

Summary
Humoral immunity depends on efficient activation of B-cells and their subsequent differentiation to antibody secreting cells (ASCs). The transcription factor NFκB cRel is critical for B-cell proliferation, but incorporating its known regulatory interactions into a mathematical model of the ASC differentiation circuit prevented ASC generation in simulations. Indeed, in experimental studies cRel was dynamically repressed during ASC differentiation, and ectopic cRel expression blocked ASC differentiation by inhibiting the transcription factor Blimp1. Conversely, after Blimp1 is induced in ASCs, it represses cRel by binding the Rel locus. Including this bi-stable circuit of mutual cRel-Blimp1 antagonism into a multi-scale model revealed that dynamic repression of cRel controls the switch from B-cell proliferation to ASC generation phases and hence the respective cell population dynamics. Our studies provide a mechanistic explanation of how dysregulation of this bi-stable circuit may result in pathologic B-cell population phenotypes and present avenues for diagnostic stratification and treatment.
Suggested revision:
Precise regulation of transcription factor NFkB mediates efficient activation of B-cells and their subsequent differentiation to antibody secreting cells (ASCs). To obtain a quantitative understanding of how specific NFκB dimers control ASC differentiation, we developed a mathematical model that investigated NFkB subunits cRel and RelA as distinct regulators. This model predicted that cRel inhibits ASC generation. Indeed, cRel was dynamically repressed during ASC differentiation, and ectopic cRel expression blocked ASC differentiation by inhibiting the transcription factor Blimp1. Conversely, Blimp1 inhibited cRel expression by binding the Rel locus. Including this bi-stable circuit of mutual cRel-Blimp1 antagonism into a multi-scale model revealed that dynamic repression of cRel controls the switch from B-cell proliferation to ASC generation phases and hence the respective cell population dynamics. Our studies provide a mechanistic explanation of how dysregulation of this bi-stable circuit may result in pathologic B-cell population phenotypes and present avenues for diagnostic stratification and treatment.
Keywords: Antibody Secreting Cells, B Cells, Proliferation, Differentiation, NFκB, Blimp1, mutual antagonism, Multi-scale Model
eTOC Blurb
Antibody production requires proper phasing of B cell proliferation and differentiation. Using an iterative systems biology approach Roy et al. reveal that while NFκB cRel enables proliferation, it must be downregulated during differentiation. Multi-scale modeling shows how coordinated cRel and RelA dynamics control B cell populations in health and disease.
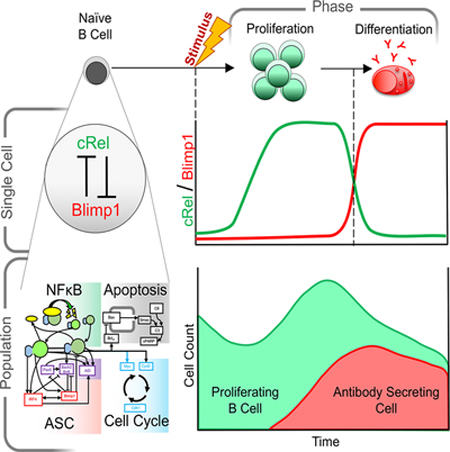
Introduction
The production of antibody is key for an effective immune response and efficacy of vaccination. Recognition of foreign antigen leads to profound changes within secondary lymphoid organs with the formation of the germinal center (GC) and extrafollicular foci that allow for the rapid expansion of antigen-specific B-cell clones to produce neutralizing antibody and memory B-cells. Indeed, T-cell independent (TI) and T-cell dependent (TD) stimulation of B cells generates rapidly proliferating cells known as activated B cells (ABCs). ABCs may differentiate into actively cycling short lived plasmablasts (PBs), which develop in the early phases of an immune response, and quiescent long-lived plasma cells (PCs), which reside in a specialized bone marrow niche. As both PBs and PCs are capable of producing antibody, they are referred to as antibody secreting cells (ASCs) (Shapiro-Shelef and Calame, 2005). The transition of ABCs to ASCs is coordinated by changes in signaling, gene expression and chromatin regulatory networks. ABC-specific transcription factors such as Pax5 and Bach2, and ASC-specific transcription factors such as Blimp1, regulate distinct genetic programs (Kallies et al., 2007; Nutt et al., 2015). Misregulation of these mutually inhibiting transcription factors, caused by common mutations, can result in B cell lymphomas with poor prognosis (Mandelbaum et al., 2010; Nutt et al., 2015; Xia et al., 2017). Transcription factor NFκB is also dysregulated in many B cell lymphomas (Shaffer et al., 2002b) and its inhibition is lethal to these transformed cells (Ceribelli et al., 2014; Staudt, 2010).
NFκB is a key inflammatory and immune transcription factor consisting of a dozen dimers made up from three activation domain-containing proteins (cRel, RelA, RelB) and two dimerization partners (p50, p52) (Hoffmann and Baltimore 2006). In ABCs the NFκB dimers RelA:p50 and cRel:p50 are induced (Kaileh and Sen, 2012). While cRel activity is required for cell survival, growth and division during B cell activation (Pohl et al., 2002; Shokhirev et al., 2015), RelA is required for the generation of GC-derived PCs by contributing to Blimp1 activation (Heise et al., 2014). Thus, both cRel and RelA are indispensable for humoral immunity but for different functional reasons. However, a recent study showed that in the genetic disease B cell expansion with NFκB and T Cell Anergy (BENTA), constitutively active NFκB results in reduced ASC generation (Arjunaraja et al., 2017), suggesting that precise regulation of each NFκB dimer is required for healthy ASC generation.
Mathematical modeling approaches have proven valuable to understand complex dynamic molecular regulatory networks. ABC population expansion dynamics are well accounted for by a multi-scale model of the intracellular molecular network of NFκB regulating apoptosis and the cell cycle (Mitchell et al., 2018; Shokhirev et al., 2015), and this model proved useful in understanding the function of cRel in cell survival, growth and division (Shokhirev et al., 2015). In the case of the ASC differentiation circuit, the scarcity of quantitative biochemical data first prompted logical models that can qualitatively recapitulate the state of regulatory networks in the terminal fates of B cells (Mendez and Mendoza, 2016), or a dynamical system of only three regulators (Martinez et al., 2012). Larger dynamical models are capable of explaining the distribution of time-spans B cells may spend undergoing somatic hypermutation (SHM) prior to terminal differentiation, but either do not include NFκB, cRel or RelA (Sciammas et al., 2011), or consider NFκB as a single regulator without distinct functions of cRel and RelA (Martinez et al., 2012; Mendez and Mendoza, 2016).
To better understand the role of NFκB within the ASC differentiation circuit we extended the work of Sciammas et al. by developing a mathematical model of ASC differentiation in which cRel and RelA are investigated as distinct regulators. Mathematical modeling, combined with mechanistic and functional experimental studies, revealed that cRel must be repressed for ASC differentiation. We showed that decreased cRel expression is mediated by Blimp1 through epigenetic remodeling of the Rel locus. This insight allowed us to integrate the extended ASC differentiation circuit model into our previously established multi-scale model to account for the cell population dynamics of ABCs and ASCs in terms of the dynamics of intra-cellular molecular signaling events.
Results
Mathematical modeling predicts that cRel inhibits ASC differentiation
The previously established ASC differentiation circuit model (Sciammas et al., 2011) includes regulatory interactions between transcription factor IRF4, Pax5, Blimp1, Bach2, and Bcl6 (Figure 1A and 1B): To explore the roles of NFκB dimers in ASC differentiation we incorporated RelA and cRel and their known dimer-specific regulatory interactions into the ordinary differential equation (ODE) model of Sciammas et al.: cRel is known to induce IRF4 (Grumont and Gerondakis, 2000), Bach2 (Hunter et al., 2016) and AID activity (Kim and Tian, 2009; Park et al., 2009); RelA induces IRF4 (Saito et al., 2007), Blimp1 (Heise et al., 2014; Morgan et al., 2009) and AID (Park et al., 2009) (Figure 1B, Table S1). The resulting “NFκB-extended ASC model v1” contains 7 molecular species controlled by 14 regulatory interactions (Figure 1B, Table S2 and Data S1).
Figure 1: Mathematical modeling predicts that cRel prevents ASC differentiation.

A) Schematic of ASC generation. Stimulation of naïve B cells produces activated B cells (ABCs). ABCs either become plasma blasts or undergo class switch recombination (CSR) and somatic hypermutation (SHM) before differentiating into plasma cells. Here, plasma blasts and plasma cells are defined as antibody secreting cells (ASCs). The regulators and genetic signature that characterize each cell type are indicated. B) Schematic of NFκB-extended ASC differentiation model v1. This model was based on a previously published model from Sciammas et al. (Sciammas et al., 2011) to which RelA and cRel were added (see text). Rela- and cRel-mediated regulation is indicated in green, genes associated with the CSR/SHM state are shown in violet, and genes associated with the ASC state are shown in red. Arrows denote activation, and barred lines denote repression. C) The trajectories of Blimp1 and AID expression kinetics in the subset of cells that expressed high Blimp1 from a simulation of 125 cells. Four states are defined by two coordinate axes, namely Blimp1lowAIDlow (Initiation); Blimp1lowAIDhigh (CSR/ SHM); Blimp1highAIDhi; Blimp1highAIDlow (ASC). The final states are in the rectangular boxes below and on right indicating Blimp1 and AID expression respectively. Green points show the trajectories and final states of the NFκB-extended ASC differentiation model v1. Blue points indicate the final state obtained by all cells in simulations using a previously published model (Sciammas et al., 2011) D) Kinetics of ASC differentiation predicted by the Sciammas et al. ASC differentiation model and the NFκB-extended ASC differentiation model v1. The percentage of ASCs is plotted with time (h). The solid line indicates the mean and shaded region indicates the standard deviation of 3 simulations of 125 cells. E) The final states (96h) reached by cells expressing high Blimp1 in simulations of 125 starting cells with the NFκB-extended ASC differentiation model v1 (phase 1, green) and a subsequent simulation in which cRel activity in those cells was reduced to 1% and trajectories were simulated for 96h (phase 2, red). F) Bar graph of the fraction of ABCs becoming ASCs when cRel was allowed to remain at 100% of its expression (phase 1, green) or when it was forcibly reduced to 1% of its peak (phase 2, red). Median (black), standard deviation (dark green) and standard error mean (light green) are indicated.
Time-course simulations showed that ASC differentiation trajectories passed through an obligate transient state of high AID followed by increased Blimp1 and subsequent AID repression to become ASCs (Figure 1C). To interpret model simulations we defined an ASC as a cell that is Blimp1hi and AIDlo (Blimp1>4, consistent with Sciammas et al. and AID <4, (Figure S1A)). However, simulations using the NFκB-extended ASC model v1 failed to produce ASCs in expected numbers (Figure 1C and 1D). AID and Blimp1 upregulation were consistent with published results, but increased Blimp1 expression was not followed by the obligate AID decrease (Figure 1C and 1D). At face value, this simulation result suggested that either RelA or cRel acts as an inhibitor of ASC differentiation. As RelA is required for ASC generation (Heise et al., 2014), we wondered whether decreased cRel expression would allow ABCs that had failed to repress AID to complete their differentiation and become ASCs (Figure 1E). We simulated the effect of decreasing cRel expression (100, 10 or 5 fold) in cells that had elevated Blimp1; the model predicted that decreased cRel expression allows for substantial downregulation of AID (Figure 1E, S1B and S1C), enabling the differentiation of ABCs to ASCs (Figure 1F). This effect was independent of the thresholds chosen to define an ASC (Figure S1A). Thus, math modeling suggested (i) that cRel may be repressed during ASC differentiation, and (ii) that this regulatory feature may be necessary for ASC generation.
Transcriptional repression of Rel in ASCs
To test the first model prediction, ABCs and ASCs were generated ex vivo by stimulating mature B cells with the TI stimulus lipopolysaccharide (LPS) (Figure S2A) (Kallies et al., 2007). The resulting ASCs showed increased expression of Blimp1 and IRF4, and decreased expression of Bach2 compared to ABCs (Figure 2A), confirming that LPS-generated ASCs (defined here as LPS-PBs) can recapitulate key regulatory events of in-vivo ASCs and characteristic of PBs (Minnich et al., 2016; Nutt et al., 2015; Shapiro-Shelef and Calame, 2005; Shi et al., 2015; Tarlinton et al., 2008). We examined cRel expression by immunoblotting and found that while both cRel and RelA expression increased during B cell activation, cRel expression decreased ∼3.5 fold in LPS-PBs, whereas RelA remained high (Figure 2A and 2B). The same results were observed when PBs were generated with the TD stimulus anti-CD40/IL-4 (defined as CD40-PB) (Figure S2B, S2C and S2D) (Kallies et al., 2007).
Figure 2: cRel repression is required for ASC differentiation.

A) Immunoblots of Blimp1, IRF4, Bach2, cRel (product of Rel gene), RelA and actin in naïve B cells, ABCs and LPS-PBs. ABCs and LPS-PBs were generated by stimulating naïve B cells with LPS for 72 h and then isolated by flow cytometry (described in Figure S2A). Data shown is representative of three biological replicates. B) Quantitative measurements of cRel and Rela immunoblots with actin as loading control. C) Quantitative measurements of mRNA expression by qPCR of Rel (encodes cRel protein) and Rela in naïve B cells, ABCs and LPS-PBs. D) Quantitative measurements of mRNA expression of Rel and Rela in naïve B cells, GC B cells and LN-ASC (Lymph node ASCs). Ubiquitin C was used as housekeeping gene to quantitate mRNA expression. E) and F) Quantitative measurements of mRNA expression of Bach2 and Blimp1 in unstimulated (Control, blank) and stimulated (IL-2+5 24 h, shaded) BCL1 (gray) and cRel-overexpressing BCL1 cells (blue, BCL1-cRel). IL-2 and IL-5 (shaded) stimulation is used throughout to differentiate BCL1 cells to ASC-like states. G) IgM production measured at 96 h by ELISA and expressed in optical density (O.D.) units. H) Quantitative measurement of mRNA expression of Blimp1 in unstimulated (control, blank) and stimulated (IL-2+5 24 h, shaded) BCL1 (gray), BCL1-cRel (blue), and Bach2-KO BCL1-cRel (red) cells. I) IgM production measured at 96 h by ELISA and expressed in optical density (O.D.) units. In all plots the mean and standard deviation of three replicates is indicated. *p < 0.05, **p< 0.01, ***p < 0.001 and not significant (ns) (Unpaired Students t-test).
Analyzing deposited RNA-seq data (Shi et al., 2015), we found that Rel mRNA was lower in splenic PBs compared to GC B cells and still lower in splenic PCs and bone marrow PCs (Figure S2E). By measuring mRNA with quantitative RT-PCR, we found that Rel expression was decreased ~3 fold in LPS-PBs whereas Rela expression remained similar (Figure 2C), and that Blimp1 and IRF4 expression were increased whereas Bach2 expression was decreased (Figure S2H). This indicated that cRel repression occurred at the transcriptional level. To validate these observations in vivo, ASCs and germinal center (GC) B cells were generated by immunization with TD antigen (NP-OVA). Lymph node-derived ASCs (LN-ASCs) and GC B cells were purified by flow cytometry sorting (Figure S2F and S2G). We found that Rel transcripts were decreased ∼20-fold in LN-ASCs compared to GC B cells whereas Rela remained similar (Figure 2D), and that Blimp1 and IRF4 were increased, whereas Bach2 was decreased (Figure S2I). We also found that cRel expression is decreased in intestinal ASCs (I-ASCs) and these cells show elevated Blimp1 compared to other B cells (Figure S2J and S2K). Taken together these data suggest that decreased cRel expression may be a general characteristic of PBs and PCs irrespective of the nature of stimulation and tissue environment.
Repression of Rel is required for ASC differentiation
To test the second model prediction that Rel repression is functionally required for ASC differentiation, we used the BCL1 cell line, which is capable of stimulus-responsive differentiation into ASCs, as evident by gene expression signature (Sciammas and Davis, 2004) and antibody production (Lin et al., 1997; Sciammas and Davis, 2004). We found that BCL1 cells stimulated with IL-2/IL-5 recapitulated the Rel repression (Figure S2L and S2M) observed in primary B cells (Figure 2 and S3). We ectopically expressed cRel in BCL1 cells (BCL1-cRel) which elevated Bach2 and diminished Blimp1 proteins (Figure S2M), and increased the proportion of cells in cell cycle (Figure S2N). Similarly, Bach2 transcript was increased (~ 2.5 fold) and Blimp1 transcript diminished (~ 1.6 fold) (Figure 2E and 2F) indicating transcriptional control. However, ectopic expression of cRel also diminished the increase in Blimp1 following stimulation (∼1.7 fold as opposed to ∼3 fold in controls) (Figure 2F). We also found that BCL1-cRel cells failed to produce IgM upon stimulation (Figure 2G) and did not show a substantial reduction in the proportion of actively cycling cells (~32% compared to ~35% in unstimulated) (Figure S2N). Thus, ectopic expression of cRel appears to block terminal differentiation of B cells.
It has been shown that Bach2−/− B cells have higher Blimp1 and a higher propensity to differentiate into ASCs (Muto et al., 2010). We wondered whether the failure to increase Blimp1 expression in BCL1-cRel cells is mediated by Bach2. We deleted Bach2 in BCL1-cRel cells (Figure S2O) using CRISPR/Cas9, which reverted Blimp1 expression to the same level as in control BCL1 cells (Figure 2H) and allowed for differentiation to ASCs as evident by a rescue of IgM production (Figure 2I). We concluded that for ASC differentiation to proceed, Rel must be repressed, so that expression of Bach2 can decay, in order to de-repress Blimp1.
Blimp1-mediates transcriptional repression of cRel
Our computational and experimental studies indicated that decreased cRel expression is required for ASC production, we wondered whether Blimp1 may transcriptionally repress Rel. In “NFκB-extended ASC model v2” (Figure 3A) we tested whether an inhibitory connection between Blimp1 and cRel production was sufficient to account for ASC generation quantitatively. Indeed, the model recapitulated experimental flow cytometry results (Figure 3B and 3C) within a broad range of Blimp1 and AID thresholds used to define ASCs (Figure S3). Thus, the mathematical model predicted that Blimp1-mediated repression of Rel may be sufficient to allow ASC differentiation.
Figure 3: Including Blimp1 inhibition of cRel expression in computational simulations enables ASC differentiation.

A) Schematic of NFκB-extended ASC differentiation model v2, in which cRel downregulation was initiated by Blimp1 (grey barred line). B) Percentage ASC production over time (h) in simulations with the model v1 (without cRel-Blimp1 inhibition, green, Figure 1B) and v2 (with cRel-Blimp1 inhibition, grey, panel A). The solid line indicates the mean and shaded region indicates the standard deviation of 3 simulations, each containing 125 founder cells. C) Experimental measures of the ASC differentiation kinetics determined by flow cytometry at 24 h, 48 h, 72 h and 96 h, with the mean and standard deviation of three replicates indicated.
To experimentally test whether Blimp1 triggers repression of Rel, we deleted Blimp1 in BCL1 cells (Figure S4A) and measured Rel transcripts upon stimulation. Whereas control BCL1 cells showed a ~4-fold decrease in Rel expression in response to stimulation with IL-2/IL-5, Blimp1-deleted BCL1 cells failed to repress Rel upon stimulation (Figure 4A), while Rela expression remained largely unchanged (Figure 4B). Blimp1-deleted BCL1 cells were unable to proceed with ASC differentiation, as evident by the lack of IgM production (Figure 4C). Further, analyzing RNA-seq data of primary B-cells (Tellier et al., 2016), we found that Blimp1−/− PCs showed increased expression of Rel (Figure S4B). This indicated that Blimp1 represses Rel expression as part of its program to coordinate ASC differentiation.
Figure 4: Blimp1 represses Rel expression and diminishes H3K9ac at the Rel locus.

A) and B) Quantitative mRNA expression by qPCR of Rel and Rela in unstimulated (control, blank) and stimulated (IL-2+5 24h, shaded) BCL1 (gray) and Blimp1−/− BCL1 (red) cells. C) IgM production measured at 96 h by ELISA and expressed in optical density (O.D.) units. D) Analysis of Blimp1 ChIP-seq data in plasmablasts (GSM1843340, Tellier et al., 2016). Blimp1 binding track (top); schematic of Rel locus (middle), consensus Blimp1 binding motif indicated in red and targeted PAM sequence is underlined (bottom). E) and F) Quantitative mRNA expression of Rel and Rela in unstimulated (control, blank) and stimulated (IL-2+5 24 h, shaded) BCL1 (gray) and RelBS-mut BCL1 cells (BCL1 cells mutated at the Blimp1 binding site in the Rel locus, blue). G) IgM production was measured at 96 h by ELISA and expressed in optical density (O.D.) units. H) and I) ChIP-qPCR yield for H3K9ac and IgG (isotype control) at the Rel locus (H) and Rela locus (I) in unstimulated (control, blank) and stimulated (IL-2+5 24h, shaded) BCL1 (gray), Blimp1−/− BCL1 (red) and RelBS-mut BCL1 cells (blue). The yield was calculated with respect to Input. The mean and standard deviation of three replicated is shown throughout. *p < 0.05, **p < 0.01, ***p < 0.001 and not significant (ns) (Unpaired Students t-test).
To establish whether Blimp1-mediated transcriptional repression of Rel is direct, we examined recently deposited Blimp1 ChIP-seq data from PBs (Minnich et al., 2016), and found a prominent Blimp1 binding peak in the Rel locus at position ∼23669376–23670485 in chromosome 11 using genome assembly mouse mm9 (Figure 4D). While BCL1 cells showed a ~4-fold transcriptional repression of Rel in response to stimuli, CRISPR/Cas9 mediated disruption of a Blimp1 binding site in the Rel locus (Figure S4C) substantially reduced this repression (Figure 4E). Rela expression remained unchanged in either cell line (Figure 4F). As expected, disruption of Blimp1 binding sites in the Rel locus rendered BCL1 cells unable to fully proceed with ASC differentiation as evident by reduced IgM production (Figure 4G). Thus, we concluded that Blimp1 directly represses Rel expression in ASCs.
Next, we sought to characterize the mechanism by which Blimp1 represses Rel transcription. Blimp1 is known to interact with histone deacetylases (e.g. HDAC2) and was shown to repress transcription by recruiting HDAC to a target promoter (Yu et al., 2000), reducing H3K9 acetylation (H3K9ac) abundance (Minnich et al., 2016). We measured H3K9ac abundance on the Rel locus in ABCs and LPS-PBs by ChIP-qPCR, and found a ∼ 1.4 ± 0.108% yield in ABCs as opposed to ∼ 0.4 ± 0.026% yield (relative to input) in LPS-PBs, whereas antibody isotype controls were low (∼ 0.05%) and unchanged (Figure S4D). This reduction in H3K9ac by more than 3-fold on the Rel locus as ABCs differentiated into LPS-PBs contrasted with unchanged H3K9ac at ∼1.9 ± 0.1% on the Rela locus (Figure S4E). Similarly, analyzing ChIP-seq data (Minnich et al., 2016), showed reduced H3K9ac at the Rel locus (Figure S4F). Upon stimulation of BCL1 cells with IL-2/IL-5 we found a ~2.6-fold decrease in H3K9ac on the Rel locus in control cells (from ∼1.7 ± 0.19% to 0.64 ± 0.1%), but no significant change in Blimp1−/− cells (from ∼ 1.84 ± 0.19% to 1.8 ± 0.2%) (Figure 4H). Similarly, mutation of the Blimp1 binding site substantially diminished the loss of H3K9ac abundance in response to stimulus (∼1.45 ± 0.11%) at the Rel locus compared to controls (0.64 ± 0.1%) (Figure 4H), but had no impact at the Rela locus (Figure 4I). Thus, we concluded that the differentiation-associated reduction of H3K9ac abundance at the Rel locus is dependent on direct binding of Blimp1 and leads to transcriptional repression of Rel in ASCs.
cRel deficiency increases the ASC differentiation propensity but results in a smaller ASC population
To further test the regulatory mechanisms encapsulated in the computational model we simulated ASC generation in the absence of cRel and found that the model predicted an increase in ASC numbers at time points after 48 hrs (Figure 5A). This prediction was independent of the threshold of Blimp1 and AID chosen to define a cell as an ASC (Figure S5A), and consistent with a previous report that Rel−/− B cells produce a higher percentage of ASCs after 3 days (Heise et al., 2014). In our experiments, we found that while Rel−/− B cells had a substantial proliferative defect and thus generated fewer cells overall compared to WT (Figure 5B), the culture contained −1%, −2%, +20% and +15% greater proportion of LPS-PBs than WT controls at 24 h, 48 h, 72 h and 96 h respectively (Figure 5C, and S5B). By measuring the cell loss associated with cRel-deficiency we found that the increased loss in the ABC population from 48 to 72 h (88 to 97%) coincided with a decreased loss of LPS-PBs from 48 to 72 h (91 to 86%) (Figure S5C). We also measured cell death propensities with Annexin V staining and found that ABCs and LPS-PBs generated from Rel−/− B cells showed a similar ~2-fold increase in cell death compared to wild type (Figure S5D and S5E). These data suggested that the increased proportion of LPS-PBs vs. ABCs in Rel−/− B cells is not due to differential death rates but rather due to an increased propensity to differentiate from ABCs into ASCs. These results showed a quantitative agreement with computational predictions (Figure 5A), except for the last timepoint, which may reflect a limitation of the in vitro culture conditions.
Figure 5: cRel-deficiency enhances ASC differentiation.

A) Time-course plot of the percentage (%) increase of ASCs in cRel-deficient (Rel−/−) B cells compared to wild type (WT) controls as predicted by mathematical modeling using the NFκB-extended ASC differentiation model v2. The percentage of LPS-PBs is plotted with time (h). The solid line indicates the mean and shaded region indicates the standard deviation of 3 replicates with 125 initial cells. B) Stacked area plot indicates experimentally determined absolute cell numbers at 0 h, 24 h, 48 h, 72 h and 96 h upon stimulation of WT (left) and Rel−/− (right) B cells with LPS. Gray plot indicates total cell number (ABCs and LPS-PBs) and the red area indicates the number of LPS-PBs. C) Bar graph showing the percentage increase of LPS-PBs in Rel−/− B cells over WT measured by flow cytometry from WT and Rel−/− B cells at 24 h, 48 h, 72 h and 96 h. The LPS-PBs percentage is determined by B220loCD138+ (described in Figure S5B). The model prediction (panel A) is reproduced as the shaded grey region behind the bar graph. D) and E) Blimp1, Bach2 and IRF4 fold change in Rel−/− B cells compared to WT B cells over time, as predicted by mathematical modeling (left) and determined experimentally by qPCR (right). The solid line indicates mean and shaded region indicates SD of fold change from three replicates (Y axis) over time (X-axis). No change over WT (Fold change = 1) is indicated with a red dashed line. The mean and standard deviation of three replicated is shown throughout. F) Representative immunoblots of Blimp1, Bach2, IRF4 and actin expression in Rel−/− and WT B cells. Immunoblots were performed at 0 h, 24 h, 36 h, 48 h, 60 h and 72 h following stimulation with LPS. G) Flow Cytometry plot of ASCs (CD138high) and ABCs (CD138-/low) at each generation (Cell Trace Red) during LPS-PB production in WT and Rel−/− B cell at 48 h and 72 h. Cell division/generation number is indicated at the top of each plot. On representative of three replicates is shown. H) Percentage of LPS-PBs at each generation in WT (red) and Rel−/− (black) B cells at 72 h. X-axis indicates generation number and Y-axis indicates LPS-PB percentage. The mean and standard deviation of three replicated is shown throughout.
To further test the regulatory connections in the model, we generated quantitative predictions of the dynamic changes of three critical regulators (Blimp1, Bach2 and IRF4) following stimulation of Rel−/− vs. WT B cells. In the mutant, we predicted a 2-fold late-phase increase of Blimp1, a 2–3 fold reduction in Bach2, and an initial decrease in IRF4 followed by a late-phase increase (Figure 5D). Experimental quantification by RT-PCR showed a quantitative match to the computational prediction with Rel−/− B cells having 2-fold higher expression of Blimp1 in the late-phase, 2–3 fold reduced expression of Bach2, and similarly dynamic regulation of IRF4 (Figure 5E and S5F). Immunoblots showed similar trends as observed by RT-PCR (Figure 5F). Thus, Rel−/− B cells had a higher propensity to differentiate into ASCs through increased expression of Blimp1.
As ASC generation propensity has been shown to increase with cell division (Hasbold et al., 2004) we wondered whether Rel−/− B cells have an increased ASC differentiation propensity in a generation-specific manner. Cell division was measured by dye dilution assay using Cell Trace Red. While both WT and Rel−/− B cells showed increasing ASC generation with each division, Rel−/− B cells showed a higher proportion of non-dividing (generation ‘0’) cells and an increased proportion of LPS-PBs at every generation (Figure 5G and 5H). Thus, Rel−/− B cells showed increased ASC differentiation propensities at all generations compared to WT B cells.
Integrating ASC differentiation into a multi-scale B-cell proliferation model reveals how cRel dynamics control the dynamics of distinct B-cell populations
The link between differentiation and division revealed in the WT and Rel−/− B cell experiments motivated us to integrate the “NFκB-extended ASC model v2” into an established multiscale model that accounts for B-cell population dynamics as a result of the molecular network in each cell (Mitchell et al., 2018; Shokhirev et al., 2015). By doing this we could discover whether our mechanistic understanding was sufficient to explain the effects we observed on differentiation, proliferation and the link between the two. This multiscale model includes an NFκB module that predicts NFκB dimer-specific dynamics as a result of the stimulus-induced regulation of multiple inhibitors of NFκB (IκBs). We added NFκB’s control over ASC differentiation, as described here in “NFκB-extended ASC model v2”, to existing NFκB-mediated control over the cell cycle and apoptosis (Figure 6A). Each cell within this multi-scale model was thus capable of undergoing apoptosis, cell-division or differentiation into an ASC (Figure 6B), with cell fate decisions being dependent on the particular parameter set sampled from previously determined distributions (see Materials & Methods (Mitchell et al., 2018)).
Figure 6: Dynamic cRel repression is initiated by Blimp1 in single cells.

A) Schematic of multi-scale modeling that determines B cell population fate dynamics with single-cell resolution. The multi-scale model is composed of the NFκB regulatory network (green box), the apoptosis gene regulatory network (grey box), the cell cycle gene regulatory network (blue box) as published previously (Mitchell et al., 2018; Shokhirev et al., 2015), and the ASC differentiation network (violet and red box) added here. B) Line plots indicate dynamics of NFκB, cPARP, Cdh1 and Blimp1 in 3 representative cells. Thresholds that trigger fate changes are indicated with red dashed lines. Exceeding the threshold of cPARP triggers apoptosis (cell 1), Cdh1 trigger mitosis (cell 2 and 3) and Blimp1 triggers differentiation into an ASC (cell 3). Thresholds must be crossed in the positive direction. Cell 1 and 2 are founder cells (generation 0) while cell 3 has undergone 4 divisions (generation 4). C) Line plots of average cRel activity (nuclear cRel:p50 concentration) within 200 simulated lineages. Each line represents the average concentration of progeny from a single founder cell. ASC generating lineages are indicated as ABCs (blue) transition to Blimp1highAIDhigh (pink) and ASCs (red). X-axis indicates time (h) and Y-axis indicates simulated nuclear cRel-p50 heterodimer in nano molar (nM). D) Stacked bar graph of the number of ASCs that will not divide (black) and those undergoing at least one division after differentiating (blue). Bars are shown for ASCs generated before and after 70 h. E) Blimp1 and cRel expression in a population of 1000 single cells simulated with the multi-scale model (panel A) at 0 h, 24 h, 48 h, 72 h and 96 h. Populations were gated into cRel–Blimp1low (black), cRelhighBlimp1low (blue), cRelhighBlimp1+ (violet) and cRellowBlimp1+ (red). Cell numbers are indicated in each quadrant. Time points at 15-minute time intervals are shown in supplemental movie 1. F) Stacked bar graph of the percentage of cells in each quadrant in the simulation (panel E) at the indicated time points. G) Blimp1 and cRel expression measured by flowcytometry with single cell resolution at 0 h, 24 h, 48 h, 72 h and 96 h. Populations were gated into cRel-Blimp1low (black), cRelhighBlimp1low (blue), cRelhighBlimp1+ (violet) and cRellowBlimp1+ (red). The percentage of cells in each gate are represent by number in each plot with the respective color code. H) Stacked bar graph of the percentage of cells in each gate at the indicated time points (panel G). The mean and standard deviation of three replicates are indicated.
A simulation was performed of 200 seeded cells stimulated with LPS. Dynamic changes of cRel activation were measured over time as cells within each lineage transition from ABCs (Blimp1low) to Blimp1highAIDhigh and subsequently to ASCs (Blimp1highAIDlow). As expected, the model showed almost immediate cRel activation and resulting proliferation. Only cells that had increased Blimp1 expression showed decreased cRel activity with further decreases at later time points as they differentiated into ASCs (Figure 6C), while RelA activity remained similar in ABCs and ASCs (Figure S6A). Based on parameter distributions, cells showed heterogeneity in cRel activation dynamics and the timing and generation/division number of ASC differentiation (Figure 6C). The model predicted that ASCs generated at early timepoints produced a higher percentage of proliferating cells compared to those generated at a later time point (Figure 6D and S6B).
To examine the time evolution of molecular markers, and generate an experimentally-testable prediction of the multi-scale modeling of signaling, proliferation, apoptosis, and differentiation, we tracked cRel and Blimp1 concentrations over time in a population of individually simulated cells (Figure 6E, supplemental movie 1). The resulting plot of the cell-population’s path through the space of cRel and Blimp1 expression revealed a distinctive trajectory that allowed us to quantify specific populations of B-cells based on their cRel and Blimp1 expression (Figure 6F). To test this prediction we measured Blimp1 and cRel abundance with single cell resolution through a flow cytometry time course and found that B cells first increased cRel expression at 24 hours, with ~ 70% of cells considered cRel high at 24 and 48 hours (Figure 6G and 6H). Increased cRel expression was followed by increased Blimp1 expression with the percentage of Blimp1high cells increasing from 20% at 24 hours to 55% at 48 hours and ~80% at 72 and 96 hours. As predicted by the model, B cells that showed increased Blimp1 subsequently reduced cRel expression, with the percentage of cells with both high Blimp1 and high cRel decreasing from 41% at 48 hrs to 29% at 72 hrs and 22% at 96 hrs, along with an increase in the population of cells with high Blimp1 and low cRel from 15% at 48 hrs to 53% at 72 and 96 hrs. This confirmed the computational prediction that proliferation and differentiation dynamics of stimulated B cells are accompanied by first high then decreased cRel expression following increased expression of Blimp1 at 72 and 96 hrs (Figure 6G and 6H).
Multi-scale modeling relates genetic perturbations to physiological or pathological B cell population dynamics
To further investigate the predictive power of the multi-scale model, we examined its ability to recapitulate population dynamics of WT and Rel−/− B cells. In the WT condition (Figure 7A left panel), the model simulations showed a characteristic transient dip in the total population due to death of a proportion of founder cells, before proliferation results in a total population expansion that then gives way to a population decline. During the proliferative burst, some cells differentiated into ASCs resulting in ASC population dynamics that are distinct but necessarily coordinated with the ABC population dynamics. In the Rel−/− simulation (Figure 7A right panel), the total population expansion was dramatically diminished (in agreement with experimental literature, (Kontgen et al., 1995; Shokhirev et al., 2015)). While the ASC population was also diminished, their proportion of the total was substantially higher, reflecting the enhanced propensity to differentiate (Figure 7A and 5B). By analyzing the simulation data not as a function of time but as a function of generation at a single timepoint (72 h), we found that the model predicted that cRel-deficiency results in an overall increase in ASC differentiation, which in fact peaks at an earlier generation than in WT cells (Figure 7B left panel). The experimental data (Figure 5G) confirmed this prediction (Figure 7B right panel), though peak ASC proportions appeared one generation later in both genotypes. In sum, the multi-scale model appeared to recapitulate key aspects of ABC and ASC population dynamics, and the intricate roles of cRel in shaping them.
Figure 7: Co-ordinated cRel and RelA dynamics control the dynamics of B cell populations.

A) Area plots from multiscale modelling simulations of 125 WT and Rel−/− cells showing total cell number (grey), and ASC populations (Blimp1highAIDlow - red). Each subsequent generation of proliferating cells is indicated with a lighter grey. B) Line graphs showing generation-specific cell counts of ABCs (black) and ASCs (red) in WT (solid line) and Rel−/− (dashed line) simulations (left) and experiment (right, Figure 5G). Cell counts are normalized to the maximum cell count of ABCs for each genotype. The shaded region shows the standard deviation of three experimental replicates. C) Bar graph of the fold change of cRel expression over WT in wild-type (WT, black), Blimp1+/− (maroon) and Blimp1−/− (red) B-cells in simulations and experiment (details of experimental data shown in Figure S7). D) Area plots from multiscale modelling simulations of 125 cRel High (left), RelBs-mut (middle) and Rela high (right) cells showing total cell number (grey), and ASC populations (Blimp1highAIDlow - red). Each subsequent generation of proliferating cells is indicated with a lighter grey. E) Illustration of cRel and RelA dynamics during the physiological B cell response and their characteristic abundances in B cell malignancies. Upper panel: during B cell differentiation naïve B cell produces ABCs, followed by GC B cells/ Extra GC B cells before differentiating into PCs. Middle panel: color graph to represent expression of cRel (green) and RelA (gray) during the stages of B cell differentiation. Color gradient represents relative expression with darker colors indicating higher expression. Lower panel: cRel and RelA expression characteristic of indicated B cell malignancies. Their putative cell of origin is indicated by aligning with the upper panel.
The concordance between model simulations and experimental observations encouraged us to use the multi-scale model as a research tool to explore the consequence of NFκB dysregulation on B cell population dynamics. Simulations of Blimp1 deficiency resulted in increased Rel expression (Figure 7C) and elevated B-cell proliferation (Figure S7) in line with experimental observation (Calado et al., 2010). Simulation of elevated cRel expression resulted in a dramatic increase in the expansion of the ABC population, and a reduction and delay in the generation of ASCs, not only proportionally but also in absolute numbers (Figure 7D left panel), reflecting experimental observations (Figure 2G). Next, we simulated the effects of disrupting the Blimp1 binding sites in the Rel locus, which did not substantially alter total ABC cell population dynamics but did result in a reduction in the generation of ASCs (Figure 7D, middle panel). This was consistent with the reduced antibody production observed experimentally (Figure 4G). Finally, we explored the effect of elevated RelA expression, which led to increased total cell population expansion, comparable to elevated cRel simulations. But, in contrast to the elevated cRel condition, elevated RelA produced predominantly ASCs (Figure 7D, right panel).
Similar misregulation of the NFκB system is associated with different B-cell lymphomas originating from distinct phases of the GC reaction (Figure 7E). Germinal Center B-cell like diffuse large cell lymphomas (GCB DLBCL) show high cRel cells, and like the cRel high simulations, GCB DLBCL cells appear trapped at the stage unable to exit the GC (Rosenwald et al., 2002; Shaffer et al., 2002b). Activated B-cell like diffuse large cell lymphomas (ABC-DLBCL) show high RelA and moderately high cRel activity with frequent Blimp1 inactivation. As seen in the Blimp1 deficiency simulation, such cells are trapped at the pre-plasmablast stage i.e. able to initiate but unable to complete plasmacytic differentiation (Davis et al., 2001). In contrast, malignant plasma cell disorder multiple myeloma (MM), is characterized by high RelA, and reflecting the highly differentiated state of cells in RelA high simulations, MM cells originate from fully differentiated plasma cells. The fact that our simulations of conditions of NFκB misregulation produced analogous in silico phenotypes of population dynamics (Figure 7C) suggests that the multi-scale model may be a useful research tool for relating genetic or molecular perturbations to the balance of proliferation and differentiation in health and disease.
Discussion
In this work, we developed a quantitative understanding of how NFκB dimers control ASC differentiation; by iterating experimental and computational modeling work we formulated an experimentally-trained kinetic model of B cell proliferation and differentiation which recapitulated experimentally observed phenotypes. While cRel activation is critical for B-cell activation, survival and ABC population expansion (Pohl et al., 2002; Shokhirev et al., 2015), we found that its repression enabled differentiation of ABCs into ASCs. Ectopic expression of cRel in B cells prevented Blimp1 expression and consequently impaired ASC differentiation. Conversely, Rel−/− B cells showed increased Blimp1 expression and increased propensity for LPS-PB differentiation, but the LPS-PB population remained diminished suggesting that the reduced antigen-specific antibody production (Grumont and Gerondakis, 2000; Heise et al., 2014; Pohl et al., 2002) associated with cRel-deficiency is not due to a defect in ASC generation but rather an indirect consequence of defective B cell expansion. Thus, cRel dynamics are a key determinant of ABC and ASC population dynamics.
Further, we established that Rel repression in ASCs is mediated by Blimp1. Blimp1 is known to repress cell growth and proliferation regulators such as Myc and Cyclin E (Lin et al., 2000; Lin et al., 1997; Shaffer et al., 2002a); Rel repression may reinforce this regulation as Myc is a cRel target gene (Duyao et al., 1990), which was shown to determine the proliferative capacity of ABCs (Heinzel and Binh Giang, 2017). A key characteristic of plasma cells is the slowing and cessation of the cell cycle and ectopic expression of the cell cycle-promoting regulator (Cyclin E) inhibits ASC differentiation (Lin et al., 2000). cRel expression also correlates with proliferative activity (Alves et al., 2014; Heinzel and Binh Giang, 2017), and cycling PBs show less cRel downregulation than quiescent PCs. Future studies may address to what extent cRel expression determines proliferative capacity of ABCs.
Our work points to a mutually antagonistic regulatory logic between cRel and Blimp1, where cRel inhibits Blimp1 expression via Bach2 and Blimp1 directly represses cRel expression. Mutual inhibition within regulatory networks can result in a bistable system, i.e. a system with two steady states (Kaplan and Glass, 2012). Here, one state was characterized by Blimp1lowcRelhigh, and another by Blimp1highcRellow. Further, our work suggests that ABCs and ASCs population dynamics are governed by how cells dynamically transition through the state space. Stimulation of B-cells does not only activate cRel but also RelA, which (directly and through IRF4) promote Blimp1 expression; this then allows a portion of cells to build up sufficient Blimp1 to transition to the Blimp1highcRellow state and differentiate into ASCs. Thus in the context of B cell proliferation and differentiation, cRel and RelA do not compensate for each other (Hoffmann et al., 2003) but they have distinct, potentially antagonistic functions. We posit that the coordinated dynamic control of these two NFκB family members, and their relative activity over time phase the GC reaction of B-cell population expansion, SHM, and ASC generation for an effective immune response.
Bistable systems may enforce differentiation decisions so that the differentiated state of the cell is maintained even when the differentiating stimulus has decayed (Wang et al., 2009). Healthy ASC differentiation is indeed irreversible, and only a significant perturbation of regulatory networks, such as the overexpression of Bcl6, may lead to deactivation of the ASC-specific gene expression program (Fujita et al., 2004). Although various ASC populations, such as PBs, PCs and ASCs from distinct niches show variable Blimp1 and cRel abundances, the mutual antagonistic regulation between Blimp1 and cRel may be conserved. Indeed, we found that as Blimp1 expression gradually increases from PBs to PCs, there is a concomitant decrease in cRel. An inverse correlation between Blimp1 and cRel expression was also observed in intestinal B cells and I-ASCs and in human pre-plasma cells exiting from the GC (Cattoretti et al., 2005). Nevertheless, the Blimp1 transcriptional repression program may differ in other ways in distinct ASC populations; for example, I-ASCs fail to repress the Blimp1 target gene CD19 (Landsverk et al., 2017), and B-cell differentiation during Salmonella infections may involve a prolonged Blimp1highAIDhigh state resulting in GC-independent affinity maturation of PBs and pathological B cell responses (Di Niro et al., 2015).
Here we presented a mathematical model (NFκB-extended ASC model v2), which demonstrated that the included gene regulatory interactions are sufficient to explain the differentiation kinetics and regulatory dynamics explored here. However, other in vivo or ex vivo experimental conditions may involve additional mechanisms. For example, recent results revealed a significant role for the complex of IRF8 and PU.1 in the propensity of B cells to undergo CSR and ASC differentiation by concurrently promoting the expression of Bcl6 and Pax5 and repressing AID and Blimp1 (Carotta et al., 2014; Willis et al., 2017). Furthermore, cytokines secreted by T-helper cells are known to play important roles in controlling B cell fate in vivo. Thus additional regulatory interactions, not represented in the current model, are likely required to account for in vivo differentiation, including IL-2 mediated activation of Bcl6 (through Stat5), IL-21 mediated activation of Blimp1 (through Stat3) and IL-4 mediated activation of AID (Mendez and Mendoza, 2016; Tangye, 2014).
The dynamics of cRel are regulated transcriptionally and through dimerization, sequestration by IκB inhibitors (Almaden et al., 2014; Alves et al., 2014; O’Dea and Hoffmann, 2009), and kinase activity (Shinohara et al., 2014). This motivated us to incorporate the NFκB-extended ASC model v2 into an established multi-scale B-cell model (Mitchell et al., 2018; Shokhirev et al., 2015) that explicitly articulates these regulatory mechanisms. The model recapitulated, with single-cell resolution, dynamical changes in cRel and Blimp1 expression, cellular proliferation and subsequent differentiation of a subset of cells as measured by flow cytometry. Interestingly, the model also recapitulated the progression of PBs, generated early in the immune response, with still substantial proliferative capacity to terminally differentiated, quiescent PCs generated late in the immune response. Thus, the multi-scale model can recapitulate the generation of heterogeneous ASC populations, however there are likely further sources of heterogeneity, including the splenic architecture’s role in extrafollicular PB and GC-derived PC generation, that remain to be investigated experimentally and incorporated into mathematical representations.
The resulting multi-scale model was also capable of recapitulating and disentangling altered proliferation and differentiation phases in systems with genetic perturbations in key regulators such as cRel, RelA and Blimp1. Thus, the model represents an in-silico laboratory that may be used iteratively with wet-lab experiments for mechanistically investigating the variety of B cell lymphomas subtypes. Our multi-scale modeing work, supported by quantitative experimental data, suggests that coordinated dynamic regulation of cRel and RelA is critical for the switch from proliferative to ASC differentiation phases, and perturbations within this network are associated with B cell malignancies. Consequently, we suggest that dynamic mis-coordination of cRel and RelA may be a more useful concept than hyper- or hypo-activity of generic NFκB when addressing the mechanisms underlying B-cell lymphoma or humoral response deficiencies.
STAR * Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alexander Hoffmann (ahoffmann@ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were maintained in environmental control facilities at the University of California, Los Angeles. Mice of both sexes were littermates and were 10–14 weeks old unless otherwise indicated. The Rel−/− (protein name cRel) mice (Kontgen et al., 1995) previously described and wild type mice were in C57BL/6 genetic background. The mice were Animal work was performed according to University of California, Los Angeles regulations under an approved protocol.
Cell Lines
HEK 293 (ATCC, CRL-1573) cells were cultured with DMEM, 10% FBS. BCL1 (ATCC, CRL-1669) were cultured with RPMI, 10% FBS, 5 mM L-glutamine, and 55 μM 2-Mercaptoethanol.
METHOD DETAILS
B cell isolation and culture
Spleens were harvested from 10–12 weeks old female C57BL/6 mice and Rel−/− mice. Homogenized splenocytes were incubated with anti-CD43 magnetic beads for 15 min at 4–8ºC, washed with MACS buffer (Phosphate buffer saline, (pH 7.4), 0.5% bovine serum albumin and 2 mM Ethylenediaminetetraacetic acid (pH 8)) and passed through LS column (Miltenyi Biotech). The purity of B cells was >95% based on B220 staining as described previously (Mitchell et al., 2018). Briefly, the enriched B cell population was stained with B220-eF450, CD3e-FITC, CD11b-FITC and Ly-6C-Alexa 488. The stained cells were gated on B220+CD3e-CD11b-Ly-6C-.
B cells were grown in fresh media with 1% penicillin streptomycin solution, 5 mM L-glutamine, 20 mM HEPES buffer, 1mM MEM non-essential amino acid, 1 mM Sodium pyruvate, 10% FBS, and 55 μM 2-Mercaptoethanol.
Sorting of B cells subtypes by flow cytometry
Mature splenic B cells were plated in a 10 cm petri-dish and treated with LPS (10 μg/mL) for 72 h in 10 mL media with 1×106 cells/mL. Cells were harvested and resuspended in 100 μL of cold media. Nonspecific binding was blocked with Fc receptor blocker by incubating for 10 min on ice and stained with B220-eF450 and CD138-APC in a total of 200 μL of cold media for 30 min in ice. The excess antibody was removed and washed by centrifugation at 500 rcf, 4ºC for 3 min, and then tubes were turned 180° and centrifugation was repeated. Cells were resuspended in media and dead cells were excluded using 7AAD. ABCs (B220hiCD138-) and ASCs (B220lowCD138+) were sorted based on expression of B220 and CD138 by flow cytometer (Figure S2A).
Inguinal lymph nodes were isolated from immunized mice. GC B cells were sorted at 7 days after primary immunization. Homogenized lymph nodes were incubated with Fc receptor blocker for 10 min in ice and stained with B220-eF450, CD3e-FITC, CD11b-FITC, CD11c-FITC, CD49b-FITC, FAS-APC and CD38-PE in a total of 100 μL of cold media for 30 min in ice. GC B cell were sorted using the gating strategy B220+CD38-FAS+ (Figure S2F). ASCs were isolated from lymph nodes 7 days after a booster dose of immunization. Homogenized lymph nodes were incubated with Fc receptor blocker for 10 min in ice and stained with B220-eF450, CD3e-FITC, CD11b-FITC, CD11c-FITC, CD49b-FITC and CD138-APC in total 100 μL of cold media for 30 min in ice. ASCs were sorted using gating strategy B220lowCD138+ (Figure S2G).
Mouse immunization
10–12 weeks old female C57BL/6 mice were immunized with 100 μg of NP-OVA emulsified in Complete Freund Adjuvant in the foot pad, followed by an equivalent booster shot after 21 days.
Isolation and characterization of intestinal ASC
Isolation of murine lymphocytes from the small-intestinal lamina propria was performed as previously described (Couter and Surana, 2016). Briefly, resected small-intestinal segments were washed, inverted, and the epithelial fraction was removed by incubating for 15 min at 37°C in RPMI with 5 mM DTT, 1 mM EDTA, and 2% FBS. The residual fraction was minced and digested for 30 min at 37°C in RPMI with collagenase II (1.5 mg/ml), dispase (0.5 mg/ml), and 1% FBS. Lamina propria single-cell suspensions were obtained by passing through 100 uM and 40 uM cell strainers, and resuspended in RPMI containing 2% FBS. Homogenized cells were incubated with fixable viability dye eF506 (Affymetrix Bioscience, #65–0866-18) in total 1 mL PBS for 30 min on ice and washed with PBS. Cells were then incubated with Fc receptor blocker for 10 min in ice and stained with B220-APC-Cy7, CD3e-FITC, CD11b-FITC, CD11c-FITC, CD49b-FITC, Ly-6G-FITC and CD138-PerCP Cy5.5 in total 100 μL of cold media for 30 min in ice. The gating strategy is similar to lymph node ASCs. B cells and ASCs were characterized using B220+ and B220-CD138+ respectively.
Genetic modification of BCL1 cells
To lentivirally transduce BCL1 cells, HEK 293 cells were transfected with all lentivirus constructs using lipofectamine (Invitrogen). 48 h post transfection, virus was harvested and used to infect murine BCL1 cells in the presence of polybrene (7.5 μg/ml). Lentivirally infected BCL1 cells were grown in the presence of puromycin for 3 days (Yoon and Boss, 2010). Plasmids used: pLX-cRel, Bach2−/− guide RNA (5’-CACCGGAACTTTCGTCCCCCTGCGC-3’ and 5’-CACCGTGCTGCAGGGACGGGCACAA-3’) and Blimp1−/− guide RNA (5’-CACCGGAATCCAGCTCACTCTGCCC-3’ and 5’-CACCGACCTGGCTGCCTGTCAGAAC −3’) cloned in the LentiV2 plasmid. The efficiency of knock in and knock out was assessed by immunoblot as indicated.
Electroporation of BCL1 cells was performed as described previously (Lin et al., 2000; Lin et al., 1997) with some modifications. Briefly, 2 ×105 cells were plated and washed with PBS (without Ca2+ and Mg2+). Cells were resuspended in Resuspension Buffer R at 2×105 cells/10 μL and 1 μg of plasmid DNA was added. Cells were gently pipetted to obtain a single cell suspension. Cells were electroporated in Neon Transfection systems (Invitrogen) and plated in 6 well plates. A pulse voltage of 1000 mW and width of 40 ms was used for electroporation. Electroporated cells were grown in the presence of puromycin for 3 days. Blimp1 binding site disruption (RelBS-mut) was performed using guide RNA (5’-CACCGTACTAGAGCATCTGAAAGCC-3’) cloned in the Lentiv2 plasmid. The efficiency of mutation was measured by Next Generation Sequencing. The targeted genomic region was PCR amplified (using the primer: ACTTAAAGCCTTTTTGTGCTTCT and CAGTTTCCTTTACAGCAGGAGTT) and PCR products were gel purified. The purified PCR products were submitted for amplicon-EZ (150–500 bp) sequencing (Genewiz). Adapter-trimmed sequencing results were processed for quality control using FASTX-Toolkit. A Python script was then used to analyze counts of unique mutated or wild type amplicons (code is available as Supplemental File 1). Piechart was plotted with Graphpad Prism (Figure S4C).
RNA and Immunoblot Analysis
For qPCR, mRNA was isolated from B cells, ABCs, ASCs, 10 μg/ml LPS-stimulated wild-type and Rel−/− B cells following 24, 36, 38, 60 and 72 hours of culture and BCL1 cells following stimulation with IL-2 (20 ng/ml) and IL-5 (20 ng/ml), using the Zymo Research kit. 1 μg mRNA was used to prepare cDNA using Oligo (dT) and quantitation was performed using the SSO syber green. Expression was normalized using ubiquitin C as a housekeeping gene. The primers used in this study are specified in Table S3.
For Immunoblot analysis, whole-cell lysates were prepared using RIPA lysis buffer. The resulting lysates were run on either 10% SDS-PAGE gels or 5–14% Criterion Tris-HCl Gel (Bio-Rad). The following antibodies were used to identify the protein of interest: Rela, cRel, Blimp1, IRF4, Bach2 and actin (all from Santa Cruz Biotechnology). The resulting proteins were detected using the Bio-Rad ChemiDoc XRS System and SuperSignal West Femto Substrate Maximum Sensitivity Substrate (Thermo Scientific) to detect chemiluminescence released by HRP-labeled secondary antibodies.
IgM production measurement by ELISA
For ELISA analysis, BCL1 and genetically modified BCL1 cells were plated in a 48 well plate with 2×105 cells/well in a 250 μl volume. The cells were stimulated with IL-2 (20 ng/ml) and IL-5 (20 ng/ml) for 96 hr and the supernatant was collected. The resulting supernatant was tested to measure secreted IgM by ELISA. ELISA was performed using mouse Ig isotyping ELISA kit according to manufacturer protocol.
Chromatin Immunoprecipitation (ChIP)
For ChIP experiments 10×106 cells were washed with 10 ml PBS at room temperature (RT) and fixed with freshly supplemented 1% methanol-free formaldehyde and incubated at RT for 10–15 min with gentle shaking. The fixation reaction was quenched with 500 mM Tris pH 8 for 2–5 min. The cells were washed twice with cold PBS followed by snap freezing with dry ice. The cell pellets were thawed and resuspended on ice in 1 ml Lysis Buffer 1 and incubated on a nutator at 4°C for 10 min before centrifugation at 800 rcf, 5 min, 4°C; supernatant was then discarded. Similar steps were performed with 1 ml Lysis Buffer 2. The nuclei were resuspended in 0.5 ml Lysis Buffer 3 Plus and incubated on ice for 10 min. The lysates were transferred into 1.5-ml TPX tubes (Diagenode #C30010010–1000, min 100 μl-max 300 μl/tube) and sonicated with a Bioruptor water bath sonicator using 25 cycles consisting of 30 sec ON/30 sec OFF at low intensity, 4°C. Sonication was stopped every 5 cycles for incubation on ice for 1 min, gentle inversion and pulse-spin. Aliquots were consolidated with the same sample into a single 1.5-ml polypropelene tube. The lysate was centrifuged at max speed for 15 min at 4°C. The supernatants were transferred to 1.5-ml no-stick microtubes (Phenix Research #MH-815S) and 3 volumes of dilution buffer were added to lysates. Unconjugated beads (25 μl/ml of diluted lysate) were washed (twice with PBS and dilution buffer) and dried up in no-stick tubes using a magnet. Lysates were mixed with dry beads and incubated for 2 hrs at 4°C on a nutator. Lysates were used for the next step and beads were discarded. 1% aliquots were taken as input and stored at 4°C with volumes noted. 2 μg anti-H3K9ac antibody and isotype IgG were incubated with the lysate overnight at 4°C on a nutator. 25 μl protein G-conjugated magnetic beads (Active Motif #53014) were washed twice with Dilution Buffer, then once with low-salt buffer. Beads were dried using a magnetic rack. Lysates were transferred to dry beads and incubated for 5 hrs at 4°C on a nutator. Beads were collected using a magnetic stand and washed twice with each of the following ice-cold buffers in order: low salt, high salt, LiCl, TE. 600 μl of buffer was used in each wash step and beads were resuspended by inversion, and placed in a high-speed shaker at RT for 5 min. Beads were resuspended in 70 μl of 250 mM NaCl. Input fractions were recovered and volumes were adjusted to 70 μl with 250 mM final concentration of NaCl. Both input and IP fractions were processed in a similar way after this. 50 μg DNAse-free RNAse A (Roche #11119915001) was added and incubated at 37°C for 1 hr with gentle mixing. SDS was added for a 1% final concentration and then 50 μg Proteinase K (Roche # 03115828001) was added for an overnight incubation at 60°C with gentle shaking. SPRI beads were resuspended by gentle vortexing, allowed to warm to RT and the supernatant was removed from Protein G beads. Protein G beads were washed with an additional 30 μl of 500 mM NaCl and combined with the first eluate. 0.9–1.0 volumes of SPRI beads were added and incubated at RT for 15 minutes on a nutator. Beads were collected using a magnetic stand and supernatants were removed. Beads were twice washed with 80% ethanol and residual ethanol from tubes was removed by air-drying beads for 5 minutes. DNA was eluted from beads with 30 μl TE buffer. Enrichment of the targeted DNA in the elution was measured by qPCR and the enrichment was calculated with respect to input.
Buffer composition:
Lysis Buffer 1: 50 mM HEPES-KOH, pH 7.6, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP-40, 0.25% Triton X-100, EDTA-free protease inhibitors.
Lysis Buffer 2: 10 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, EDTA-free protease inhibitors.
Lysis Buffer 3: 10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na Deoxycholate, 0.5% N-lauroylsarcosine, sodium salt (Sigma-Aldrich #61739), EDTA-free protease inhibitors.
Dilution Buffer: 10 mM Tris-HCl, pH 8.0, 160 mM NaCl, 1 mM EDTA, 0.01% SDS, 1.2% Triton X-100, EDTA-free protease inhibitors.
Low Salt Wash Buffer: 50 mM HEPES-KOH, pH 7.6, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na Deoxycholate, 0.1% SDS, EDTA-free protease inhibitors.
High Salt Wash Buffer: 50 mM HEPES-KOH, pH 7.6, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na Deoxycholate, 0.1% SDS.
LiCl Buffer: 20 mM Tris-HCl, pH 8.0, 250 mM LiCl, 1 mM EDTA, 0.5% Na Deoxycholate, 0.5% NP-40.
TE Buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA)
Measurement of apoptotic cells in ABC and LPS-PB
WT and Rel−/− B cells were treated with LPS (10 μg/mL) for 72 h as described above. Cells were harvested and resuspended in 100 μL of cold media. Nonspecific binding was blocked with Fc receptor blocker by incubating for 10 min on ice and stained with B220-PE and CD138-APC in a total of 200 μL of cold media for 30 min in ice. Stained cells were washed with FACS buffer (PBS with 5% FBS) and resuspended in Annexin V binding buffer. The staining of Annexin V Alexa Fluor™ 488 and Propidium Iodide (PI) was performed using Dead Cell Apoptosis Kit (ThermoFisher Scientific, # V13241) as described by manufacturer protocol. Briefly, 5 μL Alexa Fluor™ 488 annexin V and 1 μL PI (100 μg/mL PI working solution) were added to 100 μL of cell suspension. The cells were incubated at room temperature for 15 minutes. 400 μL annexin-binding buffer was added, mixed gently, and the samples were acquired in BD LSRFortessa™ flow cytometry (BD Bioscience). Data were analyzed with FlowJo. The gating strategy and ABC and LPS-PBs were characterized by as described above. ABC and LPS-PBs population were gated for Annexin V and PI positivity. The apoptotic cells were defined as AnnexinV+PI- (Figure S5C).
Measurement of generation-specific ASC
WT and Rel−/− B cells stained with Cell Trace Far Red (CTR) using CellTrace™ Far Red Cell Proliferation Kit (ThermoFisher Scientific, # C34564) as described by the manufacturer protocol. Briefly, cells were resuspended in 1 mL RT PBS and incubated with 1 μL CTR for 25 min at RT with rotation. Cells were washed followed by resuspension in RPMI with 10% FBS and incubated for 10 min at RT. The cells were washed and washing steps were repeated 1 more time. CTR labeled cells were treated with LPS (10 μg/mL) for 24 h, 48 h, 72 h and 96 h as described above. The cells were harvested at indicated time points, stained for CD138 and 7AAD as described above. The cells were acquired in CytoFlex flow cytometer. Dead (7AAD+) cells were exclude from the analysis and the data were analyzed in FlowJo. Cell generation number was defined based on dilution of CTR and ASCs were defined by expression of CD138. The generation specific percentage of ASC were calculated using the equation:
For each generation, n.
Intracellular staining
Naïve mature B cells (B cells) were isolated from spleens as described above. Cells were treated with LPS (10 μg/mL) in 12 well plates (5×105/well) for 0, 24, 48, 72 and 96 hours. Cells were washed twice with PBS, followed by permeabilization and staining with primary antibody and then secondary antibody using Fix and Perm cell permeabilization kit (ThermoFisher, # GAS003). Cell permeabilization and staining protocol as described by the manufacturer was followed. Cells were stained with cRel-PE and Blimp1-Alexa 647 antibody to measure expression of cRel and Blimp1 respectively by flowcytometry.
B cells were treated with anti-CD40 antibody (10 μg/ml) and IL-4 (5 ng/ml) for 72 h and stained for ABCs and CD40-PB as described above. Intracellular expression of cRel and Rela was measured using cRel-eF660 and Rela-PE respectively by flowcytometry using CytoFlex flow cytometry (488nm 50mW laser) (Beckman Coulter) and data were analyzed with FlowJo.
Intestinal lymphocytes were isolated and characterized as described above. Intracellular expression of cRel and Blimp1 was measured using cRel-PE and Blimp1-Alexa 647 respectively by BD LSRFortessa™ flow cytometry (BD Bioscience) and data were analyzed with FlowJo.
Cell Cycle Analysis
BCL1 and BCL1-cRel cells were plated in a 35 mm petri-dish with 1×106 cells in a 2 ml volume. The cells were stimulated with IL-2 (20 ng/ml) and IL-5 (20 ng/ml) for 24 hr. The cells were harvested and washed with cold PBS. The cell pellets were fixed by drop wise addition of 70% cold ethanol with continuous mild vortex and cells were kept overnight (16–20 hr) at 4°C. The cells were washed with PBS twice by centrifugation 850 rcf for 5 min at 4°C and turned the tube 180° to repeat centrifugation. Cells were treated with 50 μl of a 100 μg/ml sock of RNase A for 15 min at 37°C and 200 μl of propidium iodide (PI) from 50 μg/ml stock solution were incubated for 15 min at RT.
The cells were acquired in flow cytometer (CytoFLEX, Beckman Coulter). The cells were gated based on forward scattered (FSC) and side scattered to identify single cells. Doublet were excluded from analysis using FSC area and FSC width. The DNA content was measured by staining with PI.
Computational Modeling of the ABC-ASC Differentiation Circuit
Computational modeling was performed in MATLAB by re-constructing the system of ODEs, with identical parameters and reactions as described by Sciammas et al. (2011). ODE solver ODE45 was used throughout. To construct the NFκB-extended ASC model v1, cRel and RelA were included with two additional differential equations. Additional reactions were included as shown in Figure 1B and 3A with similar kinetic rate laws. Briefly, degradation was mass action with Hill equations used for activation and inhibition. Parameters and thresholds were chosen to be consistent with Sciammas et al. and maintained the same throughout (Sciammas et al., 2011). All equations, parameters and initial conditions are given in Data S1, Table S1, S2. While the previously published model sampled IRF4 activation rates to simulate cell-to-cell variability of activation strength, here we sampled cRel and Rela activation rates with the same coefficient of variation (0.4) as used for IRF4 in Sciammas et al.
cRel downregulation (Figure 1) was investigated by running a timecourse simulation of 125 individual cells for 96 hours then taking the final molecular abundances as an initial condition of a subsequent 96 hour simulation in which cRel expression was reduced as indicated.
To construct the NFκB-extended ASC model v2, Blimp1-mediated cRel downregulation was incorporated through hill kinetics consisted with the NFκB-extended ASC model model v1. Constant cRel expression ( was replaced with:
To simulate Rel−/− we set cRel expression to 0 and simulated a population of 125 individual cells. ASC generation in cRel deficiency is controlled by RelA, RT-PCR analysis showed that Rel−/− B cells reduced RelA expression at 24 h (20–40%) and had very similar expression at 72 h compared to wild type. This was incorporated into our Rel−/− simulations through reducing RelA expression rate (40% of WT) up to 24h and returning this rate to near WT (70% of WT) for the remaining simulation time.
Multiscale Modeling
The NFκB-extended ASC model v2 constructed here was combined with a published model of B-cell proliferation (Mitchell et al., 2018) to create a multiscale model capable of simulating the division, death and differentiation of a population of individual cells. All reactions and parameters within the differentiation networks were maintained as described here and all reactions and parameters within the NFκB, apoptosis and mitosis networks were maintained as described by Mitchell et al. (Mitchell et al., 2018). NFκB activation sampling as described above for NFκB-extended ASC model v1 and v2 was not performed here as cell-to-cell variability was quantified in Mitchell et al. and simulated here in the same manner resulting in realistically distributed NFκB activities. As the absolute concentration of cRel in the differentiation model and multiscale model differed (due to the arbitrary concentrations of molecular species in the previously published differentiation model (Sciammas et al., 2011)) the KD of cRel- and Rela-mediated reactions in the differentiation network were scaled to be approximately half the maximum cRel and Rela concentrations observed in simulations from Mitchell et al. A population of 1000 single cells was simulated and scatter plots of cRel vs Blimp1 were created every 15 minutes for 124 hours (Supplemental Movie 1 and Figure 6E). Rel−/− simulations were performed by removing cRel expression and reducing RelA expression as described for the ASC differentiation model. A similar approach was taken to Blimp1, with Blimp1−/− simulated by scaling Blimp1 expression by 50%. RelBS-seq cells were simulated by removing terms representing Blimp1 inhibition of cRel expression from the ODE for cRel. cRel-High simulations were performed by increasing cRel expression 5-fold. All code is available at http://www.signalingsystems.ucla.edu/models-and-code/.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details of the statistical tests applied to replicates of datasets shown in Figures can be found in the corresponding Figure legends. Statistical analyses were performed using GraphPad Prism 7 software. *p < 0.05, **p < 0.01, ***p < 0.001 and not significant (ns) (Unpaired Students t-test).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat Monoclonal anti-mouse CD16/32 (Fc receptor blocker) | BioLegend | Cat#101302 |
| Rat monoclonal anti-B220-eF450 | eBioscience | Cat#48–0452-82 |
| Rat monoclonal anti-human/mouse B220-PE | eBioscience | Cat#12–0452-83 |
| Rat monoclonal anti-mouse/human B220-FITC | BioLegend | Cat#103206 |
| Rat monoclonal anti-mouse/human B220-APC-Cy7 | BioLegend | Cat#103223 |
| Armenian hamster monoclonal anti-mouse CD3e-FITC | eBioscience | Cat#11–0031-82 |
| Rat monoclonal anti-mouse CD11b-FITC | eBioscience | Cat#11–0112-82 |
| Rat monoclonal anti-mouse Ly-6C-Alexa 488 | eBioscience | Cat#53–5932-82 |
| Armenian hamster monoclonal anti-mouse CD11c-FITC | eBioscience | Cat#11–0114-82 |
| Rat monoclonal anti-mouse CD49b-FITC | eBioscience | Cat#11–5971-82 |
| Mouse monoclonal anti-mouse FAS-APC | BioLegend | Cat#152604 |
| Rat monoclonal anti-mouse CD38-PE | BioLegend | Cat#102708 |
| Rat monoclonal anti-mouse CD138-PE | BioLegend | Cat#142504 |
| Rat monoclonal anti-mouse CD138-PerCP Cy5.5 | BioLegend | Cat#142509 |
| Rat monoclonal anti-mouse CD138-APC | BioLegend | Cat#142506 |
| Rat monoclonal anti-mouse CD138-Biotin | BioLegend | Cat#142511 |
| Rat monoclonal anti-mouse Ly-6G-FITC | BioLegend | Cat#127605 |
| Rat monoclonal anti-mouse Blimp1-Alexa 647 | BioLegend | Cat#150003 |
| Rat monoclonal anti-mouse cRel- PE | eBioscience | Cat#12–6111-80 |
| Rat monoclonal anti-mouse cRel-eF660 | eBioscience | Cat#50–6111-80 |
| Mouse monoclonal anti-Rela-PE | Cell Signaling | Cat#9460 |
| Rabbit polyclonal anti-Rela | SantaCruz | Cat#sc-372 |
| Rabbit polyclonal anti-cRel | SantaCruz | Cat#sc-71 |
| Rat monoclonal anti-Blimp1 | SantaCruz | Cat#sc-47732 |
| Goat polyclonal anti-IRF4 | SantaCruz | Cat#sc-6059 |
| Rabbit polyclonal anti-Bach2 | Abcam | Cat#ab83364 |
| Goat polyclonal anti-Actin | SantaCruz | Cat#sc-1615 |
| Rabbit polyclonal Anti-Histone H3 (acetyl K9) | Abcam | Cat#ab4441 |
| Rabbit isotype IgG | SantaCruz | Cat#sc-2027 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| fixable viability dye eF506 | Affymetrix Bioscience | Cat#65–0866-18 |
| 7 aminoacitomycine D (7AAD) | BioLegend | Cat#420404 |
| Critical Commercial Assays | ||
| Ig isotyping ELISA kit | eBioscience | Cat#88–50630-88 |
| Dead Cell Apoptosis Kit | ThermoFisher Scientific | Cat# V13241 |
| CellTrace™ Far Red Cell Proliferation Kit | ThermoFisher Scientific | Cat#C34564 |
| Fix and Perm cell permeabilization kit | ThermoFisher Scientific | Cat#GAS003 |
| Public data set | Nature Immunology, 2015,16(6):663–73 | GSE60927 |
| Public data set | Nature Immunology, 2016,17(3), 323–330 | GSE70981 |
| Public data set | Nature Immunology, 2016, 17(3), 331–343 | GSE71698 |
| Experimental Models: Cell Lines | ||
| HEK 293 | ATCC | CRL-1573 |
| BCL1 | ATCC | CRL-1669 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | JAX: 006494 |
| Mouse: C57BL/6: rel-/- | Kontgen et al., 1995, Genes & development 9, 1965–1977 | N/A |
| Oligonucleotides | ||
| pLX-cRel, Bach2 KO guide RNA (5’-CACCGGAACTTTCGTCCCCCTGCGC-3’, 5’-CACCGTGCTGCAGGGACGGGCACAA-3’) | This paper | N/A |
| Blimp1 KO guide RNA (5’-CACCGGAATCCAGCTCACTCTGCCC-3’, 5’-CACCGACCTGGCTGCCTGTCAGAAC-3’) | This paper | N/A |
| Blimp1 binding site disruption guide RNA (5’-CACCGTACTAGAGCATCTGAAAGCC-3’) | This paper | N/A |
| Blimp1 PCR primer: ACTTAAAGCCTTTTTGTGCTTCT and CAGTTTCCTTTACAGCAGGAGTT | This paper | N/A |
| Software and Algorithms | ||
| Graphpad Prism | GraphPad Software | |
| Mathworks MATLAB | MathWorks, Inc | R2014a |
| Other | ||
| All Computational Modeling Code | This paper | http://www.signalingsystems.ucla.edu/models-and-code/ |
Highlights
cRel drives B-cell proliferation but blocks antibody secreting cell differentiation
In ASCs, RelA-induced Blimp1 represses cRel via binding the Rel enhancer
NFκB dynamics transition cells across the bi-stable ABC-ASC differentiation network
Multi-scale model of single-cell fate decisions explains B cell population dynamics
Acknowledgements
We thank Sherie Morrison (UCLA) for the generous gift of BCL1 cell line, Lynn Corcoran (WEHI), Meinrad Busslinger (IMP Vienna) for depositing high quality datasets, and Dinesh Rao (UCLA) for helpful discussions, and JCCC flowcytometry core facility (UCLA) for expert support. S.L.N. was supported by a National Health and Medical Research Council of Australia grants (1054925 and 1058238). This work was supported by grants from the NIH-NIAID National Institute of Allergy and Infectious Diseases (R01AI132731 to A.H.).
Footnotes
Declaration of Interest
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Equations of the mathematical model of the ABC-ASC differentiation circuit are provided in Data S1. Matlab code for all models is available at http://www.signalingsystems.ucla.edu/models-and-code/. The analysis tool of CRISPR mutation efficiency is available as python code as Data S2.
References
- Almaden JV, Tsui R, Liu YC, Birnbaum H, Shokhirev MN, Ngo KA, Davis-Turak JC, Otero D, Basak S, Rickert RC, and Hoffmann A. (2014). A pathway switch directs BAFF signaling to distinct NFkappaB transcription factors in maturing and proliferating B cells. Cell Rep 9, 2098–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves BN, Tsui R, Almaden J, Shokhirev MN, Davis-Turak J, Fujimoto J, Birnbaum H, Ponomarenko J, and Hoffmann A. (2014). IkappaBepsilon is a key regulator of B cell expansion by providing negative feedback on cRel and RelA in a stimulus-specific manner. J Immunol 192, 3121–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjunaraja S, Nose BD, Sukumar G, Lott NM, Dalgard CL, and Snow AL (2017). Intrinsic Plasma Cell Differentiation Defects in B Cell Expansion with NF-kappa B and T Cell Anergy Patient B Cells. Front Immunol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado DP, Zhang BC, Srinivasan L, Sasaki Y, Seagal J, Unitt C, Rodig S, Kutok J, Tarakhovsky A, Schmidt-Supprian M, and Rajewsky K. (2010). Constitutive Canonical NF-kappa B Activation Cooperates with Disruption of BLIMP1 in the Pathogenesis of Activated B Cell-like Diffuse Large Cell Lymphoma. Cancer Cell 18, 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carotta S, Willis SN, Hasbold J, Inouye M, Pang SHM, Emslie D, Light A, Chopin M, Shi W, and Wang H. (2014). The transcription factors IRF8 and PU. 1 negatively regulate plasma cell differentiation. J Exp Med 211, 2169–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoretti G, Angelin-Duclos C, Shaknovich R, Zhou H, Wang D, and Alobeid B. (2005). PRDM1/Blimp-1 is expressed in human B-lymphocytes committed to the plasma cell lineage. The Journal of pathology 206, 76–86. [DOI] [PubMed] [Google Scholar]
- Ceribelli M, Kelly PN, Shaffer AL, Wright GW, Xiao W, Yang Y, Mathews Griner LA, Guha R, Shinn P, Keller JM, et al. (2014). Blockade of oncogenic IkappaB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc Natl Acad Sci U S A 111, 11365–11370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couter CJ, and Surana NK (2016). Isolation and Flow Cytometric Characterization of Murine Small Intestinal Lymphocytes. Journal of visualized experiments : JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RE, Brown KD, Siebenlist U, and Staudt LM (2001). Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 194, 1861–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Niro R, Lee SJ, Vander Heiden JA, Elsner RA, Trivedi N, Bannock JM, Gupta NT, Kleinstein SH, Vigneault F, Gilbert TJ, et al. (2015). Salmonella Infection Drives Promiscuous B Cell Activation Followed by Extrafollicular Affinity Maturation. Immunity 43, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyao MP, Buckler AJ, and Sonenshein GE (1990). Interaction of an NF-kappa B-like factor with a site upstream of the c-myc promoter. Proceedings of the National Academy of Sciences 87, 4727–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Jaye DL, Geigerman C, Akyildiz A, Mooney MR, Boss JM, and Wade PA (2004). MTA3 and the Mi-2/NuRD complex regulate cell fate during B lymphocyte differentiation. Cell 119, 75–86. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Yoshida T, Okada S, Hatano M, Miki T, Ishibashi K, Okabe S, Koseki H, Hirosawa S, Taniguchi M, et al. (1997). Disruption of the Bcl6 gene results in an impaired germinal center formation. J Exp Med 186, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont RJ, and Gerondakis S. (2000). Rel induces interferon regulatory factor 4 (IRF-4) expression in lymphocytes: Modulation of interferon-regulated gene expression by Rel/nuclear factor kappa B. J Exp Med 191, 1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbold J, Corcoran LM, Tarlinton DM, Tangye SG, and Hodgkin PD (2004). Evidence from the generation of immunoglobulin G-secreting cells that stochastic mechanisms regulate lymphocyte differentiation. Nat Immunol 5, 55–63. [DOI] [PubMed] [Google Scholar]
- Heinzel S, and Binh Giang T. (2017). A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. 18, 96–103. [DOI] [PubMed] [Google Scholar]
- Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, and Klein U. (2014). Germinal center B cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. J Exp Med 211, 2103–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Leung TH, and Baltimore D. (2003). Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. Embo J 22, 5530–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Baltimore D. (2006). Circuitry of nuclear factor kappaB signaling. Immunol Rev 210, 171–86. [DOI] [PubMed] [Google Scholar]
- Hunter JE, Butterworth JA, Zhao B, Sellier H, Campbell KJ, Thomas HD, Bacon CM, Cockell SJ, Gewurz BE, and Perkins ND (2016). The NF-kappa B subunit c-Rel regulates Bach2 tumour suppressor expression in B-cell lymphoma. Oncogene 35, 3476–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaileh M, and Sen R. (2012). NF-kappaB function in B lymphocytes. Immunol Rev 246, 254–271. [DOI] [PubMed] [Google Scholar]
- Kallies A, Hasbold J, Fairfax K, Pridans C, Emslie D, McKenzie BS, Lew AM, Corcoran LM, Hodgkin PD, Tarlinton DM, and Nutt SL (2007). Initiation of plasma-cell differentiation is independent of the transcription factor Blimp-1. Immunity 26, 555–566. [DOI] [PubMed] [Google Scholar]
- Kaplan D, and Glass L. (2012). Understanding nonlinear dynamics (Springer Science & Business Media; ). [Google Scholar]
- Kim Y, and Tian M. (2009). NF-κB family of transcription factor facilitates gene conversion in chicken B cells. Molecular immunology 46, 3283–3291. [DOI] [PubMed] [Google Scholar]
- Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, and Gerondakis S. (1995). Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes & development 9, 1965–1977. [DOI] [PubMed] [Google Scholar]
- Landsverk OJ, Snir O, and Casado RB (2017). Antibody-secreting plasma cells persist for decades in human intestine. 214, 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KI, Lin Y, and Calame K. (2000). Repression of c-myc is necessary but not sufficient for terminal differentiation of B lymphocytes in vitro. Mol Cell Biol 20, 8684–8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Wong KK, and Calame K. (1997). Repression of c-myc transcription by blimp-1, an inducer of terminal B cell differentiation. Science 276, 596–599. [DOI] [PubMed] [Google Scholar]
- Mandelbaum J, Bhagat G, Tang HY, Mo TW, Brahmachary M, Shen QO, Chadburn A, Rajewsky K, Tarakhovsky A, Pasqualucci L, and Dalla-Favera R. (2010). BLIMP1 Is a Tumor Suppressor Gene Frequently Disrupted in Activated B Cell-like Diffuse Large B Cell Lymphoma. Cancer Cell 18, 568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez MR, Corradin A, Klein U, Alvarez MJ, Toffolo GM, di Camillo B, Califano A, and Stolovitzky GA (2012). Quantitative modeling of the terminal differentiation of B cells and mechanisms of lymphomagenesis. P Natl Acad Sci USA 109, 2672–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez A, and Mendoza L. (2016). A Network Model to Describe the Terminal Differentiation of B Cells. Plos Comput Biol 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanovic M, Heise N, De Silva NS, Anderson MM, Silva K, Carette A, Orelli F, Bhagat G, and Klein U. (2017). Differential requirements for the canonical NF-B-k transcription factors c-REL and RELA during the generation and activation of mature B cells. Immunol Cell Biol 95, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnich M, Tagoh H, Bonelt P, Axelsson E, Fischer M, Cebolla B, Tarakhovsky A, Nutt SL, Jaritz M, and Busslinger M. (2016). Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol 17, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell S, Roy K, Zangle TA, and Hoffmann A. (2018). Nongenetic origins of cell-to-cell variability in B lymphocyte proliferation. P Natl Acad Sci USA 115, E2888–E2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MAJ, Magnusdottir E, Kuo TC, Tunyaplin C, Harper J, Arnold SJ, Calame K, Robertson EJ, and Bikoff EK (2009). Blimp-1/Prdm1 Alternative Promoter Usage during Mouse Development and Plasma Cell Differentiation. Mol Cell Biol 29, 5813–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto A, Ochiai K, Kimura Y, Itoh-Nakadai A, Calame KL, Ikebe D, Tashiro S, and Igarashi K. (2010). Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. Embo J 29, 4048–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Hodgkin PD, Tarlinton DM, and Corcoran LM (2015). The generation of antibody-secreting plasma cells. Nat Rev Immunol 15, 160–171. [DOI] [PubMed] [Google Scholar]
- O’Dea E, and Hoffmann A. (2009). NF-kappaB signaling. Wiley Interdiscip Rev Syst Biol Med 1, 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SR, Zan H, Pal Z, Zhang JS, Al-Qahtani A, Pone EJ, Xu ZM, Mai T, and Casali P. (2009). HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat Immunol 10, 540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl T, Gugasyan R, Grumont RJ, Strasser A, Metcalf D, Tarlinton D, Sha W, Baltimore D, and Gerondakis S. (2002). The combined absence of NF-kappa B1 and c-Rel reveals that overlapping roles for these transcription factors in the B cell lineage are restricted to the activation and function of mature cells. Proc Natl Acad Sci U S A 99, 4514–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Staudt LM, and Pr LLMP (2002). The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. New Engl J Med 346, 1937–1947. [DOI] [PubMed] [Google Scholar]
- Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, Pernis A, Pasqualucci L, and Dalla-Favera R. (2007). A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell 12, 280–292. [DOI] [PubMed] [Google Scholar]
- Sciammas R, and Davis MM (2004). Modular nature of blimp-1 in the regulation of gene expression during B cell maturation. J Immunol 172, 5427–5440. [DOI] [PubMed] [Google Scholar]
- Sciammas R, Li Y, Warmflash A, Song YQ, Dinner AR, and Singh H. (2011). An incoherent regulatory network architecture that orchestrates B cell diversification in response to antigen signaling. Molecular Systems Biology 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang LM, Zhao H, Calame K, and Staudt LM (2002a). Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 17, 51–62. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Rosenwald A, and Staudt LM (2002b). Lymphoid malignancies: the dark side of B-cell differentiation. Nat Rev Immunol 2, 920–932. [DOI] [PubMed] [Google Scholar]
- Shapiro-Shelef M, and Calame K. (2005). Regulation of plasma-cell development. Nat Rev Immunol 5, 230–242. [DOI] [PubMed] [Google Scholar]
- Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, Smyth GK, Hodgkin PD, Nutt SL, and Corcoran LM (2015). Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol 16, 663-+. [DOI] [PubMed] [Google Scholar]
- Shinohara H, Behar M, Inoue K, Hiroshima M, Yasuda T, Nagashima T, Kimura S, Sanjo H, Maeda S, Yumoto N, et al. (2014). Positive feedback within a kinase signaling complex functions as a switch mechanism for NF-kappaB activation. Science 344, 760–764. [DOI] [PubMed] [Google Scholar]
- Shokhirev MN, Almaden J, Davis-Turak J, Birnbaum HA, Russell TM, Vargas JA, and Hoffmann A. (2015). A multi-scale approach reveals that NF-kappaB cRel enforces a B-cell decision to divide. Mol Syst Biol 11, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt LM (2010). Oncogenic Activation of NF-kappa B. Csh Perspect Biol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangye SG (2014). Cytokine-mediated regulation of plasma cell generation: IL-21 takes center stage. Front Immunol 5, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlinton D, Radbruch A, Hiepe F, and Dorner T. (2008). Plasma cell differentiation and survival. Current opinion in immunology 20, 162–169. [DOI] [PubMed] [Google Scholar]
- Wang L, Walker BL, Iannaccone S, Bhatt D, Kennedy PJ, and Tse WT (2009). Bistable switches control memory and plasticity in cellular differentiation. Proc Natl Acad Sci U S A 106, 6638–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Tellier J, Liao Y, Trezise S, Light A, O’Donnell K, Garrett-Sinha LA, Shi W, Tarlinton DM, and Nutt SL (2017). Environmental sensing by mature B cells is controlled by the transcription factors PU. 1 and SpiB. Nature Communications 8, 1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Xu-Monette ZY, Tzankov A, Li X, Manyam GC, Murty V, Bhagat G, Zhang S, Pasqualucci L, Visco C, et al. (2017). Loss of PRDM1/BLIMP-1 function contributes to poor prognosis of activated B-cell-like diffuse large B-cell lymphoma. Leukemia 31, 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H, and Boss JM (2010). PU.1 Binds to a Distal Regulatory Element That Is Necessary for B Cell-Specific Expression of CIITA. J Immunol 184, 5018–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Angelin-Duclos C, Greenwood J, Liao J, and Calame K. (2000). Transcriptional repression by Blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol Cell Biol 20, 2592–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.