

Abstract
Lyme disease, also known as Lyme borreliosis, is the most common tick-transmitted disease in the Northern Hemisphere. The disease is caused by the bacterial spirochete Borrelia burgdorferi and other related Borrelia species. One of the many fascinating features of this unique pathogen is an elaborate system for antigenic variation, whereby the sequence of the surface-bound lipoprotein VlsE is continually modified through segmental gene conversion events. This perpetual changing of the guard allows the pathogen to remain one step ahead of the acquired immune response, enabling persistent infection. Accordingly, the vls locus is the most evolutionarily diverse genetic element in Lyme disease–causing borreliae. Small stretches of information are transferred from a series of silent cassettes in the vls locus to generate an expressed mosaic vlsE gene version that contains genetic information from several different silent cassettes, resulting in ∼1040 possible vlsE sequences. Yet, despite its extreme evolutionary flexibility, the locus has rigidly conserved structural features. These include a telomeric location of the vlsE gene, an inverse orientation of vlsE and the silent cassettes, the presence of nearly perfect inverted repeats of ∼100 bp near the 5′ end of vlsE, and an exceedingly high concentration of G runs in vlsE and the silent cassettes. We discuss the possible roles of these evolutionarily conserved features, highlight recent findings from several studies that have used next-generation DNA sequencing to unravel the switching process, and review advances in the development of a mini-vls system for genetic manipulation of the locus.
Keywords: bacterial genetics, bacterial pathogenesis, DNA mismatch repair, DNA recombination, genetic polymorphism, G-quadruplex, infectious disease, antigenic variation, Borrelia, Lyme disease, gene conversion
Introduction
Antigenic variation is a common pathogenic ruse employed by several bacterial, protozoan, and fungal pathogens (1–14). This process involves changes in a prominent surface antigen such that it is no longer recognized by the host acquired immune response (Fig. 1). By the time the host has assembled and produced antibodies to clear an infecting organism, new variants have appeared, which fly under the radar in terms of immune surveillance. By the time a new generation of antibody molecules has been fashioned to clear the variant pathogens, yet another collection of organisms with prominent but unrecognizable surface antigens has appeared. This cat-and-mouse game can often continue for the long haul, resulting in persistent infection by pathogenic organisms, and provides an efficient mechanism whereby they can avoid clearance by the host immune system.
Figure 1.

Cartoon of antigenic variation. Some pathogens will thwart their recognition by the host immune system by continually changing a prominent surface antigen through changes in allele expression or gene conversion events to modify the expressed allele. In the schematic, the changing surface antigen from the surface of a pathogen is depicted by the red and blue ovals.
Antigenic variation is commonly found in evolutionarily diverse obligate parasites as persistent infection imparts a distinct advantage for transmission of these organisms. A few examples are Plasmodium falciparum (malaria), Trypanosoma brucei (African sleeping sickness), Giardia lamblia (giardiasis or beaver fever), Neisseria gonorrheae (gonorrhea), Treponema pallidum (syphilis), Pneumocystis carinii (diffuse pneumonia), relapsing fever Borrelia (relapsing fever), and Lyme Borrelia (Lyme disease or Lyme borreliosis). Variable surface antigens are generated by recombination events spawning altered proteins or by changes in the allele that is expressed or both. This review will focus on the antigenic variation locus (vls) of the Lyme disease spirochete Borrelia burgdorferi and related Lyme borreliae. In particular, our primary emphases will be on new information and insights since the appearance of an excellent review on the subject by Steven Norris in 2014 (8). An increase in Borrelia genomes sequenced and recent analysis of recombinational switching at the vlsE expression locus by next-generation sequencing have taken us a step forward in understanding this complex process in Lyme Borrelia species.
Lyme disease, or Lyme borreliosis (15, 16), is a tick-transmitted infectious disease caused by several species of spirochetes or spiral-shaped bacteria (although we now know that borreliae display a flat-wave morphology rather than a corkscrew shape (17)). The disease reservoir is usually a small vertebrate, commonly the white-footed mouse. When acquiring a blood meal, larval or nymphal ticks can acquire the infection, which can then be transmitted in a subsequent blood meal. Inoculation of humans with B. burgdorferi through a tick bite first results in a localized infection in the skin, in the area surrounding the bite, often resulting in an erythema migrans (expanding bullseye) rash. Subsequently, the spirochetes invade the vasculature and traffic throughout the body to finally extravasate (escape from the vasculature) into a wide variety of potential locations. In persistent infections, they can promote a constellation of symptoms and pathologies by inducing inflammatory processes (18); these include Lyme arthritis, carditis, central and peripheral neurological manifestations, and acrodermatitis. The disease state depends upon persistence of the spirochetes, which requires antigenic variation. Lyme disease is usually effectively treated by several weeks of treatment with doxycycline or other antibiotics, although a small percentage of those infected may develop post-treatment Lyme disease syndrome (19).
B. burgdorferi, the first characterized Lyme disease agent, was discovered in 1982 (20). Since that time, about 20 related Lyme Borrelia species have been characterized, with B. burgdorferi and Borrelia mayonii being the predominant North American species and Borrelia garinii, Borrelia afzelii, B. burgdorferi, and Borrelia spielmanii being common European species. Apart from their importance as pathogens, Lyme borreliae are truly fascinating in their unusual properties. One of the most interesting is their segmented genomes (21–24). Not only are the genomes segmented (many pieces as opposed to a single chromosome), but most of the replicons, including the chromosome, are linear, a very unusual feature for bacterial genomes. The prototype strain B. burgdorferi B31 carries a linear chromosome, 10 circular and 12 linear plasmids (see Ref. 25 for a review), many of which carry essential information for the enzootic cycle (natural transmission between wild animals and vector) (26). Moreover, the linear replicons are terminated by covalently closed hairpins (27–30), an exceedingly infrequent structure in any type of DNA. These structures require a specialized enzyme, a telomere resolvase, for replication of the DNA and maintenance of the hairpin telomeres (25, 31–33). B. burgdorferi is also unusual in its lack of a role for iron (34), a unique but very useful feature for a pathogen that is an obligate parasite that must survive in mammalian hosts where iron is limiting. B. burgdorferi has lost iron-requiring proteins and uses manganese instead of iron as a component of the few metalloproteins that it encodes. Finally, Borrelia species encode an efficient antigenic variation system that is essential for persistence and has a number of distinctive features described below.
The vls locus
The vls locus is akin to a perpetual motion machine for antigenic variation in Lyme Borrelia species. It was discovered by the pioneering work of the Norris laboratory in the B. burgdorferi type strain B31 (8, 35). The name vls (VMP-like sequence) originates from the sequence relatedness between the vls locus in B. burgdorferi and the variable major protein paralogues in relapsing fever (a tick– or body louse–transmitted disease characterized by recurring febrile episodes) spirochetes (36). The vls locus in B31 is carried on linear plasmid 28-1 (lp28-1). vlsE is the expression locus (35), which encodes the outer surface–localized VlsE lipoprotein (Fig. 2A). In addition to playing a role in antigenic variation, the VlsE protein may also function as a vascular adhesin, which facilitates interaction of the spirochete with the vascular endothelium (37). In strain B31, the vlsE gene is localized near a hairpin telomere with the 15 silent cassettes located adjacent to and upstream of vlsE, but in the opposite orientation (35). A similar arrangement of vls components has been found in a variety of other Borrelia strains and species (8). The reasons for the conserved location of vlsE (∼100 bp or less from the hairpin telomere on the linear plasmid where the vls locus resides) and its orientation (always opposite to the silent cassettes) remain unknown at this time.
Figure 2.

Schematic of the vls antigenic variation locus of B. burgdorferi strain B31. A, the vls expression locus (vlsE) with its promoter (P) is located 82 bp from the right covalently closed hairpin end of the linear plasmid lp28-1. To the left of the promoter and intergenic region (gray) are 15 silent cassettes carrying information corresponding to the variable region of vlsE and situated in the opposite orientation. B, the vlsE region is shown in greater detail, with the constant regions (CR) shown in yellow and the variable region, which corresponds to the information carried in the vls cassettes, shown in blue. The variable region is flanked by 17-bp DRs. To the left of vlsE is its promoter, with the −10 and −35 sequences shown as green bars. Also shown by the bidirectional arrow is a 100-bp perfect IR that partially overlaps the promoter. C, an enlargement of the vlsE gene shows the product of multiple recombinational switching events that result in the copying of genetic information from the silent cassettes into the expression locus, producing a mosaic vlsE carrying information from a number of the silent cassettes. D, an inverted repeat found in the promoter region of the vlsE gene is shown in its normal linear configuration and as an extruded cruciform promoted by negative supercoiling or DNA unwinding from replication or transcription. Modified from Ref. 93. This research was originally published in Molecular Microbiology. Castellanos, M., Verhey, T. B., and Chaconas, G. A Borrelia burgdorferi mini-vls system that undergoes antigenic switching in mice: investigation of the role of plasmid topology and the long inverted repeat. Mol. Microbiol. 2018; 109:710–721. © John Wiley & Sons, Inc.
An intact vlsE gene is absent from most Borrelia genomes sequenced using shotgun cloning in the sequencing protocol. Cloning, PCR amplification, and genetic manipulation of the region have been fraught with difficulties, making analysis of the vlsE locus difficult (see “A mini-vls system”). In addition, analysis of the recombinational switching process has been difficult and time-consuming because switching does not occur in culture and requires mouse infections to study (8). Factor(s) that activate the recombinational switching process in the mouse remain elusive and an important unanswered question.
In the absence of lp28-1, which carries the vlsE locus in B31, low infectivity was noted (38, 39). Targeted deletion of the vls locus or expression of an unswitchable vlsE gene resulted in spirochetes that were competent for infection but were cleared by 3 weeks post-infection (40–43). The ability to vary information in vlsE has also been recently shown to be required for reinfection and is advantageous for the enzootic cycle of B. burgdorferi (44, 45).
A number of interesting features characterize the vls locus.
Segmental gene conversion underlies the antigenic variation process
The diversity-generating power of recombinational switching at vlsE (Fig. 2C) results from the segmental gene conversion events that promote diversity. In this article we define a switch as an inferred recombination tract that is generated by segmental gene conversion that contains one or more SNPs. Switching (the transfer of genetic information) is unidirectional from the silent cassettes to the vlsE gene. As first described by Zhang et al. (35), the vlsE gene in strain B31 is a mosaic of pieces derived from the 15 silent cassettes. The random mixing of genetic information from the silent cassettes into the expression locus gives rise to a theoretical possibility exceeding 1040 distinct vlsE sequence variants (46). In addition, vlsE variability can also occur through nontemplated sequence changes, as will be discussed below. The three-dimensional structure of the VlsE protein has been determined (47), revealing that the amino acid changes resulting from switching at vlsE are found in surface-exposed regions of the protein that would be accessible to antibody molecules.
Nearly perfect inverted repeats in the vlsE promoter region play an unknown role
B. burgdorferi strain B31 has a long inverted repeat (IR)2 (Fig. 2, B and D) of 100 bp, just upstream of vlsE (48). This was not observed in the original sequencing (35) of the locus, due to instability in Escherichia coli. The IR was sequenced using DNA cycle sequencing at high temperature. The IR overlaps the −35 box of the promoter region (Fig. S1). Recently, more IRs have been uncovered by targeted sequencing of several vlsE promoter regions using cycle sequencing, which does not require cloning in E. coli and by the use of next-generation sequencing technology for the second wave of sequencing of Borrelia genomes (see Ref. 49 and Table S1). At this time, the sequence of a total of nine IRs has been documented, including several new sequences reported here (Fig. S1). They comprise five unique IR sequences ranging in size from 93 to 122 bp and have thus far been found in strains from three Lyme Borrelia species: B. burgdorferi, B. garinii, and B. mayonii. The five unique inverted repeats display an average pairwise identity of 49% with a range of 40–72% compared with a pairwise identity of 44% for randomly scrambled sequences of the same base composition (49). The five distinct IRs therefore show little relatedness at the sequence level. They are, however, all found in the vlsE promoter region. Under conditions that generate negative supercoiling (transcription or replication), these IRs can be extruded as cruciform structures (50), where the inverted repeat is unwound and the bases in each repeat on a given strand hydrogen-bond with each other because of their complementarity (Fig. 2D). A role for these distinct IRs with highly variable sequences but which are capable of forming similar structures has not been conclusively demonstrated. Their conserved location in vlsE promoters and near the start of the vlsE gene positions them to play a possible role in either vlsE transcription or recombinational switching, or both. Studies on vlsE transcription when B. burgdorferi is grown in culture have shown that removal of half of the inverted repeat, precluding cruciform formation, has no effect on the level of transcription in culture (51); however, transcriptional patterns change in an animal, and the results from spirochetes grown in culture are not necessarily reflective of mouse infections. Finally, a vexing evolutionary question is how the inverted repeats are generated, given that they are found in different strains and are structurally related but have little sequence homology. All would appear to be capable of cruciform formation driven by negative supercoiling. But their lack of sequence homology would seem to indicate a mutational rate for the IRs that far surpasses any other region in the vls locus, yet this occurs through some mysterious process within the confines of maintaining nearly perfect inverted repeats.
vlsE and the silent cassettes are flanked by 17-bp direct repeats in strain B31
In addition to the IRs, direct repeats (DRs) may also be present. The original sequencing of the vls locus in strain B31 established the existence of well-conserved 17-bp direct repeats flanking the cassettes, and the variable region of vlsE (35). The direct repeats in vlsE (Fig. 1, B and C) as well as most of the repeats present between the cassettes are identical; however, the repeats at the junctions of cassettes 6–7, 8–9, 14–15, and 15–16 vary by one mismatch. The least identical repeat is between cassettes 9 and 10, with five differences found along the sequence. As discussed below, the B31 direct repeats contain G-runs that were shown to form intermolecular G-quadruplex structures in vitro (52). Such structures might possibly play a role in the gene conversion events at vlsE. Other B. burgdorferi strains like PAbe, PBoe, PBre, PKa2, and PRef1 have direct repeats closely related to those in B31. Different Lyme Borrelia species like B. garinii IP90 and B. afzelii ACAI also have the cassettes flanked by 17-bp direct repeats (53). However, these are less conserved than B31 repeats. Moreover, it has been reported that other B. burgdorferi strains like JD1 (49) do not have these flanking direct repeats at all.
The vls locus is peppered with G-runs on the coding strand
The vls locus is a relatively G-C–rich island (48% G-C) in an A-T–rich genome (29.75% G-C). In addition to the very high G-C content of the vls locus, there is a much higher than expected frequency of G-runs. Previous analysis has noted ∼20 runs/1,000 bp of G3–5 (3–5 consecutive guanine bases) on the vls coding strand in contrast to low numbers on the noncoding strand or on either strand of non-vls DNA on lp28-1 in B. burgdorferi B31, N40, and JD1 (52). This property has also been observed in a dozen different B. burgdorferi strains.3 In Fig. 3 (A–C), we now report a similar analysis for three different Lyme Borrelia species, Borrelia garinii Far04, Borrelia spielmanii A14S, and Borrelia mayonii MN14-1529. They also do not display many G-runs on either the coding or noncoding strands of non-vls DNA on the plasmids that carry the vls locus. In contrast, high numbers of G-runs are observed on the coding strand of the vls cassettes, but not on the noncoding strand. This same preference for G-runs on the coding but not noncoding strand of the adjacent expression locus vlsE is also present.
Figure 3.

Distribution of G3+-runs on the linear plasmids carrying the vls loci in Borrelia garinii Far04, Borrelia spielmanii A14S, and Borrelia mayonii MN14-1539 and levels of amino acid sequence identity in RecA, OspC, VlsE constant and VlsE variable regions in Lyme borreliae. A–C, G-runs of 3 nucleotides or more were counted in lp28–9 (CP001316.1), lp28–8 (CP001465.1) and lp28–10 (NZ_CP015805.1) in the three above species, respectively. The distribution of G-runs on both strands of non-vls DNA, in the vls silent cassettes and in the vlsE gene are plotted. The number of G-runs in a codon-optimized vlsE gene generated by reverse translation of the amino acid sequence (https://www.bioinformatics.org/sms2/rev_trans.html) using B. burgdorferi B31 codons (https://www.kazusa.or.jp/codon, species ID: 224326) is also shown. D, Sequence alignments were performed for the 25 available full length RecA sequences that were recovered in a BLAST search (see Table S1 for accession numbers) using DNASTAR MegAlign Pro. The percent identity was determined as the mean of the complete set of pairwise alignments and is shown ± the standard deviation. For OspC and the VlsE variable and N-terminal constant regions the same analysis was performed using sequences from a set of 15 Borrelia strains where both OspC and full-length or near full-length VlsE sequences were available in each strain (see Table S1 for accession numbers). (Please note that the JBC is not responsible for the long term archiving and maintenance of this site or any other third pary hosted site.)
The significance of the large number of G-runs on the coding strand of the vls cassettes and in vlsE in the A-T–rich Borrelia genomes is underscored by the number of G-runs that are found in vlsE when it is codon-optimized using the most commonly used B. burgdorferi codons (Fig. 3, A–C). Not a single G-run was observed in the codon-optimized vlsE genes from B. garinii Far04 or B. mayonii MN14-1539, and only one was found in B. spielmanii A14S vlsE. In contrast, ∼40 G-runs were found in the native vlsE genes, despite the dramatic disfavor of the resulting codons. The maintenance of the G-C content of the vls locus in general and the maintenance of the G-runs, which clearly contravene codon bias in borreliae, is perplexing, especially in a genomic region that undergoes exceedingly high mutagenic drift. This strongly argues for an essential function of the high G-C content and the G-runs based upon the preservation of these features against the strong mutagenic and translational tides.
A possible role for the G-runs is that they may be involved in the formation of G-quadruplex DNA (G4 or 4-stranded DNA stabilized by Hoogsteen hydrogen bonding of the bases) (54–58). The ability of the 17-bp DRs in B31, which carry a stretch of five G residues, to form G4 DNA in vitro has been reported (52). Although these regions can certainly form intermolecular G-quadruplex in vitro, whether such structures form in vivo and whether they influence recombinational switching or other functions is not currently known. Although G4 DNA is an important feature in recombinational switching at Neisseria pilE (5), that situation involves the formation of a specific intramolecular G-quadruplex that serves as a site for DNA nicking. The G4 structure is believed to be processed by the RecQ helicase (59), which is absent from B. burgdorferi. Nor does B. burgdorferi encode a DinG helicase, which resolves G4 structures in Mycobacterium tuberculosis (60). Quadruplex-resolving activity has not been characterized in other Borrelia helicases; however, formation of G4 DNA in Borrelia would necessitate its unwinding for DNA replication (61).
Whether there is formation of intramolecular or intermolecular G-quadruplex (54–57) in vivo is not known. G-runs themselves have not been reported to display any unusual biological properties. However, G4 DNA is a known potent inhibitor of DNA replication and may function thus with specificity for the leading or lagging strand, depending upon other factors (62, 63). The G-runs in vlsE and the silent cassettes are numerous, opening the possibility for promiscuous G4 formation between DNA at a wide variety of locations. This might act as a molecular Velcro, facilitating interaction and synapsis of DNA from distant locations. Alternatively, the nonspecific formation of G4 DNA at multiple sites may provide multiple sites for stalling of DNA replication. Paused replication sites are known to be hotspots of recombinational activity (64, 65). Pause sites might set the stage for one of a variety of replication restart activities that might provide a target for strand exchange by acting as a site for DNA cleavage or by the binding of recombination proteins.
The vls locus is characterized by framework heterogeneity and hypermutability
Lyme Borrelia species generally show a high degree of sequence conservation. As an example, 25 RecA proteins (one of the most conserved proteins in most bacterial species) from nine Borrelia species show 95% sequence identity (Fig. 3D). In contrast, the most highly variable non-VlsE protein, OspC (66), displays only about 76% sequence identity. Yet a comparison of VlsE in B. burgdorferi B31 versus B. burgdorferi 297 exhibited only 46% identity, and a similar comparison of B31 against three other Lyme species showed sequence identity levels of 35–49% (8). Norris has referred to this as “framework heterogeneity” (8) because one can infer from the data a high degree of variability outside the variable regions of VlsE and, therefore, in framework regions of the gene and protein. With more complete or nearly complete VlsE sequences now available in sequence databases, we present here an analysis of the degree of conservation of VlsE in the same 15 Borrelia strains where OspC was compared (Fig. 3D). Moreover, the VlsE N-terminal constant and variable regions were analyzed separately. The degree of divergence of each of these regions was far greater than the most variable B. burgdorferi protein OspC, with protein sequence identity values of 58 and 54% in the constant and variable regions, respectively. It is very intriguing that the VlsE constant region, which comprises the structural underpinning of VlsE (47) and most of which is not antibody-exposed, shows such a high degree of instability. It is not subject to immune selection and would not be expected to display such dramatic variability. Whatever mutagenic forces are involved in shaping the vls locus apparently act upon the entire locus and not just the region of the protein that is surface-accessible and reachable by antibody molecules.
It is tempting to speculate that the multitude of G-runs found throughout the locus that are conserved despite their lack of correspondence with optimal codon usage may play a role in directing hypermutability. Neither G-runs themselves, nor G-quadruplex DNA, nor stalling of replication forks from G-quadruplex structures has been reported to be associated with hypermutability. Nonetheless, G-runs pepper the locus and provide a series of flags that identify vls from the remainder of the A-T–rich genome and could provide a target for factors involved in hypermutation. Promiscuous quadruplex formation involving G-runs throughout the locus might result in DNA breaks and general recombination between similar but nonidentical silent cassettes and, in general, destabilize the entire locus at the nucleotide level.
Switching at vlsE is unidirectional
The unidirectionality of the gene conversion events that underpin antigenic variation remains a mystery (35). Why is genetic information always transferred only from the silent cassettes to vlsE? There are several structural features of the vls locus that might be involved in imparting such a polarity to the direction of information transfer, albeit by unknown mechanisms. One is the IRs, which vary in sequence in different strains but are structurally conserved and always located in the vlsE promoter region. Under conditions of negative supercoiling, the ∼100-bp regions can be extruded as a cruciform and provide a flag for recognition of proteins involved in the reaction and function as a distinct marker for vlsE that is not found in the silent cassettes. A number of replication, recombination, and repair proteins are known to specifically recognize cruciform structures, and cruciforms can be sites for the introduction of single- or double-stranded breaks (67, 68).
Alternatively, transcription of vlsE might provide a distinguishing feature for the expressed gene from the silent cassettes that might be involved in conferring directionality. Transcription results in DNA unwinding, which could facilitate strand invasion into the expression locus or cruciform extrusion of the IR. Moreover, the IR, when extruded as a cruciform, would reduce the level of DNA supercoiling in the promoter region, which can influence the level of transcription at B. burgdorferi promoters (69–71).
Finally, the reverse orientation of vlsE versus the silent cassettes results in a reversal of the location of the coding information from the leading strand template for the silent cassettes to the lagging strand template for the vlsE gene, resulting in Okazaki fragments originating from opposite strands and a preference for serving as a DNA donor versus recipient. Replication origins in B. burgdorferi are located near the center of the linear replicons with replication proceeding bidirectionally toward each hairpin telomere (72–74). An alternative explanation for vlsE and the silent cassettes having opposite orientations is simply that this arrangement results in entropically favored synapsis of the silent cassettes and vlsE.
There is a paucity of identified protein factors
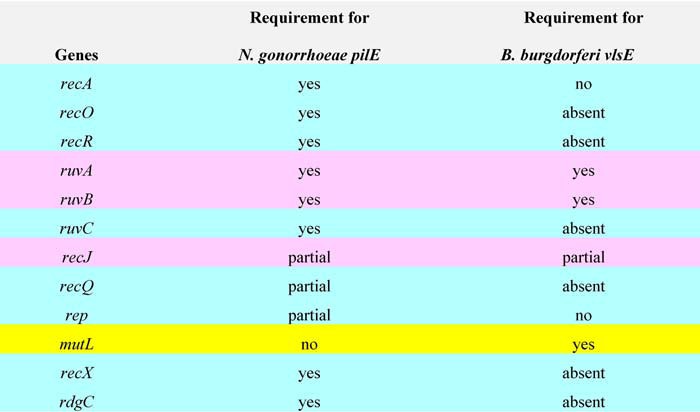
Another enigmatic feature of recombinational switching at vlsE is the protein complement required to promote the reaction, which remains largely obscure. Perhaps most surprising is the lack of a requirement for the RecA protein (75, 76) and other proteins involved in homologous recombination, which appear to be required for gene conversion in other characterized antigenic variation systems (1, 5, 10, 77). A comparison of the protein factors required for antigenic variation of PilE in N. gonorrheae (3) and B. burgdorferi VlsE (75, 76, 78) is shown in Table 1. Of the 11 genes known to play a role in switching at pilE, only three are common to both organisms: ruvA and ruvB, which encode the subunits of the RuvAB branch migrase are required in both cases, and the recJ gene encoding a 5′ to 3′ single strand–specific exonuclease is partially required in both organisms. mutL, a mismatch repair protein that is not required in Neisseria appears to be important in B. burgdorferi.3 The sparsity of identified proteins that promote switching at vlsE leaves a large gap in our understanding of this recombination process at the molecular level, especially the dispensability of RecA.
Table 1.
Recombination/replication/repair genes required for recombinational switching of pilE (3, 97) and vlsE (T.B Verhey, M. Castellanos, and G. Chaconas, unpublished results) (75, 78)
Genes highlighted in blue are required or partially required for pilE but not vlsE (absent indicates that the gene is not present in B. burgdorferi). Those highlighted in pink are common to both systems, and mutL, highlighted in yellow is required for vlsE but not pilE.
How a homology-driven process occurs in the absence of the RecA protein remains a puzzle. A possible solution to this issue may exist in the telomere resolvase, ResT (32, 33). This protein is required for the resolution of replicated telomeres (also referred to as dimer junctions) to generate the covalently closed hairpin telomeres on the linear replicons in Borrelia. In addition to its telomere resolution activity, ResT has been recently reported to have DNA single-strand annealing and strand exchange activity and ATP-dependent DNA unwinding activity (79–81). It has also been implicated in DNA replication activities other than telomere resolution (82). It is tempting to consider ResT as a possible player in switching at vlsE for the following reasons: it is a participant in DNA replication in general in B. burgdorferi, it has the types of activities involved in recombination events, its site of action (the hairpin telomeres) is less than 100 bp from vlsE, and it promotes the formation of transient double-strand DNA breaks, which might promote recombination at vlsE. Double-strand breaks have been shown to promote gene conversion events in T. brucei (83). Because of its essential nature (82, 84), a mutagenic approach to study a possible role of ResT in switching at vlsE is fraught with complexity.
It is also worthy of note that a link between switching at vlsE and the DNA replication process is certainly a feasible possibility, although no direct evidence in support of this exists at present. Related to this idea is the point that the genetic approach to identifying factors involved in switching at vlsE does suffer from the inability to identify essential proteins, such as those involved in DNA replication. Therefore, factors essential for viability, such as DNA polymerases, and other replication proteins may have thus far escaped identification. Related to the replication issue is that of plasmid copy number for lp28-1, which has not been directly determined. With a copy number greater than one, intermolecular rather than intramolecular recombination would be a possibility.
New insights, unanswered questions, and speculations
The last 2 years have seen some important technical advances in the study of the vls locus, which have resulted in an increased understanding of the antigenic variation process. These include the development of a next-generation sequencing (NGS) approach to analyze recombinational switching at vlsE and a fully automated sequencing pipeline to analyze sequencing data. They also include the development of a mini-vls system, allowing, for the first time, genetic manipulation of the component parts of the vls locus.
NGS analysis of recombinational switching
An impediment to analyzing recombinational switching at the vlsE locus has been the very high level of sequence conservation between switched vls variants and the very short length of the sequence reads generated by most NGS methods. Accurate contig assembly of the short sequence reads was therefore an impossible task. To overcome this obstacle, PacBio long read sequencing technology was used to sequence 776-bp amplicons of switched vlsE variants (46, 85). The high error frequency typical of PacBio sequencing was greatly reduced using a circular-consensus approach and filtering for base calls of high accuracy. Filtering resulted in a loss of a substantial portion of the sequencing data, but the high-stringency sequence remaining contained less than one error per 10 vlsE amplicons, a level of accuracy sufficient for a variety of analyses.
Software developments to analyze NGS switching data
To analyze the large number of switch events sequenced (45,000) with nucleotide sensitivity and to answer the unique questions associated with the recovered segmental gene conversion events, a unique fully automated sequencing pipeline was developed (46, 85). VAST (variable antigen sequence tracer) is a command-line tool for custom analysis of full-length vlsE sequences. VAST runs on a Linux platform and acts as a database manager for large libraries of sample-tagged full-length vlsE sequences. It has also been designed to consistently align ambiguous polymorphisms found in vlsE and can perform a constellation of analyses on directed groupings and data subsets. A particular strength is that VAST was designed to optimize the assignment of switched bases in vlsE genes to the silent cassettes from which they were derived. With multiple redundancies between silent cassettes and multiple gene conversion events occurring in vlsE, this is not a trivial undertaking. To be properly executed, this requires an automated and systematic analysis that is free from bias and able to analyze large data sets at nucleotide resolution, something that a manual analysis (86) cannot do.
Analysis of the switching process using long read sequencing and VAST software has revealed a variety of important new information (46, 49, 85). Analyses were performed using B. burgdorferi B31 to infect both WT and SCID (severe combined immunodeficient) mice. The SCID mice allow analysis of recombination events in the absence of immune selection, which perturbs the survival of older switch variants. The system was validated by a general agreement of findings with those reported earlier using a classical sequencing approach, a smaller data set, and manual data analysis. Those findings, including cassette usage and recombination tract length, are not discussed here, and for details, see Refs. 85 and 86.
Although switching at the vlsE locus of strain B31 has been known to exist for 2 decades, an in-depth analysis of this phenomenon in other strains has not been undertaken. A recent NGS/VAST analysis of switching at vlsE in B. burgdorferi strain JD1 was also undertaken (49). Despite differences in genetic background, a less structured vls locus, a completely different IR, and the lack of DRs, the properties of switching in this strain were found to be quite similar to those observed for B31.
New insights from the NGS sequencing of switching in strain B31 coupled with analysis by VAST are described in the sections below.
A mutational heat map and spirochete dissemination as a one-way street
A heat map showing the frequency of mutation at each position on the three-dimensional structure was derived, providing quantitative data on the most highly variable positions (Fig. 4A and Video S1). These positions (changed up to 74% of the time) are those on the surface of the molecule (46), farthest from the N terminus, which is lipidated and tethered to the outer membrane. In contrast, the N-terminal region that is juxtaposed to the membrane shows little or no variability. These results are in agreement with previous analyses (47, 86) but provide a much more detailed data set from the large number of switch events analyzed by NGS.
Figure 4.

Heat map of mutational frequencies in B31 VlsE and switching at vlsE in SCID versus WT mice. A, variant vlsE sequences were mapped onto the crystal structure of VlsE (47) (Protein Data Bank entry 1L8W) onto both protomers of the dimer structure. The frequency of mutation was depicted using a continuum of color with 0% change indicated by dark blue, ∼37% by green, ∼55% by yellow, and ∼74% by red. For a 360° rotation of the structure, see Video S1. B, the positions of purifying (red) and diversifying (green) mutations are shown on the crystal structure of B31 VlsE. Purifying residues are those with the highest SCID/WT ratios, and diversifying residues are those with the highest WT/SCID ratios. The dotted line is the axis of 180° rotational symmetry. For a 360° rotation of the structure, see Video S2. A and B are from Ref. 46. This research was originally published in Molecular Microbiology. Verhey, T. B., Castellanos, M., and Chaconas, G. Analysis of recombinational switching at the antigenic variation locus of the Lyme spirochete using a novel PacBio sequencing pipeline. Mol. Microbiol. 2018; 107:104–115. © John Wiley & Sons, Inc.
Interestingly, of the thousands of vlsE switch variants analyzed, 99.6% were uniquely found in each tissue in a given SCID mouse. Therefore, spirochetes trafficking between tissues are extremely rare, and vascular dissemination from the site of the tick bite to different tissues appears to be overwhelmingly unidirectional (46). The NGS data set has allowed a clear answer to this issue, which was not previously possible to investigate.
Dimerization of VlsE is important for function
An analysis of residues undergoing purifying versus diversifying selection was performed by comparing the frequency of mutations at each position in VlsE in WT versus SCID (no immune selection) mice (46, 49). Purifying selection results in maintenance or enrichment of amino acids at given positions, whereas diversifying selection results in diversification of the amino acids at given positions. The residues undergoing diversifying selection were located primarily in the surface loops, where changes would facilitate an escape from immune surveillance (Fig. 4B and Video S2). Conversely, amino acids undergoing purifying selection were localized internally or at the dimer interface, as might be expected for conserved structural residues. The presence of several positions undergoing purifying selection at the dimer interface strongly argues that the functional unit of VlsE is at least a dimer and not a monomer, a new piece of information not available from other analyses.
Determination of the rate of recombinational switching
It has not been previously possible to determine a rate of recombinational switching at vlsE in B. burgdorferi. Using the NGS/VAST tools, that rate (Fig. 5A) has been reported to be 0.7 switches per week per vlsE sequence (85). Assuming a copy number of one for lp28-1 (almost certainly an underestimate (69, 87)) and a doubling time of 8 h (likely also an underestimate), this translates to a switching rate of about 3.3 × 10−2 per spirochete per generation. At this rate, an infection with only 30 spirochetes would generate one new variant every 8 h at the onset of infection, with higher numbers resulting from a doubling in the number of variants generated every 8 h with each new round of DNA replication.
Figure 5.
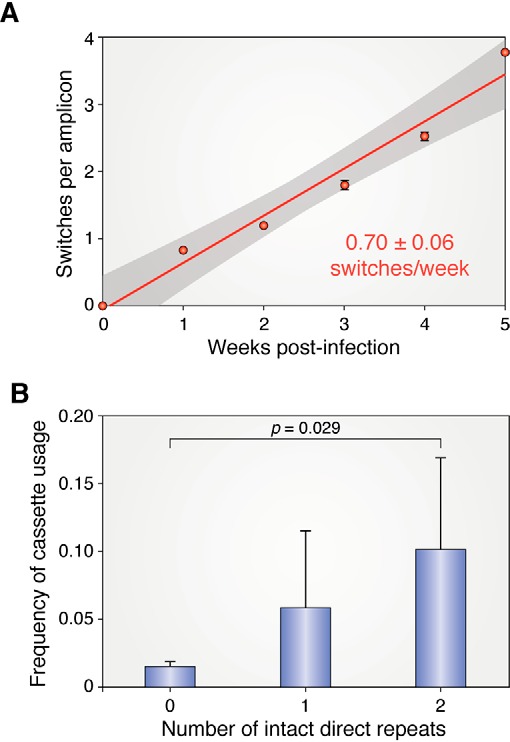
Rate of recombinational switching at vlsE and correlation of cassette usage and intact DR repeats. A, inferred switch event accumulation per read is shown from 0 to 5 weeks post-infection. Linear regression by least squares is shown, with the gray area indicating the region of 95% confidence. The switching rate was measured in SCID mice, where the variants generated are stable and not cleared by immune selection. B, the use of each cassette as a donor of genetic information to vlsE was determined and plotted against the number of completely conserved flanking 17-bp direct repeats. Error bars, S.D. A and B are from Ref. 85. This research was originally published in Cell Reports. Verhey, T. B., Castellanos, M., and Chaconas, G. Antigenic variation in the Lyme disease spirochete: new insights into the mechanism of recombinational switching with a suggested role for error-prone repair. Cell Reports. 2018; 23:2595–2605. © Elsevier Inc.
A role for the DRs and for G-runs in switching?
A possible role for the DRs was hypothesized when the B31 vlsE was first discovered (35). They were later thought to be of questionable importance based upon their nonexistence in some strains and the fact that they are not well-conserved among many strains even when present (53). However, NGS sequencing data suggest that they may in fact have a stimulatory role in gene conversion. Fig. 5B shows the frequency of silent cassette usage plotted against the number of fully conserved DRs flanking the cassette. A clear correlation was found between the number of intact cassettes and the usage of a cassette as a donor for gene conversion in B31. In contrast, an NGS analysis of switching at vlsE of B. burgdorferi strain JD1, a strain that lacks DRs, revealed switching properties similar to those found for strain B31. The possible role and mechanism of DR switching stimulation in B31 remain enigmatic at this time.
NGS analysis of switching at vlsE was also carried out to probe a possible role for G-runs and switching. The data did not reveal a correlation between the location of G-runs and the initiation of recombination (85), leaving the function of their highly elevated conserved presence a mystery. Although there was no correlation between gene conversion junctions and G-runs, this does not preclude a more subtle role for G-runs in recombinational switching.
Clustering of switching in the population and along the DNA
As noted earlier, the mechanism and factors responsible for turning on switching at vlsE after infection remain unknown. However, the recent NGS analysis has revealed two important new pieces of information related to this issue. The first is that at early time points when the dispersal of vlsE switch events in different copies of vlsE can be analyzed, there is not a stochastic distribution. Instead, there is a propensity for a second switch event in a spirochete where switching has already occurred (85). Moreover, there is a physical clustering of switch events along the DNA. In other words, second switch events occur closer to existing switch events than expected if they were to occur at random (Fig. 6A). These results suggest that switch events are clustered in space and time and preferentially occur in the same cell and in physical proximity to the last switch event, perhaps due to physical constraints, such as the position of a replication fork or the local concentration of protein factors that play a role in the reaction.
Figure 6.
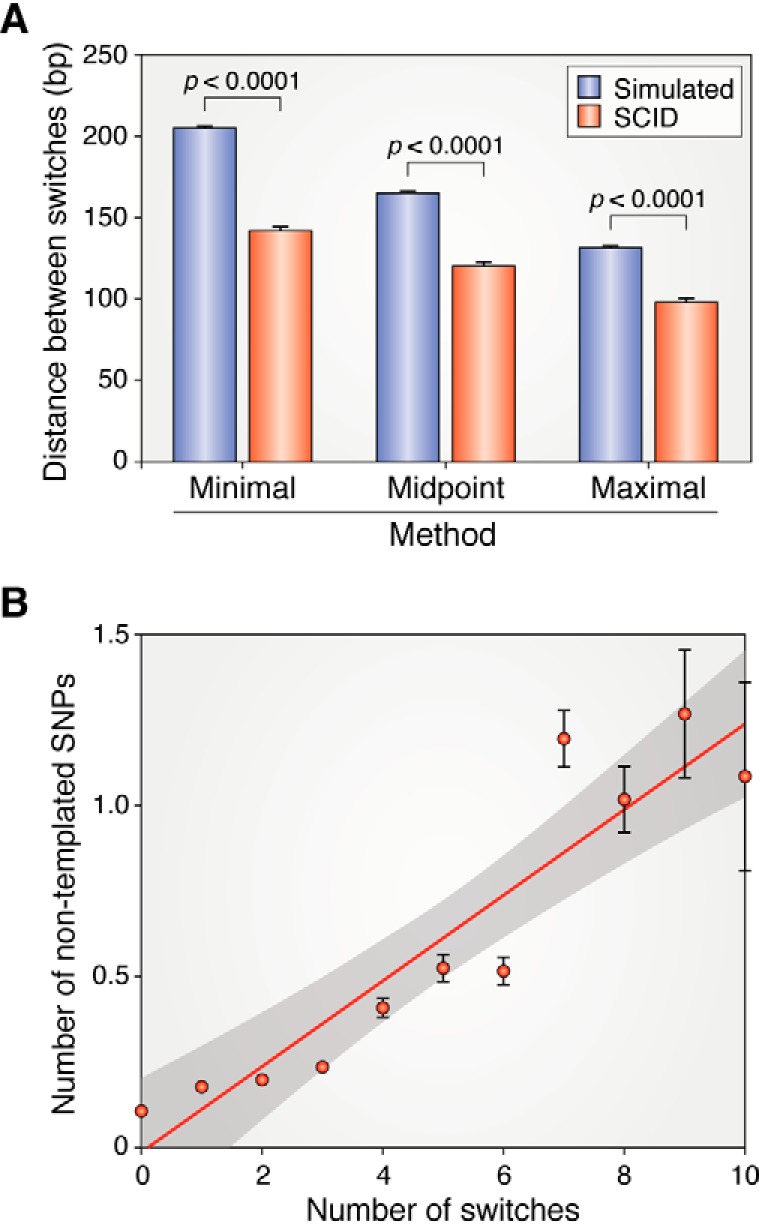
Distance between inferred switch events in spirochetes with two switches at 1 week post-infection and correlation of nontemplated SNPs with the number of switch events. A, the distance between inferred switch events was determined and plotted three ways: using the minimal possible switch lengths (which have the largest distance between switches), the maximal possible switch lengths (which have the shortest distance between the switches), or the midpoint between minimal and maximal. Each value was compared with the distance between 106 randomly generated switch variants. B, the number of nontemplated SNPs per read was enumerated and plotted against the number of templated switch events in the same read (reads with 1–10 switches were analyzed). The least-squares regression line for the data is in red, and the 95% confidence limits are indicated by gray shading. Error bars, S.E.M. A and B are from Ref. 85. This research was originally published in Cell Reports. Verhey, T. B., Castellanos, M., and Chaconas, G. Antigenic variation in the Lyme disease spirochete: new insights into the mechanism of recombinational switching with a suggested role for error-prone repair. Cell Reports. 2018; 23:2595–2605. © Elsevier Inc.
Switching is promoted by sequence homology
Although switching at vlsE is RecA-independent, the positions at which switching occurs are nonetheless correlated with the level of sequence homology (85). Moreover, switch events located 30 bp or less from the edge of a cassette boundary were frequently underrepresented, suggesting a requirement of 20–30 bp of homology for recombination to occur, despite the expendability of the RecA protein. A role for annealing of homologous sequences in switching at vlsE is apparent from these results; however, the molecular mechanism remains uncharacterized.
Nontemplated SNPs are generated by error-prone repair
Nontemplated SNPs are defined as changes in vlsE that do not correspond to the sequence found in any of the silent cassettes. These changes have been observed previously (86, 88), but their origin has remained mysterious. Analysis using the NGS/VAST system (85) has revealed some interesting properties of nontemplated SNPs. A variety of controls indicate that these SNPs are not sequencing errors and that the number of nontemplated SNPs correlates well with the number of switch events (Fig. 6B) (85). Nontemplated SNPs accumulate over time at a rate that is about 10% of the rate of templated switch events but more than 5,000-fold faster than the background mutation rate of B. burgdorferi. Analysis of about 1,000 nontemplated SNPs indicated a dramatic preference for these mutations on the 5′ side (coding strand) of switch events. Therefore, nontemplated switches appear to be the result of error-prone repair associated with recombinational switching and provide a second layer of variability to facilitate antibody avoidance. Error-prone repair can occur in regions undergoing recombination or mismatch repair in bacteria (89, 90). The compact genomes of Lyme borreliae encode a DNA polymerase III and a polymerase I (BB0548), but an error-prone DNA polymerase such as DinB is not known to exist in Lyme spirochetes. Hypermutation can also occur at a variety of steps along the mismatch repair pathway; however, this process has not been studied in any detail in B. burgdorferi. The mechanism by which hypermutability is specifically endowed to vlsE remains to be established. The error-prone repair associated with gene conversion to generate nontemplated variability is likely associated with the DNA breaks involved in the recombinational switching process (90). This would limit the mutations in time and space to regions undergoing antigenic variation, as has been observed (85).
A mini-vls system
As noted earlier, cloning, PCR amplification, and genetic manipulation of the vls locus have been difficult. The reasons for this are as follows.
(i) The physical location of vlsE genes at the end of linear plasmids makes cloning difficult due to a covalently closed DNA hairpin about 75–100 bp away from the 3′-end of the gene.
(ii) The presence of a perfect or nearly perfect inverted repeat (Fig. 2B) of 93–120 bp located about 50 bp upstream of the 5′-end of vlsE genes imparts an instability in this region when cloned in E. coli, even in sbc mutants (91) where the Sbc nuclease that cleaves at inverted repeats is absent.3
(iii) Cloning of expressed vlsE is problematic unless promoter mutations that reduce expression are present.3
(iv) The repetitive nature of the 15 silent cassettes arranged as continuous direct repeats causes cloning difficulties from recombination of directly repeated cassettes, generating deletions. PCR amplification of the vls locus is also problematic, with many false priming sites and difficulty obtaining the desired products.
Genetic analysis of the native locus has not been possible and has been limited to in vivo deletion of the entire locus (40). This was accomplished by the insertion of a replicated B. burgdorferi telomere, which is then processed by the telomere resolvase ResT (32) to generate a double-stranded break with hairpin telomeres on each side; the result is loss of the fragment lacking the origin of replication and associated replication protein genes (72, 92).
To circumvent the inability to genetically manipulate the 10-kb locus, a mini-vls system was developed by direct cloning into a high-passage, highly transformable B. burgdorferi HB19 strain (93). Constructs unclonable in E. coli were recovered in the high-passage B. burgdorferi recipient strain and subsequently transferred into a low-passage strain for mouse infections and NGS analysis of recombinational switching at vlsE. The mini-vlsE system carries vlsE, the intergenic region (between vlsE and cassette 2), and cassette 2 carried on an lp5-derived shuttle vector. This system has allowed the first genetic manipulation of several components of the vls locus, whose role in gene conversion at vlsE was then assessed as described below. One caveat on the use of the mini system is that it has a greatly reduced level of switching compared with the full-length vls locus, raising the question of whether data from the mini vls faithfully mirrors that from the intact locus. Nonetheless, it is a starting point for genetic analyses that have been stalled for 20 years.
The role of the IR and plasmid topology
The fact that the vls locus is always found on a linear plasmid has suggested the possibility that the topology of the plasmid that it is carried on is important. However, the inability to genetically manipulate the locus made it impossible to investigate this issue until now. Development of the mini-vls system on both a circular and a linear form of the same plasmid has allowed examination of this question (93). The finding from NGS-switching analysis was that when the upstream IR was present, switching frequency on the circular and linear plasmids was the same. However, removal of the IR resulted in a decrease in switching to nearly background levels on the circular plasmid, but switching was unaffected on the otherwise identical linear construct. The results point to an interrelated role for the topological form of the plasmid and the upstream IR; however, a cogent explanation for the results does not currently exist. The 100-bp IR can be spontaneously extruded as a cruciform in vitro3 and is likely to do so in vivo during DNA replication and transcription of vlsE on both topological forms of the plasmid. But why this may be more important for recombinational switching on the circular plasmid form is currently a mystery, as is the presence of perfect or nearly perfect IRs just upstream of vlsE in a hypermutable region of DNA.
Perspective
In closing this review, it is worth noting how the study of recombinational switching at the vls locus in Lyme spirochetes may contribute new understandings and potential translational information in general. As one of many pathogenic organisms using antigenic variation to avoid immune surveillance, information gained on the Lyme spirochetes may help to inform mechanisms of antigenic variation employed by other pathogens. Moreover, antigenic variation makes an attractive target for drug development for Lyme disease because switching at vlsE is a requirement for persistence. Whereas antibiotic treatment is generally effective for treatment of Lyme disease, an increasing number of reports of spirochetes that survive antibiotics in treated animals have recently appeared (94–96). Although cultivatable disease-causing spirochetes have not been recovered in these studies, these unexpected findings suggest the ability of Borrelia spirochetes to survive long-term in the presence of antibiotics. The relationship between such long-term surviving spirochetes and the disease state or post-treatment Lyme disease syndrome remains to be established. If there is a link, then the development of a drug blocking recombinational switching at vlsE would offer a promising response to drug-surviving spirochetes and would provide a direct block to the development of long-term persistence.
Apart from the clinical relevance noted above, switching at the vlsE locus presents a new recombinational paradigm that utilizes sequence homology to promote recombination in the absence of the RecA protein. The molecular mechanism of the gene conversion events will provide new information on the diversity of recombination mechanisms. Unraveling the molecular details could also potentially provide additional approaches for precise correction of mutations that cause genetic disease. In addition to recombination mechanisms, the vls locus provides a fascinating model for accelerated evolution of the entire locus as well as for the hypermutability by error-prone repair that is associated with recombinational switching. Further studies on this versatile and fascinating system for immune evasion will expand our knowledge in a variety of areas.
Supplementary Material
This work was supported by Canadian Institutes of Health Research Grant PJT-153336. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1, Fig. S1, and Videos S1 and S2.
G. Chaconas, M. Castellanos, and T. B. Verhey, unpublished results.
- IR
- inverted repeat
- DR
- direct repeat.
References
- 1. Deitsch K. W., Lukehart S. A., and Stringer J. R. (2009) Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat. Rev. Microbiol. 7, 493–503 10.1038/nrmicro2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gargantini P. R., Serradell M. D. C., Ríos D. N., Tenaglia A. H., and Luján H. D. (2016) Antigenic variation in the intestinal parasite Giardia lamblia. Curr. Opin. Microbiol. 32, 52–58 10.1016/j.mib.2016.04.017 [DOI] [PubMed] [Google Scholar]
- 3. Cahoon L. A., and Seifert H. S. (2011) Focusing homologous recombination: pilin antigenic variation in the pathogenic Neisseria. Mol. Microbiol. 81, 1136–1143 10.1111/j.1365-2958.2011.07773.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McCulloch R., Morrison L. J., and Hall J. P. (2015) DNA recombination strategies during antigenic variation in the African trypanosome. Microbiol. Spectr. 3, MDNA3-0016-2014 10.1128/microbiolspec.MDNA3-0016-2014 [DOI] [PubMed] [Google Scholar]
- 5. Obergfell K. P., and Seifert H. S. (2015) Mobile DNA in the pathogenic Neisseria. Microbiol. Spectr. 3, MDNA3-0015-2014 10.1128/microbiolspec.MDNA3-0015-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wahlgren M., Goel S., and Akhouri R. R. (2017) Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat. Rev. Microbiol. 15, 479–491 10.1038/nrmicro.2017.47 [DOI] [PubMed] [Google Scholar]
- 7. Vink C., Rudenko G., and Seifert H. S. (2012) Microbial antigenic variation mediated by homologous DNA recombination. FEMS Microbiol. Rev. 36, 917–948 10.1111/j.1574-6976.2011.00321.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Norris S. J. (2014) vls antigenic variation systems of Lyme disease Borrelia: eluding host immunity through both random, segmental gene conversion and framework heterogeneity. Microbiol. Spectr. 2, MDNA3-0038-2014 10.1128/microbiolspec.MDNA1123-0038-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barbour A. G., and Guo B. P. (2010) Pathogenesis of relapsing fever. in Borrelia: Molecular Biology, Host Interaction and Pathogenesis (Samuels D. S., and Radolf J. D., eds) pp. 327–351, Horizon Scientific Press, Norwich, UK [Google Scholar]
- 10. Devlin R., Marques C. A., and McCulloch R. (2017) Does DNA replication direct locus-specific recombination during host immune evasion by antigenic variation in the African trypanosome? Curr. Genet. 63, 441–449 10.1007/s00294-016-0662-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Prucca C. G., Rivero F. D., and Luján H. D. (2011) Regulation of antigenic variation in Giardia lamblia. Annu. Rev. Microbiol. 65, 611–630 10.1146/annurev-micro-090110-102940 [DOI] [PubMed] [Google Scholar]
- 12. Volz J. C., Bártfai R., Petter M., Langer C., Josling G. A., Tsuboi T., Schwach F., Baum J., Rayner J. C., Stunnenberg H. G., Duffy M. F., and Cowman A. F. (2012) PfSET10, a Plasmodium falciparum methyltransferase, maintains the active var gene in a poised state during parasite division. Cell Host Microbe 11, 7–18 10.1016/j.chom.2011.11.011 [DOI] [PubMed] [Google Scholar]
- 13. Glover L., Hutchinson S., Alsford S., McCulloch R., Field M. C., and Horn D. (2013) Antigenic variation in African trypanosomes: the importance of chromosomal and nuclear context in VSG expression control. Cell Microbiol. 15, 1984–1993 10.1111/cmi.12215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horn D. (2014) Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 195, 123–129 10.1016/j.molbiopara.2014.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steere A. C., Strle F., Wormser G. P., Hu L. T., Branda J. A., Hovius J. W., Li X., and Mead P. S. (2016) Lyme borreliosis. Nat. Rev. Dis. Primers 2, 16090 10.1038/nrdp.2016.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stanek G., and Strle F. (2018) Lyme borreliosis-from tick bite to diagnosis and treatment. FEMS Microbiol. Rev. 42, 233–258 10.1093/femsre/fux047 [DOI] [PubMed] [Google Scholar]
- 17. Motaleb M. A., Corum L., Bono J. L., Elias A. F., Rosa P., Samuels D. S., and Charon N. W. (2000) Borrelia burgdorferi periplasmic flagella have both skeletal and motility functions. Proc. Natl. Acad. Sci. U.S.A. 97, 10899–10904 10.1073/pnas.200221797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Norris S. J., Coburn J., Leong J. M., Hu L. T., and Hook M. (2010) Pathobiology of Lyme disease Borrelia. in Borrelia: Molecular Biology, Host Interaction and Pathogenesis (Samuels D. S., and Radolf J. D., eds) pp. 293–325, Horizon Scientific Press, Norwich, UK [Google Scholar]
- 19. Aucott J. N., Crowder L. A., and Kortte K. B. (2013) Development of a foundation for a case definition of post-treatment Lyme disease syndrome. Int. J. Infect. Dis. 17, e443–e449 10.1016/j.ijid.2013.01.008 [DOI] [PubMed] [Google Scholar]
- 20. Burgdorfer W., Barbour A. G., Hayes S. F., Benach J. L., Grunwaldt E., and Davis J. P. (1982) Lyme disease-a tick-borne spirochetosis? Science 216, 1317–1319 10.1126/science.7043737 [DOI] [PubMed] [Google Scholar]
- 21. Fraser C. M., Casjens S., Huang W. M., Sutton G. G., Clayton R., Lathigra R., White O., Ketchum K. A., Dodson R., Hickey E. K., Gwinn M., Dougherty B., Tomb J. F., Fleischmann R. D., Richardson D., et al. (1997) Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390, 580–586 10.1038/37551 [DOI] [PubMed] [Google Scholar]
- 22. Margos G., Hepner S., Mang C., Marosevic D., Reynolds S. E., Krebs S., Sing A., Derdakova M., Reiter M. A., and Fingerle V. (2017) Lost in plasmids: next generation sequencing and the complex genome of the tick-borne pathogen Borrelia burgdorferi. BMC Genomics 18, 422 10.1186/s12864-017-3804-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Casjens S. R., Gilcrease E. B., Vujadinovic M., Mongodin E. F., Luft B. J., Schutzer S. E., Fraser C. M., and Qiu W. G. (2017) Plasmid diversity and phylogenetic consistency in the Lyme disease agent Borrelia burgdorferi. BMC Genomics 18, 165 10.1186/s12864-017-3553-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Casjens S. R., Di L., Akther S., Mongodin E. F., Luft B. J., Schutzer S. E., Fraser C. M., and Qiu W. G. (2018) Primordial origin and diversification of plasmids in Lyme disease agent bacteria. BMC Genomics 19, 218 10.1186/s12864-018-4597-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chaconas G., and Kobryn K. (2010) Structure, function, and evolution of linear replicons in Borrelia. Annu. Rev. Microbiol. 64, 185–202 10.1146/annurev.micro.112408.134037 [DOI] [PubMed] [Google Scholar]
- 26. Stewart P. E., Byram R., Grimm D., Tilly K., and Rosa P. A. (2005) The plasmids of Borrelia burgdorferi: essential genetic elements of a pathogen. Plasmid 53, 1–13 10.1016/j.plasmid.2004.10.006 [DOI] [PubMed] [Google Scholar]
- 27. Barbour A. G., and Garon C. F. (1987) Linear plasmids of the bacterium Borrelia burgdorferi have covalently closed ends. Science 237, 409–411 10.1126/science.3603026 [DOI] [PubMed] [Google Scholar]
- 28. Hinnebusch J., and Barbour A. G. (1991) Linear plasmids of Borrelia burgdorferi have a telomeric structure and sequence similar to those of a eukaryotic virus. J. Bacteriol. 173, 7233–7239 10.1128/jb.173.22.7233-7239.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hinnebusch J., Bergström S., and Barbour A. G. (1990) Cloning and sequence analysis of linear plasmid telomeres of the bacterium Borrelia burgdorferi. Mol. Microbiol. 4, 811–820 10.1111/j.1365-2958.1990.tb00651.x [DOI] [PubMed] [Google Scholar]
- 30. Tourand Y., Deneke J., Moriarty T. J., and Chaconas G. (2009) Characterization and in vitro reaction properties of 19 unique hairpin telomeres from the linear plasmids of the Lyme disease spirochete. J. Biol. Chem. 284, 7264–7272 10.1074/jbc.M808918200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chaconas G. (2005) Hairpin telomeres and genome plasticity in Borrelia: all mixed up in the end. Mol. Microbiol. 58, 625–635 10.1111/j.1365-2958.2005.04872.x [DOI] [PubMed] [Google Scholar]
- 32. Kobryn K., and Chaconas G. (2002) ResT, a telomere resolvase encoded by the Lyme disease spirochete. Mol. Cell 9, 195–201 10.1016/S1097-2765(01)00433-6 [DOI] [PubMed] [Google Scholar]
- 33. Kobryn K., and Chaconas G. (2014) Hairpin telomere resolvases. Microbiol. Spectr. 2, MDNA3-0023-2014 10.1128/microbiolspec.MDNA3-0023-2014 [DOI] [PubMed] [Google Scholar]
- 34. Posey J. E., and Gherardini F. C. (2000) Lack of a role for iron in the Lyme disease pathogen. Science 288, 1651–1653 10.1126/science.288.5471.1651 [DOI] [PubMed] [Google Scholar]
- 35. Zhang J. R., Hardham J. M., Barbour A. G., and Norris S. J. (1997) Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell 89, 275–285 10.1016/S0092-8674(00)80206-8 [DOI] [PubMed] [Google Scholar]
- 36. Saint Girons I., and Barbour A. G. (1991) Antigenic variation in Borrelia. Res. Microbiol. 142, 711–717 10.1016/0923-2508(91)90085-O [DOI] [PubMed] [Google Scholar]
- 37. Antonara S., Chafel R. M., LaFrance M., and Coburn J. (2007) Borrelia burgdorferi adhesins identified using in vivo phage display. Mol. Microbiol. 66, 262–276 10.1111/j.1365-2958.2007.05924.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Purser J. E., and Norris S. J. (2000) Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. U.S.A. 97, 13865–13870 10.1073/pnas.97.25.13865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Labandeira-Rey M., and Skare J. T. (2001) Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect. Immun. 69, 446–455 10.1128/IAI.69.1.446-455.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bankhead T., and Chaconas G. (2007) The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol. Microbiol. 65, 1547–1558 10.1111/j.1365-2958.2007.05895.x [DOI] [PubMed] [Google Scholar]
- 41. Magunda P. R. H., and Bankhead T. (2016) Investigating the potential role of non-vls genes on linear plasmid 28-1 in virulence and persistence by Borrelia burgdorferi. BMC Microbiol. 16, 180 10.1186/s12866-016-0806-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lawrenz M. B., Wooten R. M., and Norris S. J. (2004) Effects of vlsE complementation on the infectivity of Borrelia burgdorferi lacking the linear plasmid lp28-1. Infect. Immun. 72, 6577–6585 10.1128/IAI.72.11.6577-6585.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rogovskyy A. S., and Bankhead T. (2013) Variable VlsE is critical for host reinfection by the Lyme disease spirochete. PLoS One 8, e61226 10.1371/journal.pone.0061226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rogovskyy A. S., Casselli T., Tourand Y., Jones C. R., Owen J. P., Mason K. L., Scoles G. A., and Bankhead T. (2015) Evaluation of the importance of VlsE antigenic variation for the enzootic cycle of Borrelia burgdorferi. PLoS One 10, e0124268 10.1371/journal.pone.0124268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bankhead T. (2016) Role of the VlsE lipoprotein in immune avoidance by the Lyme disease spirochete Borrelia burgdorferi. For. Immunopathol. Dis. Therap. 7, 191–204 10.1615/ForumImmunDisTher.2017019625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verhey T. B., Castellanos M., and Chaconas G. (2018) Analysis of recombinational switching at the antigenic variation locus of the Lyme spirochete using a novel PacBio sequencing pipeline. Mol. Microbiol. 107, 104–115 10.1111/mmi.13873 [DOI] [PubMed] [Google Scholar]
- 47. Eicken C., Sharma V., Klabunde T., Lawrenz M. B., Hardham J. M., Norris S. J., and Sacchettini J. C. (2002) Crystal structure of Lyme disease variable surface antigen VlsE of Borrelia burgdorferi. J. Biol. Chem. 277, 21691–21696 10.1074/jbc.M201547200 [DOI] [PubMed] [Google Scholar]
- 48. Hudson C. R., Frye J. G., Quinn F. D., and Gherardini F. C. (2001) Increased expression of Borrelia burgdorferi vlsE in response to human endothelial cell membranes. Mol. Microbiol. 41, 229–239 10.1046/j.1365-2958.2001.02511.x [DOI] [PubMed] [Google Scholar]
- 49. Verhey T. B., Castellanos M., and Chaconas G. (2019) Antigenic variation in the Lyme spirochete: detailed functional assessment of recombinational switching at vlsE in the JD1 strain of Borrelia burgdorferi. Mol. Microbiol. 111, 750–763 10.1111/mmi.14189 [DOI] [PubMed] [Google Scholar]
- 50. Brázda V., Coufal J., Liao J. C., and Arrowsmith C. H. (2012) Preferential binding of IFI16 protein to cruciform structure and superhelical DNA. Biochem. Biophys. Res. Commun. 422, 716–720 10.1016/j.bbrc.2012.05.065 [DOI] [PubMed] [Google Scholar]
- 51. Bykowski T., Babb K., von Lackum K., Riley S. P., Norris S. J., and Stevenson B. (2006) Transcriptional regulation of the Borrelia burgdorferi antigenically variable VlsE surface protein. J. Bacteriol. 188, 4879–4889 10.1128/JB.00229-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Walia R., and Chaconas G. (2013) Suggested role for G4 DNA in recombinational switching at the antigenic variation locus of the Lyme disease spirochete. PLoS One 8, e57792 10.1371/journal.pone.0057792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang D., Botkin D. J., and Norris S. J. (2003) Characterization of the vls antigenic variation loci of the Lyme disease spirochaetes Borrelia garinii Ip90 and Borrelia afzelii ACAI. Mol. Microbiol. 47, 1407–1417 10.1046/j.1365-2958.2003.03386.x [DOI] [PubMed] [Google Scholar]
- 54. Saranathan N., and Vivekanandan P. (2019) G-quadruplexes: more than just a kink in microbial genomes. Trends Microbiol. 27, 148–163 10.1016/j.tim.2018.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Burge S., Parkinson G. N., Hazel P., Todd A. K., and Neidle S. (2006) Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 34, 5402–5415 10.1093/nar/gkl655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Phan A. T., Kuryavyi V., and Patel D. J. (2006) DNA architecture: from G to Z. Curr. Opin. Struct. Biol. 16, 288–298 10.1016/j.sbi.2006.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sannohe Y., and Sugiyama H. (2010) Overview of formation of G-quadruplex structures. Current Protoc. Nucleic Acid Chem. Chapter 17, Unit 17.12.11–17 10.1002/0471142700.nc1702s40 [DOI] [PubMed] [Google Scholar]
- 58. Seifert H. S. (2018) Above and beyond Watson and Crick: guanine quadruplex structures and microbes. Annu. Rev. Microbiol. 72, 49–69 10.1146/annurev-micro-090817-062629 [DOI] [PubMed] [Google Scholar]
- 59. Cahoon L. A., Manthei K. A., Rotman E., Keck J. L., and Seifert H. S. (2013) The Neisseria gonorrhoeae RecQ helicase HRDC domains are essential for efficient binding and unwinding of the pilE guanine quartet structure required for pilin Av. J. Bacteriol. 195, 2255–2261 10.1128/JB.02217-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Thakur R. S., Desingu A., Basavaraju S., Subramanya S., Rao D. N., and Nagaraju G. (2014) Mycobacterium tuberculosis DinG is a structure-specific helicase that unwinds G4 DNA: implications for targeting G4 DNA as a novel therapeutic approach. J. Biol. Chem. 289, 25112–25136 10.1074/jbc.M114.563569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sauer M., and Paeschke K. (2017) G-quadruplex unwinding helicases and their function in vivo. Biochem. Soc. Trans. 45, 1173–1182 10.1042/BST20170097 [DOI] [PubMed] [Google Scholar]
- 62. Dahan D., Tsirkas I., Dovrat D., Sparks M. A., Singh S. P., Galletto R., and Aharoni A. (2018) Pif1 is essential for efficient replisome progression through lagging strand G-quadruplex DNA secondary structures. Nucleic Acids Res. 46, 11847–11857 10.1093/nar/gky1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lopes J., Piazza A., Bermejo R., Kriegsman B., Colosio A., Teulade-Fichou M. P., Foiani M., and Nicolas A. (2011) G-quadruplex-induced instability during leading-strand replication. EMBO J. 30, 4033–4046 10.1038/emboj.2011.316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kolinjivadi A. M., Sannino V., de Antoni A., Técher H., Baldi G., and Costanzo V. (2017) Moonlighting at replication forks—a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 591, 1083–1100 10.1002/1873-3468.12556 [DOI] [PubMed] [Google Scholar]
- 65. Polleys E. J., House N. C. M., and Freudenreich C. H. (2017) Role of recombination and replication fork restart in repeat instability. DNA Repair (Amst.) 56, 156–165 10.1016/j.dnarep.2017.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brisson D., Drecktrah D., Eggers C. H., and Samuels D. S. (2012) Genetics of Borrelia burgdorferi. Annu. Rev. Genet. 46, 515–536 10.1146/annurev-genet-011112-112140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brázda V., Laister R. C., Jagelská E. B., and Arrowsmith C. (2011) Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 12, 33 10.1186/1471-2199-12-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bikard D., Loot C., Baharoglu Z., and Mazel D. (2010) Folded DNA in action: hairpin formation and biological functions in prokaryotes. Microbiol. Mol. Biol. Rev. 74, 570–588 10.1128/MMBR.00026-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Beaurepaire C., and Chaconas G. (2007) Topology-dependent transcription in linear and circular plasmids of the segmented genome of Borrelia burgdorferi. Mol. Microbiol. 63, 443–453 10.1111/j.1365-2958.2006.05533.x [DOI] [PubMed] [Google Scholar]
- 70. Alverson J., Bundle S. F., Sohaskey C. D., Lybecker M. C., and Samuels D. S. (2003) Transcriptional regulation of the ospAB and ospC promoters from Borrelia burgdorferi. Mol. Microbiol. 48, 1665–1677 10.1046/j.1365-2958.2003.03537.x [DOI] [PubMed] [Google Scholar]
- 71. Alverson J., and Samuels D. S. (2002) groEL expression in gyrB mutants of Borrelia burgdorferi. J. Bacteriol. 184, 6069–6072 10.1128/JB.183.21.6069-6072.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Beaurepaire C., and Chaconas G. (2005) Mapping of essential replication functions of the linear plasmid lp17 of B. burgdorferi by targeted deletion walking. Mol. Microbiol. 57, 132–142 10.1111/j.1365-2958.2005.04688.x [DOI] [PubMed] [Google Scholar]
- 73. Picardeau M., Lobry J. R., and Hinnebusch B. J. (1999) Physical mapping of an origin of bidirectional replication at the centre of the Borrelia burgdorferi linear chromosome. Mol. Microbiol. 32, 437–445 10.1046/j.1365-2958.1999.01368.x [DOI] [PubMed] [Google Scholar]
- 74. Picardeau M., Lobry J. R., and Hinnebusch B. J. (2000) Analyzing DNA strand compositional asymmetry to identify candidate replication origins of Borrelia burgdorferi linear and circular plasmids. Genome Res. 10, 1594–1604 10.1101/gr.124000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dresser A. R., Hardy P.-O., and Chaconas G. (2009) Investigation of the role of DNA replication, recombination and repair genes in antigenic switching at the vlsE locus in Borrelia burgdorferi: an essential role for the RuvAB branch migrase. PLoS Pathog. 5, e1000680 10.1371/journal.ppat.1000680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liveris D., Mulay V., Sandigursky S., and Schwartz I. (2008) Borrelia burgdorferi vlsE antigenic variation is not mediated by RecA. Infect. Immun. 76, 4009–4018 10.1128/IAI.00027-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kirkman L. A., and Deitsch K. W. (2012) Antigenic variation and the generation of diversity in malaria parasites. Curr. Opin. Microbiol. 15, 456–462 10.1016/j.mib.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lin T., Gao L., Edmondson D. G., Jacobs M. B., Philipp M. T., and Norris S. J. (2009) Central role of the Holliday junction helicase RuvAB in vlsE recombination and infectivity of Borrelia burgdorferi. PLoS Pathog. 5, e1000679 10.1371/journal.ppat.1000679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Huang S. H., Cozart M. R., Hart M. A., and Kobryn K. (2017) The Borrelia burgdorferi telomere resolvase, ResT, possesses ATP-dependent DNA unwinding activity. Nucleic Acids Res. 45, 1319–1329 10.1093/nar/gkw1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Huang S. H., and Kobryn K. (2016) The Borrelia burgdorferi telomere resolvase, ResT, anneals ssDNA complexed with its cognate ssDNA-binding protein. Nucleic Acids Res. 44, 5288–5298 10.1093/nar/gkw344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mir T., Huang S. H., and Kobryn K. (2013) The telomere resolvase of the Lyme disease spirochete, Borrelia burgdorferi, promotes DNA single-strand annealing and strand exchange. Nucleic Acids Res. 41, 10438–10448 10.1093/nar/gkt832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bandy N. J., Salman-Dilgimen A., and Chaconas G. (2014) Construction and characterization of a Borrelia burgdorferi strain with conditional expression of the essential telomere resolvase, ResT. J. Bacteriol. 196, 2396–2404 10.1128/JB.01435-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Boothroyd C. E., Dreesen O., Leonova T., Ly K. I., Figueiredo L. M., Cross G. A., and Papavasiliou F. N. (2009) A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei. Nature 459, 278–281 10.1038/nature07982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Byram R., Stewart P. E., and Rosa P. (2004) The essential nature of the ubiquitous 26-kilobase circular replicon of Borrelia burgdorferi. J. Bacteriol. 186, 3561–3569 10.1128/JB.186.11.3561-3569.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Verhey T. B., Castellanos M., and Chaconas G. (2018) Antigenic variation in the Lyme disease spirochete: new insights into the mechanism of recombinational switching with a suggested role for error-prone repair. Cell Reports 23, 2595–2605 10.1016/j.celrep.2018.04.117 [DOI] [PubMed] [Google Scholar]
- 86. Coutte L., Botkin D. J., Gao L., and Norris S. J. (2009) Detailed analysis of sequence changes occurring during vlsE antigenic variation in the mouse model of Borrelia burgdorferi infection. PLoS Pathog. 5, e1000293 10.1371/journal.ppat.1000293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Crowder C. D., Denny R. L., and Barbour A. G. (2017) Segregation Lag in polyploid cells of the pathogen genus Borrelia: implications for antigenic variation. Yale J. Biol. Med. 90, 195–218 [PMC free article] [PubMed] [Google Scholar]
- 88. Sung S. Y., McDowell J. V., and Marconi R. T. (2001) Evidence for the contribution of point mutations to vlsE variation and for apparent constraints on the net accumulation of sequence changes in vlsE during infection with Lyme disease spirochetes. J. Bacteriol. 183, 5855–5861 10.1128/JB.183.20.5855-5861.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jolivet-Gougeon A., Kovacs B., Le Gall-David S., Le Bars H., Bousarghin L., Bonnaure-Mallet M., Lobel B., Guillé F., Soussy C. J., and Tenke P. (2011) Bacterial hypermutation: clinical implications. J. Med. Microbiol. 60, 563–573 10.1099/jmm.0.024083-0 [DOI] [PubMed] [Google Scholar]
- 90. Malkova A., and Haber J. E. (2012) Mutations arising during repair of chromosome breaks. Annu. Rev. Genet. 46, 455–473 10.1146/annurev-genet-110711-155547 [DOI] [PubMed] [Google Scholar]
- 91. DeLange A. M., Reddy M., Scraba D., Upton C., and McFadden G. (1986) Replication and resolution of cloned poxvirus telomeres in vivo generates linear minichromosomes with intact viral hairpin termini. J. Virol. 59, 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chaconas G., Stewart P. E., Tilly K., Bono J. L., and Rosa P. (2001) Telomere resolution in the Lyme disease spirochete. EMBO J. 20, 3229–3237 10.1093/emboj/20.12.3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Castellanos M., Verhey T. B., and Chaconas G. (2018) A Borrelia burgdorferi mini-vls system that undergoes antigenic switching in mice: investigation of the role of plasmid topology and the long inverted repeat. Mol. Microbiol. 109, 710–721 10.1111/mmi.14071 [DOI] [PubMed] [Google Scholar]
- 94. Hodzic E., Imai D. M., and Escobar E. (2019) The generality of post-antimicrobial treatment persistence of Borrelia burgdorferi strains N40 and B31 in genetically susceptible and resistant mouse strains. Infect. Immun. 87, e00442–19 10.1128/IAI.00442-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Embers M. E., Hasenkampf N. R., Jacobs M. B., Tardo A. C., Doyle-Meyers L. A., Philipp M. T., and Hodzic E. (2017) Variable manifestations, diverse seroreactivity and post-treatment persistence in non-human primates exposed to Borrelia burgdorferi by tick feeding. PLoS One 12, e0189071 10.1371/journal.pone.0189071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Crossland N. A., Alvarez X., and Embers M. E. (2018) Late disseminated Lyme disease: associated pathology and spirochete persistence posttreatment in rhesus macaques. Am. J. Pathol. 188, 672–682 10.1016/j.ajpath.2017.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rotman E., Webber D. M., and Seifert H. S. (2016) Analyzing Neisseria gonorrhoeae pilin antigenic variation using 454 sequencing technology. J. Bacteriol. 198, 2470–2482 10.1128/JB.00330-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Stothard P. (2000) The Sequence Manipulation Suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28, 1102–1104 10.2144/00286ir01 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.