

Abstract
Cartilage repair in clinics is a challenge owing to the limited regenerative capacities of cartilage. Synovium-derived stem cells (SDSCs) are suggested as tissue-specific stem cells for chondrogenesis. In this study, we hypothesize that decellularized extracellular matrix (dECM) deposited by SDSCs could provide a superior tissue-specific matrix microenvironment for optimal rejuvenation of adult SDSCs for cartilage regeneration. dECMs were deposited by adult stem cells with varying chondrogenic capacities; SDSCs (strong) (SECM), adipose-derived stem cells (weak) (AECM) and dermal fibroblasts (weak) (DECM), and urine-derived stem cells (none) (UECM). Plastic flasks (Plastic) were used as a control substrate. Human SDSCs were expanded on the above substrates for one passage and examined for chondrogenic capacities. We found that each dECM consisted of unique matrix proteins and exhibited varied stiffnesses, which affected cell morphology and elasticity. Human SDSCs grown on dECMs displayed a significant increase in cell proliferation and unique surface phenotypes. Under induction media, dECM expanded cells yielded pellets with a dramatically increased number of chondrogenic markers. Interestingly, SECM expanded cells had less potential for hypertrophy compared to those grown on other dECMs, indicating that a tissue-specific matrix might provide a superior microenvironment for stem cell chondrogenic differentiation.
Keywords: Chondrogenesis, Decellularized extracellular matrix, Differentiation, Preconditioning, Proliferation, Synovium-derived stem cells
1. Introduction
Adult articular cartilage displays limited capacity for intrinsic repair. Injury and joint degeneration causing minor lesions may result in progressive damage, leading to significant pain and disability. Given that it is a major clinical challenge to successfully repair articular cartilage defects, there have been numerous efforts to grow tissue-engineered grafts or patches for the repair of focal chondral and osteochondral defects, particularly for cell-based therapy which remains the most promising for articular cartilage repair [1]. Stem cells of different origins have been tested for cell therapy. As mesoderm-derived cells, mesenchymal stem cells (MSCs) have promising clinical applications due to high proliferation and multi-lineage differentiation capacity. For example, bone marrow-derived stem cells (BMSCs) exhibit robust chondrogenic potential and have been widely used in cartilage engineering and regeneration; adipose-derived stem cells (ADSCs) are also popular due to easy accessibility and availability [2]. However, stem cells isolated from adipose tissue had better differentiation potential toward the adipogenic lineage than toward the osteogenic or chondrogenic lineage and BMSCs had a tendency to undergo endochondral bone formation rather than chondrogenesis [3,4]. Recently, a search for lineage- or tissue-specific stem cells led to the isolation of synovium-derived stem cells (SDSCs), a so-called tissue-specific stem cell for chondrogenesis [5–7], which have been used in cell therapy for cartilage repair in animal studies and a recent human pilot study [8–10]. However, expansion of SDSCs on conventional tissue culture plates led to senescence and has been a challenge for large-scale expansion of SDSCs for clinical use [11,12]. We have previously investigated the role of decellularized extracellular matrix (dECM) in the expansion and maintenance of SDSCs [13,14]. dECM expansion of SDSCs ex vivo enhanced the rejuvenation of SDSCs in proliferation and differentiation capacity for the treatment of porcine cartilage defects [15].
Extracellular matrix (ECM) is the main component of the cellular niche [16], which provides the fibrous network for deposition of cell specific proteins and proteoglycans. In the case of cartilage, ECM also provides mechanical support that is required for the proper functioning of cells [17–19]. Any alterations to ECM would potentially affect cellular physiology leading to modified gene expression [20]. Mechanotransduction, cell-matrix interaction, and ECM associated growth factors play major roles in the maintenance, proliferation, and differentiation of specific cell types [21,22]. At a macromolecular level, ECM is a complex intricate structure that basically consists of collagens, proteoglycans, glycosaminoglycan (GAG), growth factors, and non-collagenous proteins. To date, a majority of studies have concentrated on individual matrix proteins to elucidate their role in cellular physiology and phenotypic modulation [23,24]. In most cases of specific ECM protein knockdown studies, the compensatory mechanisms mask the results leading to a null effect [25,26]. Apart from this, additional complexity was noticed in the tissue-specific ECM in the form of growth factor gradient [20,27]. In this context, the quantitative proteomics composition of the ECM among different cell types and transcriptomic changes of expanded cells on varied culture substrates including ECMs must be understood. It may be impossible to reproduce the exact native ECM in an artificial system, but understanding this complexity will help in elucidating the tissue-specific extracellular niche that is key for the optimal functioning of the specific cell type.
Increasing evidence shows that SDSCs are a tissue-specific stem cell for chondrogenesis [5–7] and urine-derived stem cells (UDSCs) are non-chondrogenic stem cells [28]; ADSCs and dermal fibroblasts (DFs) possess multi-lineage potential and express MSC surface markers, such as CD90, CD105, CD29, and CD44, but with less chondrogenic capacity [29–31]. In this study, we have investigated the proliferation and chondrogenic potential of human SDSCs expanded on various dECMs deposited by adult stem cells with varying chondrogenic capacities, SDSCs (strong), ADSCs and DFs (weak), and UDSCs (none). We hypothesized that dECM deposited by SDSCs could provide a superior tissue-specific matrix microenvironment for the optimal rejuvenation of adult SDSCs for cartilage regeneration.
2. Materials and Methods
2.1. Preparation of dECMs
Adult human SDSCs (4 donors, two male and two female, average 43 years old) [32], ADSCs (catalog number ASC-F-SL, from multiple donors, female, average 43 years old) and DFs (catalog number DF-F, from multiple donors, female, average 42 years old) purchased from ZenBio Inc. (Research Triangle Park, NC), and human UDSCs (4 donors, male, 20-54 years old) [28], a generous gift from Dr. Yuanyuan Zhang (Wake Forest Institute for Regenerative Medicine), were pooled to prepare dECMs, in terms of SECM, AECM, DECM, and UECM, respectively, according to our previous protocol [33]. Briefly, plastic flasks (Plastic) were precoated with 0.2% gelatin (Sigma-Aldrich, St. Louis, MO) at 37°C for 1 h and seeded with passage 4 SDSCs, ADSCs, DFs, and UDSCs at 6,000 cells per cm2. After reaching 90% confluence, cells were cultured for another 10 days with 250 μM L-ascorbic acid phosphate (Wako Chemicals USA, Inc., Richmond, VA) [34]. The cells were lysed with 0.5% Triton X-100 containing 20 mM ammonium hydroxide at 37°C for 5 min and then stored at 4°C in phosphate buffered saline (PBS) containing 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL fungizone until use.
2.2. SDSC expansion
Human SDSCs (passage 3) pooled from 4 donors were seeded onto various dECMs (SECM, AECM, DECM, and UECM) and Plastic (PL) at a density of 3,000 cells per cm2. The growth medium, containing alpha-minimum essential medium (αMEM), 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL fungizone, was changed every other day. Cell number was counted in 175 cm2 flasks (n = 6) by hemocytometer (Hausser Scientific, Horsham, PA) and morphology was photographed by phase contrast microscope.
2.3. Proliferation index assay
Before cell expansion, passage 3 SDSCs were labeled with CellVue® Claret (Sigma-Aldrich) as described by the manufacturer. After a six-day incubation, expanded cells were collected and analyzed by a BD dual laser fluorescence-activated cell sorting (FACS) Calibur™ flow cytometer (BD Biosciences, San Jose, CA). Twenty thousand events of each sample were collected using CellQuest Pro software (BD Biosciences) and cell proliferation index was analyzed by ModFit LT™ version 3.1 (Verity Software House, Topsham, ME).
2.4. FACS analysis of surface markers
The primary antibodies listed below were used to detect expanded SDSC surface markers: CD29 (Abcam, Cambridge, MA), CD90 (BD Pharmingen, San Jose, CA), CD105 (BioLegend, San Diego, CA), stage-specific embryonic antigen 4 (SSEA4) (BioLegend), and isotype matched IgGs (Beckman Coulter, Fullerton, CA). The secondary antibody was goat anti-mouse IgG (H + L) R-phycoerythrin conjugated (Life Technologies, Carlsbad, CA). Samples (n=3) of each 2 × 105 expanded cells were incubated on ice in cold PBS containing 0.1% Chrom-Pure Human IgG whole molecule (Jackson ImmunoResearch Laboratories, West Grove, PA) and 1% NaN3 (Sigma-Aldrich) for 30 min. The cells were then sequentially incubated in the dark in the primary and secondary antibodies for 30 min. Expression of cell surface markers was analyzed by a FACS Calibur (BD Biosciences) using FCS Express software (De Novo Software, Los Angeles, CA).
2.5. Atomic force microscopy (AFM)
The stiffness of dECMs alone and expanded cells on PL or dECMs was investigated using an MFP-3D-BIO AFM (Asylum Research, TE2000-U, Santa Barbara, CA) that was integrated with an inverted fluorescence microscope (Nikon Eclipse, Ti-U, Nikon Instruments Inc., Melville, NY) and Olympus TR400-PB cantilevers with spring constant of 0.09 N/m as described previously [14]. The samples were photographed in Petri dishes filled with αMEM containing 10% FBS. Each sample was mapped in five randomly selected 50 μm by 50 μm areas for a total of 2000 data points/sample. For the quantitative nanomechanical analysis, a Sneddon’s modification was employed via the Hertz model developed for a four-sided pyramid.
2.6. Scanning electron microscope (SEM)
Representative samples (n=2) were first fixed in 2.5% glutaraldehyde (Sigma-Aldrich) for 2 h, followed by secondary fixation in 2% osmium tetroxide (Sigma-Aldrich) for another 2 h. After dehydration in a gradient ethanol series, the samples were treated by hexamethyldisilazane (HMDS, Sigma-Aldrich) at 1:1 with ethanol, 1:2 with ethanol, and HMDS alone, respectively, followed by air-drying for 24 h in a desiccator and addition of gold sputter. The images were taken using SEM (Hitachi, Model S 2400).
2.7. Immunofluorescent staining of dECMs
The dECMs on tissue culture plates were fixed with 4% paraformaldehyde for 30 min. After blocking in 10% normal goat serum for 1 h, dECMs were incubated with monoclonal antibodies for type I collagen (clone COL-1, dilution 1:2000, catalog number C2456, Sigma-Aldrich) and fibronectin (EP5, dilution 1:200, catalog number sc-8422, Santa Cruz Biotechnology, Dallas, TX) overnight followed by Alexa Fluor 488 goat anti-mouse IgG (Life Technologies) for 30 min. dECMs were visualized with a Zeiss LSM 510 confocal on an AxioImager Z1 microscope using a 63× objective lens (Carl Zeiss, Jena, Germany).
2.8. Proteomics analysis of dECMs
Proteomics analysis of the dECM samples was performed as described earlier [14]. Briefly, dECM samples were scraped off the flasks and collected in 25 mM Tris-hydrochloride (pH 7.6) containing 150 mM sodium chloride and 0.5% sodium dodecyl sulfate with protease inhibitors. Total dECM proteins were precipitated in cold acetone overnight at −20°C and the pellet was solubilized with 100 μL of 0.2% ProteaseMax™ Surfactant (Promega, Madison, WI) prepared in 50 mM NH4HCO3 buffer (pH 7.9). Samples were reduced with 5 mM DTT for 15 min at 56°C and alkylated with 20 mM iodoacetamide in the dark for 0.5 h at room temperature before undergoing trypsin digestion at 37°C overnight. The digested peptide mixture was separated by high pH reversed phase chromatography on a C18 column; 40 fractions (600 μL each) were collected and pooled into eight larger fractions. Samples were concentrated under vacuum to approximately 50 μL, and 8 μL of sample was analyzed on an LTQ-FT Ultra hybrid mass spectrometer (ThermoFisher Scientific, Milford, MA). Chemical digestion was performed on ECM pellets using CNBr for 24 h. Resulting digests were separated by 1D SDS-PAGE; the entire lanes were cut and the bands were subjected to trypsin digestion (10 ng/μL of trypsin was added to each gel band and incubation at 4°C for 45 min followed by 37°C incubation for 16 h), and tandem mass spectrometry analysis was performed as described above. Data analysis was performed as described earlier [35] with protein confidence of >99% and peptide confidence of >95%.
2.9. Chondrogenic differentiation of SDSCs
2.9.1. Chondrogenic induction
0.3 × 106 SDSCs from each expansion group were centrifuged at 500 × g for 5 min in a 15-mL polypropylene tube to form a pellet. After overnight incubation (day 0) in growth medium, the pellets were cultured in a serum-free chondrogenic medium consisting of high-glucose Dulbecco’s modified Eagle’s medium (DMEM), 40 μg/mL proline, 100 nM dexamethasone, 100 U/mL penicillin, 100 μg/mL streptomycin, 0.1 mM ascorbic acid-2-phosphate, and 1× ITS™ Premix (6.25 μg/mL insulin, 6.25 μg/mL transferrin, 6.25 μg/mL selenous acid, 5.35 μg/mL linoleic acid, and 1.25 μg/mL bovine serum albumin, from BD Biosciences, Bedford, MA) with the supplementation of 10 ng/mL transforming growth factor beta3 (TGF-β3, PeproTech, Inc., Rocky Hill, NJ) in a 5% O2 incubator for up to 35 days. At days 0, 14, and 35, pellets from each group were collected for chondrogenic differentiation analysis.
2.9.2. Histochemistry and immunostaining
Representative pellets (n=2) were fixed in 4% paraformaldehyde at 4°C overnight. After embedding in paraffin blocks, 5-μm sections were stained for GAG with Alcian blue (Sigma-Aldrich, counterstained with fast red). For immunostaining, the sections were immunolabeled with primary antibodies against type II collagen (dilution 1:100, catalog number II-II6B3; Developmental Studies Hybridoma Bank, Iowa City, IA) and type X collagen (clone COL-10, dilution 1:1000, catalog number C7974, Sigma-Aldrich), followed by the secondary antibody. Immunoactivity was detected using Vectastain ABC reagent (Vector, Burlingame, CA) with 3, 3’-diaminobenzidine as a substrate.
2.9.3. Biochemical analysis for DNA and GAG content
The pellets (n=4) were digested for 4 h at 60°C with 125 μg/mL papain in PBE buffer (100 mM phosphate, 10 mM ethylenediaminetetraacetic acid, pH 6.5) containing 10 mM cysteine. To quantify cell density, the amount of DNA in the papain digestion was measured using the QuantiT™ PicoGreen® dsDNA assay kit (Invitrogen, Carlsbad, CA) with a CytoFluor® Series 4000 (Applied Biosystems, Foster City, CA). GAG was measured using dimethylmethylene blue dye and a Spectronic™ BioMate™ 3 Spectrophotometer (ThermoFisher Scientific) with bovine chondroitin sulfate (Sigma-Aldrich) as a standard.
2.9.4. Real-time polymerase chain reaction (PCR)
Total RNA was extracted from samples (n=4) using an RNase-free pestle in TRIzol® (Invitrogen). Two μg of mRNA was used for reverse transcription (RT) with High-Capacity cDNA Archive Kit (Applied Biosystems) at 37°C for 120 min. Chondrogenic marker genes, including type II collagen (COL2A1) (Assay ID Hs00156568_m1), SRY (sex determining region Y)-box 9 (SOX9) (Assay ID Hs00165814_m1), and aggrecan (ACAN) (Assay ID AIQJAP5), and hypertrophic marker genes, including type I collagen (COL1A1) (Assay ID Hs00164004_m1) and type X collagen (COL10A1) (Assay ID Hs00166657_m1), were customized by Applied Biosystems as part of the Custom Taqman® Gene Expression Assays. Eukaryotic 18S RNA (Assay ID HS99999901_s1) was carried out as the endogenous control gene. Real-time PCR was performed with the iCycler iQ™. Relative transcript levels were calculated as χ=2−ΔΔCt.
2.10. Western blot
Total protein was extracted using lysis buffer (Cell Signaling, Danvers, MA) with protease inhibitors and quantified using BCA™ Protein Assay Kit (Thermo Fisher Scientific). Thirty micrograms of protein from each sample were denatured and separated using NuPAGE® Novex® Bis-Tris Mini Gels (Invitrogen) in the XCell SureLock™ Mini-Cell (Invitrogen) for 3 h at 120 V at 4°C. The resolved proteins were transferred onto a nitrocellulose membrane using an Xcell II™ Blot module (Life Technologies) at 15 V at 4°C overnight. The membrane was probed with primary antibodies followed by the horseradish peroxidase-conjugated secondary antibody. SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) was used to detect the presence of specific proteins of interest. The primary antibodies included the mitogen-activated protein kinase (MAPK) family antibody sampler kit [extracellular signal-regulated protein kinases 1 and 2 (Erk1/2), Jun N-terminal kinase (Jnk), and p38], phosphorylated (p−) MAPK family antibody sampler kit, and the Wnt signaling antibody sampler kit (Cell Signaling). Sample loading was normalized with β-actin. This experiment was repeated three times.
2.11. Microarray analysis
Global gene expression was evaluated in human SDSCs after expansion on dECMs followed by chondrogenic induction as described previously [36]. Total RNAs were isolated from expanded cells and corresponding day 35 pellets using Trizol (Invitrogen) followed by additional purification and hybridization. The raw data were normalized by the Quantile algorithm (Genespring Gx 12.6 Software) for initial analysis. Briefly, raw intensity was background-subtracted, robust multi-array analysis (RMA) was normalized and log-transformed, and then fold changes were determined. Heatmaps representing differentially regulated genes were generated using the pheatmap function from the R package pheatmap. Enrichr was used for gene ontology (GO) analysis (https://amp.pharm.mssm.edu/Enrichr/) [37,38].
2.12. Statistics
The Kruskal-Wallis test was used to determine significant differences among groups. For a comparison between two groups in biochemistry analysis and real-time PCR, the Mann-Whitney U test was used instead. All statistical analyses were performed with SPSS 13.0 statistical software (SPSS Inc., Chicago, IL). P < 0.05 were considered statistically significant.
3. Results
The matrix microenvironment associated with tissue-specific stem cells has an important role in the determination of stem cell differentiation efficiency and efficacy. In this work, we evaluated the rejuvenation role of SDSC specific dECM and non-SDSC dECM on chondrogenic potential of expanded human SDSCs, including but not limited to expanded cell stiffness, proliferation, and chondrogenic differentiation. Compared with dECM deposited by SDSCs with strong chondrogenic capacity, non-SDSC dECMs were deposited by adult stem cells with less or no chondrogenic capacity, ADSCs and DFs (weak), and UDSCs (none).
3.1. Evaluation of dECMs’ microstructure, composition, and elasticity alone or with SDSCs seeded on different culture substrates
SEM was used to confirm the morphology of dECMs alone (w/o cells) and SDSCs grown on PL or dECMs (w/ cells). Human SDSCs expanded on Plastic exhibited a well-spread morphology (Fig. 1A); in contrast, SDSCs expanded on dECMs showed a spindle shaped structure (Fig. 1B). We also noticed that cells seeded onto SECM and AECM were embedded in the matrix (Fig. 1B). Immunostaining using specific antibodies against fibronectin and type I collagen revealed the presence of these macromolecules in the dECMs deposited by respective cells (Fig. 1C). AFM data (Fig. 1D) showed that the elasticity of Plastic was infinite. The elasticity of the DECM was the highest among the dECMs with a value of 88.29 ± 49.9 kPa, while UECM was the lowest with a value of 21.41 ± 11.30 kPa. AECM had an elasticity close to UECM with a value of 25.65 ± 9.38 kPa. On the other hand, SECM was less than DECM but higher than AECM. The value of SECM was 61.82 ± 35.86 kPa. However, the elasticity of SDSCs after seeding onto different dECMs reduced drastically upon the attachment of the cells (Fig. 1E). SDSCs grown on DECM showed the highest elasticity (4.87 ± 2.16 kPa) among different dECMs, while SECM maintained a slightly lower elasticity (3.58 ± 1.94 kPa). The plastic surface recorded the highest value of elasticity (9.07 ± 4.56 kPa) in seeded cells compared with all dECM substrates. All groups were significantly different from each other (P < 0.05) in matrix stiffness (Fig. 1D) and cell stiffness (Fig. 1E).
Fig. 1.
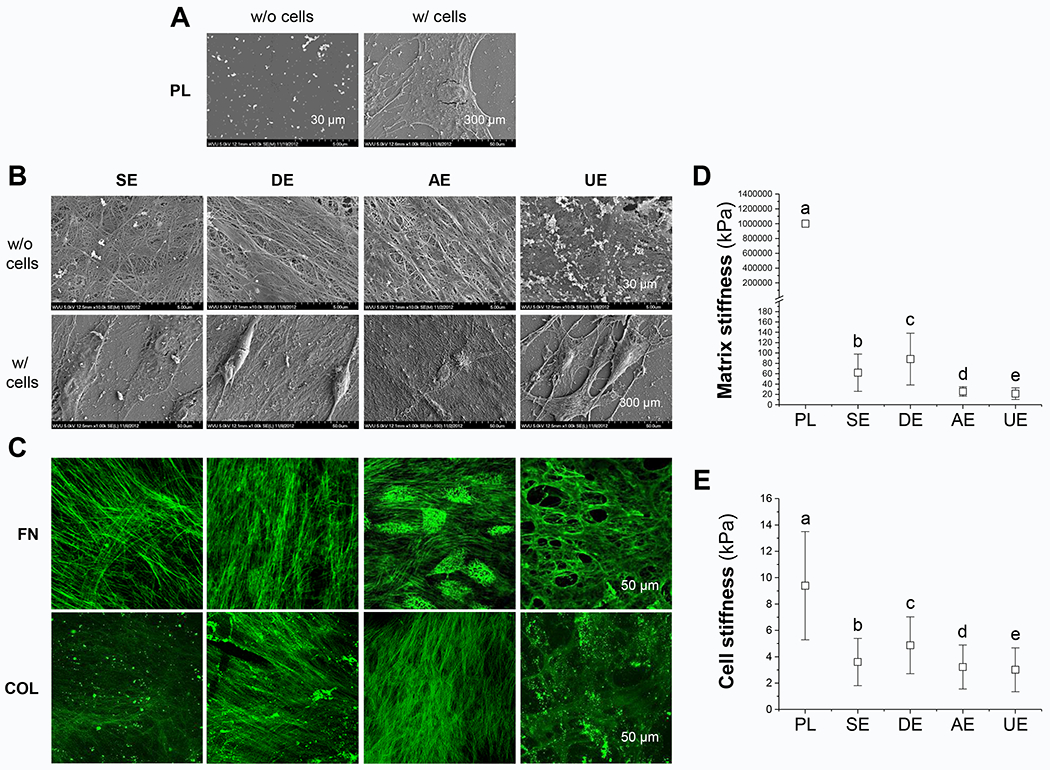
Effect of biochemical and biophysical properties of dECMs on expanded cells’ morphology and stiffness. Scanning electron microscopy (SEM) was used to observe SDSC morphology during expansion on Plastic (PL, A) and dECMs (B), including SECM (SE), DECM (DE), AECM (AE), and UECM (UE). Scale bars: 30 μm for dECMs and 300 μm for expanded cells on different culture substrates. Immunostaining was used to identify typical matrix protein expression including fibronectin (FN) and type I collagen (COL) in dECMs (C). Scale bar: 50 μm. Atomic force microscopy (AFM) was used to measure stiffness of both cell substrates (D) and expanded SDSCs (E). Data are shown as average ± standard deviation (SD) for n=1881-2001 (D) and n=1778-1981 (E). Groups not connected by the same letter are significantly different (P < 0.05).
3.2. dECM expanded SDSCs exhibited an enhanced proliferation capacity and unique surface marker expression
We examined the effect of dECMs on human SDSC morphology and proliferation. Under light microscopy, the cells cultured on a plastic surface showed a random distribution and a flattened and irregular shape with enlarged size; on the other hand, cells grown on dECMs were organized and spindle shaped (Fig. 2A). The cell counts increased considerably in all dECM groups compared with the Plastic group, with the highest cell counts recorded in the UECM group and the lowest cell counts in the SECM group (Fig. 2a). The number of generations of the cell division on different substrates was measured using flow cytometry with fluorescent dyes. SDSCs grown on the plastic substrate showed minimal cell division across four generations, while other dECM substrates showed a higher number of cell divisions. SDSCs grown on the SECM substrate showed cell generations up to six while non-SDSC dECM substrates had a higher number of cell generations (7-8) (Fig. 2B). The rate of proliferation index analysis after eight days of culture with Plastic as a control (Fig. 2b) indicated that the cells grown on UECM displayed the highest proliferation index at eleven-fold. Cells grown on DECM and AECM showed a proliferation index at five-fold and six-fold, respectively. Cells grown on SECM displayed a proliferation index at 1.08-fold. In addition, we examined the stemness of SDSCs grown on different substrates. Flow cytometry was used to examine the expression of surface markers (CD29, CD90, CD105, and SSEA4) specific for MSCs in SDSCs. FACS results indicated that all substrates including Plastic showed consistent expression of cell surface markers (Fig. 2C). However, the median fluorescence intensity (MFI) of CD29, CD90, and CD105 were significantly reduced in the non-SDSC dECM groups (DECM, AECM, and UECM) (Fig. 2c).
Fig. 2.

dECM expanded SDSCs exhibited an enhanced proliferative capacity and unique mesenchymal stem cell surface phenotype changes. SDSCs were expanded on Plastic (PL) and dECMs including SECM (SE), DECM (DE), AECM (AE), and UECM (UE) for one passage. Cell morphology was shown using phase-contrast microscope. Scale bars: 400 μm (A). Cell number was counted in 175 cm2 flasks (n=6) using a hemocytometer (a). Flow cytometry was used to measure proliferation index of expanded SDSCs (B/b) and (C) median fluorescence intensity (MFI) of MSC surface markers (CD29, CD90, and CD105) and SSEA4 (C/c).
3.3. Proteomics analysis of dECM substrates
dECM from each cell type (SECM, DECM, AECM, and UECM) was prepared and run in duplicate for the proteomics study (Run1 and Run2). Principal Component Analysis (PCA) revealed that these cell types produced unique protein expression patterns based on their ability to discriminate from one another in this unsupervised clustering analysis (Supplementary Fig. 1). UECM samples appeared most distinct from dECMs deposited by other cell types based on the polarization in principal component 1 from other cell types. A Venn diagram illustrated the number of proteins uniquely presented and shared with other cell-type derived dECMs (Supplementary Fig. 2). Interestingly, 25.5% of dECM proteins were shared among all cell types; 5.3% of SECM proteins shared homology with UECM, followed by 1.4% with DECM, and 0.3% with AECM. Also, there were 12 proteins uniquely present in SECM. Global dECM protein expressions clearly represented the similarity between duplicates and the unique patterns in the dECM samples, especially in UECM (Supplementary Table 1).
The top 30 proteins that expressed differentially were presented as a heatmap (Fig. 3A). A heatmap of dECM proteins, especially collagens, basement membrane associated proteins, matrix-stiffness associated proteins, and growth factors, was also presented (Fig. 3B). The collagen heatmap showed that COL6A2 (1:0.10 for SE:AE and 1:0.18 for SE:DE) and COL6A3 (1:0.35 for SE:AE and 1:0.48 for SE:DE) had the highest expression in SECM and the lowest in UECM (non-detectable), basement membrane associated collagens COL4A1 (non-detectable in SECM) and COL4A2 (1:9.03 for SE:UE) were lower in SECM but higher in UECM, while fibril-associated collagens with interrupted triple helices (FACIT collagens) also had unique expression in each dECM, such as the highest expression of COL14A1 in SECM (1:0.02 for SE:AE, 1:0.10 for SE:DE, and non-detectable in UE) but the lowest expression of COL12A1 in UECM (1:0.02 for SE:UE) (Fig. 3B/a). Among other basement membrane associated proteins, SECM expressed fewer or no laminins (LAMAs), heparan sulfate proteoglycan 2 (HSPG2) (1:59.46 for SE:UE), and nidogen 1/2 (NID1/2) (1:7.16 for SE:UE), while UECM showed high expression of these proteins (Fig. 3B/b).
Fig. 3.
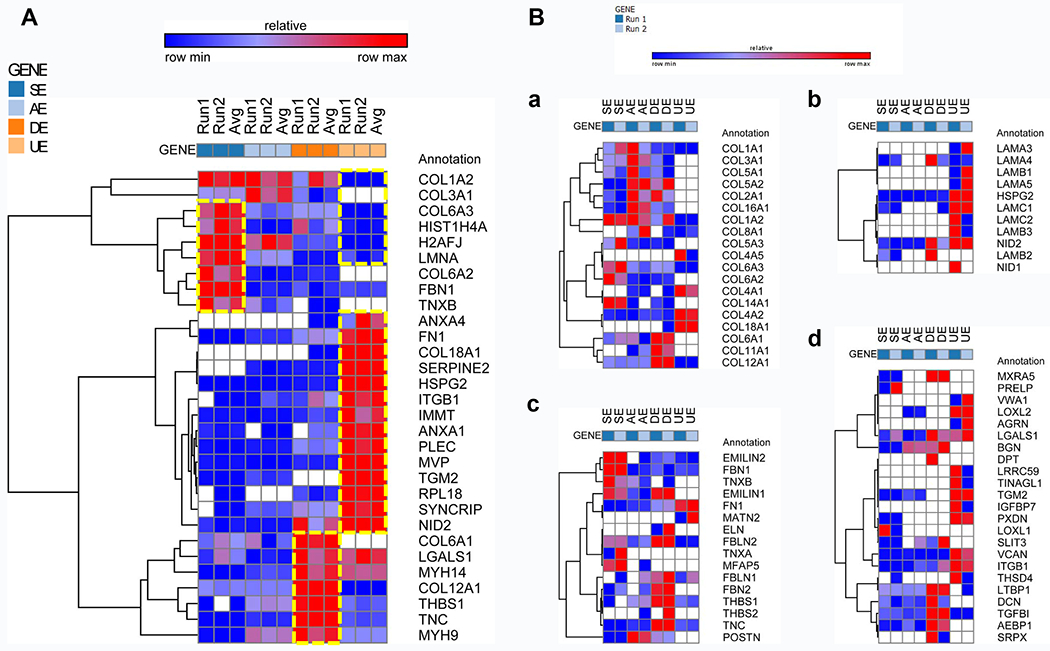
Heatmap from proteomics analysis of dECMs, including (A) top 30 proteins that expressed differentially in dECMs [SECM (SE), DECM (DE), AECM (AE), and UECM (UE)] and (B) some unique protein clustering in terms of collagens (a), basement membrane associated proteins (b), matrix-stiffness associated proteins (c), and growth factors (d). Each type of dECM was prepared and run in duplicate (Run1 and Run2). The cell-type specificity among the 30 proteins was represented with a boxed dotted line.
Matrix stiffness is controlled by multiple groups of proteins. Interestingly, we were able to specifically identify some of these proteins by proteomics analysis. The expression profiles of matrix stiffness regulatory proteins, such as elastin microfibrillar interface proteins (EMILINs) (1:0.45 for SE:AE and non-detectable in UE), tenascin-X (TNX) (1:0.41 for SE:AE, 1:0.06 for SE:DE and non-detectable in UE), and fibulin (FBN1) (1:0.24 for SE:AE, 1:0.29 for SE:DE and 1:0.34 for SE:UE), were exhibited by heatmap. Higher expressions of matrix stiffness regulators were found in SECM (Fig. 3B/c). Many of the growth factors analyzed, such as lysyl oxidase like 2 (LOXL2), transglutaminase 2 (TGM2) (1:27.37 for SE:UE), and peroxidasin (PXDN) (1:148.59 for SE:UE), were found to be present at high concentrations in UECM samples, while these growth factors were found at low concentrations or non-detectable in SECM samples (Fig. 3B/d).
3.4. dECM mediated enhancement of SDSC chondrogenic capacity
To determine the rejuvenating effects of dECMs on SDSC chondrogenic potential, expanded SDSCs were chondrogenically induced for up to 35 days. The evaluation of chondrogenesis consisted of biochemical, histological, and real-time PCR for chondrogenic markers. At 14 days after chondrogenic induction (Fig. 4A), gross observation demonstrated little change in pellet size among the groups except for the UECM group. Interestingly, the histology data showed an intensified expression of sulfated GAG and type II collagen in parallel in the periphery of pellets from the cells after expansion on AECM and UECM. Interestingly, no visible expression of type X collagen was noticed in the pellets from any group. At day 35 of induction (Fig. 4B), all dECM expanded cells yielded pellets with a larger size (Fig. 4C) and intensified staining of sulfated GAG and types II and X collagen, particularly for the DECM, AECM, and UECM groups, compared with the Plastic group.
Fig. 4.
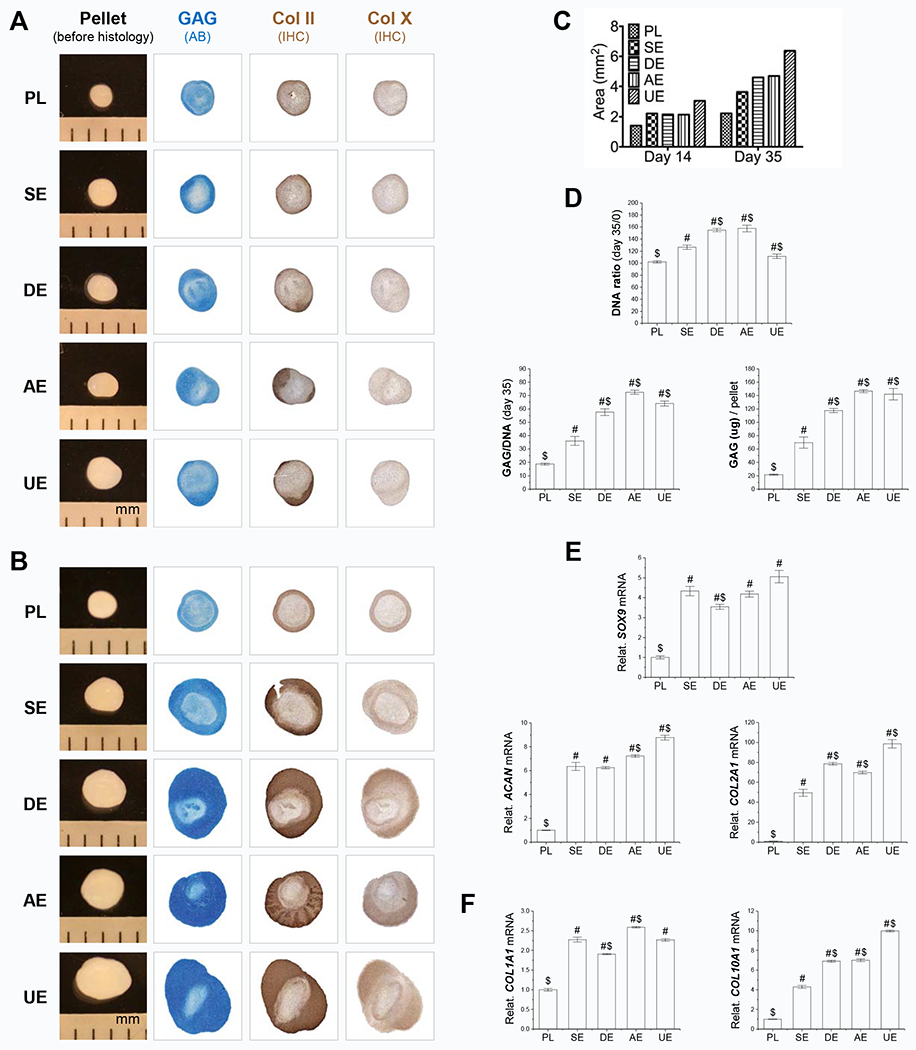
dECM expanded SDSCs exhibited enhanced chondrogenic potential. After expansion on different culture substrates for one passage, expanded cells were chondrogenically induced in a pellet culture system. Pellet samples were collected for data analysis at two time points: day 14 (earlier stage) (A/E/F) and day 35 (late stage) (B/C/D). Before histological staining, pellet size was measured with a scale bar as mm; Alcian blue (AB) was used to stain sulfated GAGs and immunohistochemistry staining (IHC) was used to detect type II collagen (Col II) and type X collagen (Col X) (A/B). Both day 14 and day 35 pellet sizes were quantified by ImageJ (C). Biochemical analysis was used for DNA and GAG amounts in pellets; cell viability in chondrogenic induction was evaluated using DNA ratio (DNA amount adjusted by that at day 0); a ratio of GAG to DNA indicated chondrogenic index (D). Real-time PCR was used to evaluate expression of chondrogenic marker genes (SOX9, ACAN, and COL2A1) (E) and hypertrophic genes (COL1A1 and COL10A1) (F) in pellets after chondrogenic induction. Data are shown as average ± standard deviation (SD) for n=4. P < 0.05 indicates a statistically significant difference compared with the PL group (#) and/or the SE group ($).
In order to quantitatively measure chondrogenesis, we estimated the amount of DNA as a reference for cell number and compared DNA ratio among groups at each time point by normalizing DNA amount by the day 0 value. At 14 days after chondrogenic induction (Supplementary Fig. 3A), we found that, compared with the Plastic group, SDSCs grown on dECMs yielded pellets with a decrease in DNA ratio except in the SECM group and an increase of GAG amount per pellet except in the AECM group. At day 35 of induction (Fig. 4D), interestingly, all pellets from the dECM groups had significantly higher DNA ratios than those of the Plastic group, indicating that dECM preconditioning promoted cell viability during chondrogenic induction. All dECM expanded cells yielded pellets with significantly higher GAG amount per pellet and GAG/DNA ratio compared with the Plastic group, especially for cells expanded on DECM, AECM, and UECM.
At the mRNA level, we found that pellets collected from the Plastic group at both day 14 (Fig. 4E) and day 35 (Supplementary Fig. 3B) after chondrogenic induction showed the lowest levels of chondrogenic gene expression, in terms of SOX9, ACAN, and COL2A1, compared with the corresponding dECM groups. At day 14, among the dECM groups, UECM expanded cells yielded pellets with the highest expression of chondrogenic genes ACAN and COL2A1, followed by the AECM group in ACAN expression and the DECM group in COL2A1 expression (Fig. 4E). Of note, all dECM groups had similar SOX9 expression except the DECM group which had a lower level of SOX9. With continuing induction for up to day 35, DECM expanded cells yielded pellets with the highest expression of chondrogenic genes including SOX9, ACAN, and COL2A1, followed by the AECM and UECM groups in SOX9 and ACAN expression (Supplementary Fig. 3B). The SECM group had similar COL2A1 expression as the AECM and UECM groups (Supplementary Fig. 3B). Interestingly, DECM expanded cells yielded pellets with the highest expression of COL1A1 and COL10A1, whereas SECM expanded cells yielded pellets with the lowest expression of hypertrophic gene COL10A1 at both day 14 (Fig. 4F) and day 35 (Supplementary Fig. 3C) after chondrogenic induction.
3.5. Signaling mechanisms on different substrates
In the case of Erk, Jnk, and p38, the ratio of the respective intensities of phosphorylated over total proteins was measured and fold activation was calculated with Plastic as a control (one-fold) (Fig. 5A). The phosphorylation studies indicated higher Erk activity (phosphorylation) in cells grown on Plastic surfaces, while cells grown on dECM showed reduced activity. We noticed minimal Erk phosphorylation in AECM. Interestingly, the total Erk protein expression remained high in all groups. In the SECM group, the phosphorylation was moderate; on the other hand, the expression of p38 protein in all groups varied widely. We detected higher levels of p38 protein (total) in the cells grown on softer dECM (UECM), while the expression was faint in the stiffer dECM (DECM). However, the phosphorylation activity was higher in stiffer ECM (DECM), while softer ECM (UECM) recorded lower phosphorylation activity. We studied the expression of Jnk protein (total) in cells grown on different substrates and noticed an extremely low expression level in SECM, while the expression among other groups was similar. Distinctly, the phosphorylation of Jnk protein had the highest activity in UECM samples, while both DECM and AECM showed similar phosphorylation levels; more interestingly, the Plastic group showed negligible phosphorylation activity. The phosphorylation activity of Erk, p38, and Jnk was quantitatively examined by intensity scanning of the immunoblot bands (Fig. 5a). Interestingly, the Erk phosphorylation pathway was relatively suppressed in all dECM groups, while Jnk and p38 phosphorylation activities were specifically activated in UECM and DECM, respectively (Fig. 5a). However, SECM and AECM showed balanced phosphorylation activity related to p38 and Jnk pathways. As a canonical Wnt signal, Wnt3a exhibited higher expression in SDSCs after expansion on SECM, DECM, and AECM (Fig. 5B). Similarly, Wnt5a had higher expression in SDSCs after expansion on dECMs compared with the Plastic group, particularly for those from the UECM, DECM, and AECM groups (Fig. 5B/b).
Fig. 5.
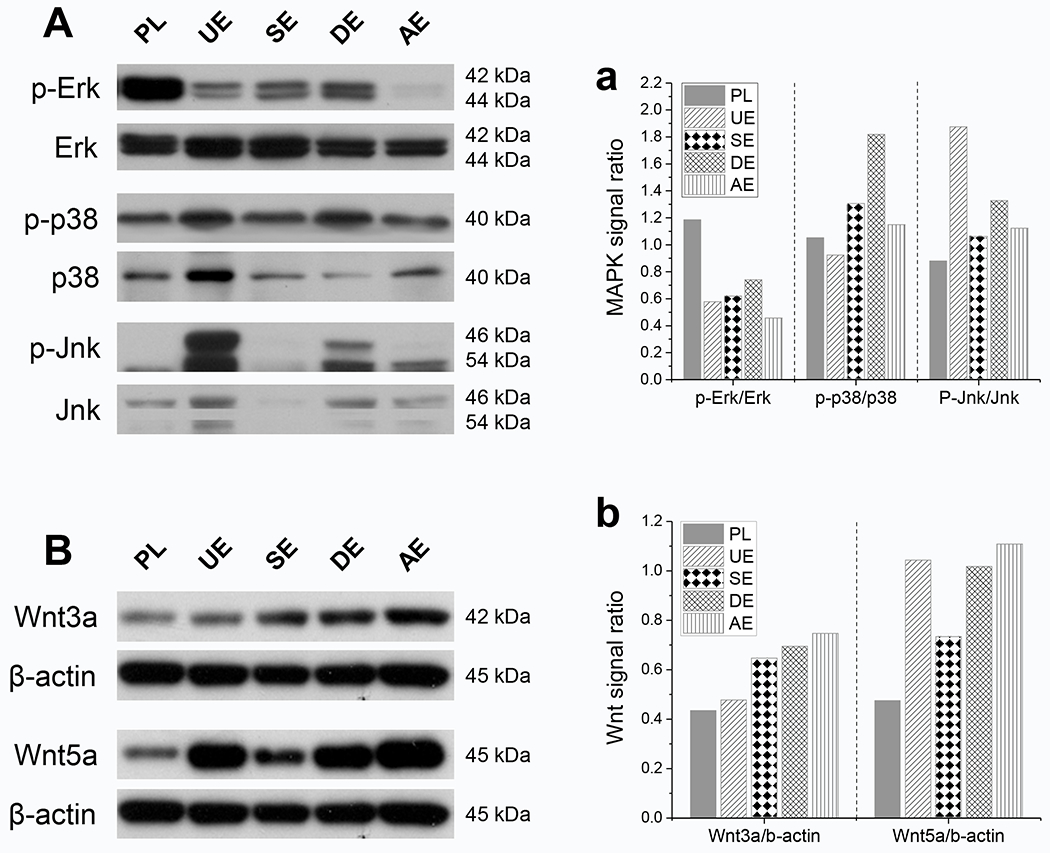
Potential mechanisms involved in the rejuvenation of SDSCs after expansion on dECMs [SECM (SE), DECM (DE), AECM (AE), and UECM (UE)]. Western blot was used to detect the mitogen-activated protein kinase (MAPK) signals (A), including Erk, p38, and Jnk, and the Wnt signals (B), including Wnt3a and Wnt5a. The intensity of immunoblotting bands was measured using ImageJ software and MAPK signals were presented as a ratio of phosphorylated protein to non-phosphorylated protein (a). The expression of Wnt signals was quantitated and normalized with β-actin (b). This experiment was repeated three times.
3.6. dECM expansion mediated SDSCs’ proliferation gene profiles detected by microarray
To gain additional insight into the molecular mechanisms underlying the effect of dECM expansion on human SDSCs, we performed microarray analysis to analyze the gene expression profiles in the SDSCs after expansion on different dECMs and plastic substrate. A total of 482, 722, 760, and 860 genes was upregulated or downregulated (>2 fold) in SECM, DECM, AECM, and UECM expanded SDSCs (SSC, SDC, SAC, and SUC) compared to plastic substrate expanded cells (SPC). The top 100 differentially expressed genes in SSC were clustered with SUC before clustering with the SAC and SDC groups (Fig. 6A) (Supplementary Table 2). A Venn diagram was generated to show the common genes within upregulated or downregulated genes among all groups. We found a significant percentage (44.19% in SSC, 29.50% in SDC, 28.03% in SAC, and 24.77% in SUC) of these genes was shared among all groups. Gene ontology (GO) analysis showed that 93 overlapping and upregulated genes among all groups (37.5% of SSC, 33.33% of SDC, 30.10% of SAC, and 27.19% of SUC) were associated with positive regulation of cell proliferation, ECM organization, regulation of the Erk1 and Erk2 cascade as well as positive regulation of the non-canonical Wnt signaling pathway (Fig. 6B). GO terms associated with 120 overlapping and downregulated genes (51.28% of SSC, 27.09% of SDC, 26.61% of SAC, and 23.17% of SUC) included regulation of cell migration, cell-cell junction organization, ECM organization, and regulation of the p38 MAPK cascade (Fig. 6C).
Fig. 6.

Gene expression profile of SDSCs expanded on dECMs by microarray analysis. A heatmap showing relative expression patterns of the top 100 differentially expressed genes changed in SSC (A). Venn diagram of upregulated (B) or downregulated (C) genes in all dECM expanded cells compared to plastic substrate expanded cells which were then compared to each other, followed by gene ontology analysis of the overlapping 93 upregulated (B) or 120 downregulated (C) genes among all groups. Gene ontology analysis was also performed for the upregulated and downregulated genes in UE expanded cells (SUC) (D).
Since UECM expansion resulted in the highest proliferation index and chondrogenic potential in SDSCs as shown in Figs. 2 and 4, we further investigated the differentially expressed genes in SUC compared to SPC. Interestingly, 342 genes upregulated in UECM expanded cells (SUC) were related to GO terms including positive regulation of cell proliferation, regulation of ossification, regulation of cartilage development, negative regulation of the Erk1 and Erk2 cascade, and regulation of the p38 MAPK cascade; on the other hand, 518 genes downregulated in SUC were associated with GO terms, such as ECM organization, regulation of cell migration, negative regulation of cell proliferation, and positive regulation of MAPK cascade (Fig. 6D).
3.7. dECM expansion promoted SDSCs’ chondrogenic potential detected by microarray
To further characterize gene expression changes following chondrogenic induction of SDSCs after expansion on varied substrates, we also performed microarray analysis on the day 35 pellets. By comparing gene expression profiles between varied substrate expanded cells and their corresponding pellets, we found 853 genes were upregulated in the SPC-derived pellets (SPP), 907 for SSC-derived pellets (SSP), 1233 for SDC-derived pellets (SDP), 1094 for SAC-derived pellets (SAP), and 979 for SUC-derived pellets (SUP) (Fig. 7A). A significant number of genes (46.57% for SPP, 43.39% for SSP, 36.29% for SDP, 34.81% for SAP, 46.05 for SUP) were common among these groups. Interestingly, the overlapping 461 and downregulated genes among all groups were associated with GO terms including ECM organization, GAG biosynthetic process, cartilage development, and chondrocyte differentiation (Fig. 7B) (Supplementary Table 3). Hierarchical clustering analysis of these 461 genes showed that SSC and SPC shared a more similar gene expression profile during chondrogenic induction rather than SUC. Similarly, SAC and SDC also exhibited a more similar gene expression pattern than with the other groups (Fig. 7C).
Fig. 7.

Venn diagram of upregulated genes in chondrogenically induced day 35 pellets compared to their respective cells (A). Gene ontology analysis of 461 overlapping genes among all groups (B). A heatmap showing relative expression patterns of the 461 genes (C). Specifically, gene ontology analysis was performed in the 69 genes that overlapped among all dECM expanded cell groups but not the plastic substrate group (D) and 80 genes that overlapped among SDP, SAP, and SUP groups (E).
To explore the mechanisms underlying enhanced chondrogenic capacity in dECM expanded cells, we performed GO analysis on 69 genes that overlapped among all dECM expanded cells but not SPC. Our results showed that these genes were associated with regulation of smooth muscle cell migration, cartilage development, and skeletal system development (Fig. 7D). Interestingly, the 80 overlapping genes among SDC-, SAC-, and SUC-derived pellets showed that these genes were related to the collagen fibril organization, chondroitin sulfate biosynthetic process, negative regulation of ossification, and regulation of Wnt signaling pathway (Fig. 7E).
4. Discussion
One of the common disadvantages of adult cell-based therapy for cartilage repair is the expansion of stem cells without sabotaging functional and genetic expressions of the differentiated cells [12]. Implanted cells changed their phenotypes leading to partial or hypertrophic chondrogenesis. Biophysical factors and bio-macromolecular factors affect chondrogenesis phenotypes. SDSCs, isolated from the synovium, are potentially a good source of cells for cartilage repair [3,5–7]. A large quantity of high quality cells is required for successful cartilage regeneration. Our previous studies have indicated that dECM deposited by stem cells could serve as a better expansion substrate for promoting proliferative and chondrogenic potentials [13,22,39]. In order to bring out the role of lineage-specific matrix in stem cell chondrogenesis, we were interested in demonstrating the specificity of SECM as a culture substrate for SDSC expansion for chondrogenic application. We also evaluated dECMs derived from other easily available cell sources such as ADSCs, UDSCs, and DFs for their roles in SDSC expansion and chondrogenic potential.
Substrate stiffness and topography affect proliferation and differentiation potentials of the cultured stem cells [19]. The mechanotransduction via integrin signaling pathways and focal adhesion kinases dominate lineage specification [40–42]. We demonstrated that the mechanical stiffness of dECMs differs considerably and SDSCs exhibited comparable stiffness with the substrate on which they grew. These findings are in accordance with earlier observations that substrate stiffness contributes to cellular phenotype. BMSCs, when cultured on a stiff substrate, were well spread with high cytoskeletal tension, and expressed high levels of osteogenic lineage markers [40]. In contrast, stem cells cultured on soft substrates were less spread, had low cytoskeletal tension, and became quiescent [43]. In this study, we found that the morphology of the SDSCs varied on different substrates and their stiffness. The SDSCs exhibited a morphology with less spreading on lower elastic substrates (UECM and AECM) compared to more spreading on higher elastic substrates (DECM and Plastic). Furthermore, SDSCs grown on substrates with soft stiffness (UECM and AECM) had a higher rate of proliferation compared to a stiffer substrate (Plastic).
Proteomics of different dECM substrates suggested the presence of unique combinatorial matrix proteins in each of the specific cell types. The global heatmap and top 30 candidate heatmap showed clear differences in the dECMs between cell types. Several proteins associated with extracellular matrix structure such as collagen were ubiquitously present in all dECMs. However, some collagen subunits had a unique expression profile indicating cell specificity. Non-collagenous proteins such as laminins also had a clear distinction among dECMs. Interestingly, we observed differences in the expression of matrix stiffness (EMILIN and tenascin) indicating an important role in the establishment of matrix stiffness and cell adhesion via integrin binding [44]. Furthermore, the tenascin family of proteins plays a critical role in collagen fibrillogenesis and has been implicated in osteoarthritis (OA) indicating phenotype modulation by matrix stiffness regulators [45].
Next, we examined whether stemness and proliferation of SDSCs were modulated by different deposited matrices. Both proliferation index and the stem cell marker SSEA4 expression dramatically increased in expanded cells grown on dECMs compared with the Plastic group, particularly for those cells grown on non-SDSC dECMs. However, the expression of MSC surface markers, including CD29, CD90, and CD105, exhibited an opposite trend compared with proliferation index and SSEA4 expression. dECM expansion of SDSCs dramatically decreased the MSC surface marker expressions compared with the Plastic group, especially for those grown on non-SDSC dECMs. These findings are in line with previous observations indicating that preconditioning of human SDSCs with basic fibroblast growth factor (FGF2) [46] and dECM [34] increased their proliferation and SSEA4 expression but decreased the expression of surface markers (CD29, CD90, and CD105). In addition, our previous results demonstrated that preconditioning improved proliferation and chondrogenic potential of SDSCs [47]. In the current study, we evaluated and compared the preconditioning effect of SDSC and non-SDSC dECMs. Results presented in Figure 2 suggest that a balanced expression of SSEA4 and MSC surface markers are key in identifying the chondrogenic potential of SDSCs. This further necessitates evaluating the role of the current predicted MSC surface markers (CD29, CD90, and CD105) in SDSCs stemness including proliferation and differentiation capacity.
One important observation we made in this study was that the kinetics of differentiation of SDSCs toward chondrogenesis do not remain the same on different dECM substrates. It is known that a delicate balance between proliferation and differentiation remains during chondrogenesis [48]. After a transient reduction in DNA ratio at day 14 following chondrogenic induction, dECM expanded cells yielded pellets with higher DNA ratio at day 35 indicating cell viability and proliferation after dECM expansion was increased. Moreover, dECM expanded cells also exhibited enhanced chondrogenic potential, particularly those grown on non-SDSC dECMs. These results indicated that the speed or kinetics at which the differentiation happens in the UECM, DECM, and AECM groups is higher than in the SECM group, while the Plastic group has the lowest differentiation kinetics. Interestingly, we also observed the robust expression of hypertrophic chondrogenic markers in the non-SDSC dECM groups compared with the SECM group, suggesting that the matrix microenvironment in SECM was finely tuned by its components and matrix stiffness to favor SDSC chondrogenesis with limited hypertrophic potential [49].
Increasing evidence suggests the implication of both MAPK and Wnt signals in chondrogenesis [50]. With regard to chondrogenesis, Erk and p38 MAPK were reported to play a critical role in mediating cell proliferation [51,52]. It was demonstrated that stiffer microenvironments increased Erk activity [53,54]. In this study, the phosphorylation of Erk varied widely among the dECM and Plastic groups. The ratio of p-Erk/Erk showed higher levels in the Plastic group followed by the DECM group. This pattern of phosphorylation complemented the matrix stiffness trend observed for dECMs. In line with previous reports [14,36], we found that dECM expansion decreased the p-Erk1/2/Erk1/2 ratio but increased ratios of p-p38/p38 and p-Jnk/Jnk. Our differentiation study demonstrated a downregulation of the p-Erk1/2/Erk ratio and/or an upregulation of p-p38/p38 that promoted chondrogenesis of mesenchymes [55–59]. Despite previous findings that Jnk phosphorylation was not affected during chondrogenesis, meaning that Jnk signals had a limited role in chondrogenesis [58,60], the upregulation of both p-Jnk and total Jnk in the cells expanded on non-SDSC dECMs might lead to a new hypothesis regarding the potential roles of Jnk signals in stem cell proliferation and chondrogenic differentiation, which warrants further investigation.
In this study, we also found that both canonical (Wnt3a) and non-canonical Wnt signals (Wnt5a) were increased in SDSCs grown on dECMs. Wnt signaling plays an essential role in the chondrogenesis [61,62]. Increasing evidence indicates canonical Wnt signals’ role in promoting stem cell proliferation [63] and inhibiting the differentiation of MSCs into skeletal precursors [64]. The crosstalk between Wnt and Jnk pathways might upregulate Jnk signals in the non-SDSC dECM groups. Wnt3a treatment was reported to stimulate the phosphorylation of c-Jun and Jnk; inhibition of Jnk with SP600125 could block the activation of Wnt3a via activator protein-1 (AP-1) [65]. Wnt5a specifically promoted entry into the prehypertrophic phase but contrarily blocked chondrocyte hypertrophy [66,67]. Interestingly, non-canonical Wnt signaling more frequently take effect through the crosstalk between Wnt and Jnk signaling pathways [68]. Triggering Wnt5a signaling by interleukin 1 beta resulted in an upregulation of matrix metalloproteinases (MMPs) via the Jnk pathway in rabbit temporomandibular joint (TMJ) condylar chondrocytes, while blockage of Jnk signals impaired Wnt5a-induced upregulation of MMPs [69].
5. Conclusion
Taken together, we found that dECM deposited by adult stem cells with varying chondrogenic capacities [SDSCs (strong) (SECM), ADSCs (weak) (AECM) and DFs (weak) (DECM), and UDSCs (none) (UECM)], could significantly promote SDSC proliferation and chondrogenic potential compared with non-ECM coated Plastic. Compared with SECM, surprisingly, the non-SDSC dECM groups exhibited a better rejuvenation effect in both proliferation and chondrogenic capacity but also, unfortunately, higher expression of hypertrophic markers. On the other hand, SECM provided a superior tissue-specific matrix microenvironment for the optimal rejuvenation of adult SDSCs for cartilage regeneration. Biochemical and biophysical cues in dECMs might contribute to rejuvenation signals for stem cell proliferation and lineage-specific differentiation.
Supplementary Material
Acknowledgements
We thank Suzanne Danley for editing the manuscript. We also thank Drs. Chengbo Dong and Cerasela Zoica Dinu for their help with the AFM instrument. This project was supported by Research Grants from the National Institutes of Health (1R01AR067747-01A1) and the Musculoskeletal Transplant Foundation (MTF). We also would like to acknowledge the WVU Flow Cytometry & Single Cell Core Facility and the grants that support the facility, TME CoBRE grant P20GM131322 and the WV CTS grant GM104942.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
None declared.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also forms part of an ongoing study.
References
- 1.Karnes J, Zhang Y, Pei M, Cell Therapy for the Creation of Cartilage and Related Clinical Trials. N. S. Templeton Gene and Cell Therapy: Therapeutic Mechanisms and Strategies. 4th Edition Taylor & Francis/CRC Press, 2014. pp. 1123–1135. (K16570_C048). [Google Scholar]
- 2.Fellows CR, Matta C, Zakany R, Khan IM, Mobasheri A, Adipose, Bone Marrow and Synovial Joint-Derived Mesenchymal Stem Cells for Cartilage Repair, Front. Genet 7 (2016) 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pizzute T, Lynch K, Pei M, Impact of tissue-specific stem cells on lineage specific differentiation: a focus on musculoskeletal system, Stem Cell Rev. Rep. 11 (2015) 119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshimura H, Muneta T, Nimura A, Yokoyama A, Koga H, Sekiya I, Comparison of rat mesenchymal stem cells derived from bone marrow, synovium, periosteum, adipose tissue, and muscle, Cell Tissue Res. 327 (2007) 449–462. [DOI] [PubMed] [Google Scholar]
- 5.Jones BA, Pei M M, Synovium-derived stem cells: a tissue-specific stem cell for cartilage engineering and regeneration, Tissue Eng. Part B Rev. 18 (2012) 301–311. [DOI] [PubMed] [Google Scholar]
- 6.Kubosch EJ, Lang G, Furst D, Kubosch D, Izadpanah K, Rolauffs B, et al. , The Potential for Synovium-derived Stem Cells in Cartilage Repair, Curr. Stem Cell Res. Ther 13 (2018) 174–184. [DOI] [PubMed] [Google Scholar]
- 7.Zupan J, Drčbnic M, Stražar K, Synovium-Derived Mesenchymal Stem/Stromal Cells and their Promise for Cartilage Regeneration, Adv. Exp. Med. Biol (2019) doi: 10.1007/5584_2019_381. [DOI] [PubMed] [Google Scholar]
- 8.Pei M, He F, Boyce BM, Kish VL, Repair of full-thickness femoral condyle cartilage defects using allogeneic synovial cell-engineered tissue constructs, Osteoarthritis Cartilage 17 (2009) 714–722. [DOI] [PubMed] [Google Scholar]
- 9.Pei M, Yan ZQ, Shoukry M, Boyce BM, Failure of xenoimplantation using porcine synovium-derived stem cell-based cartilage tissue constructs for the repair of rabbit osteochondral defects, J. Orthop. Res 28 (2010) 1064–1070. [DOI] [PubMed] [Google Scholar]
- 10.Shimomura K, Yasui Y, Koizumi K, Chijimatsu R, Hart DA, Yonetani Y, et al. , First-in-Human Pilot Study of Implantation of a Scaffold-Free Tissue-Engineered Construct Generated From Autologous Synovial Mesenchymal Stem Cells for Repair of Knee Chondral Lesions, Am. J. Sports Med. 46 (2018) 2384–2393. [DOI] [PubMed] [Google Scholar]
- 11.Benya PD, Padilla SR, Nimni ME, Independent regulation of collagen types by chondrocytes during the loss of differentiated function in culture, Cell 15 (1978) 1313–1321. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Pei M, Cell senescence: a challenge in cartilage engineering and regeneration, Tissue Eng. Part B Rev. 18 (2012) 270–287. [DOI] [PubMed] [Google Scholar]
- 13.He F, Chen X, Pei M, Reconstruction of an in vitro tissue-specific microenvironment to rejuvenate synovium-derived stem cells for cartilage tissue engineering, Tissue Eng. Part A 15 (2009) 3809–3821. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Hansen KC, Zhang Y, Dong C, Dinu CZ, Dzieciatkowska M, et al. , Rejuvenation of chondrogenic potential in a young stem cell microenvironment, Biomaterials 35 (2014) 642–653. [DOI] [PubMed] [Google Scholar]
- 15.Pei M, He F, Li JT, Tidwell J, Jones A, McDonough EB, Repair of large animal partial-thickness cartilage defects using matrix rejuvenated synovium-derived stem cells, Tissue Eng. Part A 19 (2013) 1144–1154. [DOI] [PubMed] [Google Scholar]
- 16.Gattazzo F, Urciuolo A, Bonaldo P, Extracellular matrix: a dynamic microenvironment for stem cell niche, Biochim. Biophys. Acta 1840 (2014) 2506–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ozbek S, Balasubramanian PG, Chiquet-Ehrismann R, Tucker RP, Adams JC, The evolution of extracellular matrix, Mol. Biol. Cell 21 (2010) 4300–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watt FM, Huck WT, Role of the extracellular matrix in regulating stem cell fate, Nat. Rev. Mol. Cell Biol. 14 (2013) 467–473. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Chen S, Pei M, Biomechanical signals guiding stem cell cartilage engineering: from molecular adaption to tissue functionality, Eur. Cell Mater 31 (2015) 59–78. [DOI] [PubMed] [Google Scholar]
- 20.Hynes RO, The Extracellular Matrix: Not Just Pretty Fibrils, Science 326 (2009) 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DuFort CC, Paszek MJ, Weaver VM, Balancing forces: architectural control of mechanotransduction, Nat. Rev. Mol. Cell Biol. 12 (2011) 308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pei M, Li JT, Shoukry M, Zhang Y, A Review of Decellularized Stem Cell Matrix: a Novel Cell Expansion System for Cartilage Tissue Engineering, Eur. Cell Mater 22 (2011)333–343. [DOI] [PubMed] [Google Scholar]
- 23.Berrier AL, Yamada KM, Cell-matrix adhesion J Cell Physiol. 213 (2007) 565–573. [DOI] [PubMed] [Google Scholar]
- 24.Kresse H, Schonherr E, Proteoglycans of the extracellular matrix and growth control, J. Cell Physiol. 189 (2001) 266–274. [DOI] [PubMed] [Google Scholar]
- 25.Garcion E, Faissner A, ffrench-Constant C, Knockout mice reveal a contribution of the extracellular matrix molecule tenascin-C to neural precursor proliferation and migration, Development 128 (2001) 2485–2496. [DOI] [PubMed] [Google Scholar]
- 26.Sabatelli P, Bonaldo P, Lattanzi G, Braghetta P, Bergamin N, Capanni C, et al. , Collagen VI deficiency affects the organization of fibronectin in the extracellular matrix of cultured fibroblasts, Matrix Biol. 20 (2001) 475–486. [DOI] [PubMed] [Google Scholar]
- 27.Discher DE, Mooney DJ, Zandstra PW, Growth factors, matrices, and forces combine and control stem cells, Science 324 (2009) 1673–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pei M, Li JT, Zhang Y, Liu GH, Wei L, Zhang YY, Expansion on matrix deposited by nonchondrogenic urine stem cells strengthens repeated passage bone marrow stromal cells’ chondrogenic capacity, Cell Tissue Res. 356 (2014) 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brohem CA, de Carvalho CM, Radoski CL, Santi FC, Baptista MC, Swinka BB, et al. , Comparison between fibroblasts and mesenchymal stem cells derived from dermal and adipose tissue, Int. J. Cosmet. Sci 35 (2013) 448–457. [DOI] [PubMed] [Google Scholar]
- 30.Lorenz K, Sicker M, Schmelzer E, Rupf T, Salvetter J, Schulz-Siegmund M, et al. , Multilineage differentiation potential of human dermal skin-derived fibroblasts, Exp. Dermatol 17 (2008) 925–932. [DOI] [PubMed] [Google Scholar]
- 31.Mildmay-White A, Khan W, Cell Surface Markers on Adipose-Derived Stem Cells: A Systematic Review, Curr. Stem Cell Res. Ther. 12 (2017) 484–492. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Pizzute T, Li J, He F, Pei M, sb203580 preconditioning recharges matrix-expanded human adult stem cells for chondrogenesis in an inflammatory environment - A feasible approach for autologous stem cell based osteoarthritic cartilage repair, Biomaterials 64 (2015) 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li JT, Pei M, A protocol to prepare decellularized stem cell matrix for rejuvenation of cell expansion and cartilage regeneration, Methods Mol. Biol. 1577 (2018) 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pizzute T, Zhang Y, He F, Pei M, Ascorbate-dependent impact on cell-derived matrix in modulation of stiffness and rejuvenation of infrapatellar fat derived stem cells toward chondrogenesis, Biomed. Mater 11 (2016) 045009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Brien JH, Vanderlinden LA, Schedin PJ, Hansen KC, Rat mammary extracellular matrix composition and response to ibuprofen treatment during postpartum involution by differential GeLC-MS/MS analysis, J. Proteome Res. 11 (2012) 4894–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Li JT, Davis ME, Pei M, Delineation of in vitro chondrogenesis of human synovial stem cells following preconditioning using decellularized matrix, Acta Biomaterialia 20 (2015) 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. , Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool, BMC Bioinformatics 14 (2013) 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. , Enrichr: a comprehensive gene set enrichment analysis web server 2016 update, Nucleic Acids Res. 44 (2016) W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li JT, Pei M, Optimization of an in vitro three-dimensional microenvironment to reprogram synovium-derived stem cells for cartilage tissue engineering, Tissue Eng. Part A 17 (2011) 703–712. [DOI] [PubMed] [Google Scholar]
- 40.Engler AJ, Sen S, Sweeney HL, Discher DE, Matrix elasticity directs stem cell lineage specification, Cell 126 (2006) 677–689. [DOI] [PubMed] [Google Scholar]
- 41.Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, et al. , Harnessing traction-mediated manipulation of the cell/matrix interface to control stem-cell fate, Nat. Mater 9 (2010) 518–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ross TD, Coon BG, Yun S, Baeyens N, Tanaka K, Ouyang M, et al. , Integrins in mechanotransduction, Curr. Opin. Cell Biol. 25 (2013) 613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winer JP, Janmey PA, McCormick ME, Funaki M, Bone marrow-derived human mesenchymal stem cells become quiescent on soft substrates but remain responsive to chemical or mechanical stimuli, Tissue Eng. Part A 15 (2009) 147–154. [DOI] [PubMed] [Google Scholar]
- 44.Colombatti A, Spessotto P, Doliana R, Mongiat M, Bressan G, Esposito G, The EMILIN/Multimerin Family, Front. Immunol 2 (2012) 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balague F, Do not dismiss intense pain, Lancet 363 (2004) 2021. [DOI] [PubMed] [Google Scholar]
- 46.Pizzute T, Li JT, Zhang Y, Pei M, FGF ligand dependent proliferation and multi-differentiation of synovium-derived stem cells and concomitant adaptation of Wnt/MAPK signals during chondrogenesis, Tissue Eng. Part A 22 (2016) 1036–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pei M, Environmental preconditioning rejuvenates stem cells’ chondrogenic potential, Biomaterials 117 (2017) 10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dexheimer V, Frank S, Richter W, Proliferation as a requirement for in vitro chondrogenesis of human mesenchymal stem cells, Stem Cells Dev. 21 (2012) 2160–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen S, Fu PL, Cong RJ, Wu HS, Pei M, Strategies to minimize hypertrophy for cartilage regeneration, Genes Dis. 2 (2015) 76–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Pizzute T, Pei M, A review of crosstalk between MAPK and Wnt signals and its impact on cartilage regeneration, Cell Tissue Res. 358 (2014) 633–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krens SF, Spaink HP, Snaar-Jagalska BE, Functions of the MAPK family in vertebrate-development, FEBS Lett. 580 (2006) 4984–4990. [DOI] [PubMed] [Google Scholar]
- 52.Yoon S, Seger R, The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions, Growth Factors 24 (2006) 21–44. [DOI] [PubMed] [Google Scholar]
- 53.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. , Tensional homeostasis and the malignant phenotype, Cancer Cell 8 (2005) 241–254. [DOI] [PubMed] [Google Scholar]
- 54.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ, Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage, Oncogene 28 (2009) 4326–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bobick BE, Kulyk WM, MEK-ERK signaling plays diverse roles in the regulation of facial chondrogenesis, Exp. Cell Res. 312 (2006) 1079–1092. [DOI] [PubMed] [Google Scholar]
- 56.Chang SH, Oh CD, Yang MS, Kang SS, Lee YS, Sonn JK, et al. , Protein kinase C regulates chondrogenesis of mesenchymes via mitogen-activated protein kinase signaling, J. Biol. Chem 273 (1998) 19213–19219. [DOI] [PubMed] [Google Scholar]
- 57.Oh CD, Chang SH, Yoon YM, Lee SJ, Lee YS, Kang SS, et al. , Opposing role of mitogen-activated protein kinase subtypes, erk-1/2 and p38, in the regulation of chondrogenesis of mesenchymes, J. Biol. Chem 275 (2000) 5613–5619. [DOI] [PubMed] [Google Scholar]
- 58.Stanton LA, Underhill TM, Beier F, MAP kinases in chondrocyte differentiation. Dev. Biol 263 (2003) 165–175. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe H, de Caestecker MP, Yamada Y, Transcriptional crosstalk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells, J. Biol. Chem 276 (2001) 14466–14473. [DOI] [PubMed] [Google Scholar]
- 60.Nakamura K, Shirai T, Morishita S, Uchida S, Saeki-Miura K, Makishima F, p38 mitogen-activated protein kinase functionally contributes to chondrogenesis induced by growth/differentiation factor-5 in ATDC5 cells, Exp. Cell Res. 250 (1999) 351–363. [DOI] [PubMed] [Google Scholar]
- 61.Church V, Nohno T, Linker C, Marcelle C, Francis-West P, Wnt regulation of chondrocyte differentiation, J. Cell Sci. 115 (2002) 4809–4818. [DOI] [PubMed] [Google Scholar]
- 62.Tuan RS, Cellular signaling in developmental chondrogenesis: N-cadherin, Wnts, and BMP-2, J. Bone J. Surg. Am 85-A Suppl 2 (2003) 137–141. [DOI] [PubMed] [Google Scholar]
- 63.Boland GM, Perkins G, Hall DJ, Tuan RS, Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells, J. Cell Biochem. 93 (2004) 1210–1230. [DOI] [PubMed] [Google Scholar]
- 64.Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C, Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes, Dev. Cell 8 (2005) 727–738. [DOI] [PubMed] [Google Scholar]
- 65.Hwang SG, Yu SS, Lee SW, Chun JS, Wnt-3a regulates chondrocyte differentiation via c-Jun/AP-1 pathway, FEBS Lett. 579 (2005) 4837–4842. [DOI] [PubMed] [Google Scholar]
- 66.Kawakami Y, Wada N, Nishimatsu SI, Ishikawa T, Noji S, Nohno T, Involvement of Wnt-5a in chondrogenic pattern formation in the chick limb bud, Dev. Growth Differ. 41 (1999) 29–40. [DOI] [PubMed] [Google Scholar]
- 67.Yang Y, Topol L, Lee H, Wu J, Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation, Development 130 (2003) 1003–1015. [DOI] [PubMed] [Google Scholar]
- 68.Logan CY, Nusse R, The Wnt signaling pathway in development and disease, Annu. Rev. Cell Dev. Biol. 20 (2004) 781–810. [DOI] [PubMed] [Google Scholar]
- 69.Ge X, Ma X, Meng J, Zhang C, Ma K, Zhou C, Role of Wnt-5A in interleukin-1 beta-induced matrix metalloproteinase expression in rabbit temporomandibular joint condylar chondrocytes, Arthritis Rheum. 60 (2009) 2714–2722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.