

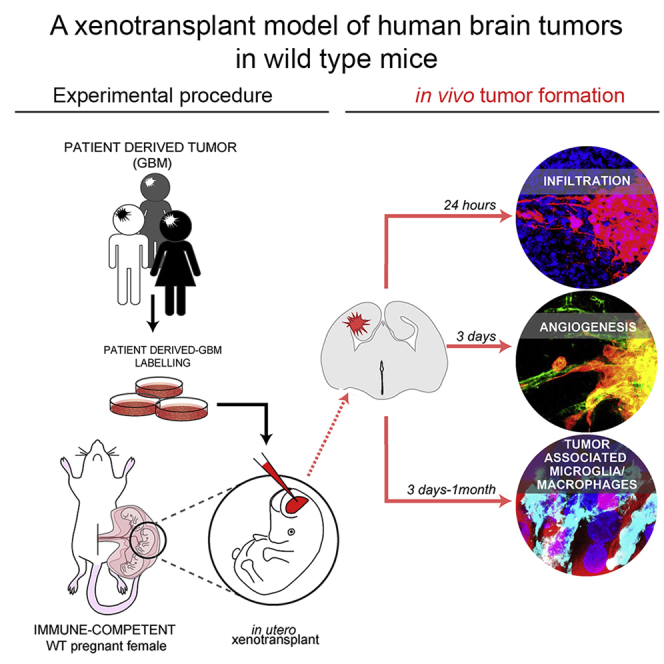
Summary
The development of adequate model systems to study human malignancies is crucial for basic and preclinical research. Here, we exploit the “immune-privileged” developmental time window to achieve orthotopic xenotransplantation of human brain tumor cells in wild-type (WT) mice. We find that, when transplanted in utero, human glioblastoma (GBM) cells readily integrate in the embryonic mouse brain mirroring key tumor-associated pathological features such as infiltration, vascularization, and complex tumor microenvironment including reactive astrocytes and host immune cell infiltration. Remarkably, activation of the host IBA1 tumor-associated microglia/macrophages depends on the type of glioma cell transplanted, suggesting our approach allows one to study human GBM interactions with the immune system of WT host mice. The embryonic engraftment model complements existing ones, providing a rapid and valuable alternative to study fundamental biology of human brain tumors in immune competent mice.
Subject Areas: Biological Sciences, Cancer, Biological Sciences Research Methodologies, Model Organism
Graphical Abstract
Highlights
-
•
Human glioblastoma cells quickly form tumors in brain of WT mouse embryos
-
•
Glioblastoma xenografts mirror several key characteristics of the original tumor
-
•
Host IBA1+ microglia/macrophages associate with glioblastoma xenografts
-
•
Activation of IBA1+ tumor-associated cells depends on the type of xenograft
Biological Sciences; Cancer; Biological Sciences Research Methodologies; Model Organism
Introduction
The development of adequate model systems to study human malignancies is crucial for basic and preclinical research. For over 30 years animal (rodent) models of cancers have provided tremendous insights into the underlying mechanisms of oncogenesis and tumor growth. However, the existing models do not fully recapitulate the complex and interconnected human tumor microenvironment (TME) and the role played by the immune system in tumor progression, thus being questioned for preclinical studies (Arrieta et al., 2018, Muldoon et al., 2013, Brown et al., 2018). Moreover, existing models often do not allow one to study the immune-escape features of several malignancies (Brown et al., 2018, Quail and Joyce, 2017).
Glioblastoma (GBM) is the most common and lethal primary human brain malignancy (Ostrom et al., 2017, Touat et al., 2017). In vivo, GBM has been mostly modeled in immune-compromised mice engrafted with human glioma cells (Candolfi et al., 2007). However, these models are very susceptible to side effects triggered by anticancer therapy and are not suitable for testing immune-based therapies (Fulop and Phillips, 1990, Biedermann et al., 1991, Ishii-Ohba et al., 2007). On the other hand, syngeneic GBM models in immune-competent rodents allow one to study tumorigenesis and the interplay between gliomas and the host immune system, but these models often fail to recapitulate many of the human-specific TME features (Candolfi et al., 2007, Oh et al., 2014, Chongsathidkiet et al., 2018). Recently, “humanized mice” have been developed to study tumors in clinically relevant settings (Patrizii et al., 2018, Ashizawa et al., 2017, Williams, 2018). However, this approach is costly and complicates the use of existing genetically modified mice. Finally, pharmacological blockade of T cell activation during engraftment of human GBM neurospheres into brains of adult wild-type (WT) mice has been used to prevent xenograft rejection (Semenkow et al., 2017). Although this approach is relatively simple and potentially applicable to genetically modified mice, it does not allow excluding that chemical immunosuppression may alter the role of the immune system on tumor progression and/or escape properties of the neoplasm.
Here, we exploited the “immune-privileged” developmental time window to develop an efficient in vivo human brain tumor engraftment model within the physiological tissue environment of WT mice. Human GBM cells, injected in the telencephalic ventricle of WT mouse embryos, readily invaded the host brain tissue and developed tumors exhibiting complex TME such as functional vasculature, reactive astrocytes, and host immune cell infiltration. Importantly, upon embryonic engraftment, patient-derived GBM xenografts persisted in postnatal brains of immune-competent mice. Our model is a valuable tool that can be used to study fundamental biology of human brain tumors and potentially applicable to clinically relevant tests, such as intratumor accumulation of therapeutic molecules upon intravascular delivery and immune-escape properties of the neoplasm in the context of an intact immune system.
Results
WT Embryonic Mouse Brain Supports the Engraftment and Growth of a Human GBM Cell Line (Tumor Xenografts)
In mice, the innate/non-specific immune system (e.g., microglia) is already active in the embryonic brain (Kaur et al., 2017), whereas the adaptive immune response (e.g., lymphocytes) matures postnatally (Holladay and Smialowicz, 2000). Hence, we hypothesized that the immature immune environment in WT embryonic mouse brains could tolerate the engraftment of human GBM cells.
As an entry point to test our hypothesis, we first transduced immortalized grade IV human GBM cell line U87MG (Pontén, 1975) with a lentiviral vector encoding Discosoma sp. red fluorescent protein (dsRed) to generate a dsRed+ U87MG GBM cell line. Then, we microinjected U87MG GBM single-cell suspensions in the lateral ventricles of embryonic day 12.5 (E12.5) WT mice developing in utero (Figure 1A). Of note, to exclude aspecific effects due to dsRed overexpression, or clonal evolution of transduced GBM clones, in the initial experiments we microinjected equal ratio of naive and dsRed+ GBM cells (see Figure 1H). More than 90% of embryos undergoing injection of U87MG cells survived the procedure (an efficiency that was similar to the in utero electroporation [Saito and Nakatsuji, 2001, Hoffmann et al., 2018]), and 92% of them presented tumor foci at E18.5 (Figure 1B, tumor xenografts [TX]). The number of dsRed+ human U87MG TX per brain (Figure 1C) increased until E18.5 (mean 2 at E13.5; 4 at E15.5, and 6 at E18.5 Figure 1C, red dots), whereas it decreased after birth (mean 5 at postnatal day 7 [P7], Figure 1C, red dot). Next, we analyzed TX by immunofluorescence (Figures 1D–1H). Human cells in TX were identified by intrinsic dsRed fluorescence and by immunostaining for human-specific nuclear antigen (HuNu). Measurements of TX revealed an exponential growth of their volume in both embryonic and postnatal mouse brains, but the highest volumetric increment occurred between E18.5 and P7 (Figures 1D and 1E, mean tumor volume 0.0038 ± 0.0009 mm³ at E13.5; 0.0351 ± 0.0081 mm³ at E15.5; 0.3339 ± 0.1276 mm³ at E18.5; and 3.7902 ± 1.0249 mm³ at P7). In contrast, nuclear density in TX (Figure 1F, nuclear DNA staining with Hoechst) did not change between E18.5 (429 ± 26 nuclei/field) and P7 (451 ± 4 nuclei/field). These results indicated that increase in tumor cell number likely accounted for expansion of TX volume. dsRed+ and naive GBM cells (HuNu + dsRed- cells) showed similar distributions in TX (Figure 1D), and their numbers increased proportionally across all the time points analyzed (Figures 1G and 1H; Number of total Hoechst + nuclei in TX per brain 2∗103 ± 0.2∗103 at E13.5, 1.8∗104 ± 0.8∗104 at E15.5, and 7∗104 ± 1.1∗104 at E18.5; proportion of HuNu + cells over total nuclei in TX per brain 77.64 ± 1.78% at E13.5, 71.53 ± 1.83% at E15.5, and 65.47 ± 1.15% at E18.5; proportion of dsRed+ cells over total nuclei in TX per brain 32.29 ± 3.48% at E13.5, 32.31 ± 2.37% at E15.5, and 31.15 ± 0.75% at E18.5), indicating that viral transduction did not affect growth of GBM cells. Together, these results demonstrated that WT embryonic mouse brain supports the engraftment and growth of a human GBM cell line.
Figure 1.

WT Embryonic Mouse Brain Supports the Engraftment and Growth of a Human GBM Cell Line (TX)
(A) Experimental procedure: (I) Preparation of single cell suspension of dsRed-labeled tumor cells, (II) surgery, and (III–V) tumor cell injection in the lateral ventricle (LV) of E12.5 mouse embryos; Cx, cortex; LGE/MGE, lateral/medial ganglionic eminence.
(B) Dorsal view at E18.5 and P7 mouse brains with TX; white arrowheads: dsRed+ tumors; red arrowheads: tumors in higher magnification (insets); OB, olfactory bulb; dT, dorsal telencephalon; M, midbrain; H, hindbrain; dashed lines indicate brain compartment boundaries. Scale bar: 2 mm; insets: 500 μm.
(C) Number of tumors per brain at the indicated time points, n = number of brains with tumors; light gray: N°/single brains, red: mean N°/age.
(D) Immunofluorescence images of cryosections through mouse TX at indicated ages; blue: Nuclei (Hoechst+), red: dsRed+ U87MG cells, and green: HuNu; T, tumor; H, host tissue; *, ventricle (same nomenclature in all the figures); scale bar: 200 μm.
(E) Tumor volume at the indicated time points [log10 scale]; light gray: single tumors, red: mean volume/age, n = number of brains with tumors.
(F) Nuclear density at E18.5 (n = 3) and P7 (n = 3).
(G) Number of total Hoechst + nuclei within TX per brain at E13.5 (n = 7 tumors in 4 brains), E15.5 (n = 4 tumors in 3 brains), and E18.5 (n = 6 tumors in 4 brains).
(H) Proportion of dsRed+ or HuNu + cells at E13.5 (n = 4), E15.5 (n = 3), and E18.5 (n = 4) over total Hoechst + nuclei in TX.
Data are represented as mean ± SEM; statistical significance revealed by using one-way ANOVA and Tukey's multiple comparisons test. * p value < 0.05; ** p value < 0.01; *** p value < 0.001; **** p value < 0.0001; n.s., not significant.
WT Embryonic Mouse Brain Supports Growth of Patient-Derived GBM Xenografts
To validate our engraft model, we repeated the above-described procedure with patient-derived GBM cells, which are considered a better standard in cancer research compared with tumor-derived cell lines (Patrizii et al., 2018, Williams, 2018, Allen et al., 2016). The patient-derived GBM (proneural subtype) cells were previously demonstrated to maintain cancer stem cell properties when cultured in the absence of serum and tumor-initiating properties in vivo (Gangemi et al., 2009). dsRed-labeled patient-derived GBM cells were injected in brains of E12.5 WT mouse embryos. Animals were analyzed at E18.5 and P7 to ascertain the presence of patient-derived GBM xenografts (PDX) (Figure 2A). We found an average of five to six dsRed+ PDX per brain (Figure 2B). This result was similar to the number of TX observed at the same time post-injection (Figure 1). Volumetric analysis (Figure 2A and Video S1 and S2) revealed a significant increase in PDX volume and in the percentage of host brain occupancy from E18.5 to P7 (Figure 2B and Video S1 and S2), although at E18.5 mean volume of PDX was significantly smaller than TX at the same time post-injection (Figure 2B PDX: 0.017 ± 0.01 mm³ versus TX: 0.33 ± 0.13 mm³ E18.5). In contrast, nuclear density in PDX did not change between E18.5 and P7 (Figure 2C), being comparable with TX at respective times (Figure 1). Histological analysis of tumors confirmed the highly infiltrative structure of the PDX (Figure 2D). Immunofluorescence analysis of tumor masses identified expression of human-specific antigen HuNu, as well as the proliferative marker KI67, GFAP, and S100β (calcium-binding protein B) (Figure 2D) in the PDX, which are typical GBM diagnostic markers (Bian et al., 2018). Quantification of proliferation indicated that not all the dsRed+ tumor cells were KI67+ (Figure 2E), suggesting the presence of a quiescent stem cell compartment in human GBM PDX. These results confirmed that WT embryonic mouse brain supports the engraftment and growth of patient-derived tumors.
Figure 2.

WT Embryonic Mouse Brain Supports Growth of Patient-Derived GBM Xenografts (PDX)
(A) Left image: dorsal view at E18.5 embryonic mouse brain with PDX; arrowheads: dsRed+ tumors; dashed lines indicate brain compartment boundaries. Center and right: 3D reconstruction of mouse brains at the indicated age showing PDX in red. Note the dramatic increase in tumor volume between the two time points. Scale bar: 1 mm.
(B) Graphs showing (from left to right) Number of tumors per brain (brains n = 3); tumor volume [log10 scale] (tumors n = 10 in three different brains per condition), and percentage of brain volume occupied by tumors (brains n = 3), at the indicated time points.
(C) Immunofluorescence images of PDX in coronal cryosections through embryonic mouse brains at E18.5 and P7; blue: Nuclei (Hoechst+) and graphical representation of nuclear density in PDX tumors at the indicated ages (tumors n = 7 in three different brains per condition).
(D) Immunofluorescence images of PDX in coronal cryosections through embryonic mouse brains at E18.5; blue: Nuclei (Hoechst+), red: dsRed+ patient-derived cells, green: HuNu, cyan: KI67; white: GFAP (center panel) or S100β (bottom panel).
(E) Graphical representation of (top) Total number of Hoechst + nuclei within PDX at E18.5 per brain (n = 12 tumors in 5 brains); (center) proportion of dsRed+ or HuNu + cells at E18.5 (n = 5) over total nuclei in PDX; (bottom) proportion of KI67 + +dsRed+ cells over total dsRed+ cells in PDX at E18.5 (n = 3).
Data are represented as mean ± SEM; statistical significance revealed by using unpaired Student's t test. * p value < 0.05; ** p value < 0.01; n.s., not significant.
3D reconstruction of PDX tumors (Red) in serial images of coronal sections through the brain of an E18.5 mouse embryo. Scale bar: 1 mm.
3D reconstruction of PDX tumors (Red) in serial images of coronal sections through the brain of a P7 mouse, showing 3D reconstruction of PDX tumors (Red). Scale bar: 1 mm.
TX and PDX Show Invasive Growth Pattern and Mesenchymal-like Features in Brains of WT Mice
Next, we analyzed the invasion pattern of human TX (Figure S1) and PDX (Figure 3) within WT embryonic mouse tissue. At E13.5 (i.e., 24 h post injection, Figure S1A) the gross majority of human U87MG cells, identified by dsRed, by immunostaining for HuNu, and by the astroglial cell marker GLAST (glial high-affinity glutamate transporter or SLC1A3), clustered in the lateral ventricle of the developing embryos (Figure S1A’). However, at the same time, we found some tumor cells adjacent to the apical membrane of the host GLAST + radial glial cells (Figure S1A’’, arrow) and few “leader tumor cells” that started to invade the host neuroepithelium, as revealed by discontinuity of the apical GLAST + staining in radial glial cells (Figure S1A’’', red arrows). Consistent with previous studies on cancer cell invasion (Denais et al., 2016), we postulated that infiltration of GBM cells in the host tissue might occur through active pressure exerted by leader tumor cells on the host ventricular zone, as indicated by the appearance of poly-nucleated tumor cells (Figure S1A’’', white arrows), which is a hallmark of nuclear rupture.
Figure 3.

PDX Show Invasive Growth Pattern and Mesenchymal-like Features in Brains of WT Mice
(A–E) Immunofluorescence images of PDX in cryosections of embryonic mouse brains; blue: Nuclei (Hoechst+), red: patient derived GBM cells. (A) Cross section through a PDX at E18.5; in high magnification showing invading tumor cells (T) inside the host tissue (H); dashed line indicates the host ventricular line border, red arrows: broken apical membrane after tumor cell invasion; scale bar: 100 μm. (B) Cross section through a PDX at E18.5 in high magnification of invading tumor cells (T) inside the host tissue (H), showing infiltrative growth pattern of GBMs. Hollow arrow and hollow arrowheads indicate different routes of invasion; scale bars: 50 μm. (C) Cross section through a PDX at E18.5 showing tumor cells (T) inside the host tissue (H); dashed line indicates the tumor line border in high magnification. Immunostaining for CD44 (C’, green) and Snail + Slug (C’’, white). (D and E) Green: E-CADHERIN (D) or N-CADHERIN (E); cross sections through TX at indicated times showing downregulation of E-CADHERIN and constant expression of N-CADHERIN; scale bars (D and E): 50 μm.
At E18.5 (i.e., 6 days post injection) both TX and PDX largely invaded the host tissue (Figures S1 and 3). Interestingly, several patterns of tumor cells infiltration could be observed in our model, ranging from collective cell migration (Figures S1B and 3A) to mesenchymal-like migration of individual cells (Figure 3B, hollow arrows). These patterns are consistent with those observed in brain malignancies (Friedl and Alexander, 2011, Lefranc et al., 2018). To corroborate these observations, we investigated markers of epithelial to mesenchymal transition (EMT), a reversible process characterized by loss of cell polarity and cell-cell adhesion that is known to increase motility and migratory capacity of tumor cells thus enhancing their invasiveness and aggressiveness (Kahlert et al., 2017). As expected, we found that PDX expressed mesenchymal markers such as CD44, Snail, and Slug (Figure 3C), consistent with previous observation in glioma (Mooney et al., 2016, Yang et al., 2010, Caja et al., 2018). Moreover, we observed a progressive downregulation of epithelial Cadherin (E-CAD, i.e., epithelial marker) during development of both TX and PDX (Figures S1C and 3D), whereas expression of the mesenchymal marker neural Cadherin (N-CAD, Figures S1D and 3E) remained constant (Figures S1D and 3E). Although further research is needed to demonstrate whether epithelial to mesenchymal transition governs the infiltration of GBM cells, these results suggest that our model can be used to address this question.
TX and PDX Develop Functional Vasculature and Complex TME in Brains of WT Mice
Blood vessels are essential for tumor growth and an important component of the GBM TME (Quail and Joyce, 2017, Wesseling et al., 1994, Holash et al., 1999). We asked whether human GBM xenografts develop functional vasculature in our model (Figure 4).
Figure 4.

TX and PDX Develop Functional Vasculature in Brains of WT Mice
(A and B) Immunofluorescence images of TX (left panels) and PDX (right panels) in cryosections of embryonic mouse brains at E18.5 (blue: Nuclei (Hoechst+), red: dsRed+ cells) showing (A) white: immune reactivity for VEGF-A; (B) white: immune reactivity for the endothelial marker CD31 inside TX (left panels) and PDX (right panels).
(C–E) (C) Experimental procedure of FITC-Dextran injection, (D and E) Immunofluorescence images of coronal cryosections through embryonic brains at E18.5 showing green: 4 kDA FITC-Dextran, red: dsRed+ U87MG (TX) or Patient-derived GBM (PDX) cells; (D) low magnification image of FITC-Dextran labeled blood vessels in embryonic mouse brain and tumor (T); (E) high magnification of inset in (D) (left panels) and similar example on a vascularized tumor formed by patient-derived GBM cells (right panels) showing co-localization (arrows) of FITC-Dextran with dsRed + tumor cells. Scale bars: 100 μm in (A) and (B), 500 μm in (D), and 50 μm in (E).
Vascular endothelial growth factor (VEGF) is an essential growth factor for blood vessel formation, required for both vasculogenesis and angiogenic sprouting during embryogenesis (Yancopoulos et al., 2000). In gliomas, VEGF-A plays a pivotal role of tumor-induced angiogenesis and increased VEGF-A levels correlate with higher tumor microvessel density and are linked to reduced response to treatment and poor prognosis (Hendriksen et al., 2009). We found VEGF-A immunoreactivity in both TX and PDX at E18.5 (Figure 4A). This result confirms previous reports of neovascularization occurring in both xenogeneic and syngeneic GBM tumors upon engraftment in adult rodents (Candolfi et al., 2007). To corroborate this finding, we performed immunofluorescence for CD31 (Cluster of differentiation 31), a marker of endothelial cells. Indeed, we found CD31 + cells within and surrounding TX and PDX at E18.5 (Figure 4B). To ascertain whether GBM xenografts develop a functional vascular system, we injected 4 kDA fluorescein isothiocyanate-conjugated Dextran (FITC-Dextran) into the tail vain of the pregnant dam 18.5 days postcoitum (Figure 4C). Remarkably, 4 h after tail vein injection, we found a strong FITC signal colocalizing with dsRed+ cells and vessel-like structures associated to TX and PDX (Figures 4D and 4E, arrowheads) in brains of E18.5 embryos. These results indicate that tumors develop a functional vascular system in our model. Moreover, the enrichment of FITC signal in tumors, in contrast to its low levels in the host brain parenchyma consistent with the notion that embryonic blood brain barrier (BBB) becomes functional around E15.5 in mouse (Ben-Zvi et al., 2014), suggests an increased permeability of the vessels associated with GBM xenografts.
Another important feature of GBM TME is astrocyte reactivity. Characterized by altered morphology and high expression of glial fibrillary acid protein (GFAP), astrocyte reactivity plays crucial functions for GBM and other brain tumors, such as regulation of BBB permeability and immune-modulation (Quail and Joyce, 2017). However, owing to the presence of GFAP + astrocytes in both glioma and non-neoplastic tissue, ontogeny of tumor-associated reactive astrocytes remains controversial in studies performed in adult mice. We explored this issue taking advantage of our embryonic engraftment model in which host GFAP + astrocytes mostly arise postnatally (Kriegstein and Alvarez-Buylla, 2009, Ge et al., 2012). We performed staining with a human-specific GFAP antibody at E18.5 in the brains of embryos subject to transplantation and found a high immunoreactivity in the xenografts and nearby regions but not in the host tissue devoid of engrafted human GBM cells (Figure S2A). This result suggests that human GBM can give rise to reactive astrocytes, as shown by GFAP expression, consistent with the glial origin of this tumor (Laug et al., 2018).
Human GBM cells can form large and functional syncytia by cell-to-cell-connections (i.e., tumor microtubes [Osswald et al., 2015]), which have been proposed as possible mechanism of GBM therapy resistance (Weil et al., 2017). These connections include CONNEXIN 43 (CX43), which is the predominant connexin expressed in GBM cells (Osswald et al., 2015). Interestingly, we observed CX43 + immunostaining in TX as well as in the host tissue at P7 (Figure S2B) suggesting intra-tumoral (i.e., tumor cell-to-tumor cell) and/or tumor cell-to-host cell communication might occur in our model.
Collectively, these results demonstrate that our engraftment model in the brain of WT mouse embryos efficiently recapitulates several of the major features of GBM, such as tumor cell infiltration and formation of a complex TME, including vascularization and reactive gliosis. Further studies will be required to investigate whether GFAP expression surrounding the xenograft is a mechanism of defense toward the host immune system and/or plays a structural role for tumor infiltration/growth, as well as its relevance for vascular permeability.
Embryonic Engraftment of Human GBM Cells in WT Mice Allows Investigation of Tumor Interaction with the Host Immune System
We wanted to ascertain whether our model is suitable to study the role of the immune system on tumor progression and/or escape properties of the neoplasm. We first quantified total lymphocytes by flow cytometry forward and side scattering in bone marrow (BM) and peripheral blood from control mice at P7, P15, P21, and adult (Figures 5A and S3). As expected, we found increasing proportions of lymphocytes that, at P21, reached a similar level to the BM of adult mice (Figures 5A, red line and S3), suggesting that the immune system at P21 was likely mature, with respect to lymphocytes. This result is consistent with the notion that adaptive immune response in mice matures postnatally, reaching a plateau around 2 months (Muldoon et al., 2013, Holladay and Smialowicz, 2000). Second, embryonic engraftment of human TX and PDX did not alter proportions of bulk lymphocyte populations in BM and peripheral blood at P21 compared with control mice (Figures 5B, 5C, and S3), suggesting that embryonic engraftment of human tumor cells likely did not impair the development of the host immune system. We then quantified the number of human TX and PDX in brains of P7 to P28 WT mice (Figures 5D and 5E). Remarkably, nearly 60% of brains supported human PDX at P28, whereas the number of human TX (U87MG cell line) dramatically decreased postnatally (Figure 5E), in agreement with the known immune-escape properties of GBM in patients (Arrieta et al., 2018, Daga et al., 2011).
Figure 5.

Embryonic Engraftment of Human GBM Cells in WT Mice Allows Investigation of Tumor Interaction with the Host Immune System
(A) Lymphocytes in bone marrow (BM) of WT control mice at P7 (n = 2), P15 (n = 2), and P21 (n = 6) relative to BM of adult (20–29 weeks) WT females (n = 4; red line).
(B) Lymphocytes in BM of P21 mice (TX n = 5 and PDX n = 11), normalized for CTL mice (n = 4).
(C) Lymphocytes in peripheral blood of P21 mice (TX n = 5 and PDX n = 14) normalized for CTL (n = 6).
(D) Ventral view at P21 and P28 mouse brains with TX or PDX; arrowheads: dsRed+ tumors; OB, olfactory bulb; Cx, cortex; Hy, hypothalamus; dashed lines: brain compartment boundaries. Scale bar: 2 mm.
(E) Number of tumor-positive brains as % of total brains dissected at P7 (TX n = 10, PDX n = 7), P15 (n = 23; n = 8), P21 (n = 24; n = 25), and P28 (n = 29; n = 19).
(F) Cell composition in tumors at P7 and P28 in TX (n = 3; n = 1) or PDX (n = 4; n = 2) (proportion of dsRed+ [red] or IBA1+ [cyan] or Hoechst + dsRed-IBA1- [blue]) over total Nuclei/field.
(G) Immunofluorescence images of TX in cryosections through E18.5 brains as example of activated IBA1+CD68+ (G’’, arrow) and non-activated IBA1+CD68- (G’); blue: Nuclei (Hoechst+), red: dsRed+ U87MG cells, cyan: IBA1, and white: CD68; scale bar: 20 μm.
(H) Proportion of IBA1+ cells at E18.5, P7, P21, and P28 in TX (n = 3, n = 3, n = 1, n = 1) or PDX (n = 5, n = 4, n = 6, n = 2) over total nuclei/field.
(I) Proportion of activated IBA1+CD68 + cells at E18.5, P7, P21, and P28 in TX or PDX over total IBA1+ cells/field (value of n as presented in [H]).
Data are represented as mean ± SEM; statistical significance revealed by using unpaired Student's t test and one-way ANOVA followed by Tukey's multiple comparison test. * p value < 0.05; n.s., not significant.
Microglia and tumor-associated macrophages (TAMs) constitute up to 30% of the tumor mass with a crucial role in tumorigenesis and are the main challenge that needs to be overcome for successful application of immune therapy (Brown et al., 2018, Daga et al., 2011). TAMs comprise tissue-resident microglia, other non-parenchymal macrophages, as well as bone marrow-derived macrophages (BMDMs) (Brown et al., 2018, Quail and Joyce, 2017, Kaur et al., 2017). Of note, microglia and BMDMs express very similar markers and genetic lineage tracing mice cannot be directly translated to current human glioma xenograft models. Hence, the relative contributions of microglia versus BMDMs to different glioma phenotypes remain controversial (Quail and Joyce, 2017). To understand whether our model is suitable to investigate infiltration of TAMs, we performed immunofluorescence for IBA1 (Ionized calcium binding adaptor molecule 1) marker of both microglia and macrophages (Quail and Joyce, 2017, Ito et al., 1998). We found infiltration of IBA1+ cells in TX and PDX from E13.5 up to P28 (Figures 5F–5I and S4). We also observed some IBA1+ cells that were HuNu+, suggesting phagocytosis (or fusion) of tumor cells by host TAMs (Figures S4A’’’ and S4B’’’), consistent with known phagocytic function of microglia and macrophages.
Quantification of tumor cell composition at P7 and P28 revealed a similar decrease in proportions of dsRed+ cells in both TX and PDX (Figures 5F and S4; proportions of total dsRed+ or IBA1+ cells over total nuclei in TX or PDX per brain). Regarding TAMs, we observed a trend of increasing proportions of IBA1+ cells over total cells in TX, whereas this proportion remained constant in PDX (Figures 5F, 5H and S4). Finally, we quantified activated TAMs in TX and PDX from E18.5 to P28 by co-immunostaining for IBA1 and CD68 (Cluster of Differentiation 68), a marker of reactive TAMs (Yoshida et al., 2018) (Figures 5G, 5I, and S4). We found in TX a significant two-fold increase in the proportion of IBA1+CD68 + cells over total IBA1+ cells at P7, compared with E18.5, remaining then constant up to P28 (Figures 5I and S4). In contrast, the proportion of IBA1+CD68 + cells over total IBA1+ cells in PDX significantly decreased from E18.5 to P21 and was significantly lower at P7 compared with TX, consistent with the higher number of PDX compared with TX tumors in postnatal brains of WT mice (Figure 5E). Because PDX persisted longer in postnatal brains and a lower proportion of reactive microglia/macrophages was associated with PDX, compared with TX, these results indicate that our model allows one to study tumor-specific interactions with the immune system of the WT host.
Discussion
Orthotopic xenotransplantation of human tumor cells in immune-compromised animals has been considered the in vivo “gold standard” in brain cancer field for decades. However, results obtained with this approach warrants careful considerations for preclinical research (Williams, 2018). Indeed, most oncology drugs fail late in clinical trials owing to lack of efficacy. Hence, conducting parallel preclinical studies on biologically diverse models is potentially a more cost-effective alternative. Our model, complementing existing ones, is a valuable alternative that potentially will increase the repertoire of approaches to test the efficacy of immune-based therapies, which are promising in several cancers but not yet fully exploited in brain malignancies (Arrieta et al., 2018, Muldoon et al., 2013, Brown et al., 2018).
The “immune-privileged” developmental time window is a great opportunity. Here we exploit it with the orthotopic transplantation of human brain tumor cells in WT mice. Remarkably, when transplanted in utero, human PDX persist in postnatal brains of immune-competent mice. Hence, we postulate that our model is potentially suitable to study immune-escape properties of human GBM and possibly of other types of brain tumors.
Our model allows investigation of important aspects of TME such as tumor cell infiltration, astrogliosis, and intratumor accumulation of exogenous compounds through intravascular administration. Few drug molecules cross the BBB, and several GBM clinical trials have failed because the compounds did not reach therapeutically relevant concentrations at the target (i.e., gefitinib and erlotinib, Shergalis et al., 2018). Hence, although we did not formally prove whether tumors develop a BBB in our model, its exploitation could contribute moving the field forward for future drug development and efficacy studies.
Unlike immune-compromised or humanized mice our model can be readily applied to genetically modified mice. For instance, by taking advantage of existing lineage-tracing mice, it could be further exploited to infer the relative contributions of tumor versus host cells in the TME or autocrine versus paracrine signaling, which are highly debated topics in brain cancer field (Quail and Joyce, 2017, Barcia et al., 2013, Bowman et al., 2016).
Finally, our model is also easy to implement as it requires a surgical procedure very similar to in utero electroporation, very efficient and widely used in developmental neuroscience (Tabata and Nagata, 2016).
Despite great advancement in treating other cancer types, the median survival of patients with GBM remained nearly the same over the last decades. Problems that remain unsolved are extensive infiltration of GBM cells in brain parenchyma, lack of therapies capable of efficiently overcoming the BBB, and the various immune-escape strategies used by GBM. Future exploitation of the embryonic xenograft model will lead to increase our understanding of brain tumor biology and therapeutic development.
Limitations of the Study
In this study, we provide fast qualitative and quantitative measurements of human tumor development in WT mice, thus paving the way toward applications in basic cancer research. However, it should be noted that we did not formally determine the survival of tumor-bearing mice or monitor tumor development upon manipulation or drug administration in the same animal. Therefore, our model warrants further confirmation of its applicability to clinical research.
Although we noted a progressive reduction in the number of tumor-bearing brains after birth of transplanted animals, at present it is difficult to forecast whether the human GBM xenografts will be rejected or some of the transplanted mice will become tolerant to tumors. It will be interesting for researchers to experimentally address these questions in our model upon engraftment of more aggressive malignancies, such as GBM mesenchymal subtypes or tumors metastasizing in brain. Moreover, as our model is potentially applicable to genetically modified mice, another possibility to address these questions would be to explore whether the genetic background of the host mice influences their survival upon tumor transplantation.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors wish to thank A. Bajetto, T. Florio, and A. Daga (University of Genoa, Italy) for providing patient-derived GBM cell clones. We thank E. Albanesi and G. Bardi (IIT, Genoa, Italy) for advice and assistance on cell flow cytometry assays and M. Pesce (IIT and Nikon Imaging Center, Genoa, Italy) for advice and assistance on microscopy. We thank the staff of IIT Animal Facility and M. Morini for excellent support. We are grateful to P. Decuzzi (Nano Nanotechnology for Precision Medicine Lab, IIT, Genoa, Italy) for advice and discussion and to P. Malatesta (University of Genoa, Italy) for critical reading of the manuscript. This study was funded by Fondazione Istituto Italiano di Tecnologia (IIT) and partly by the Investigator grant (AIRC-IG 2017 #20106) from Fondazione AIRC per la Ricerca sul Cancro to D.D.P.T.
Author Contributions
D.D.P.T. conceived and supervised the project. D.D.P.T. and N.H. designed the experiments and wrote the manuscript. N.H. and V.F. performed most of the experiments, analyzed the data, and prepared the figures. R.C.P. performed the FACS experiments for Figures 5A–5C and S4 and analyzed the data. S.R. and R.P. virally transduced and maintained cultures of human GBM cells for transplantation. All authors read, edited and approved the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: January 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.100813.
Data and Code Availability
Original samples, data, and transduced cells are available upon request.
Supplemental Information
References
- Allen M., Bjerke M., Edlund H., Nelander S., Westermark B. Origin of the U87MG glioma cell line: good news and bad news. Sci. Transl. Med. 2016;8:354re3. doi: 10.1126/scitranslmed.aaf6853. [DOI] [PubMed] [Google Scholar]
- Arrieta V.A., Cacho-Díaz B., Zhao J., Rabadan R., Chen L., Sonabend A.M. The possibility of cancer immune editing in gliomas. A critical review. Oncoimmunology. 2018;7:e1445458. doi: 10.1080/2162402X.2018.1445458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashizawa T., Iizuka A., Nonomura C., Kondou R., Maeda C., Miyata H., Sugino T., Mitsuya K., Hayashi N., Nakasu Y. Antitumor effect of programmed death-1 (PD-1) blockade in humanized the NOG-MHC double knockout mouse. Clin. Cancer Res. 2017;23:149–158. doi: 10.1158/1078-0432.CCR-16-0122. [DOI] [PubMed] [Google Scholar]
- Barcia C., Sr., Mitxitorena I., Carrillo-de Sauvage M.A., Gallego J.M., Pérez-Vallés A., Barcia C., Jr. Imaging the microanatomy of astrocyte-T-cell interactions in immune-mediated inflammation. Front. Cell. Neurosci. 2013;7:58. doi: 10.3389/fncel.2013.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A., Lacoste B., Kur E., Andreone B.J., Mayshar Y., Yan H., Gu C. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. 2014;509:507–511. doi: 10.1038/nature13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian S., Repic M., Guo Z., Kavirayani A., Burkard T., Bagley J.A., Krauditsch C., Knoblich J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods. 2018;15:631–639. doi: 10.1038/s41592-018-0070-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biedermann K.A., Sun J.R., Giaccia A.J., Tosto L.M., Brown J.M. Scid mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair. Proc. Natl. Acad. Sci. U S A. 1991;88:1394–1397. doi: 10.1073/pnas.88.4.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman R.L., Klemm F., Akkari L., Pyonteck S.M., Sevenich L., Quail D.F., Dhara S., Simpson K., Gardner E.E., Iacobuzio-Donahue C.A. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 2016;17:2445–2459. doi: 10.1016/j.celrep.2016.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N.F., Carter T.J., Ottaviani D., Mulholland P. Harnessing the immune system in glioblastoma. Br. J. Cancer. 2018;119:1171–1181. doi: 10.1038/s41416-018-0258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caja L., Tzavlaki K., Dadras M.S., Tan E.J., Hatem G., Maturi N.P., Moren A., Wik L., Watanabe Y., Savary K. Snail regulates BMP and TGFbeta pathways to control the differentiation status of glioma-initiating cells. Oncogene. 2018;37:2515–2531. doi: 10.1038/s41388-018-0136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candolfi M., Curtin J.F., Nichols W.S., Muhammad A.G., King G.D., Pluhar G.E., McNiel E.A., Ohlfest J.R., Freese A.B., Moore P.F. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J. Neurooncol. 2007;85:133–148. doi: 10.1007/s11060-007-9400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chongsathidkiet P., Jackson C., Koyama S., Loebel F., Cui X., Farber S.H., Woroniecka K., Elsamadicy A.A., Dechant C.A., Kemeny H.R. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018;24:1459–1468. doi: 10.1038/s41591-018-0135-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daga A., Bottino C., Castriconi R., Gangemi R., Ferrini S. New perspectives in glioma immunotherapy. Curr. Pharm. Des. 2011;17:2439–2467. doi: 10.2174/138161211797249206. [DOI] [PubMed] [Google Scholar]
- Denais C.M., Gilbert R.M., Isermann P., McGregor A.L., te Lindert M., Weigelin B., Davidson P.M., Friedl P., Wolf K., Lammerding J. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352:353–358. doi: 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P., Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- Fulop G.M., Phillips R.A. The scid mutation in mice causes a general defect in DNA repair. Nature. 1990;347:479–482. doi: 10.1038/347479a0. [DOI] [PubMed] [Google Scholar]
- Gangemi R.M., Griffero F., Marubbi D., Perera M., Capra M.C., Malatesta P., Ravetti G.L., Zona G.L., Daga A., Corte G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009;27:40–48. doi: 10.1634/stemcells.2008-0493. [DOI] [PubMed] [Google Scholar]
- Ge W.P., Miyawaki A., Gage F.H., Jan Y.N., Jan L.Y. Local generation of glia is a major astrocyte source in postnatal cortex. Nature. 2012;484:376–380. doi: 10.1038/nature10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriksen E.M., Span P.N., Schuuring J., Peters J.P., Sweep F.C., van der Kogel A.J., Bussink J. Angiogenesis, hypoxia and VEGF expression during tumour growth in a human xenograft tumour model. Microvasc. Res. 2009;77:96–103. doi: 10.1016/j.mvr.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Hoffmann N., Weise S.C., Marinaro F., Vogel T., De Pietri Tonelli D. DGCR8 promotes neural progenitor expansion and represses neurogenesis in the mouse embryonic neocortex. Front. Neurosci. 2018;12:281. doi: 10.3389/fnins.2018.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holash J., Maisonpierre P.C., Compton D., Boland P., Alexander C.R., Zagzag D., Yancopoulos G.D., Wiegand S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- Holladay S.D., Smialowicz R.J. Development of the murine and human immune system: differential effects of immunotoxicants depend on time of exposure. Environ. Health Perspect. 2000;108:463–473. doi: 10.1289/ehp.00108s3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii-Ohba H., Kobayashi S., Nishimura M., Shimada Y., Tsuji H., Sado T., Ogiu T. Existence of a threshold-like dose for gamma-ray induction of thymic lymphomas and no susceptibility to radiation-induced solid tumors in SCID mice. Mutat. Res. 2007;619:124–133. doi: 10.1016/j.mrfmmm.2007.02.028. [DOI] [PubMed] [Google Scholar]
- Ito D., Imai Y., Ohsawa K., Nakajima K., Fukuuchi Y., Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res. Mol. Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Kahlert U.D., Joseph J.V., Kruyt F.A.E. EMT- and MET-related processes in nonepithelial tumors: importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017;11:860–877. doi: 10.1002/1878-0261.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur C., Rathnasamy G., Ling E.A. Biology of microglia in the developing brain. J. Neuropathol. Exp. Neurol. 2017;76:736–753. doi: 10.1093/jnen/nlx056. [DOI] [PubMed] [Google Scholar]
- Kriegstein A., Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laug D., Glasgow S.M., Deneen B.A. Glial blueprint for gliomagenesis. Nat. Rev. Neurosci. 2018;19:393–403. doi: 10.1038/s41583-018-0014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc F., Le Rhun E., Kiss R., Weller M. Glioblastoma quo vadis: will migration and invasiveness reemerge as therapeutic targets? Cancer Treat. Rev. 2018;68:145–154. doi: 10.1016/j.ctrv.2018.06.017. [DOI] [PubMed] [Google Scholar]
- Mooney K.L., Choy W., Sidhu S., Pelargos P., Bui T.T., Voth B., Barnette N., Yang I. The Role of CD44 in glioblastoma multiforme. J. Clin. Neurosci. 2016;34:1–5. doi: 10.1016/j.jocn.2016.05.012. [DOI] [PubMed] [Google Scholar]
- Muldoon L.L., Alvarez J.I., Begley D.J., Boado R.J., Del Zoppo G.J., Doolittle N.D., Engelhardt B., Hallenbeck J.M., Lonser R.R., Ohlfest J.R. Immunologic privilege in the central nervous system and the blood-brain barrier. J. Cereb. Blood Flow Metab. 2013;33:13–21. doi: 10.1038/jcbfm.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh T., Fakurnejad S., Sayegh E.T., Clark A.J., Ivan M.E., Sun M.Z., Safaee M., Bloch O., James C.D., Parsa A.T. Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med. 2014;12:107. doi: 10.1186/1479-5876-12-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osswald M., Jung E., Sahm F., Solecki G., Venkataramani V., Blaes J., Weil S., Horstmann H., Wiestler B., Syed M. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528:93. doi: 10.1038/nature16071. [DOI] [PubMed] [Google Scholar]
- Ostrom Q.T., Gittleman H., Truitt G., Boscia A., Kruchko C., Barnholtz-Sloan J.S. CBTRUS Statistical Report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19:v1–v88. doi: 10.1093/neuonc/nox158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrizii M., Bartucci M., Pine S.R., Sabaawy H.E. Utility of glioblastoma patient-derived orthotopic xenografts in drug discovery and personalized therapy. Front. Oncol. 2018;8:23. doi: 10.3389/fonc.2018.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontén J. Neoplastic human glia cells in culture. In: Fogh J., editor. Human Tumor Cells in Vitro. Springer; 1975. [Google Scholar]
- Quail D.F., Joyce J.A. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31:326–341. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T., Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 2001;240:237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Semenkow S., Li S., Kahlert U.D., Raabe E.H., Xu J., Arnold A., Janowski M., Oh B.C., Brandacher G., Bulte J.W.M. An immunocompetent mouse model of human glioblastoma. Oncotarget. 2017;8:61072–61082. doi: 10.18632/oncotarget.17851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shergalis A., Bankhead A., 3rd, Luesakul U., Muangsin N., Neamati N. Current challenges and opportunities in treating glioblastoma. Pharmacol. Rev. 2018;70:412–445. doi: 10.1124/pr.117.014944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata H., Nagata K.I. Decoding the molecular mechanisms of neuronal migration using in utero electroporation. Med. Mol. Morphol. 2016;49:63–75. doi: 10.1007/s00795-015-0127-y. [DOI] [PubMed] [Google Scholar]
- Touat M., Idbaih A., Sanson M., Ligon K.L. Glioblastoma targeted therapy: updated approaches from recent biological insights. Ann. Oncol. 2017;28:1457–1472. doi: 10.1093/annonc/mdx106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil S., Osswald M., Solecki G., Grosch J., Jung E., Lemke D., Ratliff M., Hänggi D., Wick W., Winkler F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017;19:1316–1326. doi: 10.1093/neuonc/nox070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling P., van der Laak J.A., de Leeuw H., Ruiter D.J., Burger P.C. Quantitative immunohistological analysis of the microvasculature in untreated human glioblastoma multiforme. Computer-assisted image analysis of whole-tumor sections. J. Neurosurg. 1994;81:902–909. doi: 10.3171/jns.1994.81.6.0902. [DOI] [PubMed] [Google Scholar]
- Williams J.A. Using PDX for preclinical cancer drug discovery: the evolving field. J. Clin. Med. 2018;7 doi: 10.3390/jcm7030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancopoulos G.D., Davis S., Gale N.W., Rudge J.S., Wiegand S.J., Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- Yang H.W., Menon L.G., Black P.M., Carroll R.S., Johnson M.D. SNAI2/Slug promotes growth and invasion in human gliomas. BMC Cancer. 2010;10:30. doi: 10.1186/1471-2407-10-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y., Sejimo Y., Kurachi M., Ishizaki Y., Nakano T., Takahashi A. X-ray irradiation induces disruption of the blood-brain barrier with localized changes in claudin-5 and activation of microglia in the mouse brain. Neurochem. Int. 2018;119:199–206. doi: 10.1016/j.neuint.2018.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
3D reconstruction of PDX tumors (Red) in serial images of coronal sections through the brain of an E18.5 mouse embryo. Scale bar: 1 mm.
3D reconstruction of PDX tumors (Red) in serial images of coronal sections through the brain of a P7 mouse, showing 3D reconstruction of PDX tumors (Red). Scale bar: 1 mm.
Data Availability Statement
Original samples, data, and transduced cells are available upon request.