

Abstract
The neuromuscular junction is the synapse between a motor neuron of the spinal cord and a skeletal muscle fiber in the periphery. Reciprocal interactions between these excitable cells, and between them and others cell types present within the muscle tissue, shape the development, homeostasis and plasticity of skeletal muscle. An important aim in the field is to understand the molecular mechanisms underlying these cellular interactions, which include identifying the nature of the signals and receptors involved but also of the downstream intracellular signaling cascades elicited by them. This review focuses on work that shows that skeletal muscle fiber-derived extracellular signal-regulated kinases 1 and 2 (ERK1/2), ubiquitous and prototypical intracellular mitogen-activated protein kinases, have modulatory roles in the maintenance of the neuromuscular synapse and in the acquisition and preservation of fiber type identity in skeletal muscle.
Keywords: MAP kinase, ERK1/2, Agrin, neuromuscular junction, synapsespecific transcription
1. ERK1/2.
Mitogen-activated protein kinases (MAPKs) are part of intracellular signaling modules that control multiple cellular processes. MAPK modules consist of 3 core protein kinases. The most downstream enzyme, the actual MAPK, is the S/T (Ser/Thr) kinase that phosphorylates the transcription factors, cytoskeletal elements or other kinases, that are the targets of regulation by signaling pathways launched at the cell surface by activation of receptor tyrosine kinases, integrins and ion channels. A MAPK is activated by an upstream MAPK kinase (MAP2K), which in turn is activated by a MAP2K kinase (MAP3K). MAP3Ks generally integrate signals derived from small, monomeric GTPases such as the Ras family or from other more elaborate mechanisms [1]. In mammalian cells the prototypical MAPK module includes the MAPKs ERK1/2, the MAP2Ks MEK1/2 and the MAP3K Raf (Figure 1). ERK1/2 regulate normal cellular responses to numerous growth factors and cytokines in proliferation, differentiation and apoptosis [2].
Figure 1. Schematic diagram of canonical ERK1/2 signaling.
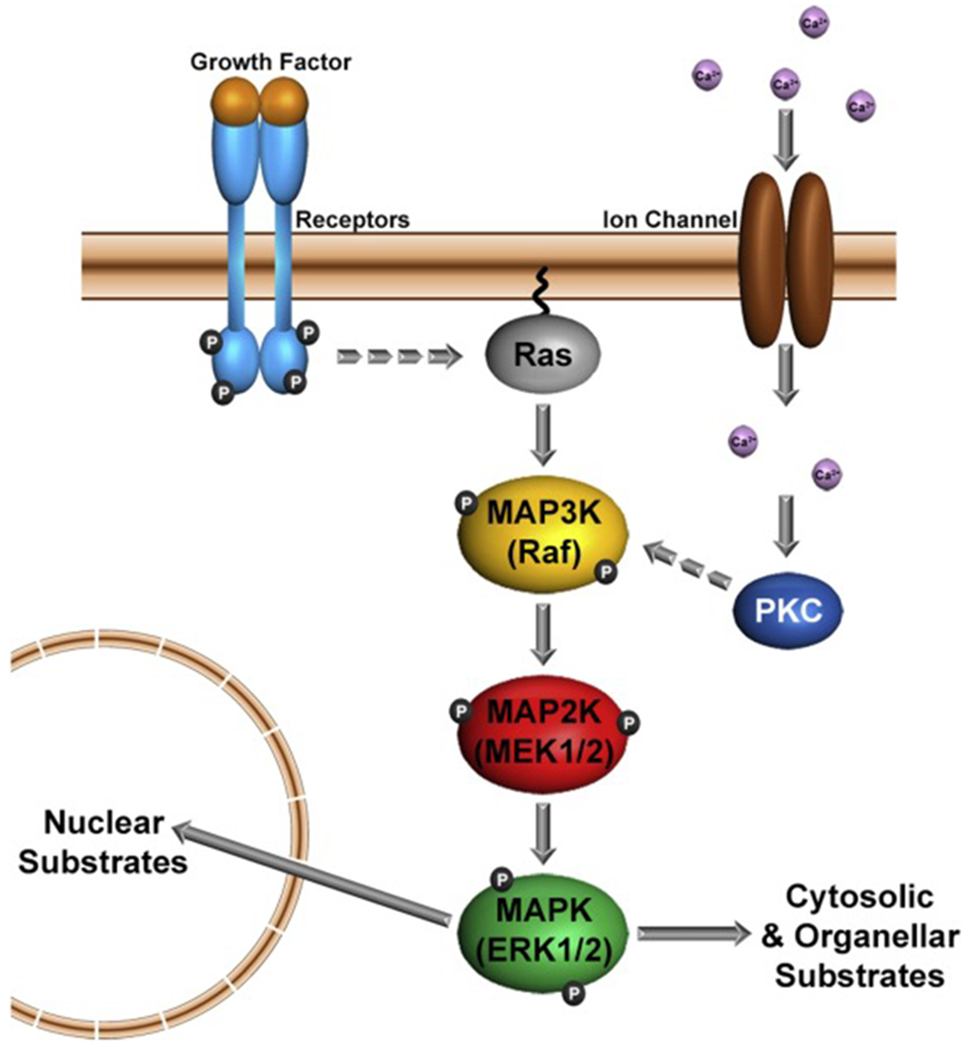
Ligand activation of growth factor receptors at the plasma membrane leads to phosphorylation of their cytoplasmic domains. Adaptor proteins (not depicted) bind to these phosphorylated sites and transduce the signal to small GTP-binding proteins such as Ras (dashed arrow). GTP-Ras, activates the 3-tier MAP kinase module of Raf (MAP3K), MEK1/2 (MAP2K), ERK1/2 (MAPK). Active ERK1/2 (a.k.a. pERK1/2) regulate many cellular processes by phosphorylating target proteins in the nucleus, cytosol and subcellular organelles. A rise in intracellular Ca2+ (lavender spheres) through plasma membrane Ca2+-channels can also lead to activation of the ERK1/2-MAP kinase module via protein kinase C (PKC). For simplicity, other pathways for ERK1/2 activation via integrins or G-protein-coupled receptors are not presented.
2. ERK1/2 and Muscle Fiber Mass and Type.
In adult muscle, four major myofiber types can be distinguished based on the expression of myosin heavy chain (MyHC) protein isoforms: type 1 (expressing MyHC-β/slow encoded by Myh7), type 2A (MyHC-2A encoded by Myh2), type 2X (MyHC-2X encoded by Myh1) and type 2B (MyHC-2B encoded by Myh4) [3, 4]. The contractile properties of these myofiber types are controlled by different classes of motoneurons, which impose on them specific patterns of activity from the slow (type 1), to the fast fatigue resistant (type 2A) and fast fatigable (2B). These patterns are paired with different metabolic properties that match the strength and persistence of the contractile forces involved: type 1 and 2A are more oxidative while type 2B are more glycolytic.
Table 1 summarizes results of studies on regulation of fiber size and specification by ERK signaling covered in this review. Pharmacological inhibition of ERK signaling induced atrophy in myotubes derived from the mouse muscle cell line C2C12. This result suggested that ERK1/2 might have a role in maintaining myofiber mass in vivo [5]. Consistent with these results, developmental selective abrogation of Erk1 and Erk2 in myofibers induced muscle fiber loss and atrophy, with stronger effects in the type1-fiber-rich soleus (SOL) than in type 2-fiber-rich tibialis anterior (TA) and sternomastoid (STN) [6, 7]. However, sustained ERK activation via selective myofiber expression of an MEK1 constitutively active mutant [8] or a Ras mutant that only activates the MEK-ERK1/2 pathway and not the PI3K-Akt pathway [9], failed to induce fiber hypertrophy. Together, these results suggest that ERK1/2 signaling is necessary, but not sufficient, to maintain normal fiber size.
Table 1.
Summary of studies on ERK1/2 signaling and myofiber size and specification discussed in this reviewa.
| Study [Reference] | Experimental Model | Manipulation of ERK1/2 pathway | Major Findings |
|---|---|---|---|
| [5] | C2C12 cells | Pharmacological block of ERK1/2 activation | Reduced myotube size |
| [6] | Mouse | Muscle conditional, loss-of-function Erk1/2 mutant. Focus on slow fibers | Dramatic fiber loss and atrophy. Marginal effect on fiber identity |
| [7] | Mouse | Muscle conditional, loss-of-function Erk1/2 mutant. Focus on fast fibers | Mild muscle fiber loss and atrophy. Marginal effect on fiber identity |
| [8] | Mouse | Muscle conditional, gain-of-function Mek1 mutant | No effect on fiber size. Fast-to-slow fiber switch |
| [9] | Rat | Electroporation of Ras constitutively-active (CA) & dominant-negative (DN) mutants Electrical stimulation of denervated SOL with slow/fast patterns |
No effect on fiber size. MyHC-β/ slow expression: Induced with CARas; suppressed with DNRas ERK activation: strong with slow stimulation; weak with fast stimulation |
| [13] | C2C12 cells and mouse SOL | How ERK1/2 activation and calcineurin-NFATc1 regulate MyHC-β/ slow transcription | pERK1/2 enhances NFATc1 transcriptional activity |
| [10] | Rat and mouse | Electroporation of: CAERK2 MKP-1 |
Slow-to-fast fiber switch Fast-to-slow fiber switch |
| [12] | Rat | pERK1/2 levels vis-a-vis fiber type | High pERK1/2 linked to fast fibers |
For abbreviations see text
ERK1/2 have also been implicated in the acquisition and maintenance of fiber type identity. Seemingly paradoxical, both the type 1 and type 2 phenotypes have been reported to be regulated by ERK signaling (Table 1). Thus, Shi and colleagues concluded that ERK signaling was critical to promote the fast type 2 fiber phenotype [10] based on the effects of overexpressing (i) a constitutively active ERK2 in slow rat SOL muscle, which induced the fast fiber type program; and (ii) MAPK phosphatase 1 (MKP-1) to inactivate ERK in fast adult mouse and rat skeletal muscle, which induced the slow fiber program. However, MKP-1 not only inactivates ERK1/2 but actually shows substrate preference for other MAPKs such as JNK and p38 [11]. Thus, the in vivo effects on fiber type expression reported in this study could have been unspecific to the inactivation of ERK1/2. Nevertheless, recent studies in the rat continue to support a role for ERK signaling in the promotion of the fast fiber phenotype [12]. On the other hand, compelling evidence supports a role for ERK1/2 in the promotion of slow fiber development and maintenance. Thus, a constitutively active form of Ras that selectively activates ERK1/2 induced MyHC-β/slow expression in regenerating denervated rat SOL muscle, while a dominant-negative form of Ras had the opposite effect [9]. In the same study, a low frequency/high amount impulse pattern (20 Hz), typical of slow motoneurons that innervate type 1 fibers, elicited the highest increase in ERK phosphorylation when applied to adult rat denervated SOL muscle. Firing patterns more similar to those for type 2B or type 2A fibers failed to stimulate, or more modestly induced ERK activation, respectively [9]. Thus, these results suggested innervation-dependent, higher basal levels of ERK activation in type 1 than type 2 myofibers. Consistent with the above results, selective myofiber expression of a constitutively active MEK1 led to a fast-to-slow-fiber type switch both in adult skeletal muscle as well as during development [8]. A molecular mechanism that may underlie induction of MyHC-β/slow by ERK activation involves increases in intracellular Ca2+ downstream of motoneuron-elicited electrical activity, which activate ERK1/2. Active ERK1/2, in turn, is proposed to phosphorylate the transcriptional co-activator p300 within the nucleus, which acetylates the transcription factor nuclear factor of activated T cells c1 (NFATc1) stimulating its DNA binding activity and resulting in up-regulation of Myh7 transcription [13]. Prior studies showed that the nuclear localization of NFATc1, which is regulated by the Ca2+-dependent phosphatase calcineurin, is critical for the promotion and maintenance of the slow fiber phenotype in vivo [14–16]. Thus both the calcineurin-NFAT and the ERK1/2-NFAT pathways are thought as slow nerve activity sensors [17]. Developmental loss-of-function manipulation of Erk1 and Erk2 in myofibers failed to induce slow-to-fast fiber-type switching [6], however it yielded preferential dramatic atrophy of type 1 fibers, with reduction in Myh7 mRNA expression consistent with a role for ERK signaling in the postnatal growth of slow fibers. The persistence of slow-fibers in ERK-deficient muscles, albeit atrophied, suggest that the calcineurin-NFAT signaling axis may still work in these muscles, and that consistent with results in C2C12 cells [13], it does not require active ERK in vivo, it is just potentiated by ERK activity. Although this possibility needs further investigation, it could explain the striking ability of gain-of-function manipulations of ERK signaling to induce a fast-to-slow fiber type switch [8, 9], and the inability of loss-of-function manipulations to generate a robust slow-to-fast fiber type switching. Thus although results are conflicting, perhaps due to particulars of experimental systems and methodologies, most in vitro and in vivo studies reviewed above favor an important role for ERK1/2 in type 1 fiber specification.
3. ERK1/2 at the Neuromuscular Junction.
3.1. Key Cellular and Molecular Players at the NMJ.
During embryonic development motor neurons send out axons that synapse onto skeletal muscle fibers to form the neuromuscular junction (NMJ) [18]. Three different cell types contribute the components of the NMJ. Synaptic vesicles containing the neurotransmitter acetylcholine (ACh) reside within the motoneuron’s presynaptic axon terminal; nicotinic acetylcholine receptors (AChRs) cluster on the myofiber’s postsynaptic sarcolemma, and the nerve terminal is capped by the non-myelinating glia terminal Schwann cells (tSCs), a.k.a. perisynaptic Schwann cells [19]. A narrow extracellular space, the synaptic cleft, filled with a specialized extracellular matrix (ECM), the synaptic basal lamina, separates pre- and postsynaptic components. Basal laminae also encapsulate tSCs and muscle fibers extrasynaptically. Cells called kranocytes, currently of unknown nature and function, that lie outside basal laminae but that cap the NMJ, have been proposed as a fourth cellular component of the NMJ [20]. Neuromuscular synaptic transmission is terminated by the hydrolysis of ACh by acetylcholinesterase (AChE), which is concentrated at the synaptic basal lamina. The formation and maintenance of the NMJ involves reciprocal interactions among these cell types. The best characterized molecular signaling pathway in NMJ formation and maintenance is the Agrin-Lrp4/MuSK-Dok7-Rapsyn cascade responsible for the clustering of AChRs on the postsynaptic apparatus [21]. The fundamental role of these proteins in the formation of the NMJ is underscored by the absence of synapses in loss-of-function mutant mice for all their corresponding coding genes [22–26]. Conditional gene targeting and siRNA knockdown have also demonstrated an essential role for Agrin, Lrp4 and MuSK in NMJ maintenance [27–30]. Agrin was purified from extracts of the electric organ of Torpedo californica, a source rich in NMJ proteins, based on its ability to induce aggregates or clusters of AChRs on the membrane of cultured myotubes [31]. Agrin is a heparan sulfate proteoglycan that localizes to the basal lamina. It is encoded by a large gene that through alternative splicing and differential promoter usage generates multiple isoforms with differential subcellular localization and biological activity [32, 33]. Binding of Agrin to basal lamina is mediated by an N-terminal domain [34], while the C-terminal domain harbors sequences encoded by exon Z (so named in mammals), of either 8, 11, or 19 amino acids, which are critical for Agrin’s AChR clustering activity [35]. Agrin is expressed by motoneurons, skeletal muscle fibers and tSCs, but only motoneurons express and secrete Z+ Agrin, the isoforms able to cluster AChRs (and many other synaptic proteins [36, 37]). Z-Agrin isoforms are inactive in AChR clustering assays. Although it was later found in other tissues, notably brain [38], the Muscle-Specific Kinase (MuSK) is a receptor tyrosine kinase first cloned from Torpedo electric organ [39], but shortly after shown to be highly concentrated at mammalian NMJs, induced by muscle denervation [40] and essential for neuromuscular synaptogenesis [24]. Albeit it does not bind MuSK, Z+ Agrin acts through MuSK by inducing its tyrosine phosphorylation [41]. The Low Density Lipoprotein (LDL)-receptor-related protein 4 (Lrp4) associates with MuSK and selectively binds Z+ Agrin [42, 43]. It is required for the assemply of the postsynaptic apparatus but has also been proposed as an early retrograde signal for presynaptic differentiation [44]. Downstream of tyrosine kinase 7 (Dok7) was identified as a member of the Dok family of phosphotyrosine-binding adaptor proteins that selectively binds to the tyrosine-phosphorylated NPXY (Gln-Pro-Any Amino acid-Tyr) intracellular motif of MuSK. Tyrosine-phosphorylation of this MuSK domain is induced by Z+ Agrin and is critical for Agrin’s AChR clustering activity [45, 46]. Thus, Dok7 binds and stabilizes Agrin-induced NPXY MuSK phosphorylation, is concentrated at synaptic sites and is essential for NMJ formation [25]. The AChR-associated protein at the synapse (Rapsyn) was also purified from Torpedo, even before Agrin, as a 43 kDa peripheral membrane protein that binds to intracellular domains of the AChR [47]. Although it is critical for AChR clustering in vivo [26], exactly how it is involved in the process is still a mystery. Initially proposed as a purely structural anchoring link between AChR clusters and the underlying synaptic cytoskeleton [48], recent findings indicate that Rapsyn is also endowed with enzymatic activity, specifically neddylation [49], that seems important for AChR clustering.
3.2. Experiments that Initially Suggested a Role for ERK Signaling at the NMJ.
The high concentration of AChRs at the NMJ is the result of both their clustering at the postsynaptic sarcolemma and their localized synthesis, which is mainly driven by the high rate of transcription for their coding genes at the synaptic myonuclei relative to that at extrasynaptic nuclei [50]. Synapse-specific transcription extends to the genes encoding many other synaptic proteins (e.g. MuSK) [18] and its physiological significance is underscored by the cases of human congenital myasthenias caused by mutation of the specific cis-regulatory promoter elements involved [51, 52]. Nerve-derived inductive signals were traditionally thought as responsible for clustering and synapse-specific transcription of synaptic proteins and their encoding genes. However, until around the turn of the century, separate signals, and signaling pathways, were believed to be involved. While Z+ Agrin was the clustering signal [53], the favorite nerve-derived signal for synapse-specific transcription was ARIA, or acetylcholine receptor-inducing activity [54]. Whereas soluble Z+ Agrin was unable to stimulate AChR gene transcription in cultured muscle cells [55]. ARIA was capable of inducing AChR synthesis without causing AChR clustering in cultured myotubes. Molecular cloning of the ARIA gene revealed that ARIA belongs to the family of ligands of the neu protooncogene. This gene was later renamed the Neuregulin 1 gene. It encodes multiple isoforms differentially expressed by motoneurons, muscle fibers and tSCs. Neuregulin-1 (Nrg-1) is a growth factor-like protein, that acts via tyrosine phosphorylation of the EGF-like receptors ErbB2, -3 and -4 [56]. Several labs showed that Nrg-1-induced AChR gene transcription in vitro was mediated, at least in part, by ERK signaling, which were among the first experiments that suggested a possible role for ERK1/2 at the NMJ [57–60]. Although multiple lines of evidence supported a critical role for Nrg-1 as the key neural signal driving synapse-specific AChR transcription (reviewed in [61]), ultimately loss- and gain-of-function gene targeting experiments showed that Nrg-1 was mostly dispensable for synapse-specific AChR gene transcription in vivo [62–64].
Because ACh and ATP are co-released from synaptic vesicles at the NMJ, and ATP can stimulate AChR- and AChE gene transcription in cultured myotubes via purinergic receptors P2RY1 and P2RY2 and downstream ERK activation [65], ATP was also investigated as a candidate for a neural signal for synapse-specific transcription. However, in P2ry1−/− mice, which also showed a large reduction in P2RY2 expression, AChE mRNA levels were very modestly decreased and an expected reduction in AChR gene transcription, consistent with prior in vitro experiments [65], was not even reported [66].
Genetic approaches surprisingly demonstrated that Z+ Agrin, acting through Lrp4/MuSK-Dok7, was necessary and apparently sufficient to mediate both AChR clustering and synapse-specific gene expression in vivo [22–25, 36, 67]. Rapsyn was found essential for AChR clustering but dispensable for synapse-specific transcription [26]. Because Z+ Agrin could induce the aggregation of muscle-derived Nrg-1 and its ErbB receptors in vivo [67, 68], a tantalizing mechanism by which Agrin could induce AChR gene transcription was to aggregate an autocrine Nrg-1/ErbB receptor signaling complex on the sarcolemma [69, 70], which would have implicated local downstream activation of ERK signaling. However, this potential mechanism was also shown unlikely in vivo by the aforementioned Nrg-1/ErbB mouse genetics experiments [62, 63].
Soluble Z+ Agrin was shown to activate ERK1/2 phosphorylation in myotubes derived from the C2 mouse muscle cell line [45, 71]. Within 5-10 min after application, Z+ Agrin induced a rapid and transient ERK1/2 activation in wild-type cells that was absent in myotubes from muscle cell lines genetically lacking either the Lrp4 or MuSK genes [71]. Beyond 30 min after application, Z+ Agrin-treated cultures exhibited ERK1/2 phosphorylation levels below baseline. This latter response was observed in WT cells and in the Lrp4- or MuSK-deficient myotubes [71]. Thus, Z+ Agrin induces both Lrp4/MuSK-dependent and –independent changes in ERK1/2 activation in the muscle cell lines studied thus far. Z+ Agrin-induced Lrp4/MuSK-dependent ERK1/2 activation is not necessary for Z+ Agrin-induced AChR clustering on C2 myotubes. Actually, pharmacological inhibition of ERK1/2 activation had either no effect [45] or potentiated [71] Z+ Agrin induced AChR clustering in these cells. Experiments that test the effects of soluble or substrate-bound Z+ Agrin treatment on ERK1/2 activation, and its role in AChR clustering or gene expression in the more physiological primary myotubes have yet to be done. In this context, it is important to mention here that soluble Z+ Agrin can stimulate ERK1/2 phosphorylation in myeloid cells [72] and primary cardiomyocytes [73]. In both settings, ERK1/2 activation is dependent on Agrin binding to α-dystroglycan, a receptor for both Z+ and Z− Agrin [74]. Dystroglycan is a transmembrane α–β heterodimer glycoprotein, β-dystroglycan appears to be a scaffold for MEK2 and ERK1 signaling [75], and interacts with the adaptor protein Grb2 [76], which usually links plasma membrane receptors to the canonical RAF-MEK-ERK pathway [2]. Duchene muscular dystrophy (DMD) is caused by loss of dystrophin protein due to mutation of the gene encoding it. Dystrophin depletion also leads to the loss of the dystrophin glycoprotein complex (DGC) from the sarcolemma, which contains dystroglycan and other proteins and contributes to the dystrophic phenotype. The DGC links the cytoskeleton to the ECM and provides mechanical stability to the sarcolemma during forceful contraction or stretch. At the NMJ, the dystrophin homolog utrophin binds AChRs and links them to the DGC [77]. Dystrophin is also concentrated at the NMJ but does not interact directly with AChRs [78]. Possible roles for α– and β–dystroglycan in the maintenance of the NMJ have been documented (e.g. [79, 80]).
3.3. Recent Experiments that Implicate ERK Signaling at the NMJ.
In a seminal study, William Snider and collaborators were the first to use gene targeting to study the developmental role of ERK1/2 signaling in Schwann cells, motor and sensory neurons [81]. Abrogation of Erk1 and Erk2 in Schwann cells or their precursors revealed essential roles for ERK1/2 signaling in glial cell survival and their ability to support and myelinate axons, by acting downstream of the Nrg1-ErbB2/3 interaction. On the other hand, loss of ERK signaling in motoneurons resulted surprisingly dispensable for the motor axons to grow out and form NMJs during embryonic development [81]. Potential roles for motoneuronal ERK signaling in the maintenance of the NMJ after birth or in the adult are yet to be studied using mouse genetics. It is not currently possible to perform gene targeting experiments selectively on tSCs without also perturbing Schwann cells that myelinate axons. Thus, mouse genetic experiments that specifically test the role of tSC ERK signaling in NMJ differentiation and maintenance remain to be done. Mice with selective developmental abrogation of Erk1 and Erk2 in skeletal muscle fibers formed NMJs as embryos but displayed postnatal defects in NMJ maintenance. Lack of ERK1/2 in myofibers caused reduction in mRNA and protein expression of Chrne, the gene encoding the ε-subunit of the pentameric AChR, typical of mature NMJs [82], specifically transcribed at synaptic myonuclei [50]. This effect was fiber-type independent and only required the abrogation of Erk2 to be observed [6, 7]. This result is consistent with prior experiments by Lin Mei and colleagues, who showed that dominant negative mutants forms of Ras, Raf, and MEK1 selectively inhibited synapse-specific expression of Chrne-luciferase reporters that were expressed in adult TA muscle following plasmid DNA injection [60]. These findings suggest that in vivo ERK signaling regulates AChRε expression at the transcriptional level.
NMJs in myofibers lacking ERK1/2 tended to be fragmented, with discontinuous domains of AChRs resembling endplates in aged muscles [83, 84], or in muscles from mammalian models of DMD [85, 86]. Synaptic fragmentation in DMD muscle is linked to persistent cycles of degeneration/regeneration of muscle fibers due to contraction-induced damage. Unlike normal fibers that have myonuclei in their periphery, close to the sarcolemma, regenerating fibers accumulate myonuclei in the center of the fiber, so called central nuclei [87]. Thus, central myonuclei are a hallmark for regenerating muscle tissue that has undergone damage. Sternomastoid muscle in mice lacking myofiber ERK2 exhibited NMJs as fragmented as those in muscles lacking both ERK1 and ERK2. However, the fraction of fibers with central nuclei in the former was no different than in WT control (1-2%), and in the double knockout Erk1/2 mutant the percentage of fragmented endplates (~70%) was much higher that the percentage of myofibers with central nuclei (~4%) [7]. Thus, synaptic fragmentation in ERK-deficient muscle is not tightly linked to muscle damage and is, perhaps, a consequence of disruption of local mechanisms that control the expression of synaptic components such as the AChRε subunit. Although this was not examined quantitatively, dystrophin is present on the surface of myofibers lacking ERK1/2 both synaptically and extrasynaptically [7]. This result suggests that α-dystroglycan and the rest of the DGC are also likely to be present on ERK1/2-less fibers. Whether the NMJ maintenance phenotype in ERK1/2-deficient fibers stems from DGC defects on composition, assembly or downstream signaling remains an open question.
In addition to synaptic fragmentation, evidence of at least partial denervation was also observed on a muscle group-dependent manner in the Erk1/2 double knockout mice [6, 7]. The SOL and STN muscles, but not the TA, displayed cellular and/or molecular signs of denervation such as terminal nerve sprouting and induction of denervation markers such as Musk, Chrng, the gene encoding the γ-subunit of the AChR, typical of fetal and denervated NMJs [88], the transcription factors Runxl [89] and myogenin [90], and the embryonic myosin heavy chain Myh3 [89]. Mitochondrial biogenesis and functional defects, consistent with denervation, were also observed in the ERK-deficient SOL [6]. In addition, in the mutant SOL a defective AChR γ/ε-subunit switch at innervated endplates preceded muscle fiber atrophy. This seemingly synaptic maturation defect was most prominent in type 1 than in type 2 muscle fibers [6]. This dramatic phenotype is unlikely the result of a simple reduction in AChRε expression and upregulation and persistence of AChRγ expression. Mice harboring a deletion of Chrne show persistent postnatal AChRγ expression and some synaptic fragmentation but were not reported to undergo bona fide denervation or muscle atrophy [91, 92]. It will be important to figure out what are the molecular mechanisms by which anomalies in synaptic maturation apparently lead to denervation and subsequent muscle atrophy in ERK1/2-deficient myofibers of specific muscle groups.
4. ERK1/2 in Satellite Cells and Potential Role in Synaptic Regeneration after Nerve Injury and Age-related NMJ Degeneration.
Following muscle damage, satellite cells (SCs), the resident myogenic stem cells in skeletal muscle [93], are activated to divide and give rise to myoblasts and these, in turn, fuse to produce myotubes that then become myofibers. Satellite cells are thus the principal source of myofiber nuclei during regeneration of adult muscle. Recent studies also revealed detectable levels of continuous SC activation, differentiation, fusion and turnover even in noninjured adult muscles [94]. Genetic depletion of SCs in young adults (3-6 month-old) [95] does not lead to synaptic myonuclei loss. Reduction of synaptic myonuclei in such SC-depleted muscles only is observed following transient denervation, which suggests that SCs have a direct role in regeneration of NMJs after nerve injury [95]. Thus from these data SCs are not required for normal NMJ homeostasis, at least in young mice. However, if SCs are depleted in young muscle and tissue examined at middle age [96], NMJs from SC-depleted animals show much more age-induced-like degeneration than controls. Thus, onset of age-related degeneration at NMJs seems accelerated by depletion of SCs. Age-related loss of NMJ myonuclei and NMJ integrity was rescued by SC-specific overexpression of Spry-1, a suppressor of receptor tyrosine kinase/FGF (RTK/FGF)-induced ERK signaling [96]. Conversely, Spry-1 deletion in SCs, which presumably raises levels of ERK activation, is linked to the loss of the SC pool in aged muscles [97]. Together, these results suggest that reducing ERK activity in SCs may lead to a delay in age-related degeneration of NMJs. Elevated ERK activity has been reported in muscle biopsies from old sedentary individuals, which presumably have associated NMJ degeneration [98]. This finding would be consistent with higher ERK signaling in SCs with aging. However, it is unclear from these data whether extrafusal fibers also contribute to this increase. Furthermore, as described above developmental deletion of Erk1/2 in extrafusal fibers leads to postnatal aging-like NMJ fragmentation and even denervation depending on the muscle group examined [6, 7], which suggest that reduced extrafusal ERK signaling can cause NMJ degeneration. The literature regarding changes in basal ERK phosphorylation with aging in rodent muscles is highly variable, with reports of increases, no changes or decreases depending on the muscle group studied [99]. Additional studies are needed to confirm and further characterize a role for SC- and myofiber-ERK signaling both in NMJ regeneration after nerve injury and in age-related synaptic degeneration.
5. Conclusions and Future Directions.
The initial in vitro studies that suggested a role for ERK signaling in AChR gene transcription seem now borne out by the more recent in vivo studies that detected a decrease in Chrne expression in myofibers lacking ERK1/2 genetically. Two major mechanistic questions remain open: what key molecules are downstream of ERK1/2 activation and what is the signal that activates ERK1/2 at the synapse in vivo. In regards to the first question, it will be important to investigate whether ERK1/2 can regulate the expression, localization and activity of Erm/ETV5, a E26 transformation-specific (ETS) transcription factor that is necessary for the synapse-specific expression of many postsynaptic genes, including Chrne and Musk [100]. This seems likely as transcription factors of the ETS family, such as Erm/ETV5, are known mediators and effectors of MAPK signaling in other cells [101–104]. If ERK signaling were to regulate Erm/ETV5 at the NMJ, its influence would extend to the transcription of many other postsynaptic genes besides Chrne, which might explain the larger impact of muscle ERK1/2-deficiency on NMJ integrity. Regarding the second question, Z+ Agrin has been shown to induce ERK activation but this has only been done in muscle cell lines and under conditions where Z+ Agrin fails to activate transcription. Future experiments need to determine whether Z+ Agrin, applied on an experimental condition where it stimulates AChR gene transcription (i.e. bound to ECM), can activate ERK1/2 phosphorylation, and whether that activation is Lrp4/MuSK- and/or α-dystroglycan-dependent. ERK1/2 can also be activated by increases in intracellular Ca2+ [1], which is critical to transduce patterns of electrical activity at the plasma membrane into changes in gene expression in nuclei of excitable cells such as muscle fibers (e.g. type 1 slow fibers) and neurons. About 4.1% of the ACh-evoked current passing through the ε-subunit-containing AChR at adult synapses is carried by Ca2+ ions [105]. This localized Ca2+ flux has putative physiological significance in that it activates Ca2+-dependent K+ channels that accelerate endplate repolarization [106]. It is tempting to speculate that this ACh-dependent influx of Ca2+ could also support high transcriptional levels of Chrne at subsynaptic myonuclei by inducing localized ERK1/2 activation. Figure 2 summarizes potential mechanisms by which ERK1/2 could regulate synapse-specific transcription of Chrne and perhaps additional postsynaptic genes. It is important to emphasize that regardless of the mechanism(s) ultimately involved, ERK1/2 activation is not essential for Chrne synapse-specific transcription. It just seems to enhance it perhaps to ensure the high rate of transcription required to maintain the high density of AChRs at the mature NMJ. Additional experimentation is also needed to account for the effects on NMJ integrity due to alterations of ERK activation in adult and aging muscle, which may as well involve transcriptional and posttranscriptional mechanisms.
Figure 2. Potential mechanisms for ERK signaling involvement in synapse-specific transcription.
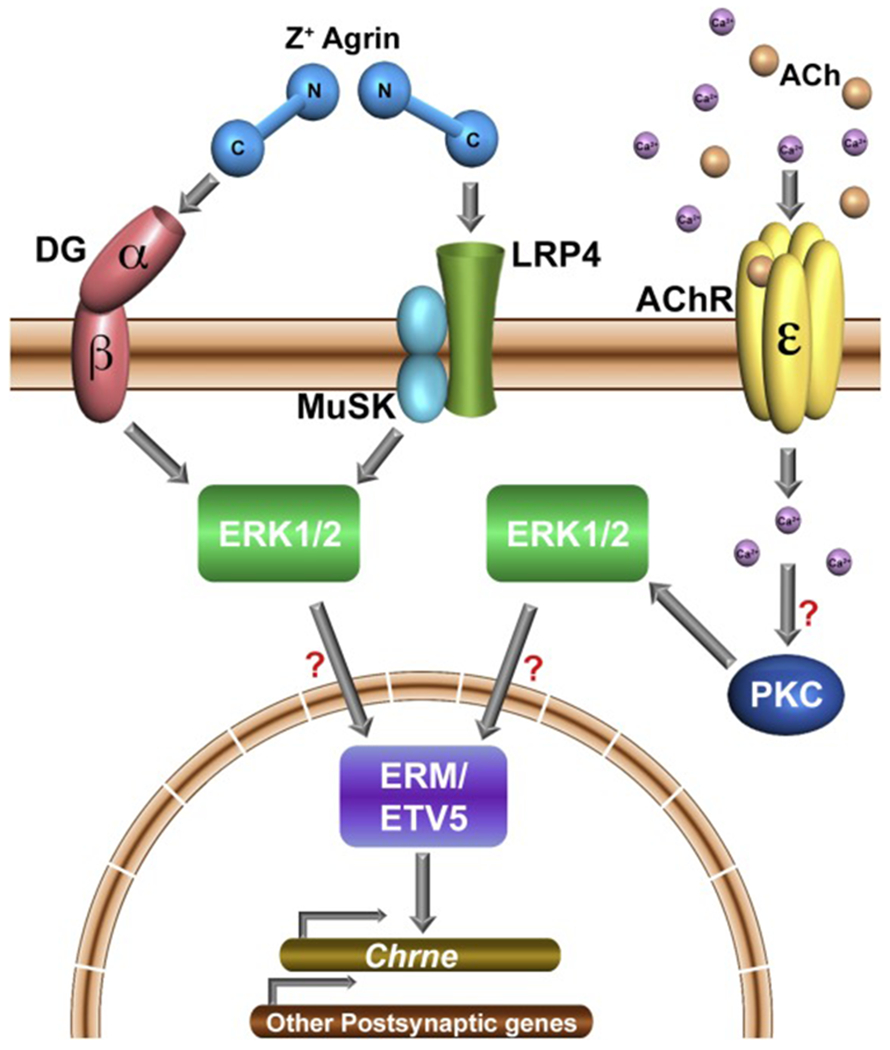
Z+ Agrin binding to either dystroglycan (DG) or to the Lrp4/MuSK complex can lead to ERK1/2 activation, which in turn would activate the Erm/ETV5 transcription factor. Erm/ETV5 would stimulate transcription of Chrne and other synapse-specific genes within synaptic myonuclei. ACh gating of the ε-subunit-containing AChR (α2βδε pentamer) would lead to Ca2+ entry that may activate PKC, which in turn would activate ERK1/2 and lead to transcriptional stimulation of synapse specific genes via Erm/ETV5. (?): Steps without direct experimental evidence currently.
The recent loss- and gain-of-function gene targeting experiments reviewed here [6–8] strongly support a role for ERK signaling in the determination and maintenance of the type 1 muscle fiber phenotype. Further experiments are needed to flesh out the mechanisms involved in vivo, in particular, the interactions of ERK-signaling with the Calcineurin-NFAT pathway, critical for type 1 muscle fiber identity. These studies may have translational relevance as the ability of constitutively active ERK1/2 to induce a fast-to-slow fiber switch has be invoked as a potential therapeutic approach to protect muscle fibers from the effects of lack of dystrophin [8]. Caution and further studies on ERK signaling in muscle are warranted though as excessive ERK activation is a hallmark of autosomal Emery Dreifuss muscular dystrophy (EDMD-A) [107], another muscular dystrophy due to mutation in the Lamin A/C encoding gene. Indeed, inhibition of ERK activation ameliorates skeletal and cardiac muscle symptoms of EDMD-A in a mouse model for the disease [108, 109].
Highlights.
ERK1/2 signaling regulate neuromuscular synapse-specific transcription
ERK1/2 signaling regulate acquisition and maintenance of slow myofiber phenotype
ERK1/2 signaling may control muscle progenitor role after nerve injury and aging
Acknowledgements
I thank Bonnie Seaberg for generating the cartoons in Figs 1 and 2, and Matt Lee for critically reading the manuscript. Research on ERK signaling in my lab was supported in part by a grant from NIH (NS077177).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References.
- [1].Yoon S, Seger R, The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions, Growth Factors 24 (2006) 21–44. [DOI] [PubMed] [Google Scholar]
- [2].Roskoski R, ERK1/2 MAP kinases: Structure, function, and regulation, Pharmacol Res 66 (2012) 105–143. [DOI] [PubMed] [Google Scholar]
- [3].Schiaffino S, Reggiani C, Fiber types in mammalian skeletal muscles., Physiological reviews 91 (2011) 1447–1531. [DOI] [PubMed] [Google Scholar]
- [4].Schiaffino S, Muscle fiber type diversity revealed by anti-myosin heavy chain antibodies, FEBS J 285 (2018) 3688–3694. [DOI] [PubMed] [Google Scholar]
- [5].Shi H, Scheffler JM, Zeng C, Pleitner JM, Hannon KM, Grant AL, Gerrard DE, Mitogen-activated protein kinase signaling is necessary for the maintenance of skeletal muscle mass., American journal of physiology. Cell physiology 296 (2009) C1040–1048. [DOI] [PubMed] [Google Scholar]
- [6].Wang S, Seaberg B, Paez-Colasante X, Rimer M, Defective Acetylcholine Receptor Subunit Switch Precedes Atrophy of Slow-Twitch Skeletal Muscle Fibers Lacking ERK1/2 Kinases in Soleus Muscle, Scientific Reports 6 (2016) 38745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Seaberg B, Henslee G, Wang S, Paez-Colasante X, Landreth GE, Rimer M, Muscle-derived extracellular signal-regulated kinases 1 and 2 are required for the maintenance of adult myofibers and their neuromuscular junctions, Molecular and Cellular Biology 35 (2015) 1238–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Boyer JG, Prasad V, Song T, Lee D, Fu X, Grimes KM, Sargent MA, Sadayappan S, Molkentin JD, ERK1/2 signaling induces skeletal muscle slow fiber-type switching and reduces muscular dystrophy disease severity, JCI Insight 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Murgia M, Serrano AL, Calabria E, Pallafacchina G, Lomo T, Schiaffino S, Ras is involved in nerve-activity-dependent regulation of muscle genes., Nat Cell Biol 2 (2000) 142–147. [DOI] [PubMed] [Google Scholar]
- [10].Shi H, Scheffler JM, Pleitner JM, Zeng C, Park S, Hannon KM, Grant AL, Gerrard DE, Modulation of skeletal muscle fiber type by mitogen-activated protein kinase signaling, FASEB J 22 (2008) 2990–3000. [DOI] [PubMed] [Google Scholar]
- [11].Flach RJR, Bennett AM, MAP kinase phosphatase-1--a new player at the nexus between sarcopenia and metabolic disease., Aging 2 (2010) 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Oishi Y, Ogata T, Ohira Y, Roy RR, Phosphorylated ERK1/2 protein levels are closely associated with the fast fiber phenotypes in rat hindlimb skeletal muscles, Pflugers Archiv - European Journal of Physiology (2019). [DOI] [PubMed] [Google Scholar]
- [13].Meissner JD, Freund R, Krone D, Umeda PK, Chang KC, Gros G, Scheibe RJ, Extracellular signal-regulated kinase 1/2-mediated phosphorylation of p300 enhances myosin heavy chain I/beta gene expression via acetylation of nuclear factor of activated T cells c1, Nucleic Acids Res 39 (2011) 5907–5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chi ER, N OE, Richardson JA, Yang Q, Humphries C, Shelton JM, Wu H, Weiguang Z, Bassel-Duby R, Williams RS, A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type, Genes & development 12 (1998) 2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].McCullagh KJ, Calabria E, Pallafacchina G, Ciciliot S, Serrano AL, Argentini C, Kalhovde JM, Lomo T, Schiaffino S, NFAT is a nerve activity sensor in skeletal muscle and controls activity-dependent myosin switching, Proc Natl Acad Sci U S A 101 (2004) 10590–10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Serrano AL, Murgia M, Pallafacchina G, Calabria E, Coniglio P, Lomo T, Schiaffino S, Calcineurin controls nerve activity-dependent specification of slow skeletal muscle fibers but not muscle growth, Proc Natl Acad Sci U S A 98 (2001) 13108–13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pallafacchina G, Calabria E, Serrano AL, Kalhovde JM, Schiaffino S, A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification, Proc Natl Acad Sci U S A 99 (2002) 9213–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tintignac LA, Brenner HR, Ruegg MA, Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting, Physiol Rev 95 (2015) 809–852. [DOI] [PubMed] [Google Scholar]
- [19].Ko CP, Robitaille R, Perisynaptic Schwann Cells at the Neuromuscular Synapse: Adaptable, Multitasking Glial Cells, Cold Spring Harb Perspect Biol 7 (2015) a020503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Court FA, Gillingwater TH, Melrose S, Sherman DL, Greenshields KN, Morton AJ, Harris JB, Willison HJ, Ribchester RR, Identity, developmental restriction and reactivity of extralaminar cells capping mammalian neuromuscular junctions, J Cell Sci 121 (2008) 3901–3911. [DOI] [PubMed] [Google Scholar]
- [21].Burden SJ, Huijbers MG, Remedio L, Fundamental Molecules and Mechanisms for Forming and Maintaining Neuromuscular Synapses, Int J Mol Sci 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR, Defective neuromuscular synaptogenesis in agrin-deficient mutant mice, Cell 85 (1996) 525–535. [DOI] [PubMed] [Google Scholar]
- [23].Weatherbee SD, Anderson KV, Niswander LA, LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction, Development 133 (2006) 4993–5000. [DOI] [PubMed] [Google Scholar]
- [24].DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD, The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo, Cell 85 (1996) 501–512. [DOI] [PubMed] [Google Scholar]
- [25].Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, S K, Shiraishi H, Eguchi K, Motomura M, Akiyama T, Iwakura Y, Higuchi O, Yamanashi Y, Kubo S, Shiraishi H, Eguchi K, Motomura M, Akiyama T, Iwakura Y, Higuchi O, Yamanashi Y, The Muscle Protein Dok-7 Is Essential for Neuromuscular Synaptogenesis, Science 312 (2006) 1802–1806. [DOI] [PubMed] [Google Scholar]
- [26].Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP, Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice, Nature 377 (1995) 232–236. [DOI] [PubMed] [Google Scholar]
- [27].Samuel MA, Valdez G, Tapia JC, Lichtman JW, Sanes JR, Agrin and synaptic laminin are required to maintain adult neuromuscular junctions., PloS one 7 (2012) e46663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Barik A, Lu Y, Sathyamurthy A, Bowman A, Shen C, Li L, Xiong W-C, Mei L, LRP4 Is Critical for Neuromuscular Junction Maintenance, Journal of Neuroscience 34 (2014) 13892–13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kong XC, Barzaghi P, Ruegg MA, Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference, EMBO Rep 5 (2004) 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hesser BA, Henschel O, Witzemann V, Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK, Mol Cell Neurosci 31 (2006) 470–480. [DOI] [PubMed] [Google Scholar]
- [31].Nitkin RM, Smith MA, Magill C, Fallon JR, Yao YM, Wallace BG, McMahan UJ, Identification of agrin, a synaptic organizing protein from Torpedo electric organ, J Cell Biol 105 (1987) 2471–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rupp F, Ozcelik T, Linial M, Peterson K, Francke U, Scheller R, Structure and chromosomal localization of the mammalian agrin gene, J Neurosci 12 (1992) 3535–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ruegg MA, Tsim KW, Horton SE, Kroger S, Escher G, Gensch EM, McMahan UJ, The agrin gene codes for a family of basal lamina proteins that differ in function and distribution, Neuron 8 (1992) 691–699. [DOI] [PubMed] [Google Scholar]
- [34].Denzer AJ, Gesemann M, Schumacher B, Ruegg MA, An amino-terminal extension is required for the secretion of chick agrin and its binding to extracellular matrix, J Cell Biol 131 (1995) 1547–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Burgess RW, Nguyen QT, Son YJ, Lichtman JW, Sanes JR, Alternatively spliced isoforms of nerve- and muscle-derived agrin: their roles at the neuromuscular junction, Neuron 23 (1999) 33–44. [DOI] [PubMed] [Google Scholar]
- [36].Cohen I, Rimer M, Lømo T, McMahan UJ, Lomo T, Agrin-induced postsynaptic-like apparatus in skeletal muscle fibers in vivo [published erratum appears in Mol Cell Neurosci 1997;10(3-4):208], Mol Cell Neurosci 9 (1997) 237–253. [DOI] [PubMed] [Google Scholar]
- [37].Jones G, Meier T, Lichtsteiner M, Witzemann V, Sakmann B, Brenner HR, Induction by agrin of ectopic and functional postsynaptic-like membrane in innervated muscle, Proc Natl Acad Sci U S A 94 (1997) 2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Garcia-Osta A, Tsokas P, Pollonini G, Landau EM, Blitzer R, Alberini CM, MuSK expressed in the brain mediates cholinergic responses, synaptic plasticity, and memory formation, J Neurosci 26 (2006) 7919–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jennings CG, Dyer SM, Burden SJ, Muscle-specific trk-related receptor with a kringle domain defines a distinct class of receptor tyrosine kinases, Proc Natl Acad Sci U S A 90 (1993) 2895–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nunez L, Park JS, Stark JL, Gies DR, et al. , Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury, Neuron 15 (1995) 573–584. [DOI] [PubMed] [Google Scholar]
- [41].Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD, Agrin acts via a MuSK receptor complex, Cell 85 (1996) 513–523. [DOI] [PubMed] [Google Scholar]
- [42].Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ, Lrp4 is a receptor for Agrin and forms a complex with MuSK, Cell 135 (2008) 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L, LRP4 serves as a coreceptor of agrin, Neuron 60 (2008) 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yumoto N, Kim N, Burden SJ, Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses., Nature 489 (2012) 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Herbst R, Burden SJ, The juxtamembrane region of MuSK has a critical role in agrin-mediated signaling [published erratum appears in EMBO J 2000 Mar 1;19(5):1167], EMBO J 19 (2000) 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhou H, Glass DJ, Yancopoulos GD, Sanes JR, Distinct domains of MuSK mediate its abilities to induce and to associate with postsynaptic specializations, J Cell Biol 146 (1999) 1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Neubig RR, Krodel EK, Boyd ND, Cohen JB, Aceltylcholine and local anesthetic binding to Torpedo nicotinic postsynaptic membranes after removal of nonreceptor peptides, Proc Natl Acad Sci U S A 76 (1979) 690–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Apel ED, Roberds SL, Campbell KP, Merlie JP, Rapsyn may function as a link between the acetylcholine receptor and the agrin-binding dystrophin-associated glycoprotein complex, Neuron 15 (1995) 115–126. [DOI] [PubMed] [Google Scholar]
- [49].Li L, Cao Y, Wu H, Ye X, Zhu Z, Xing G, Shen C, Barik A, Zhang B, Xie X, Zhi W, Gan L, Su H, Xiong WC, Mei L, Enzymatic Activity of the Scaffold Protein Rapsyn for Synapse Formation, Neuron (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Merlie JP, Sanes JR, Concentration of acetylcholine receptor mRNA in synaptic regions of adult muscle fibres, Nature 317 (1985) 66–68. [DOI] [PubMed] [Google Scholar]
- [51].Ohno K, Anlar B, Engel AG, Congenital myasthenic syndrome caused by a mutation in the Ets-binding site of the promoter region of the acetylcholine receptor epsilon subunit gene, Neuromuscul Disord 9 (1999) 131–135. [DOI] [PubMed] [Google Scholar]
- [52].Nichols P, Croxen R, Vincent A, Rutter R, Hutchinson M, Newsom-Davis J, Beeson D, Mutation of the acetylcholine receptor epsilon-subunit promoter in congenital myasthenic syndrome, Ann Neurol 45 (1999) 439–443. [PubMed] [Google Scholar]
- [53].McMahan UJ, The agrin hypothesis, Cold Spring Harb Symp Quant Biol 55 (1990) 407–418. [DOI] [PubMed] [Google Scholar]
- [54].Martinou JC, Falls DL, Fischbach GD, Merlie JP, Acetylcholine receptor-inducing activity stimulates expression of the epsilon-subunit gene of the muscle acetylcholine receptor, Proc Natl Acad Sci U S A 88 (1991) 7669–7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jones G, Herczeg A, Ruegg MA, Lichtsteiner M, Kroger S, Brenner HR, Substrate-bound agrin induces expression of acetylcholine receptor epsilon-subunit gene in cultured mammalian muscle cells, Proc Natl Acad Sci U S A 93 (1996) 5985–5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Falls DL, Rosen KM, Corfas G, Lane WS, Fischbach GD, ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family, Cell 72 (1993) 801–815. [DOI] [PubMed] [Google Scholar]
- [57].Altiok N, Altiok S, Changeux JP, Heregulin-stimulated acetylcholine receptor gene expression in muscle: requirement for MAP kinase and evidence for a parallel inhibitory pathway independent of electrical activity, EMBO J 16 (1997) 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tansey MG, Chu GC, Merlie JP, ARIA/HRG regulates AChR epsilon subunit gene expression at the neuromuscular synapse via activation of phosphatidylinositol 3-kinase and Ras/MAPK pathway, J Cell Biol 134 (1996) 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Si J, Luo Z, Mei L, Induction of acetylcholine receptor gene expression by ARIA requires activation of mitogen-activated protein kinase, J Biol Chem 271 (1996) 19752–19759. [DOI] [PubMed] [Google Scholar]
- [60].Si J, Mei L, ERK MAP kinase activation is required for acetylcholine receptor inducing activity-induced increase in all five acetylcholine receptor subunit mRNAs as well as synapse-specific expression of acetylcholine receptor epsilon-transgene, Brain Res Mol Brain Res 67 (1999) 18–27. [DOI] [PubMed] [Google Scholar]
- [61].Rimer M, Neuregulins at the neuromuscular synapse: past, present, and future, J Neurosci Res 85 (2007) 1827–1833. [DOI] [PubMed] [Google Scholar]
- [62].Escher P, Lacazette E, Courtet M, Blindenbacher a., Landmann L, Bezakova G, Lloyd KC, Mueller U, Brenner HR, Synapses form in skeletal muscles lacking neuregulin receptors., Science (New York, N.Y.) 308 (2005) 1920–1923. [DOI] [PubMed] [Google Scholar]
- [63].Jaworski A, Burden SJ, Neuromuscular synapse formation in mice lacking motor neuron- and skeletal muscle-derived Neuregulin-1, J Neurosci 26 (2006) 655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ponomareva ON, Ma H, Vock VM, Ellerton EL, Moody SE, Dakour R, Chodosh LA, Rimer M, Defective neuromuscular synaptogenesis in mice expressing constitutively active ErbB2 in skeletal muscle fibers, Mol Cell Neurosci 31 (2006) 334–345. [DOI] [PubMed] [Google Scholar]
- [65].Tung EK, Choi RC, Siow NL, Jiang JX, Ling KK, Simon J, Barnard EA, Tsim KW, P2Y2 receptor activation regulates the expression of acetylcholinesterase and acetylcholine receptor genes at vertebrate neuromuscular junctions, Mol Pharmacol 66 (2004) 794–806. [DOI] [PubMed] [Google Scholar]
- [66].Xu ML, Bi CW, Cheng LK, Mak S, Yao P, Luk WK, Lau KK, Cheng AW, Tsim KW, Reduced Expression of P2Y2 Receptor and Acetylcholinesterase at Neuromuscular Junction of P2Y1 Receptor Knock-out Mice, J Mol Neurosci 57 (2015) 446–451. [DOI] [PubMed] [Google Scholar]
- [67].Meier T, Hauser DM, Chiquet M, Landmann L, Ruegg MA, Brenner HR, Neural agrin induces ectopic postsynaptic specializations in innervated muscle fibers, J Neurosci 17 (1997) 6534–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rimer M, Cohen I, Lomo T, Burden SJ, McMahan UJ, Neuregulins and erbB receptors at neuromuscular junctions and at agrin-induced postsynaptic-like apparatus in skeletal muscle, Mol Cell Neurosci 12 (1998) 1–15. [DOI] [PubMed] [Google Scholar]
- [69].Meier T, Masciulli F, Moore C, Schoumacher F, Eppenberger U, Denzer AJ, Jones G, Brenner HR, Agrin can mediate acetylcholine receptor gene expression in muscle by aggregation of muscle-derived neuregulins, J Cell Biol 141 (1998) 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Moore C, Leu M, Muller U, Brenner HR, Müller U, Induction of multiple signaling loops by MuSK during neuromuscular synapse formation, Proc Natl Acad Sci U S A 98 (2001) 14655–14660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Rimer M, Modulation of Agrin-induced Acetylcholine Receptor Clustering by Extracellular Signal-regulated Kinases 1 and 2 in Cultured Myotubes, J Biol Chem 285 (2010) 32370–32377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mazzon C, Anselmo A, Soldani C, Cibella J, Ploia C, Moalli F, Burden SJ, Dustin ML, Sarukhan A, Viola A, Agrin is required for survival and function of monocytic cells, Blood 119 (2012) 5502–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch-Umansky K, Yifa O, Kain D, Rajchman D, Leach J, Bassat DR, Udi Y, Sarig R, Sagi I, Martin JF, Bursac N, Cohen S, Tzahor E, The extracellular matrix protein Agrin promotes heart regeneration in mice, Nature (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bowe MA, Deyst KA, Leszyk JD, Fallon JR, Identification and purification of an agrin receptor from Torpedo postsynaptic membranes: a heteromeric complex related to the dystroglycans, Neuron 12 (1994) 1173–1180. [DOI] [PubMed] [Google Scholar]
- [75].Spence HJ, Dhillon AS, James M, Winder SJ, Dystroglycan, a scaffold for the ERK-MAP kinase cascade, EMBO Rep 5 (2004) 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP, SH3 domain-mediated interaction of dystroglycan and Grb2, J Biol Chem 270 (1995) 11711–11714. [DOI] [PubMed] [Google Scholar]
- [77].Banks GB, Fuhrer C, Adams ME, Froehner SC, The postsynaptic submembrane machinery at the neuromuscular junction: requirement for rapsyn and the utrophin/dystrophin-associated complex, J Neurocytol 32 (2003) 709–726. [DOI] [PubMed] [Google Scholar]
- [78].Bewick GS, Young C, Slater CR, Spatial relationships of uthrophin, dystrophin, beta-dystroglycan and beta-spectrin to acetylcholine receptor clusters during postnatal maturation of the rat neuromuscular junction. , J Neurocytol 25 (1996) 367–379. [PubMed] [Google Scholar]
- [79].Jacobson C, Montanaro F, Lindenbaum M, Carbonetto S, Ferns M, alpha-Dystroglycan functions in acetylcholine receptor aggregation but is not a coreceptor for agrin-MuSK signaling, J Neurosci 18 (1998) 6340–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kahl J, Campanelli JT, A role for the juxtamembrane domain of beta-dystroglycan in agrin-induced acetylcholine receptor clustering, J Neurosci 23 (2003) 392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Newbern JM, Li X, Shoemaker SE, Zhou J, Zhong J, Wu Y, Bonder D, Hollenback S, Coppola G, Geschwind DH, Landreth GE, Snider WD, Specific Functions for ERK/MAPK Signaling during PNS Development, Neuron 69 (2011) 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S, Methfessel C, Sakmann B, Molecular distinction between fetal and adult forms of muscle acetylcholine receptor, Nature 321 (1986) 406–411. [DOI] [PubMed] [Google Scholar]
- [83].Valdez G, Tapia JC, Kang H, Clemenson GD, Gage FH, Lichtman JW, Sanes JR, Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise., Proc Natl Acad Sci U S A 107 (2010) 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Li Y, Lee YI, Thompson WJ, Changes in Aging Mouse Neuromuscular Junctions Are Explained by Degeneration and Regeneration of Muscle Fiber Segments at the Synapse, J Neurosci 31 (2011) 14910–14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Lyons PR, Slater CR, Structure and function of the neuromuscular junction in young adult mdx mice., Journal of neurocytology 20 (1991) 969–981. [DOI] [PubMed] [Google Scholar]
- [86].Haddix SG, Lee YI, Kornegay JN, Thompson WJ, Cycles of myofiber degeneration and regeneration lead to remodeling of the neuromuscular junction in two mammalian models of Duchenne muscular dystrophy, PLoS One 13 (2018) e0205926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Carlson BM, The regeneration of skeletal muscle - a review, Am J Anat 137 (1973) 119–149. [DOI] [PubMed] [Google Scholar]
- [88].Witzemann V, Barg B, Nishikawa Y, Sakmann B, Numa S, Differential regulation of muscle acetylcholine receptor gamma- and epsilon-subunit mRNAs., FEBS letters 223 (1987) 104–112. [DOI] [PubMed] [Google Scholar]
- [89].Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, Burden SJ, Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle., Genes & development 19 (2005) 1715–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Moresi V, Williams AH, Meadows E, Flynn JM, Potthoff MJ, McAnally J, Shelton JM, Backs J, Klein WH, Richardson JA, Bassel-Duby R, Olson EN, Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases, Cell 143 (2010) 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Witzemann V, Schwarz H, Koenen M, Berberich C, Villaroel A, Werning A, Brenner HR, Sakmann B, Acetylcholine receptor epsilon -subunit deletion causes muscle weakness, Proc Natl Acad Sci U S A 93 (1996) 13286–13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Missias AC, Mudd J, Cunningham JM, Steinbach JH, Merlie JP, Sanes JR, Deficient development and maintenance of postsynaptic specializations in mutant mice lacking an ‘adult’ acetylcholine receptor subunit, Development 124 (1997) 5075–5086. [DOI] [PubMed] [Google Scholar]
- [93].Mauro A, Satellite cell of skeletal muscle fibers., The Journal of biophysical and biochemical cytology 9 (1961) 493–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Pawlikowski B, Pulliam C, Betta ND, Kardon G, Olwin BB, Pervasive satellite cell contribution to uninjured adult muscle fibers, Skeletal Muscle 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Liu W, Wei-LaPierre L, Klose A, Dirksen RT, Chakkalakal JV, Inducible depletion of adult skeletal muscle stem cells impairs the regeneration of neuromuscular junctions, eLife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Liu W, Klose A, Forman S, Paris ND, Wei-LaPierre L, Cortes-Lopez M, Tan A, Flaherty M, Miura P, Dirksen RT, Chakkalakal JV, Loss of adult skeletal muscle stem cells drives age-related neuromuscular junction degeneration, eLife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chakkalakal JV, Jones KM, Basson MA, Brack AS, The aged niche disrupts muscle stem cell quiescence, Nature 490 (2012) 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Williamson D, Gallagher P, Harber M, Hollon C, Trappe S, Mitogen-activated protein kinase (MAPK) pathway activation: effects of age and acute exercise on human skeletal muscle., The Journal of physiology 547 (2003) 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Rahnert JA, Luo Q, Balog EM, Sokoloff AJ, Burkholder TJ, Changes in growth-related kinases in head, neck and limb muscles with age., Experimental gerontology 46 (2011) 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Hippenmeyer S, Huber RM, Ladle DR, Murphy K, Arber S, ETS Transcription Factor Erm Controls Subsynaptic Gene Expression in Skeletal Muscles, Neuron 55 (2007) 726–740. [DOI] [PubMed] [Google Scholar]
- [101].Znosko WA, Yu S, Thomas K, Molina GA, Li C, Tsang W, Dawid IB, Moon AM, Tsang M, Overlapping functions of Pea3 ETS transcription factors in FGF signaling during zebrafish development, Developmental Biology 342 (2010) 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zhang Z, Newton K, Kummerfeld SK, Webster J, Kirkpatrick DS, Phu L, Eastham-Anderson J, Liu J, Lee WP, Wu J, Li H, Junttila MR, Dixit VM, Transcription factor Etv5 is essential for the maintenance of alveolar type II cells, Proc Natl Acad Sci U S A 114 (2017) 3903–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Oh S, Shin S, Janknecht R, ETV1, 4 and 5: An oncogenic subfamily of ETS transcription factors, Biochimica et Biophysica Acta - Reviews on Cancer 1826 (2012) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Janknecht R, Monte D, Baert J-L, Yvan d.L., The ETS-related transcription factor ERM is a nuclear target of signaling cascades involving MAPK and PKA, Oncogene 13 (1996) 1745–1754. [PubMed] [Google Scholar]
- [105].Grassi F, Fucile S, Calcium influx through muscle nAChR-channels: One route, multiple roles, Neuroscience (2019). [DOI] [PubMed] [Google Scholar]
- [106].Allard B, Bernengo J-C, Rougier O, Jacquemond V, Intracellular Ca2+ changes and Ca2+ - activated K+ channel activation induced by acetylcholine at the endplate of mouse skeletal muscle fibers J Physiol 494 (1996) 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacène E, Fromes Y, Toussaint M, Mura A-M, Keller DI, Amthor H, Isnard R, Malissen M, Schwartz K, Bonne G, Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies., Human molecular genetics 14 (2005) 155–169. [DOI] [PubMed] [Google Scholar]
- [108].Muchir A, Kim YJ, Reilly SA, Wu W, Choi JC, Worman HJ, Inhibition of extracellular signal-regulated kinase 1/2 signaling has beneficial effects on skeletal muscle in a mouse model of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutation., Skeletal muscle 3 (2013) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Wu W, Chordia MD, Hart BP, Kumarasinghe ES, Ji MK, Bhargava A, Lawlor MW, Shin JY, Sera F, Homma S, Muchir A, Khire UR, Worman HJ, Macrocyclic MEK1/2 inhibitor with efficacy in a mouse model of cardiomyopathy caused by lamin A/C gene mutation, Bioorg Med Chem 25 (2017) 1004–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]