proceeding with statistical analysis. We also quantified airway protection for each video using the standardized 8-point Penetration–Aspiration Scale (PAS) that ranges from 1 (no penetration or aspiration) to 8 (silent aspiration) [37].
Table 1.
VFSS metrics for objective quantitation of dysphagia in mice
| VFSS metrics | Operational definitions | Stage of swallowing |
|---|---|---|
| Lick rate | The number of lick cycles per second (30 frames) during uninterrupted drinking, starting with the jaw at maximum open position | Oropharyngeal |
| Swallow rate | The number of swallows occurring during each 2-s (60 frames) episode of uninterrupted drinking, beginning at the rest frame immediately preceding bolus transit from the valleculae (i.e., anatomic swallow trigger point in mice) | |
| Inter-swallow interval | The time (ms) between two successive, uninterrupted swallows during uninterrupted drinking | |
| Lick–swallow ratio | The number of licks between two successive, uninterrupted swallows | |
| Pharyngeal transit time | The time (ms) between swallow onset and when the bolus tail enters the esophagus | |
| Esophageal transit time | The time (ms) it takes the bolus to transit from the proximal esophagus into the stomach | Esophageal |
Postmortem Assays
Within 24 h after VFSS testing, mice were euthanized by sodium pentobarbital overdose and randomly allocated to either fresh tissue (i.e., tongue morphometry, n = 61) or fixed tissue (i.e., hypoglossal nucleus histology; n = 36) assays, as described below.
Tongue Morphometry
After obtaining whole body weight (grams), the entire tongue was removed from fresh (nonperfused) specimens. However, 7 tongues were damaged during dissection, leaving 54 tongues for analysis: 12 HCN-SOD1 and 14 HCN colony controls; 17 LCN-SOD1 and 11 LCN colony controls. Immediately after collection, each tongue was weighed (grams; wet weight) and the dorsal surface was imaged on a Leica M80 dissecting microscope with a Leica EC4HD camera. Images were analyzed using ImageJ (NIH public domain) to quantitate the following morphometric measurements [38, 39] (Fig. 2): surface area of the dorsal tongue, total tongue length, median eminence width, rostral tongue length, rostral tongue width, caudal tongue length, and caudal tongue width. Tongue length and width measurements were recorded in millimeters (mm), surface area in square mm (mm2), and tongue weight in milligrams (mg).
Fig. 2.

Tongue weight and morphometry. Tongue weight (mg; wet weight of the entire tongue) and seven morphometric measurements were obtained from the dorsal tongue surface of individual mice. a Area (mm2; green outline); b length (mm), divided at the median eminence into anterior (rostral) and posterior (caudal) regions, as well as total length; and c width (mm), measured separately for the rostral and caudal regions, as well as the median eminence. Scale bar = 2 mm
Hypoglossal Nucleus Histology
After transcardial perfusion with 0.9% NaCl (saline) followed by 4% paraformaldehyde (PFA; in 0.1 M phosphate-buffered saline, pH ~ 7.4), the entire brain was gently removed and post fixed in 4% PFA for 1–3 days at 4 °C with rotation. Samples were then randomly allocated to either thin (10 μm, paraffin) or thick (40 μm, frozen) sectioning protocols for visualization of vacuolar pathology and neuronal counts, respectively, in the hypoglossal nucleus, as described below.
Vacuolar Pathology
Fixed brains (n = 24; 6 for each of the 4 study groups) underwent paraffin processing and embedding using our standard laboratory protocols. The entire brainstem was transversely sectioned at 10 μm using a microtome (Leica RM 2155, Germany) and collected as ribbons onto positively charged (silane-coated) glass slides. Every other slide from each animal was subjected to Gill’s hematoxylin and eosin (H&E) staining for gross assessment of brainstem morphology, using our standard laboratory protocols. For visualization of mutant SOD1 aggregates, adjacent slides were subjected to SOD1 immunostaining, which entailed citrate antigen retrieval, antihuman SOD1 (hSOD1) antibody (1:1000 dilution; rabbit polyclonal, ab13498; Abcam, Cambridge, MA), biotinylated secondary antibody (VEC-TASTAIN Elite ABC HRP kit, rabbit IgG; Vector Laboratories, Inc., Burlingame, CA), DAB (3,3′-diaminobenzidine) substrate (SK-4100; Vector Laboratories), and DAB-enhancing solution (H-2200; Vector Laboratories), according to associated manufacturer’s protocols. Counterstaining with Gill’s hematoxylin was applied for anatomic landmark identification. Sections incubated without primary or secondary antibodies served as negative controls. H&E-and SOD1-stained slides were cover slipped using Permount (Fisher Scientific) and air dried flat at room temperature for 3 days before proceeding with microscopy. The hypoglossal nucleus was identified in each section by referencing a mouse brain atlas [40, 41]. Images of the hypoglossal nucleus were captured using a brightfield microscope (Leica DM4000) and imaging software (Leica Application Suite v4.3). Images were scored for vacuolar pathology (present/absent) by a blinded investigator.
Neuronal Counts
As hypoglossal neuron counts have been previously published for HCN-SOD1 and age-matched colony controls [42, 43], we chose to apply this labor-intensive methodology to only LCN colony mice to acquire novel data. Fixed brains (n = 12; 6 LCN-SOD1 and 6 age-matched colony controls) were cryoprotected with graded solutions of sucrose (20 and 30%) at 4 °C until sinking. Each medulla was transversely sectioned at 40 pm using a freezing-sliding microtome (Leica SM 2010R) and stored in antifreeze (30% ethylene glycol, 30% glycerol and 1X PBS) at − 20 °C until all samples were processed and ready for immunohistochemistry (IHC).
For IHC batch staining, eight evenly distributed sections from each mouse were selected throughout from the entire length of the hypoglossal motor nucleus, identified using a mouse brain reference atlas [40, 41]. The tissue from each animal was placed in a separate well of a 12-well plate and washed with 1 × PBS 3 times for 5 min each. To prevent nonspecific antibody binding, the tissues were incubated in a blocking solution consisting of 5% normal donkey serum (NDS), 1 × PBS, and 0.2% Triton for 1 h on a shaker at room temperature. The blocking solution was then replaced with a primary antibody solution consisting of 5% NDS, 1X PBS, 0.1% Triton, and an antibody against neuronal nuclei (NeuN; Millipore, catalog #MAB377, RRID:AB_2298772; 1:500) and incubated overnight on a shaker at 4 °C. The next day, the tissue was washed with 1X PBS (3 × for 5 min each) and then incubated in a secondary antibody solution consisting of 5% NDS, 1X PBS, 0.1% Triton, and secondary antibody (donkey antimouse Alexa-Fluor 555, 1:1000; Molecular Probes, Eugene, OR, catalog #A31570) on a shaker for 2 h in the dark at room temperature. The tissue underwent one final series of three 5-min washes before being mounted on positively charged (silane-coated) glass slides, covered with ProLong® Gold Antifade Reagent (Molecular Probes, Eugene, OR) to prevent quenching of fluorescence, cover-slipped, and air-dried in the dark. Slides were stored at 4 °C except while hypoglossal motor neuron quantitation was being performed. Sections incubated without primary or secondary antibodies served as negative controls.
For hypoglossal motor neuron counts, NeuN(+) cells were manually counted at 20 × by a blinded investigator using an epifluorescence microscope (Leica DM4000). Stereo Investigator software (MBF Bioscience, Williston, VT) was used to outline the hypoglossal motor nucleus bilaterally, identified using a mouse brain reference atlas [40]. Neurons within this outlined region of interest were counted live in each section by using the software’s manual marking tool, and the corresponding marked images were saved for future reference. To increase the likelihood that counted cells were alpha motor neurons, only those larger than 15 pm were counted, as described elsewhere [42]. In addition, counted cells had to have a prominent nucleolus and visible projections. Once the counts were completed for both sides of the hypoglossal nucleus in all 8 sections per mouse, the data were extrapolated to estimate the number of hypoglossal motor neurons in the entire length of the nucleus (960 μm) for individual mice [40]. The extrapolated neuronal counts (8 sections × 40 μm/section = 320 μm; entire nucleus = 960 μm/320 μm = 3; therefore, we multiplied the counted neurons by 3) were averaged within each group of mice (LCN-SOD1 versus controls), from which the percentage of hypoglossal motor neuron loss was calculated for LCN-SOD1 mice. Findings (% neuronal loss) were compared to previously published data for HCN-SOD1 mice and age-matched colony controls.
Statistics
A nonrepeated measures design was used for this study because it focused on group comparisons at a single time point (i.e., advanced disease stage). Basic summary statistics, boxplots, and Q–Q plots revealed that data from all three assays (VFSS, tongue weight/morphometry, and hypoglossal nucleus histology) violated the assumptions of normality and homogeneity of variance required for parametric statistical tests. Therefore, only nonparametric tests were used for statistical analyses, as described in the Results section. For all outcome measures, outliers were identified but not removed from the dataset. All statistical analyses were performed using IBM SPSS Statistics 24, and two-sided p values of less than 0.05 were considered significant. Data visualization software (BioVinci version 1.1.5; BioTuring, Inc., San Diego, CA) was used to create side-by-side boxplots and dotplots for complete presentation of data where appropriate.
Results
Genotyping
Quantitative PCR accurately differentiated HCN-SOD1 and LCN-SOD1 mice and nontransgenic controls, as shown in Fig. 3. Based on threshold cycle (Ct) detection values, mice in this study fell into three distinct genotypes: HCN-SOD1 (Ct ~ 19–21), LCN-SOD1 (Ct ~ 25–27), and controls (Ct ~ 32–35). The Ct detection value was ~ 30% higher for LCN-SOD1 compared to HCN-SOD1 mice, which translates to an approximate 30% reduction in transgene copy number for LCN-SOD1 mice. This finding is consistent with published studies for these mouse models [27, 30], thus demonstrating our success in maintaining reproducibility and rigor in our colonies.
Fig. 3.

Genotype and colony determinations. Quantitative PCR (qPCR) was used to identify mice carrying the mutant human SOD1-G93A gene, as well as to distinguish HCN-versus LCN-expressing mice from controls. In this qPCR amplification plot, each line represents the transgene concentration for individual mice from our HCN and LCN colonies. Fluorescent signal detection at lower PCR cycle numbers signifies higher transgene copy numbers in the sample. As expected, the threshold cycle (Ct) was lower for HCN-SOD1 (red) compared to LCN-SOD1 (blue) mice. Control (green) mice do not carry the SOD1-G93A transgene; however, a late amplification signal was detected because the fluorescent reporter (SYBR green) binds to any double-stranded DNA, including the primer dimers formed during qPCR. LCN low copy number, HCN high copy number, SOD1 SOD1-G93A transgene
As expected, all HCN-SOD1 mice developed stereotypical paralysis in both hindlimbs. In contrast, LCN-SOD1 mice displayed wide variability in limb involvement, ranging from only bilateral hindlimb paralysis (similar to HCN-SOD1 mice) to only bilateral forelimb paralysis, as well as various combinations of hindlimb and forelimb paralysis that affected one or both sides. Examples of some of the observed limb phenotypes are shown in Fig. 4. The limb phenotype variability in LCN-SOD1 mice was unexpected because it was not a focus of prior studies. In fact, after careful review of the literature, we identified only one published study that briefly reported impaired forelimb function (with minor hindlimb involvement) in a small subset of LCN-SOD1 mice [29].
Fig. 4.

Limb phenotypes. Control mice from both colonies (HCN and LCN) maintained normal hindlimb and forelimb functions (a). All HCN-SOD1 transgenic mice developed bilateral hindlimb paralysis (b), whereas LCN-SOD1 transgenic mice displayed a variety of limb phenotypes ranging from bilateral hindlimb paralysis (b) to bilateral forelimb paralysis (c), and combined hindlimb and forelimb paralysis affecting one or both sides (d). Note the bedding on the face of the forelimb-affected mouse (c), which demonstrates reduced grooming ability. Also note the lateral forelimb posture during ambulation (d), demonstrating altered weight-bearing capacity of the affected forelimb. As expected, both SOD1 strains displayed a variety of coat colors due to the mixed background strains (i.e., C57BL/6 and SJL) that result in random segregation of coat color alleles. Possible coat colors include white bellied agouti, albino, black, light gray, and tan, some of which are depicted here
As objective characterization of limb phenotype was not an a priori goal of this study, we elected to incorporate a descriptive limb assessment after recognizing an increasing number of mice with forelimb involvement as the study progressed. Thus, for the remaining LCN-SOD1 mice (n = 10), we observed individual mice exploring within the home cage as well as walking on a paper-lined benchtop, and then classified each mouse as displaying only hindlimb paralysis (n = 5) versus forelimb ± hindlimb paralysis (n = 5) at the study endpoint. We then used this dichotomous classification scheme to explore differences in swallowing function (described for VFSS results, below).
Survival Time
The age at which transgenic mice reached the humane endpoint criterion for this study served as survival time, which was analyzed using a Kaplan–Meier log rank test. As expected, survival time was significantly different between genotypes (X2(1) = 52.114, p < 0.0001), with LCN-SOD1 mice living approximately twice as long as HCN-SOD1 mice. For both genotypes, there was a trend for females to live longer than males; however, this finding was not statistically significant: HCN-SOD1: X2(1) = 0.866, p = 0.352; LCN-SOD1: X2(1) = 3.739, p = 0.053. Based on Kaplan–Meier survival curves (Fig. 5), the estimated median survival time was 4.2 months for HCN-SOD1 mice (males = 4.13 months; females = 4.23 months) and 8.3 months for LCN-SOD1 mice (males = 7.93 months; females = 8.50 months), which is consistent with previous reports [22, 27, 29, 30].
Fig. 5.

Extended survival time for LCN-SOD1 mice. Kaplan–Meier plot showing the age at which HCN-SOD1 (left) and LCN-SOD1 (right) mice reached advanced disease stage (i.e., obvious limb paralysis but still able to forage for food and water in the home cage). LCN-SOD1 mice survived approximately two times longer than HCN-SOD1 mice. Although there was a trend that females (green line) in both groups survived longer than males (blue line), this finding was not statistically significant. HCN-SOD1 = high copy number SOD1-G93A mice (n = 24; 14 males and 10 females); LCN-SOD1 low copy number SOD1-G93A mice (n = 25; 11 males and 14 females)
VFSS Analysis
All mice voluntarily drank the oral contrast agent during VFSS testing, resulting in a total of 97 VFSS studies (one per mouse) for manual, frame-by-frame analysis to objectively quantitate the 6 outcome measures (VFSS metrics) described in Table 1. Differences between the two control groups (i.e., nontransgenic mice from the HCN and LCN colonies) and sex differences within each genotype (i.e., HCN-SOD1, LCN-SOD1, and colony controls) were explored using the Mann–Whitney U test. This step was necessary to determine whether it was acceptable to collapse data across groups (i.e., form a single control variable and a single gender variable) to derive more meaningful results from subsequent statistical analyses. Results revealed that the two control groups (i.e., nontransgenic mice from the HCN and LCN colonies) did not differ significantly for any of the VFSS metrics (Table 2); therefore, data from controls in the HCN (n = 25) and LCN (n = 23) colonies were combined into a single control group (n = 48) for each VFSS metric for subsequent comparison with the HCN-SOD1 and LCN-SOD1 groups. Similarly, there were no significant sex differences within each genotype for any of the VFSS metrics; therefore, males and females were combined within each genotype (HCN: n = 24; LCN-SOD1: n = 25; combined control group: n = 48) for subsequent statistical analyses of VFSS metrics.
Table 2.
Pairwise comparisons to determine collapsibility of sexes within each group for analysis of VFSS metrics
| VFSS metrics | Pairwise comparisons (p-values) |
|||
|---|---|---|---|---|
| Controls from HCN and LCN colonies |
Sex differences |
|||
| HCN-SOD1 | LCN-SOD1 | Controls | ||
| Lick rate | 0.395 | 0.796 | 1.000 | 0.383 |
| Swallow rate | 0.335 | 0.472 | 0.572 | 0.753 |
| Inter-swallow interval | 0.421 | 0.403 | 0.809 | 0.630 |
| Lick–swallow ratio | 0.612 | 0.546 | 0.893 | 0.622 |
| Pharyngeal transit time | 0.811 | 0.192 | 0.572 | 0.635 |
| Esophageal transit time | 0.660 | 0.808 | 0.331 | 0.216 |
Differences in VFSS data between genotypes (i.e., HCN-SOD1, LCN-SOD1, and combined control group) were investigated using the Kruskal–Wallis test. Results showed that 4 of the 6 VFSS metrics were significantly different between groups: lick rate [X2(2) = 66.616, p < 0.0001], swallow rate [X2(2) = 39.409, p < 0.0001], inter-swallow interval [X2(2) = 35.988, p < 0.0001], and pharyngeal transit time [X2(2) = 8.410, p = 0.015]. The two nonsignificant VFSS metrics were lick–swallow ratio, a measure not dependent on time, and esophageal transit time, typically a late emerging symptom in human ALS patients [3] (both p ≥ 0.05). Bonferroni post hoc comparisons revealed that only 3 of the 4 statistically significant VFSS metrics were significantly altered for both HCN-SOD1 and LCN-SOD1 mice compared to controls (p < 0.05). Specifically, both models had slower lick and swallow rates and longer inter-swallow intervals; however, symptoms were worse for HCN-SOD1 compared to LCN-SOD1 mice. Pharyngeal transit time was significantly longer only for HCN-SOD1 mice. These results are summarized in Fig. 6.
Fig. 6.

Videofluoroscopic evidence of dysphagia. The data distribution for the four statistically significant VFSS metrics is visually displayed by boxplots (median, quartiles, and whiskers, mean = X) with adjacent dotplots (individual data points) for the three groups of mice (control, HCN-SOD1, and LCN-SOD1). Compared to controls, both HCN-SOD1 and LCN-SOD1 mice had significantly altered swallow function at advanced disease stage, characterized by significantly slower lick (a) and swallow (b) rates, and significantly longer time between successive swallows (d; i.e., inter-swallow interval). In addition, pharyngeal transit time (c) was significantly longer for HCN-SOD1 but not LCN-SOD1 mice. However, none of the VFSS metrics were significantly different between HCN-SOD1 and LCN-SOD1 mice, thus highlighting the similarity in their dysphagia phenotype. Data points outside the whiskers are considered mild outliers; the star indicates an extreme outlier. Significance levels: p ≥ 0.05 (ns; nonsignificant), p< 0.05 (*), p< 0.01 (**), p < 0.001 (***), and p < 0.0001 (****). HCN-SOD1 high copy number SOD1-G93A mice (n = 24), LCN-SOD1 low copy number SOD1-G93A mice (n = 25), controls (n = 48) combined controls from HCN (n = 25) and LCN (n = 23) colonies
The final VFSS metric under investigation was airway protection. All mice in this study had a PAS score of 1, indicating no evidence of laryngeal penetration or aspiration. Therefore, statistical analysis was not performed for this metric because there was no variability in the data (i.e., standard deviation = 0).
Based on our unexpected observation of marked variability in limb involvement for LCN-SOD1 mice, we explored whether dysphagia severity differed as a function of limb phenotype. As we did not analytically characterize limb phenotypes for this study, we chose to focus this preliminary inquiry on a simple dichotomous classification scheme: LCN-SOD1 mice with only hindlimb paralysis (n = 5) versus those with obvious forelimb ± hindlimb paralysis (n = 5). Although there was a trend for differences in VFSS metrics between limb phenotypes, particularly swallow rate as shown in Fig. 7, an independent samples Mann–Whitney U test failed to detect significant differences (p ≥ 0.05).
Fig. 7.

Swallow rate differs between hindlimb-and forelimb-affected LCN-SOD1 mice. Although the data distribution for swallow rate (a) was not statistically different between LCN-SOD1 mice with predominant hindlimb (b) versus forelimb (c) paralysis, there was a trend for forelimb-affected mice to have slower swallow rates compared to hindlimb-affected mice. Specifically, forelimb-affected mice had relatively tight clustering of the data, which spanned only half the distribution range of hindlimb-affected mice. This finding suggests additional phenotype classifications may exist beyond the basic dichotomy used in this preliminary study. Radiographic images from two different mice are shown in (b, c)—one with hindlimb paralysis (b) and the other with forelimb paralysis (c). Radiographic images from a single control mouse (d, e) are shown for comparison. Boney structures are labeled for the hindlimbs (F femur, f fibula, t tibia, hp hindpaw), forelimbs (H humerus, r radius, u ulna, fp forepaw), and tail. Note the caudally extended hindlimbs and inverted hindpaws (i.e., soles facing upward) in the hindlimb-affected mouse (b). In the forelimb-affected mouse (c), one forepaw is curled under and the other is inverted (i.e., palm facing upward), with both forepaws located in close proximity to the hindpaws. Also note the “dropped” head and torso, which demonstrates the reduced weight-bearing capacity of the forelimbs in this mouse. Sample size (n) = 5 mice per group; ns nonsignificant (p = 0.340); X mean
Tongue Weight and Morphometry
Tongue weight plus 7 measures of tongue morphometry were compared between the 4 study groups (i.e., HCN-SOD1 transgenic mice, LCN-SOD1 transgenic mice, and age-matched controls from each colony) using the Kruskal–Wallis test. In this case, the two control groups were not combined into a single control group because it was unknown if the large age difference (~ 4 months) and corresponding significant difference in body weight [T(44) = 3.445, p = 0.001] between the two control groups [HCN mean (± standard error) = 29.2 (0.85) g, LCN mean (± standard error) = 35.4 (1.58) grams] would have an influence on tongue weight. Further, only a subsample of mice (n = 61 minus 7 damaged during dissection = 54 mice) was included in this postmortem assay of fresh tongue tissue; therefore, the group sample sizes were insufficient to permit meaningful exploration of sex differences. Results showed that 5 of the 8 tongue variables were significantly reduced between SOD1 mice versus controls: tongue weight [X2(3) = 14.530, p < 0.002], total tongue area [X2(3) = 19.423, p < 0.0001], total tongue length [X2(3) = 21.183, p < 0.0001], rostral tongue length [X2(3) = 14.630, p < 0.002], and caudal tongue length [X2(3) = 20.325, p < 0.0001]. The three nonsignificant metrics were median eminence width, rostral tongue width, and caudal tongue width (all p ≥ 0.05).
Bonferroni post hoc comparisons (Fig. 8) showed that 4 of the 5 significant tongue variables were significantly reduced for LCN-SOD1 mice compared to age-matched colony controls: tongue weight (p = 0.015), tongue dorsum surface area (p = 0.012), total tongue length (p = 0.004), and caudal tongue length (p = 0.001). Although there was a trend for these indicators of tongue atrophy to be reduced in HCN-SOD1 mice compared to age-matched colony controls, they did not reach statistical significance (p ≥ 0.05). Rostral tongue length was not statistically different for HCN-SOD1 or LCN-SOD1 mice compared to age-matched colony controls (p > 0.05). However, rostral tongue length was significantly greater for controls from the LCN colony compared to controls from the HCN colony (p = 0.041), suggesting an aging effect (i.e., controls from the LCN colony were approximately twice as old as controls from the HCN colony).
Fig. 8.

Evidence of tongue atrophy at advanced disease stage. Adjacent boxplots and dotplots show the data distribution of the four tongue-based measurements that were significantly reduced for LCN-SOD1 mice compared to age-matched controls: tongue weight (a), tongue surface area (b), total tongue length (c), and posterior (caudal) tongue length (d). These indicators of tongue atrophy were also apparent in HCN-SOD1 mice, but did not reach statistical significance. Data points outside the whiskers are considered mild outliers; there were no extreme outliers. Group sample sizes: HCN colony [controls: n = 14; 8 males and 6 females; HCN-SOD1: n = 12; 5 males and 7 females] and LCN colony [controls: n = 11; 5 males and 6 females; LCN-SOD1: n = 17; 8 males and 9 females]. Significance levels: p≥ 0.05 (ns nonsignificant), p< 0.05 (*), p< 0.01 (**), p < 0.001 (***), and p < 0.0001 (****), X mean
Brain Histology
Vacuolar Pathology
All HCN-SOD1 and LCN-SOD1 mice (100%) had vacuolar pathology within the hypoglossal nucleus and tract, whereas none of the age-matched colony controls had vacuolar pathology in these same neuroanatomic regions. Representative images of H&E and SOD1 staining are shown in Fig. 9. Although not objectively quantified, the vacuoles appeared visibly larger and less abundant in advanced disease stage LCN-SOD1 compared to HCN-SOD1 mice, a finding that has not been previously reported to our knowledge. Moreover, the vacuoles in LCN-SOD1 mice were more visible in the hypoglossal tract than the hypoglossal nucleus. This finding is consistent with findings for ventral horn cells of the spinal cord in this same model, and suggests that axonal transport may be more affected (i.e., physically blocked) in LCN-SOD1 than HCN-SOD1 mice [44].
Fig. 9.
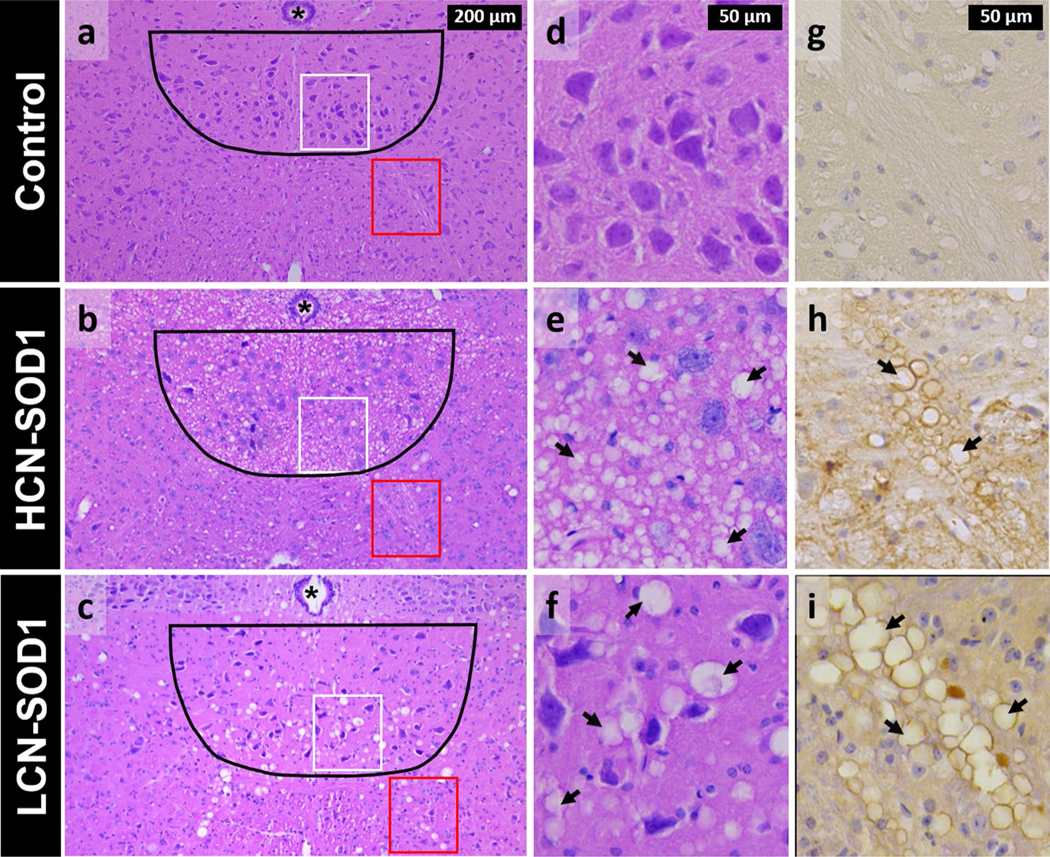
Histological evidence of degeneration within the hypoglossal nucleus and tract in advanced disease stage HCN-SOD1 and LCN-SOD1 mice. Left panel Representative hematoxylin-and eosin-stained sections from the hypoglossal nucleus region (black outline) in the brainstem of HCN-SOD1 (b) and LCN-SOD1 (c) mice at advanced disease stage, as well as an age-matched control (a) from our LCN colony. Axons from motor neurons in the hypoglossal nucleus project ventrally (red boxes) to form the hypoglossal tract and ultimately the hypoglossal nerve that innervates the tongue. Middle panel Higher magnification images of the hypoglossal nucleus within the white boxed regions in the left panel (a–c). Compared to controls (d), the hypoglossal nucleus of HCN-SOD1 (e) and LCN-SOD1 (f) mice had marked vacuolation (arrows) throughout the neuropil, a hallmark feature of mitochondrial pathology in SOD1 models of ALS. Right panel Higher magnification images of the hypoglossal tract within the red boxed regions in the left panel (a–c). hSOD1(+) lined vacuoles are abundant in the hypoglossal tracts of HCN-SOD1 (h) and LCN-SOD1 (i) mice but not controls (g). Note the larger size of the vacuoles for LCN-SOD1 mice compared to HCN-SOD1 mice in both the hypoglossal nucleus and tract. Asterisk = central canal
Neuronal Counts
Two mice from the control group were excluded from statistical analysis because they lacked one or more tissue sections due to loss/damage during processing. The mean and standard error (SE) for hypoglossal neuron counts were 1615 ± 166 for LCN-SOD1 mice (n = 6) versus 2370 ± 159 for controls (n = 4). This equates to a 32% loss of hypoglossal motor neurons for LCN-SOD1 mice at advanced disease stage, which is within the ~ 28% [43] to ~ 64% [42] range reported for this same brainstem nucleus in HCN-SOD1 mice. The Mann–Whitney U test revealed that there were significantly fewer hypoglossal neurons in LCN-SOD1 mice versus age-matched controls (U =24, p = 0.01), as shown in Fig. 10.
Fig. 10.

Quantitation of hypoglossal motor neuron death in LCN-SOD1 mice at advanced disease stage. Images (a–d) are representative NeuN-stained sections of the hypoglossal motor nucleus in the brainstem of advanced disease stage LCN-SOD1 mice (n = 6) and age-matched colony controls (n = 4). In a and b, the yellow outlines indicate the regions within which motor neurons were counted, and the white boxes highlight the areas shown at higher magnification in (c) and (d). White arrows denote viable/healthy neurons, whereas dead/dying neurons are indicated by yellow arrows. Long arrows point to the neurons shown at higher magnification in (e) and (f). Images (a, b) and (e, f) were captured with an epifluorescence microscope (Leica DM4000) using either the 10 × or 100 × objective. For improved visualization of the regions of interest depicted here, stacked images (c, d) were taken through the 20 × objective of a Leica confocal microscope using LAS-AF software. g Adjacent boxplots and dotplots show the significant reduction of hypoglossal neurons for LCN-SOD1 versus control mice (*p = 0.01)
Correlations
Given our finding that only LCN-SOD1 mice had significantly altered tongue weight and tongue morphometry, in addition to VFSS evidence of dysphagia, we limited our investigation of correlations to only LCN-SOD1 mice to further characterize dysphagia in this understudied model of ALS. Spearman’s rank order correlation was used to determine the strength and direction of the association between pairs of rank-ordered variables for advanced stage LCN-SOD1 mice. Results showed that no significant correlations existed between survival age and any of the previously identified significant tongue variables (i.e., weight, surface area, total length, and caudal length; all p ≥ 0.05) or significant VFSS metrics (i.e., lick rate, swallow rate, pharyngeal transit time, and inter-swallow interval; all p ≥ 0.05). However, a significant positive correlation was identified between tongue weight and body weight of LCN-SOD1 mice (rs = 0.565 p = 0.016), as shown in Fig. 11a. We also explored correlations between survival age and VFSS metrics for the two different limb phenotypes (i.e., hindlimb versus forelimb involvement). For forelimb-affected mice, there was only a significant positive correlation between survival age and lick rate (rs = 0.900, p = 0.037; Fig. 11b); however, no significant correlations existed for the other VFSS metrics (i.e., swallow rate, pharyngeal transit time, and inter-swallow interval; all p ≥ 0.05). For hindlimb-affected mice, no significant correlations were identified between survival age and VFSS metrics (i.e., lick rate, swallow rate, pharyngeal transit time, and inter-swallow interval; all p ≥ 0.05).
Fig. 11.

Correlations for advanced disease stage LCN-SOD1 mice. There was a significant positive correlation between body weight and tongue weight in LCN-SOD1 mice (a). Body weight loss in LCN-SOD1 mice was moderately associated with tongue weight loss. For forelimb-affected LCN-SOD1 mice, there was a significant positive correlation between survival age and lick rate (b). Slower lick rate in forelimb-affected LCN-SOD1 mice was very strongly associated with shorter survival. p p value, n sample size
Discussion
The goal of this study was to optimize the translational value of transgenic mouse models of ALS for use in dysphagia research and therapeutic discovery. We specifically focused on two mouse models of ALS: the widely used high copy number SOD1-G93A (HCN-SOD1) model [19, 20] versus the rarely studied low copy number SOD1-G93A (LCN-SOD1) model [19, 27, 30]. To prevent potential confounding effects of repeated X-ray exposure on swallowing function, we limited our investigation to a single humane endpoint corresponding to an advanced stage of ALS (20% reduction from maximum body weight), To characterize the clinicopathological features of dysphagia, we utilized the VFSS protocol we previously adapted for use in mice [31, 32], in conjunction with postmortem assays of tissues relevant to swallowing. Although numerous muscles, cranial nerves, and synaptic connections are involved in swallowing, we limited our investigation to the tongue and its peripheral innervation (i.e., hypoglossal neurons). Our rationale was that tongue weakness due to lower motor neuron degeneration is a common clinical sign in ALS patients, regardless of onset phenotype (spinal or bulbar) [16]. Further, impaired tongue strength is an independent prognostic indicator of earlier mortality in ALS patients [16, 17]. Thus, investigating the tongue and hypoglossal neurons may provide insight into the etiology and pathogenesis of dysphagia in ALS, thereby paving the way for targeted therapeutics.
The main finding of this study was that the LCN-SOD1 model is better suited for translational studies aimed at exploring the mechanisms leading to dysphagia than the HCN-SOD1 model. Whereas both models showed videofluoroscopic evidence of dysphagia (i.e., slower lick and swallow rates, and longer inter-swallow interval) compared to controls, only LCN-SOD1 mice developed significant tongue atrophy (i.e., reduced tongue weight, length, and dorsal surface area). Moreover, we found significant positive correlations between tongue and body weights and between lick rate and survival in LCN-SOD1 mice. Specifically, LCN-SOD1 mice with lower body weights had smaller/lighter tongues, and forelimb-affected mice with slower lick rates died at a younger age. In addition, LCN-SOD1 mice had a 32% loss of hypoglossal motor neurons, which is within the ~ 28% [43] to ~ 64% [42] range reported for the same region in HCN-SOD1 mice. These collective findings for LCN-SOD1 mice are congruent with reports of dysphagia [3, 45], associated tongue atrophy [46, 47], and hypoglossal nucleus pathology [48] in the human ALS literature, thus highlighting the translational potential of this model in ALS research.
For reasons that remain unclear, the gold standard dysphagia diagnostic test (i.e., VFSS) used in this study has been remarkably underutilized in human ALS research. The few published studies reported a variety of dysphagic signs/symptoms in patients with ALS, including delayed bolus transfer from the oral cavity to the pharynx [3, 45], delayed triggering of the pharyngeal swallow [47], reduced pharyngeal constriction [3, 45, 49], longer pharyngeal swallow duration [47], postswallow residue in the oral and pharyngeal cavities [45, 49, 50], reduced elevation of the hyoid and larynx [45], and laryngeal aspiration [50]. The majority of these outcome measures were not possible to quantitate in mice, due to a combination of anatomic and technological limitations. For example, we have previously shown that mice swallow approximately 10 times faster than humans [31, 32]. Thus, the 30 frame per second limitation of our camera provides insufficient temporal resolution for quantitation of some swallow kinematics in this small animal. As a result, our VFSS metrics were based predominantly on measures of bolus flow during drinking rather than movement of anatomic structures. Moreover, there was no evidence of laryngeal penetration or aspiration in either model (HCN-SOD1 or LCN-SOD1), which was expected because the larynx in mice resides in the nasopharynx (similar to human infants), which inherently protects the larynx from the path of the bolus [31, 32]. However, we have identified two VFSS metrics that readily detect dysphagia in transgenic SOD1 mice: swallow rate and inter-swallow interval, both potential functional biomarkers of dysphagia in human ALS. We envision these two VFSS metrics could be easily incorporated into clinical VFSS testing, or included in acoustic-and EMG-based swallowing assessments in patients with ALS.
Impaired lick rate, a general indicator of tongue dysmotility, was also identified as a robust functional biomarker of dysphagia in both transgenic SOD1 mouse models; although this finding may not directly translate to human swallowing kinematics. Nonetheless, in ALS patients the tongue is affected early in the disease, regardless of spinal or bulbar onset phenotype [13]. We previously identified tongue dysmotility at weaning (P21–P23) in HCN-SOD1 mice [23] that worsened with advancing disease [51]. Thus, our finding of lick rate impairment in HCN-SOD1 mice by 21–23 days of age validated the advantage of using a manual lick rate assay over an automated lickometer, which did not detect changes in lingual function until 100 days of age [52]. Earlier postnatal (i.e., P4–P10) evidence of aberrant neuronal activity has also been identified in the hypoglossal nucleus of HCN-SOD1 mice [53]. Collectively, these early findings of tongue dysfunction and pathology in HCN-SOD1 mice raise serious concerns regarding the translatability of this model to human ALS, which is an adult-onset rather than infantile disease. For this reason, we propose the LCN-SOD1 model with delayed clinical disease onset is better suited for dysphagia research.
An unexpected finding of this study was, compared to the stereotypical bilateral hindlimb paralysis of HCN-SOD1 mice [19, 20, 22], LCN-SOD1 mice displayed marked variability in limb involvement, ranging from only hindlimb paralysis to only forelimb paralysis, as well as various combinations affecting one or both sides. This limb phenotype variability in LCN-SOD1 mice resembles the heterogeneous clinical disease course in human ALS, which typically begins in one body region and spreads to other regions with no known pattern [11, 54–56]. This finding for LCN-SOD1 mice has not been well described in the literature; we identified only one study that briefly reported impaired forelimb function (with minor hindlimb involvement) in a small subset of LCN-SOD1 mice [29]. We have therefore begun incorporating analytical methods in our ongoing research with this model to characterize the various limb phenotypes relative to the anatomic site (specific limb versus bulbar muscles) of onset, age at onset, pattern of disease spread, and survival age. Our goal is to use this model to elucidate the mechanisms driving the heterogeneous clinical disease course in human ALS, which currently remain largely unknown.
Interestingly, we found that LCN-SOD1 mice with forelimb involvement appear to develop a more severe dysphagia phenotype resembling patients with bulbar-onset ALS, which is known to result in more rapid progression and worse survival rates compared to spinal-onset ALS [11, 12, 57–61]. However, larger-scale studies with this model are essential to validate this preliminary finding. This model may ultimately provide an ideal translational platform for identifying which clinical phenotypes are most responsive to therapeutic interventions, which may guide appropriate allocation of ALS patients into treatment arms in clinical trials.
Despite the substantial negative outcomes associated with dysphagia in ALS patients, few studies have addressed the responsible mechanisms for progressive bulbar deterioration affecting the vast majority of ALS patients. As a result, effective therapeutic targets to delay, slow, or prevent the onset and progression of dysphagia have not been identified [62–65]. This is indeed the case for the two existing FDA-approved pharmacological interventions for ALS—riluzole (Rilutek©, approved in 1995) and edaravone (Radicava©, approved in 2017). While riluzole has been shown to extend the median survival of ALS patients by 2–3 months [66–68], this antiglutamatergic oral medication has no beneficial effect on muscle weakness and does not improve functional outcomes, including those pertaining to swallowing [69, 70]. Edaravone, a free radical scavenging drug has been shown to slow the progression of general ALS symptoms in a small subset (~ 7%) of patients, but not for swallowing dysfunction; dysphagia was reported as a serious adverse event in ~ 12% of cases, regardless of treatment group assignment (edaravone versus placebo) [71]. This finding not only highlights the negative impact of dysphagia in ALS, but also demonstrates the critical need for new ALS treatments that specifically target swallowing function.
In summary, dysphagia is an understudied symptom of ALS that significantly contributes to poor outcomes, including malnutrition, major depression, and early mortality. We propose that utilizing the LCN-SOD1 mouse model of ALS, which emulates clinicopathological features of dysphagia at advanced stages of the disease, may facilitate our understanding of the mechanisms contributing to dysphagia in ALS. Addressing this scientific knowledge gap is an essential precursor to the development of targeted and effective therapeutics. Thus, we are currently conducting clinicopathological investigations of LCN-SOD1 mice across the life span that may guide the selection and timing of dysphagia interventions in clinical trials. Our ultimate goal is earlier detection of dysphagia in ALS, which would provide an opportunity for earlier intervention and better functional outcomes for prolonged survival and improved quality of life for ALS patients.
Acknowledgements
We graciously thank the numerous students affiliated with the Lever Lab who assisted with VFSS analysis (Kaitlin Flynn, Dana Aleman, Mitchell Allen, Victoria Caywood, Loren Littrell, Rebecca Harris Healy, Mollie Ulsas, and Elizabeth White) and histology (Iyas Daghlas, Abby Lind, Chandler DeJonge, Victoria Rufo, and Michaela Thomson).
Funding This study was funded in part by two grants from the National Institute on Deafness and Other Communication Disorders of the National Institutes of Health (Award Numbers R03 DC0110895 and R21 DC016071; T.E. Lever), University of Missouri Research Board Grant (T.E. Lever and N.L. Nichols), and Mizzou Advantage Seed Funding (T.E. Lever and J.R. Coates). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Research involving Human Participants and/or Animals No human participants were included in this study; only animals. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Footnotes
Compliance with Ethical Standards
Conflict of interest The authors declare that they have no conflict of interest.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ertekin C, Aydogdu I, Yuceyar N, Kiylioglu N, Tarlaci S, Uludag B. Pathophysiological mechanisms of oropharyngeal dysphagia in amyotrophic lateral sclerosis. Brain. 2000;123(Pt 1):125–40. [DOI] [PubMed] [Google Scholar]
- 2.Hadjikoutis S, Wiles CM. Respiratory complications related to bulbar dysfunction in motor neuron disease. Acta Neurol Scand. 2001;103(4):207–13. [PubMed] [Google Scholar]
- 3.Higo R, Tayama N, Watanabe T, Nitou T. Videomanofluorometric study in amyotrophic lateral sclerosis. Laryngoscope. 2002;112(5):911–7. [DOI] [PubMed] [Google Scholar]
- 4.Hillel AD, Miller RM. Management of bulbar symptoms in amyotrophic lateral sclerosis. Adv Exp Med Biol. 1987;209:201–21. [DOI] [PubMed] [Google Scholar]
- 5.Kawai S, Tsukuda M, Mochimatsu I, Enomoto H, Kagesato Y, Hirose H, Kuroiwa Y, Suzuki Y. A study of the early stage of dysphagia in amyotrophic lateral sclerosis. Dysphagia. 2003;18(1):1–8. [DOI] [PubMed] [Google Scholar]
- 6.Tayama N. Dysphagia in amyotrophic lateral sclerosis—the mechanism and managements. Rinsho Shinkeigaku. 1995;35(12):1557–9. [PubMed] [Google Scholar]
- 7.Kuhnlein P, Gdynia HJ, Sperfeld AD, Lindner-Pfleghar B, Ludolph AC, Prosiegel M, Riecker A. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat Clin Pract Neurol. 2008;4(7):366–74. [DOI] [PubMed] [Google Scholar]
- 8.Shigemitsu H, Afshar K. Aspiration pneumonias: under-diagnosed and under-treated. Curr Opin Pulm Med. 2007;13(3):192–8. [DOI] [PubMed] [Google Scholar]
- 9.Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942–55. 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 10.Leder SB, Novella S, Patwa H. Use of fiberoptic endoscopic evaluation of swallowing (FEES) in patients with amyotrophic lateral sclerosis. Dysphagia. 2004;19(3):177–81. [DOI] [PubMed] [Google Scholar]
- 11.Sandyk R. Serotonergic mechanisms in amyotrophic lateral sclerosis. Int J Neurosci. 2006;116(7):775–826. 10.1080/00207450600754087. [DOI] [PubMed] [Google Scholar]
- 12.Wright R, Jordan C. Videofluoroscopic evaluation of dysphagia in motor neurone disease with modified barium swallow. Palliat Med. 1997;11(1):44–8. [DOI] [PubMed] [Google Scholar]
- 13.DePaul R, Abbs JH, Caligiuri M, Gracco VL, Brooks BR. Hypoglossal, trigeminal, and facial motoneuron involvement in amyotrophic lateral sclerosis. Neurology. 1988;38(2):281–3. [DOI] [PubMed] [Google Scholar]
- 14.DePaul R, Brooks BR. Multiple orofacial indices in amyotrophic lateral sclerosis. J Speech Hear Res. 1993;36(6):1158–67. [DOI] [PubMed] [Google Scholar]
- 15.Dworkin JP. Tongue strength measurement in patients with amyotrophic lateral sclerosis: qualitative vs quantitative procedures. Arch Phys Med Rehabil. 1980;61(9):422–4. [PubMed] [Google Scholar]
- 16.Weikamp JG, Schelhaas HJ, Hendriks JC, de Swart BJ, Geurts AC. Prognostic value of decreased tongue strength on survival time in patients with amyotrophic lateral sclerosis. J Neurol. 2012;259(11):2360–5. 10.1007/s00415-012-6503-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umemoto G, Furuya H, Tsuboi Y, Fujioka S, Arahata H, Sugahara M, Sakai M. Characteristics of tongue and pharyngeal pressure in patients with neuromuscular diseases. Degener Neurol Neuromuscul Dis. 2017;7:71–8. 10.2147/DNND.S132745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hillel AD, Miller R. Bulbar amyotrophic lateral sclerosis: patterns of progression and clinical management. Head Neck. 1989;11(1):51–9. [DOI] [PubMed] [Google Scholar]
- 19.Gurney ME, Pu H, Chiu AY, Canto Dal MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–5. [DOI] [PubMed] [Google Scholar]
- 20.Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA. 1996;93(7):3155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39(2):147–57. 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- 22.Pfohl SR, Halicek MT, Mitchell CS. Characterization of the contribution of genetic background and gender to disease progression in the SOD1 G93A mouse model of amyotrophic lateral sclerosis: a meta-analysis. J Neuromuscul Dis. 2015;2(2):137–50. 10.3233/JND-140068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daghlas I, Schmittdiel T, Martin H, Kadosh K, Goding G, Mancini S, Dougherty D, Harris K, Robbins K, Lever T. Effects of gene copy number on dysphagia onset in SOD1-G93A transgenic mice. Amyotroph Later Scler Frontotemp Degener. 2015;16:217–26. 10.3109/21678421.2015.1098818/0016. [DOI] [Google Scholar]
- 24.Acevedo-Arozena A, Kalmar B, Essa S, Ricketts T, Joyce P, Kent R, Rowe C, Parker A, Gray A, Hafezparast M, Thorpe JR, Greensmith L, Fisher EM. A comprehensive assessment of the SOD1G93A low-copy transgenic mouse, which models human amyotrophic lateral sclerosis. Dis Model Mech. 2011;4(5):686–700. 10.1242/dmm.007237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Canto Dal M, Gurney M. A low expressor line of transgenic mice carrying a mutant human Cu, Zn superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis. Acta Neuropathol. 1997;93:537–50. [DOI] [PubMed] [Google Scholar]
- 26.Shibata N. Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Neuropathology. 2001;21(1):82–92. [DOI] [PubMed] [Google Scholar]
- 27.Alexander GM, Erwin KL, Byers N, Deitch JS, Augelli BJ, Blankenhorn EP, Heiman-Patterson TD. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res Mol Brain Res. 2004;130(1–2):7–15. [DOI] [PubMed] [Google Scholar]
- 28.JAX Mice Database. 2014. https://www.jax.org/strain/002300. Accessed 3 Nov 2019.
- 29.Jaarsma D, Teuling E, Haasdijk E, De Zeeuw C, Hoogenraad C. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J Neurosci. 2008;28(9):2075–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gurney ME. The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J Neurol Sci. 1997;152(Suppl 1):S67–73. [DOI] [PubMed] [Google Scholar]
- 31.Lever T, Brooks R, Thombs L, Littrell L, Harris R, Allen M, Kadosh M, Robbins K. Videofluoroscopic validation of a translational murine model of presbyphagia. Dysphagia. 2015;30:328. [DOI] [PubMed] [Google Scholar]
- 32.Lever TE, Braun SM, Brooks RT, Harris RA, Littrell LL, Neff RM, Hinkel CJ, Allen MJ, Ulsas MA. Adapting human videofluoroscopic swallow study methods to detect and characterize dysphagia in murine disease models. J Vis Exp. 2015;97:e52319. 10.3791/52319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Achilli F, Boyle S, Kieran D, Chia R, Hafezparast M, Martin JE, Schiavo G, Greensmith L, Bickmore W, Fisher EM. The SOD1 transgene in the G93A mouse model of amyotrophic lateral sclerosis lies on distal mouse chromosome 12. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6(2):111–4. 10.1080/14660820510035351. [DOI] [PubMed] [Google Scholar]
- 34.The Jackson Laboratory, editor. Breeding strategies for maintaining colonies of laboratory mice. Bar Harbor: TJ Laboratory; 2009. [Google Scholar]
- 35.Ginzinger DG. Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp Hematol. 2002;30(6):503–12. [DOI] [PubMed] [Google Scholar]
- 36.D’Haene B, Vandesompele J, Hellemans J. Accurate and objective copy number profiling using real-time quantitative PCR. Methods (San Diego, Calif). 2010;50(4):262–70. 10.1016/j.ymeth.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Rosenbek JC, Robbins JA, Roecker EB, Coyle JL, Wood JL. A penetration-aspiration scale. Dysphagia. 1996;11(2):93–8. [DOI] [PubMed] [Google Scholar]
- 38.Reiner DJ, Jan TA, Boughter JD Jr, Li CX, Lu L, Williams RW, Waters RS. Genetic analysis of tongue size and taste papillae number and size in recombinant inbred strains of mice. Chem Sens. 2008;33(8):693–707. 10.1093/chemse/bjn025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salas M, Rubio L, Torrero C, Carreon M, Regalado M. Effects of perinatal undernutrition on the circumvallate papilla of developing Wistar rats. Acta Histochem. 2016;118(6):581–7. 10.1016/j.acthis.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. 4th ed Cambridge: Academic Press; 2013. [Google Scholar]
- 41.Paxinos G, Franklin K. The mouse brain in sterotaxic coordinates. 2nd ed Sidney: Academic Press; 2001. [Google Scholar]
- 42.Ferrucci M, Spalloni A, Bartalucci A, Cantafora E, Fulceri F, Nutini M, Longone P, Paparelli A, Fornai F. A systematic study of brainstem motor nuclei in a mouse model of ALS, the effects of lithium. Neurobiol Dis. 2010;37(2):370–83. [DOI] [PubMed] [Google Scholar]
- 43.Haenggeli C, Kato AC. Differential vulnerability of cranial motoneurons in mouse models with motor neuron degeneration. Neurosci Lett. 2002;335(1):39–43. [DOI] [PubMed] [Google Scholar]
- 44.Sasaki S, Warita H, Abe K, Iwata M. Slow component of axonal transport is impaired in the proximal axon of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2004;107(5):452–60. 10.1007/s00401-004-0838-y. [DOI] [PubMed] [Google Scholar]
- 45.Higo R, Tayama N, Nito T. Longitudinal analysis of progression of dysphagia in amyotrophic lateral sclerosis. Auris Nasus Larynx. 2004;31(3):247–54. 10.1016/j.anl.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 46.Cha CH, Patten BM. Amyotrophic lateral sclerosis: abnormalities of the tongue on magnetic resonance imaging. Ann Neurol. 1989;25(5):468–72. 10.1002/ana.410250508. [DOI] [PubMed] [Google Scholar]
- 47.Morimoto N, Yamashita T, Sato K, Kurata T, Ikeda Y, Kusuhara T, Murata N, Abe K. Assessment of swallowing in motor neuron disease and Asidan/SCA36 patients with new methods. J Neurol Sci. 2013;324(1–2):149–55. 10.1016/j.jns.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 48.Kiernan JA, Hudson AJ. Changes in sizes of cortical and lower motor neurons in amyotrophic lateral sclerosis. Brain. 1991;114(Pt 2):843–53. [DOI] [PubMed] [Google Scholar]
- 49.Waito AA, Tabor-Gray LC, Steele CM, Plowman EK. Reduced pharyngeal constriction is associated with impaired swallowing efficiency in amyotrophic lateral sclerosis (ALS). Neurogastroenterol Motil. 2018;30(12):e13450. 10.1111/nmo.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paris G, Martinaud O, Petit A, Cuvelier A, Hannequin D, Roppeneck P, Verin E. Oropharyngeal dysphagia in amyotrophic lateral sclerosis alters quality of life. J Oral Rehabil. 2013;40(3):199–204. 10.1111/joor.12019. [DOI] [PubMed] [Google Scholar]
- 51.Lever TE, Gorsek A, Cox KT, O’Brien KF, Capra NF, Hough MS, Murashov AK. An animal model of oral dysphagia in amyotrophic lateral sclerosis. Dysphagia. 2009;24(2):180–95. 10.1007/s00455-008-9190-z. [DOI] [PubMed] [Google Scholar]
- 52.Smittkamp SE, Brown JW, Stanford JA. Time-course and characterization of orolingual motor deficits in B6SJL-Tg(SOD1-G93A)1Gur/J mice. Neuroscience. 2008;151(2):613–21. 10.1016/j.neuroscience.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH Jr, Constantine-Paton M, Bellingham MC. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci. 2008;28(43):10864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–49. 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 55.Ravits J, Appel S, Baloh RH, Barohn R, Brooks BR, Elman L, Floeter MK, Henderson C, Lomen-Hoerth C, Macklis JD, McCluskey L, Mitsumoto H, Przedborski S, Rothstein J, Trojanowski JQ, van den Berg LH, Ringel S. Deciphering amyotrophic lateral sclerosis: what phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph Later Scler Frontotemp Degener. 2013;14(Suppl 1):5–18. 10.3109/21678421.2013.778548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walhout R, Verstraete E, van den Heuvel MP, Veldink JH, van den Berg LH. Patterns of symptom development in patients with motor neuron disease. Amyotroph Later Scler Frontotemp Degener. 2018;19(1–2):21–8. 10.1080/21678421.2017.1386688. [DOI] [PubMed] [Google Scholar]
- 57.Christensen PB, Hojer-Pedersen E, Jensen NB. Survival of patients with amyotrophic lateral sclerosis in 2 Danish counties. Neurology. 1990;40(4):600–4. [DOI] [PubMed] [Google Scholar]
- 58.del Aguila MA, Longstreth WT Jr, McGuire V, Koepsell TD, van Belle G. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60(5):813–9. [DOI] [PubMed] [Google Scholar]
- 59.Magnus T, Beck M, Giess R, Puls I, Naumann M, Toyka KV. Disease progression in amyotrophic lateral sclerosis: predictors of survival. Muscle Nerve. 2002;25(5):709–14. 10.1002/mus.10090. [DOI] [PubMed] [Google Scholar]
- 60.Rosen AD. Amyotrophic lateral sclerosis. Clinical features and prognosis. Arch Neurol. 1978;35(10):638–42. [DOI] [PubMed] [Google Scholar]
- 61.Testa D, Lovati R, Ferrarini M, Salmoiraghi F, Filippini G. Survival of 793 patients with amyotrophic lateral sclerosis diagnosed over a 28-year period. Amyotroph Later Scler Other Motor Neuron Disord. 2004;5(4):208–12. [PubMed] [Google Scholar]
- 62.Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH. Amyotrophic lateral sclerosis. Nat Rev Dis Prim. 2017;3:17071. 10.1038/nrdp.2017.71. [DOI] [PubMed] [Google Scholar]
- 63.Corcia P, Pradat PF, Salachas F, Bruneteau G, Forestier N, Seilhean D, Hauw JJ, Meininger V. Causes of death in a post-mortem series of ALS patients. Amyotroph Later Scler. 2008;9(1):59–62. [DOI] [PubMed] [Google Scholar]
- 64.Sejvar JJ, Holman RC, Bresee JS, Kochanek KD, Schonberger LB. Amyotrophic lateral sclerosis mortality in the United States, 19792001. Neuroepidemiology. 2005;25(3):144–52. [DOI] [PubMed] [Google Scholar]
- 65.Spataro R, Re Lo M, Piccoli T, Piccoli F, La Bella V. Causes and place of death in Italian patients with amyotrophic lateral sclerosis. Acta Neurol Scand. 2010;122(3):217–23. [DOI] [PubMed] [Google Scholar]
- 66.Brooks BR, Sanjak M. Disease-modifying drug therapies. Amyotroph Later Scler Other Motor Neuron Disord. 2004;5(Suppl 1):68–75. 10.1080/17434470410019898. [DOI] [PubMed] [Google Scholar]
- 67.Lacomblez L, Bensimon G, Leigh PN, Guillet P, Powe L, Durrleman S, Delumeau JC, Meininger V. A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole study group-II. Neurology. 1996;47(6 Suppl 4):S242–50. [DOI] [PubMed] [Google Scholar]
- 68.Orsini M, Oliveira AB, Nascimento OJ, Reis CH, Leite MA, de Souza JA, Pupe C, de Souza OG, Bastos VH, de Freitas MR, Teixeira S, Bruno C, Davidovich E, Smidt B. Amyotrophic lateral sclerosis: new perpectives and update. Neurol Int. 2015;7(2):5885 10.4081/ni.2015.5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miller RG, Mitchell JD, Lyon M, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2007;1:CD001447. 10.1002/14651858.cd001447.pub2. [DOI] [PubMed] [Google Scholar]
- 70.Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;3:001447. 10.1002/14651858.cd001447.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Group Writing, Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16(7):505–12. 10.1016/S1474-4422(17)30115-1. [DOI] [PubMed] [Google Scholar]