

Abstract
Rationale: Differences in cystic fibrosis (CF) airway microbiota between periods of clinical stability and exacerbation of respiratory symptoms have been investigated in efforts to better understand microbial triggers of CF exacerbations. Prior studies have often relied on a single sample or a limited number of samples to represent airway microbiota. However, the variability in airway microbiota during periods of clinical stability is not well known.
Objectives: To determine the temporal variability of measures of airway microbiota during periods of clinical stability, and to identify factors associated with this variability.
Methods: Sputum samples (N = 527), obtained daily from six adults with CF during 10 periods of clinical stability, underwent sequencing of the V4 region of the bacterial 16S ribosomal RNA gene. The variability in airway microbiota among samples within each period of clinical stability was calculated as the average of the Bray-Curtis similarity measures of each sample to every other sample within the same period. Outlier samples were defined as samples outside 1.5 times the interquartile range within a baseline period with respect to the average Bray-Curtis similarity. Total bacterial load was measured with droplet digital polymerase chain reaction.
Results: The variation in Bray-Curtis similarity and total bacterial load among samples within the same baseline period was greater than the variation observed in technical replicate control samples. Overall, 6% of samples were identified as outliers. Within baseline periods, changes in bacterial community structure occurred coincident with changes in maintenance antibiotics (P < 0.05, analysis of molecular variance). Within subjects, bacterial community structure changed between baseline periods (P < 0.01, analysis of molecular variance). Sample-to-sample similarity within baseline periods was greater with fewer interval days between sampling.
Conclusions: During periods of clinical stability, airway bacterial community structure and bacterial load vary among daily sputum samples from adults with CF. This day-to-day variation has bearing on study design and interpretation of results, particularly in analyses that rely on single samples to represent periods of interest (e.g., clinical stability vs. pulmonary exacerbation). These data also emphasize the importance of accounting for maintenance antibiotic use and granularity of sample collection in studies designed to assess the dynamics of CF airway microbiota relative to changes in clinical state.
Keywords: cystic fibrosis, microbiome, airway infection
Cystic fibrosis (CF) pulmonary exacerbations are generally defined as increases in respiratory signs and symptoms relative to a person’s baseline state of health (1). These events are common in persons with CF and are associated with both short- and long-term decreases in lung function (2–4). In a significant minority of cases, treatment with antibiotics and airway clearance fails to result in lung function recovery to preexacerbation baseline levels (2, 4).
In efforts to improve understanding of the microbiological triggers of exacerbations, culture-independent profiling of CF respiratory samples has been used to compare airway microbiota between periods of clinical stability and pulmonary exacerbation. Such comparisons have typically relied on a single sample or a limited number of samples to represent airway microbiota during these respective clinical states. However, it is not clear how representative a single sample is of airway microbial communities over the broader time periods of each state, because community structure variability during these periods is largely unknown. In a recent study of daily sputum samples from four adults with CF, we identified day-to-day variation in measures of sputum microbiota at baseline clinical state that exceeded variation of DNA sequencing controls (5). This biological variation suggests that reliance on a limited number of samples has the potential to mischaracterize bacterial communities during periods of clinical stability.
Failure to recognize variability of airway microbiota during periods of clinical stability could lead to mistaken conclusions about differences between baseline and exacerbation states, and it may have contributed to the conflicting findings of prior studies. Although some culture-independent studies have identified microbial community differences between clinical stability and exacerbation, including increases in the relative abundance of facultative anaerobic taxa (6, 7) (e.g., Streptococcus milleri group [8], Rothia [9], and Gemella [10]) at exacerbation, other studies have not identified significant differences between these clinical states (5, 11, 12).
A better understanding of airway microbiota variability during periods of clinical stability, as well as the identification of factors that impact this variation, would inform sampling strategies for studies seeking to identify predictable changes in microbiota between clinical states. We sought to assess CF airway microbiota variability during periods of clinical stability. Additional objectives included identifying clinical variables associated with variation in airway microbiota, determining within-subject changes in microbiota over time, and determining the impact of sampling interval on measures of microbiota.
Methods
Subject and Baseline Period Sample Selection
Subjects and baseline period samples were selected from among a cohort of persons with CF enrolled in a long-term study of airway microbiota. Under approval of the Michigan Medicine Institutional Review Board, subjects collected daily sputum samples for a period of 1–2 years. Samples were refrigerated for up to 27 days before being shipped on ice to the investigator’s laboratory for subsequent aliquoting and storage at −80°C. We have previously shown that storage of sputum at 4°C for this length of time does not significantly impact measures of bacterial community structure (13). Study subjects completed a daily brief questionnaire reporting respiratory symptoms and antibiotic use, including both maintenance (i.e., chronic inhaled antibiotics or chronic azithromycin) and episodic (i.e., oral or intravenous, prescribed to treat pulmonary exacerbation) antibiotics (14). Electronic medical records were reviewed for subject demographic and clinical data. Disease stage was assigned on the basis of percent predicted forced expiratory volume in 1 second (%FEV1) at study enrollment: early (%FEV1, >70%), intermediate (70% ≥ %FEV1 ≥ 40%), or advanced (%FEV1, <40%) (10, 15).
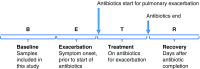
Ten periods of baseline clinical stability (“baseline period”) in six adult subjects were identified using the “BETR” framework (baseline, exacerbation, treatment, recovery) as we have done previously (16). Dates of episodic antibiotic treatment for pulmonary exacerbation were identified from the subjects’ medical records and daily antibiotic use questionnaires. For this study, baseline periods were defined as starting 21 days after the end of an episodic antibiotic treatment course and ending 14 days before the next episodic treatment course. This window of time was selected to allow recovery of airway microbiota after treatment of an exacerbation and to exclude the 2 weeks before the diagnosis and treatment of the next exacerbation, during which symptoms of exacerbation would be expected to begin (17, 18). Medical records and questionnaire data were reviewed to verify that subjects did not report sustained increases in symptoms suggestive of pulmonary exacerbation during the identified baseline periods. Antibiotic questionnaire data were reviewed to verify that subjects did not report taking unprescribed oral antibiotics during the baseline periods (14). To allow for the maximum number of measures of day-to-day changes in microbiota during baseline clinical state, inclusion criteria were set for adherence to daily sample collection and questionnaire completion. Specific inclusion and exclusion criteria for baseline periods are listed in Table 1.
Table 1.
Criteria for periods of baseline clinical state
| Inclusion Criteria |
|---|
|
| Exclusion Criteria |
|---|
|
Sputum DNA Extraction
Sputum samples were thawed on ice, then homogenized with 10% SPUTOLYSIN (MilliporeSigma). Samples were treated with bacterial lysis buffer (Roche Diagnostics Corp.), lysostaphin (MilliporeSigma), and lysozyme (MilliporeSigma) as previously described (19), followed by mechanical disruption by glass bead beating and digestion with proteinase K (Qiagen). DNA was extracted and purified using a MagNA Pure nucleic acid purification platform (Roche Diagnostics Corp.) according to the manufacturer’s protocol. DNA extraction was similarly performed on reagent control samples, with UltraPure DNase/RNase-free distilled water (Life Technologies Corp.) substituted for the sputum sample.
Technical Replicates and DNA Sequencing Control Samples
DNA extractions from approximately 5% of the total sample set (n = 28 samples) were randomly selected for repeat sequencing to evaluate variation between sequencing runs. Aliquots of DNA extracted from “generous donor” CF sputum (i.e., large-volume sputum samples from patients not in this study used as CF technical replicate control samples) were also sequenced to determine variation between sequencing runs. Reagent control samples were sequenced to assess their potential contributions to sputum sample DNA sequencing results. Mock bacterial community DNA standards and water control samples were included in the sequencing runs by the University of Michigan Microbial Systems Molecular Biology Laboratory.
16S Ribosomal RNA Gene Sequencing
The DNA libraries were prepared by the University of Michigan Microbial Systems Molecular Biology Laboratory as described previously (20). Briefly, the V4 region of the bacterial 16S ribosomal RNA (rRNA) gene was amplified using touchdown polymerase chain reaction (PCR) with barcoded dual-index primers. The touchdown PCR cycles consisted of 2 minutes at 95°C; followed by 20 cycles of 95°C for 20 seconds, 60°C (starting from 60°C, the annealing temperature was decreased 0.3°C each cycle) for 15 seconds, and 72°C for 5 minutes; followed by 20 cycles of 95°C for 20 seconds, 55°C for 15 seconds, and 72°C for 5 minutes; and a final 72°C for 10 minutes. The amplicon libraries were normalized and sequenced on an Illumina sequencing platform using a MiSeq Reagent Kit V2 (Illumina). The final load concentration was 4.0–5.5 pM with a 15% PhiX spike to add diversity.
DNA Sequence Analyses
The raw DNA sequences were analyzed using mothur version 1.41.3 (21) and the mothur MiSeq standard operating procedure. Briefly, after discarding low-quality and chimeric reads, sequences were assigned to taxonomy against the SILVA database (release 132) using the RDP Bayesian Classifier and clustered into operational taxonomic units (OTUs) based on 97% similarity using the optiClust algorithm (22). Sputum samples with fewer than 1,000 reads were excluded from analyses (n = 1). To limit the effect of sequencing depth, each sample was rarefied to the lowest number of reads in the sample set (n = 1,066). α-Diversity of the subsampled data was measured using the inverse Simpson index. β-Diversity measures were calculated on the basis of the Bray-Curtis dissimilarity coefficient and, for ease of interpretation, are reported as Bray-Curtis similarity (i.e., 1 − Bray-Curtis dissimilarity).
Droplet Digital PCR
Total bacterial load was quantified on DNA extractions from all sputum samples and reagent control samples by 16S rRNA droplet digital polymerase chain reaction (ddPCR) [23] on a QX200 AutoDG Droplet Digital PCR System (Bio-Rad Laboratories). Primer sequences used were 5′-TCCTACGGGAGGCAGCAGT-3′ and 5′-GGACTACCAGGGTATCTAATCCTGTT-3′ (final concentration, 900 nM each), and the probe sequence used was (6-FAM)-5′-CGTATTACCGCGGCTGCTGG-3′-(IBFQ) (final concentration, 250 nM). Before amplification, DNA was diluted by a factor of 1:250. Diluted DNA (2 μl) was used as the template. All reactions, including reagent and negative control samples, were run in duplicate. Reactions were transferred to the Automated Droplet Generator (Bio-Rad Laboratories), followed by gene amplification in a C1000 Touch Thermal Cycler (Bio-Rad Laboratories). Cycling conditions consisted of 10 minutes at 95°C, followed by 40 cycles at 94°C for 30 seconds and 58°C for 2 minutes, and a final 98°C for 10 minutes, with a ramp rate of 2°C/s per step. DNA quantification was performed with the QX200 Droplet Reader (Bio-Rad Laboratories), and data analysis was performed with QuantaSoft Analysis Pro (Bio-Rad Laboratories) using default parameters for threshold setting. Reactions with less than 10,000 droplets were omitted from analysis. DNA concentrations between replicates were averaged, adjusted for dilution factor, reported in copies of target gene per microliter of DNA, then converted to copies of target gene per milliliter of sputum sample based on the DNA extraction steps.
Data Analyses
To determine variation in microbiota within each baseline period, the average of the pairwise Bray-Curtis similarity measures of each sample to every other sample within the same baseline period was calculated (“β-diversity”). Similarly, the average Bray-Curtis similarity of each technical replicate (technical replicates of sputum samples and generous donor samples) to the relevant technical replicate sample(s) was calculated. Outlier samples were defined as samples outside 1.5 times the interquartile range within a baseline period (or within a collection of technical replicates). To compare β-diversity between baselines and technical replicate controls, a linear mixed model (R packages lme4 [24] and lmerTest [25]) was used with β-diversity as the dependent variable, the baseline period as a fixed effect, and the intercept for subjects as a random effect (thereby accounting for repeated measures). A linear mixed model was used to compare total bacterial load between outlier and nonoutlier groups, with total bacterial load as the dependent variable, outlier status as a fixed effect, and intercepts for subjects and baseline periods as random effects. A linear mixed model was also used to assess the association of variation of community structure within each baseline period with the length of each period, with β-diversity as the dependent variable, the length of baseline period as a fixed effect, and the intercept for subject as a random effect. A one-sided binomial exact test using the binomial distribution functions (pbinom, qbinom) from the default R package stats (26) was used to test the power to identify nonoutlier (i.e., majority) samples within a sample set.
Bray-Curtis–based nonmetric multidimensional scaling plots were used to visualize the changes in bacterial community structure between different maintenance antibiotic regimens within baseline periods and between baseline periods within subjects. Analysis of molecular variance (AMOVA) was used to compare differences in centroids of bacterial communities between different maintenance antibiotic regimens within baseline periods and between baseline periods within individual subjects (mothur version 1.41.3). Because of autocorrelation of time series data, a generalized least-squares autoregressive model (R nlme package [27]) was used for comparison of total bacterial load between different maintenance antibiotic regimens within baseline periods, with total bacterial load as the dependent variable and antibiotic regimen and number of days as independent variables, and included the first-order autoregressive covariance structure by days.
Variation in sample-to-sample similarity due to changes in interval sampling days was determined within each baseline period by calculating all pairwise Bray-Curtis similarity values using a sliding window of sampling intervals. For example, interval day = 1 represents all day-to-the-next-day Bray-Curtis similarity values. Interval day = 2 represents the pairwise Bray-Curtis similarity values of Day 1 to Day 3, Day 2 to Day 4, Day 3 to Day 5, and so forth. When the sliding window of interval days fell on a day on which a sample was not collected, no value was assigned for the pairwise comparison. A linear mixed model was used for analyzing the impact of interval days on pairwise sample similarity, with Bray-Curtis similarity as the dependent variable, interval days and antibiotics as fixed effects, and the intercepts for subjects, baselines, and the random slope for the effect of interval days by subjects as random effects.
All P values were adjusted for multiple comparisons using the Holm-Bonferroni method. All figures were plotted using the R ggplot2 package (28). The 95% confidence intervals (CIs) were calculated using the confint and confint.merMod function in R stats and lme4 packages.
Reproducibility
Raw sequencing data have been deposited with the National Center for Biotechnology Information (Sequence Read Archive accession no. PRJNA520924). The daily antibiotic questionnaire template, detailed DNA extraction protocol, mothur command file, deidentified subject data, 16S rRNA gene sequencing data, 16S ddPCR data, and reproducible code for data analyses and figures are available https://github.com/caverlyl/LiPuma_baselineCFmicrobiome.
Results
Subjects and Samples
Ten baseline periods meeting inclusion criteria were identified in six subjects. Subjects ranged in age from 30 to 51 years at enrollment. The number of sputum samples per subject ranged from 21 to 228. Three subjects had early stage lung disease, two had intermediate disease, and one had advanced disease (Table 2).
Table 2.
Subjects and samples
| Subject | Sex | Age (yr) at Enrollment | No. of Baseline Periods* | No. of Samples | Disease Stage |
|---|---|---|---|---|---|
| 1 | M | 39 | 1 | 21 | Intermediate |
| 2 | F | 37 | 4 | 228 | Early |
| 3 | M | 40 | 1 | 42 | Early |
| 4 | F | 44 | 1 | 21 | Advanced |
| 5 | F | 30 | 2 | 172 | Early |
| 6 | F | 51 | 1 | 43 | Intermediate |
| Total | 10 | 527 |
Definition of abbreviations: F = female; M = male.
The number of days in each baseline period is shown in Figure E2. In subjects with multiple baseline periods, the number of days per period and between periods is shown in Figure 4.
DNA Sequencing Control Samples
The median sequencing error rate, based on mock community analyses, was 0.036% (range, 0.013–0.69%). OTUs with the highest relative abundances in reagent and water control samples had minimal overlap with those in sputum samples (see Figure E1 in the online supplement). The median log10-transformed bacterial load of reagent control samples was 4.36 copies/ml (range, 4.25–4.48 copies/ml), which was greater than 104 less than the median bacterial load of sputum samples (9.13 copies/ml; range, 7.14–10.18 copies/ml).
Variation in Microbiota within Baseline Periods
The variation of bacterial community structure within each baseline period was greater than the variation in technical replicates (repeat sputum samples and generous donor samples) in all but one baseline period (baseline 6; P < 0.001, linear mixed model fixed effects contrast) (Figure 1). The variation of bacterial community structure within subjects was less than the variation between subjects (average intrasubject Bray-Curtis similarity of 0.686 vs. average intersubject Bray-Curtis similarity of 0.362; P < 0.001; 95% CI, 0.322–0.325; linear model). The variation of bacterial community structure within each baseline period was not associated with increasing length of the baseline period (P = 0.62; 95% CI, −0.0002, 0.0004; linear mixed model).
Figure 1.

Sample-to-sample similarity of airway microbiota within baseline periods compared with control samples. Ten baseline periods in six subjects (labeled 1–6) are shown, with multiple baseline periods from the same subject (subjects 2 and 5) labeled a–d. Each point represents the average pairwise Bray-Curtis similarity of that sample to all other samples within that baseline period. Medians and interquartile ranges of all samples within each baseline period and within DNA sequencing control samples (technical replicates of sputum samples [TR1] and generous donor samples [TR2]) are shown. Average pairwise Bray-Curtis similarity of samples is significantly less than average pairwise Bray-Curtis similarity of DNA sequencing control samples within all but one baseline period (P < 0.001 for all baseline periods except baseline 6 [P = 0.262], linear mixed model).
Outlier samples in β-diversity were observed in 7 of the 10 baseline periods (periods 2a, 2b, 2d, 3, 5a, 5b, and 6), accounting for 6.5% (34 of 527) of samples overall. When outliers were present, they accounted for 4–13% of the samples within the respective baseline period. In investigating whether outlier samples shared common features that could facilitate their a priori identification (i.e., before sequence analysis), we observed no clear temporal patterns in the occurrence of outlier samples within each baseline period (Figure E2), and we found that total bacterial load of outlier samples did not differ significantly from that of nonoutlier samples (Figure E3). Visualization of all samples (outliers and nonoutliers) from each subject on Bray-Curtis–based non-metric multidimensional scaling (nMDS) ordination plots showed no clustering of outlier samples, suggesting that they did not share common bacterial community structures within subjects (Figure E4). Biplots for each within-subject ordination did not identify common bacterial community structures in outlier samples across subjects (i.e., taxa driving the separation of outliers from nonoutlier samples were not shared across subjects) (Figure E4). Outlier samples did not, as a group, differ from nonoutlier samples with respect to time stored at 4°C (Figure E5). A subset of the outlier samples underwent repeat 16S rRNA gene sequencing. Community structures of these outlier samples were reproducible across repeated sequencing runs, with an average pairwise Bray-Curtis similarity of 0.86 (range, 0.77–0.91), consistent with the variation observed in the other technical replicates (Figure 1).
Given the proportions of outlier samples we observed (range, 0–13%; mean, 6.5%), we calculated the power to identify the majority (i.e., nonoutlier) population on the basis of analysis of various sample sizes. As expected, the power to identify nonoutlier samples in a mixed sample set increased as the sample set size increased and the proportion of outlier samples in the population decreased (Figure E6).
Variation in Total Bacterial Load within Baseline Periods
The total bacterial load of sputum samples (log10 transformed) ranged from 7.14 to 10.18 copies/ml (median, 9.13 copies/ml) (Figure 2). The average range of bacterial loads among samples within each baseline period was 1.6 ± 0.43 copies/ml. This variation in bacterial load among samples within baseline periods was not attributable to technical variation, because the variation among samples was 100-fold greater (two units greater on the log10-transformed scale) than the within-sample variation between repeated ddPCR measurements (n = 36; average range between repeated ddPCR measurements, 0.018 ± 0.017 copies/ml). Outlier samples of total bacterial load (samples outside 1.5 times the interquartile range from the median total bacterial load within baseline periods) represented 1.9% (10 of 527) of the samples overall (Figure 2). Only two of these outliers in total bacterial load were also outliers in β-diversity.
Figure 2.

Total bacterial load within baseline periods. Subjects and baseline periods are labeled as in Figure 1. Each sample point represents the average of droplet digital polymerase chain reaction (ddPCR) measurements run in duplicate. The medians and interquartile ranges of total 16S rRNA gene copy ddPCR measurements of samples in each baseline period are shown.
Changes in Microbial Community Structure Associated with Changes in Maintenance Antibiotics
A routine change in maintenance antibiotic regimen (chronic inhaled antibiotics and/or oral azithromycin) occurred during 6 of the 10 baseline periods included in this study. Given the long half-life of azithromycin, subjects receiving azithromycin three times weekly were considered “on azithromycin” all days of that week and were not considered “off azithromycin” until more than 10 days from the last azithromycin dose. Within each of these six baseline periods, the change in maintenance antibiotic regimen was associated with a significant change in bacterial community structure (P < 0.05, AMOVA) (Figure 3). Changes in maintenance antibiotic regimen were not associated with changes in total bacterial load in the majority of cases, although a trend in decreasing bacterial load was observed with the addition of inhaled antibiotics (Figure E7).
Figure 3.

Bacterial community structure and changes in maintenance antibiotics. Bray-Curtis–based nonmetric multidimensional scaling (nMDS) plots of samples from each baseline period during which the subject had a change in maintenance antibiotic use are shown. Points are colored by maintenance antibiotic regimen. The centroids of the clusters (black points) significantly differ between maintenance antibiotic regimens (P < 0.05 for all plots, analysis of molecular variance).
Within-Subject Changes in Bacterial Community Structure between Baseline Periods
Significant changes in bacterial community structure (P < 0.01, AMOVA) were observed between baseline periods in the two subjects for whom more than a single baseline period was included in the study (Figure 4). Within-subject changes in community structure between baseline periods were not attributable to variation between sequencing runs, based on analyses of samples that underwent repeat sequencing (Figure E8).
Figure 4.

Bacterial community structure changes between baseline periods within the same subjects. Bray-Curtis–based nonmetric multidimensional scaling (nMDS) plots of samples from multiple baseline periods are shown for (A) subject 2 and (B) subject 5. Below each plot is a timeline of the duration (days) of each baseline period (above the line) and the duration separating the baseline periods (below the line). Baseline periods are color coded, and centroids of each baseline period are designated by letter. The centroid of the clusters within each subject significantly differs between baseline periods (P < 0.01, analysis of molecular variance).
Impact of Sampling Interval on Sample-to-Sample Similarity
On the basis of all pairwise measures of sample-to-sample Bray-Curtis similarity from 1 to 30 interval days, measures of sample-to-sample similarity significantly decreased as interval days of sampling increased (Figure 5) (P = 0.048; 95% CI for the change in similarity with increasing sampling interval by 1 day, −0.002, −0.0003; linear mixed model). Sample-to-sample similarity measures were significantly lower in pairwise comparisons of samples from different maintenance antibiotic regimens than in pairwise comparisons of samples from the same maintenance antibiotic regimen (P < 0.001; 95% CI, −0.013, −0.024; linear mixed model). Individual subject sample-to-sample similarity at select sampling intervals are presented in Figure E9.
Figure 5.

Sample-to-sample similarity measures based on sampling interval. Medians and interquartile ranges of all possible pairwise comparisons of Bray-Curtis similarity within each baseline period are shown at select pairwise intervals. Pairwise comparisons of samples from different maintenance antibiotic regimens (yellow points) are shown separately from comparisons of samples from the same maintenance antibiotic regimens (gray points). Bray-Curtis similarity significantly decreases with increasing sampling intervals (P = 0.048; 95% confidence interval, −0.002, −0.0003; linear mixed model).
Discussion
Identifying differences in airway microbiota between periods of clinical stability compared with exacerbation of respiratory symptoms has the potential to elucidate microbiological triggers of CF pulmonary exacerbations, which, in turn, might suggest improved treatment strategies in CF. However, recognition of meaningful changes in airway bacterial community structures associated with clinical signs and symptoms depends on a more thorough appreciation of the day-to-day variability in these communities observed during periods clinical stability. Because CF airway microbiota are most commonly assessed in expectorated sputum, careful characterization of variability within serial sputum samples from clinically stable individuals is important.
The variation we observed in serial sputum samples exceeded that measured in control samples, demonstrating that bacterial community variability in serial samples is not solely a function of technical artifact. These findings provide greater context for our previous observations suggesting changes in bacterial community structure coincident with onset of pulmonary exacerbation (5). Perhaps the most striking and relevant finding was the occurrence of outlier samples, which showed significant deviation in community structure relative to samples obtained from the same individual around the same time. Repeat sequence analysis of these samples provided reproducible results, and our DNA sequencing control data indicate that the large variances in community structure observed in the outlier samples did not result from DNA sequencing error or from reagent DNA contamination. We noted no temporal pattern to the occurrence of these samples, nor were outliers uniformly characterized by common community structural motifs. Rather than the appearance of a new taxon, outlier samples typically showed large variation in the relative abundance of one or more OTUs that were present, albeit at relatively lower abundance, in other samples from the same individual. In an effort to facilitate a priori identification of outliers (i.e., before sequence analysis), we investigated total bacterial load but found no overall differences in bacterial density between outlier and nonoutlier samples.
The presence of at least one outlier sample within the majority of periods of clinical stability highlights the limitation of using a single sample to represent baseline bacterial community structure during a longer period of time. Clearly, inadvertent selection of a sample that poorly represents the bacterial community during a period of interest (in this case, clinical stability) would have a major impact on interpretation of results when comparing microbiota between these periods. Although only 6% of the several hundred samples analyzed in this study were found to poorly represent the bacterial community structures of the majority of samples in the respective baseline periods, the range of outliers was large (0–13%). On the basis of this observation, we calculated the power to confidently identify the majority (nonoutlier) community within a mixed sample set under various conditions of sample set size and proportion of outliers within the larger population (Figure E6). Assuming that outlier samples comprised 15% of the samples from a baseline period, analysis of five samples would provide greater than 80% probability of correctly discerning the majority (nonoutlier) community with a significance level of 0.2. Analysis of five samples would provide greater than 95% probability of discerning the majority community if the proportion of outliers in the baseline sample set was only 5%.
Previous studies have identified patient-specific variables, including age, lung disease stage, disease aggressiveness, and clinical state, that have bearing on measures of airway microbiota and must be taken into account in study design and interpretation of results in studies exploring the dynamics of airway microbiota in CF (29). Our study identifies changes in maintenance antibiotic use (e.g., chronic oral macrolides or inhaled antipseudomonal antibiotics) as additional potential confounders in studies characterizing airway microbiota during clinically relevant periods of interest. Although the impact of episodic antibiotic use (i.e., to treat pulmonary exacerbation) on measures of airway microbiota over both the short and long terms has been described (30, 31), the relevance of changes in maintenance antibiotics on these measures is less clear. Importantly, between 60% and 80% of persons with CF are prescribed chronic oral azithromycin and/or at least one chronic inhaled antibiotic (32). We observed significant changes in bacterial community structures in each of the six baseline periods during which a change in maintenance antibiotic use occurred. This observation is consistent with a recent study of ivacaftor-associated changes in CF airway microbiota in which significant changes were identified only after controlling for maintenance antibiotic use between the comparator groups (33). Our study is also consistent with a recent study of CF microbiota at baseline state before and after a single 28-day course of inhaled aztreonam (34), in which within-subject changes in relative abundances of certain taxa were observed.
This study also highlights the importance of granularity of sample collection in studies of CF airway microbiota dynamics. We observed that sample-to-sample similarity in community structure decreased with increasing interval days of sampling, as well as that sample-to-sample similarity was lower when the interval days of sampling crossed maintenance antibiotic regimens, again suggesting that observed sample-to-sample differences in structures within individuals were not entirely attributable to technical artifact. Thus, both sampling frequency and changes in maintenance antibiotic use are important considerations in studies assessing longitudinal changes in airway microbiota relative to changes in patient clinical state.
Our study was limited to adults with CF capable of daily sputum expectoration during periods of clinical stability. As such, it is not clear how these findings apply to persons with CF more broadly, including to children or to adults during other clinical states (e.g., pulmonary exacerbation or during episodic antibiotic treatment). We also note that of the subjects whose maintenance antibiotics were changed during a baseline period, and of those in whom more than one baseline period was available for analyses, all had early-stage lung disease. It is not clear from our data how the variation in bacterial community structures we observed across maintenance antibiotic regimens and across baseline periods may compare with that found in persons with more advanced lung disease, although we suspect that the low-diversity bacterial communities typically observed with advanced lung disease would be more resistant to antibiotic-induced perturbations.
To maximize measurements of microbiota in daily sputum samples, subjects and baseline periods were selected for which adherence to daily sample collection and survey completion was high. Although this study did not capture adherence to daily therapies other than antibiotics (e.g., airway clearance), it is possible that adherence to the study protocol was positively associated with adherence to CF therapies in general and that results may differ with varying degrees of adherence to prescribed CF therapies.
Finally, our study used expectorated sputum samples to measure airway microbiota. Noninvasive sampling of airway microbiota (with sputum or oropharyngeal swabs) is the standard of clinical care for routine monitoring of CF airway microbiota and guiding clinical decision making (35). Noninvasive sampling is also the only feasible strategy for analyses of daily variation of airway microbiota. Because sputum sample expectoration requires passage of the sample through the oral cavity, expectorated sputum samples can be assumed to contain a mixture of both sputum and saliva. Recent data, obtained via carefully controlled bronchoscopic sampling and controlling for DNA contamination from reagents, have confirmed that oral microbiota-associated taxa (e.g., Streptococcus, Prevotella, and Veillonella) are present in the CF lower airways as early as childhood (in addition to the typical CF pathogens) (36), providing further support for the use of sputum samples to represent airway microbiota.
In summary, appreciation of the range of variation in measures of CF airway microbiota during periods of clinical stability provides context for the degree of variation that could be considered significant in association with a change in clinical state. The occurrence of outlier samples (in which bacterial community structure deviates significantly from others around the same time), changes in maintenance antibiotic use, and frequency of sample collection and analysis have potential to confound interpretation of studies aimed at assessing bacterial community dynamics in CF and provide guidance for future studies designed to assess the dynamics of airway microbiota relative to changes in clinical state in persons with CF.
Supplementary Material
Acknowledgments
Acknowledgment
The authors are grateful for the dedication of the study subjects, whose generous participation made this study possible.
Footnotes
Supported by National Institutes of Health (NIH) grant K23HL136934 and Cystic Fibrosis Foundation grant CAVERL17A0 (L.J.C.) and by NIH grants R01HL136647 and R56HL126754 and Cystic Fibrosis Foundation grants LIPUMA13I0 and LIPUMA15P0 (J.J.L.). The views expressed in this article do not communicate official positions of the funding sources.
Author Contributions: L.J.C., J.L., L.A.C., L.M.K., R.H.S., and J.J.L. were involved in the conception and design of the work and in acquisition, analysis, and/or interpretation of data. K.S., K.O., M.A., S.C., B.F., and D.R.V. were involved in acquisition, analysis, and/or interpretation of data. All authors were involved in drafting and/or critically revising the work for important intellectual content, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Goss CH, Burns JL. Exacerbations in cystic fibrosis. 1: Epidemiology and pathogenesis. Thorax. 2007;62:360–367. doi: 10.1136/thx.2006.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanders DB, Bittner RC, Rosenfeld M, Hoffman LR, Redding GJ, Goss CH. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am J Respir Crit Care Med. 2010;182:627–632. doi: 10.1164/rccm.200909-1421OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanders DB, Bittner RC, Rosenfeld M, Redding GJ, Goss CH. Pulmonary exacerbations are associated with subsequent FEV1 decline in both adults and children with cystic fibrosis. Pediatr Pulmonol. 2011;46:393–400. doi: 10.1002/ppul.21374. [DOI] [PubMed] [Google Scholar]
- 4.West NE, Beckett VV, Jain R, Sanders DB, Nick JA, Heltshe SL, et al. STOP investigators. Standardized Treatment of Pulmonary Exacerbations (STOP) study: physician treatment practices and outcomes for individuals with cystic fibrosis with pulmonary exacerbations. J Cyst Fibros. 2017;16:600–606. doi: 10.1016/j.jcf.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmody LA, Zhao J, Kalikin LM, LeBar W, Simon RH, Venkataraman A, et al. The daily dynamics of cystic fibrosis airway microbiota during clinical stability and at exacerbation. Microbiome. 2015;3:12. doi: 10.1186/s40168-015-0074-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carmody LA, Caverly LJ, Foster BK, Rogers MAM, Kalikin LM, Simon RH, et al. Fluctuations in airway bacterial communities associated with clinical states and disease stages in cystic fibrosis. PLoS One. 2018;13:e0194060. doi: 10.1371/journal.pone.0194060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Layeghifard M, Li H, Wang PW, Donaldson SL, Coburn B, Clark ST, et al. Microbiome networks and change-point analysis reveal key community changes associated with cystic fibrosis pulmonary exacerbations. NPJ Biofilms Microbiomes. 2019;5:4. doi: 10.1038/s41522-018-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sibley CD, Parkins MD, Rabin HR, Duan K, Norgaard JC, Surette MG. A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc Natl Acad Sci USA. 2008;105:15070–15075. doi: 10.1073/pnas.0804326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whiteson KL, Meinardi S, Lim YW, Schmieder R, Maughan H, Quinn R, et al. Breath gas metabolites and bacterial metagenomes from cystic fibrosis airways indicate active pH neutral 2,3-butanedione fermentation. ISME J. 2014;8:1247–1258. doi: 10.1038/ismej.2013.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmody LA, Zhao J, Schloss PD, Petrosino JF, Murray S, Young VB, et al. Changes in cystic fibrosis airway microbiota at pulmonary exacerbation. Ann Am Thorac Soc. 2013;10:179–187. doi: 10.1513/AnnalsATS.201211-107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coburn B, Wang PW, Diaz Caballero J, Clark ST, Brahma V, Donaldson S, et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci Rep. 2015;5:10241. doi: 10.1038/srep10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whelan FJ, Heirali AA, Rossi L, Rabin HR, Parkins MD, Surette MG. Longitudinal sampling of the lung microbiota in individuals with cystic fibrosis. PLoS One. 2017;12:e0172811. doi: 10.1371/journal.pone.0172811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao J, Li J, Schloss PD, Kalikin LM, Raymond TA, Petrosino JF, et al. Effect of sample storage conditions on culture-independent bacterial community measures in cystic fibrosis sputum specimens. J Clin Microbiol. 2011;49:3717–3718. doi: 10.1128/JCM.01189-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caverly LJ, Caverly TJ, Kalikin LM, Foster BK, Simon RH, LiPuma JJ. Episodic oral antibiotic use in CF: discordance between the electronic medical record and self-report. J Cyst Fibros. 2016;15:630–633. doi: 10.1016/j.jcf.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konstan MW, Wagener JS, VanDevanter DR. Characterizing aggressiveness and predicting future progression of CF lung disease. J Cyst Fibros. 2009;8:S15–S19. doi: 10.1016/S1569-1993(09)60006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci USA. 2012;109:5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenfeld M, Emerson J, Williams-Warren J, Pepe M, Smith A, Montgomery AB, et al. Defining a pulmonary exacerbation in cystic fibrosis. J Pediatr. 2001;139:359–365. doi: 10.1067/mpd.2001.117288. [DOI] [PubMed] [Google Scholar]
- 18.Sanders DB, Solomon GM, Beckett VV, West NE, Daines CL, Heltshe SL, et al. STOP Study Group. Standardized Treatment of Pulmonary Exacerbations (STOP) study: observations at the initiation of intravenous antibiotics for cystic fibrosis pulmonary exacerbations. J Cyst Fibros. 2017;16:592–599. doi: 10.1016/j.jcf.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao J, Carmody LA, Kalikin LM, Li J, Petrosino JF, Schloss PD, et al. Impact of enhanced Staphylococcus DNA extraction on microbial community measures in cystic fibrosis sputum. PLoS One. 2012;7:e33127. doi: 10.1371/journal.pone.0033127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seekatz AM, Theriot CM, Molloy CT, Wozniak KL, Bergin IL, Young VB. Fecal microbiota transplantation eliminates Clostridium difficile in a murine model of relapsing disease. Infect Immun. 2015;83:3838–3846. doi: 10.1128/IAI.00459-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westcott SL, Schloss PD. OptiClust, an improved method for assigning amplicon-based sequence data to operational taxonomic units. mSphere. 2017;2:e00073-17. doi: 10.1128/mSphereDirect.00073-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 2002;148:257–266. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- 24.Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1–48. [Google Scholar]
- 25.Kuznetsova A, Brockhoff PB, Christensen RHB. lmerTest package: tests in linear mixed effects models. J Stat Softw. 2017;82:1–26. [Google Scholar]
- 26.R Core Team R: a language and environment for statistical computing. R Foundation for Statistical Computing; 2017 [accessed 2019 Jul 18]. Available from: https://www.R-project.org/
- 27.Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team nlme: linear and nonlinear mixed effects models. 2019 [accessed 2019 Jul 18]. Available from: https://CRAN.R-project.org/package=nlme.
- 28.Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016 [accessed 2019 Jul 18]. Available from: https://www.springer.com/gp/book/9780387981413.
- 29.Caverly LJ, LiPuma JJ. Cystic fibrosis respiratory microbiota: unraveling complexity to inform clinical practice. Expert Rev Respir Med. 2018;12:857–865. doi: 10.1080/17476348.2018.1513331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith DJ, Badrick AC, Zakrzewski M, Krause L, Bell SC, Anderson GJ, et al. Pyrosequencing reveals transient cystic fibrosis lung microbiome changes with intravenous antibiotics. Eur Respir J. 2014;44:922–930. doi: 10.1183/09031936.00203013. [DOI] [PubMed] [Google Scholar]
- 31.Zhao J, Murray S, LiPuma JJ. Modeling the impact of antibiotic exposure on human microbiota. Sci Rep. 2014;4:4345. doi: 10.1038/srep04345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry 2017 annual data report. Bethesda, MD: Cystic Fibrosis Foundation; 2018. [Google Scholar]
- 33.Peleg AY, Choo JM, Langan KM, Edgeworth D, Keating D, Wilson J, et al. Antibiotic exposure and interpersonal variance mask the effect of ivacaftor on respiratory microbiota composition. J Cyst Fibros. 2018;17:50–56. doi: 10.1016/j.jcf.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Heirali AA, Acosta N, Storey DG, Workentine ML, Somayaji R, Laforest-Lapointe I, et al. The effects of cycled inhaled aztreonam on the cystic fibrosis (CF) lung microbiome. J Cyst Fibros. 2019;19:30028–1. doi: 10.1016/j.jcf.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 35.Saiman L, Siegel JD, LiPuma JJ, Brown RF, Bryson EA, Chambers MJ, et al. Cystic Fibrous Foundation; Society for Healthcare Epidemiology of America. Infection prevention and control guideline for cystic fibrosis: 2013 update. Infect Control Hosp Epidemiol. 2014;35(Suppl 1):S1–S67. doi: 10.1086/676882. [DOI] [PubMed] [Google Scholar]
- 36.Jorth P, Ehsan Z, Rezayat A, Caldwell E, Pope C, Brewington JJ, et al. Direct lung sampling indicates that established pathogens dominate early infections in children with cystic fibrosis. Cell Rep. 2019;27:1190–1204.e3. doi: 10.1016/j.celrep.2019.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.