

ABSTRACT
Cancer immunotherapy continues to make headway as a treatment for advanced stage tumors, revealing an urgent need to understand the fundamentals of anti-tumor immune responses. Noteworthy is a scarcity of data pertaining to the breadth and specificity of tumor-specific T cell responses in metastatic breast cancer. Autochthonous transgenic models of breast cancer display spontaneous metastasis in the FVB/NJ mouse strain, yet a lack of knowledge regarding tumor-bound MHC/peptide immune epitopes in this mouse model limits the characterization of tumor-specific T cell responses, and the mechanisms that regulate T cell responses in the metastatic setting. We recently generated the NetH2pan prediction tool for murine class I MHC ligands by building an FVB/NJ H-2q ligand database and combining it with public information from six other murine MHC alleles. Here, we deployed NetH2pan in combination with an advanced proteomics workflow to identify immunogenic T cell epitopes in the MMTV-PyMT transgenic model for metastatic breast cancer. Five unique MHC I/PyMT epitopes were identified. These tumor-specific epitopes were confirmed to be presented by the class I MHC of primary MMTV-PyMT tumors and their T cell immunogenicity was validated. Vaccination using a DNA construct encoding a truncated PyMT protein generated CD8 + T cell responses to these MHC class I/peptide complexes and prevented tumor development. In sum, we have established an MHC-ligand discovery pipeline in FVB/NJ mice, identified and tracked H-2Dq/PyMT neoantigen-specific T cells, and developed a vaccine that prevents tumor development in this metastatic model of breast cancer.
KEYWORDS: Tumor immunology, breast cancer, MMTV-PyMT, MHC Class I, peptide prediction, NetH2pan
Introduction
Breast cancer is the most common cancer in women, with a lifetime risk of 1 in 8.1 Unfortunately, despite state-of-the-art treatment, 40,000 deaths are attributed to breast cancer every year in the U.S., largely due to metastatic recurrence and therapeutic resistance. Development of new targeted therapies is challenging because the ‘drivers’ of metastatic breast cancer and drug resistance are mostly unknown, and breast tumors are extremely heterogeneous.2,3 Therefore, alternative approaches designed to stimulate the immune system to selectively identify and eliminate tumor cells are gaining traction. Once activated, the immune system could theoretically recognize and kill tumor cells, independent from knowledge of the cancer drivers, through a process known as epitope spreading.4 Harnessing the power of the host immune system has induced disease remission in patients with other cancer types where traditional chemotherapy drugs have failed. Similar vaccine strategies and immune checkpoint blockade therapies have been tested in breast cancer patients, but so far have shown limited efficacy.4,5 A greater understanding of the delicate interplay between the host immune system and tumors is required to optimize immunotherapy strategies for metastatic breast cancer.
Essential to the success of immunotherapy is the cognate interaction between a tumor cell’s class I Major Histocompatibility Complex (MHC) and the recipient T cell receptor (TCR). The class I MHC serves to alarm surveilling CD8 + T cells by sampling intracellular pathogens/cancer neoantigens for display on the cell surface. If a unique TCR succeeds in its peptide:MHC recognition, it will trigger a series of epigenetic and transcriptional changes that convert the T cell to a cytotoxic T lymphocyte (CTL) with full capacity to eliminate the target cell. As such, TCR recognition of tumor-specific peptides represents a significant bottleneck in the functionality of current immunotherapies. In melanoma patients treated with checkpoint inhibitor therapy, only 40% respond.6 One explanation for the failure of immunotherapy is that a large portion of non-responders intrinsically lack T cells specific to the peptides presented by the tumor. This is likely the case in breast cancer where tumors harbor low mutational burden, have decreased infiltration of T cells into the tumor, and patients in general see poor responses to immunotherapy.7 Because of this, methods for augmenting antigen-specific CTL responses against metastatic breast cancer are needed.
For T cell studies such as these, representative models are required. The Mouse Mammary Tumor Virus-Polyoma Middle T (MMTV-PyMT) transgenic mouse (The Jackson Laboratory, stock # 002374) is a widely used model for metastatic breast cancer because tumors arise with high penetrance, exhibit clinically relevant histology, activate relevant oncogenic signaling pathways, and spontaneously metastasize to lungs (one of the most common sites of human breast cancer metastasis).8 In addition to MMTV-PyMT, several other clinically relevant models of breast cancer are found on the FVB/NJ genetic background.9–11 Unfortunately, studying tumor immunology in FVB mice is difficult due to lack of characterization of the MHC class I alleles in this H-2q haplotype strain. Backcrossing to the C57/BL6 background (The Jackson Laboratory, stock # 000664), which has well characterized MHC alleles, significantly reduces tumor penetrance and almost eliminates metastasis.12 Therefore, the FVB/NJ strain is required. In order to enable the study of tumor immunity in this mouse model, we recently defined the MHC class I alleles of the FVB/NJ strain, characterized their peptide binding properties, and developed the NetH2pan prediction tool.13 Here, we use these new tools to predict immunogenic epitopes in MMTV-PyMT mice with high fidelity. We then validate these tumor antigens via immune-proteomic analysis of primary tumors, test a DNA vaccine that successfully abolishes MMTV-PyMT tumor growth, and identify key populations of antigen-specific CD8 + T cells associated with anti-tumor immunity. This study provides an enhanced method for tracking tumor-specific T cells in a FVB mouse model of metastatic breast cancer.
Materials & methods
Cell lines, PyMT transfection, and production of soluble MHC for elution studies
HeLa cells were purchased from the American Type Culture Collection (ATCC), and cultured according to ATCC protocol in DMEM-F12K (Wisent) with 10% fetal bovine serum (Serum Source International). Routine authentication of cultured cells was completed with sequence-based HLA-typing. All cells were maintained at 37°C in a 5% CO2 incubator. Soluble MHC (sMHC) constructs were generated with a truncation at the junction of the trans-membrane and cytoplasmic domains with the addition of a VLDLr purification tag and cloned into pcDNA3.1(-).14 HeLa cells were stably transfected with the sMHC by nucleofection (Lonza), followed by G418 drug selection and subcloning. sMHC-producing clones were identified using a capture ELISA with anti-VLDLr as the capture antibody and an anti-β2-microglobulin (DAKO) as a detection antibody. To co-express PyMT, sMHC-producing clones were co-transfected with pcDNA3.1-Hygro-PyMT-FLAG, selected with hygromycin (Roche), and sub-cloned. PyMT protein expression was confirmed via flow cytometry with an anti-FLAG tag antibody (Biolegend, clone L5). PyMT and sMHC co-transfected cells were seeded into 48 roller bottles and sMHC-containing supernatant was collected within one week. sMHC was purified from the supernatant using affinity chromatography with an anti-VLDLr antibody on CNBr-activated Sepharose 4 Fast Flow Matrix (GE Healthcare). Complexes were eluted from the column in 0.2 N acetic acid and immediately processed for isolation of the peptide ligands.
H-2Dq-PyMT peptides
Peptide ligands were eluted as described previously.14,15 Briefly, purified MHC was denatured with an acid boil and the peptides were separated from α- and β-chains by filtration using a Millipore 3-kDa molecular mass limit ultrafiltration membrane (Merck Millipore). Resultant peptides were separated into 39 fractions with off-line RP-HPLC with the mobile phase at pH 10. Peptide-containing fractions were analyzed individually with liquid chromatography mass spectrometry (LC-MS). Nano-scale liquid chromatography was performed with an Eksigent nano-LC-4000 with autosampler (Sciex). Fractions were combined with internal Retention Time (iRT) peptides (Biognosys), loaded on a C18 trap (350 μm [i.d.] × 0.5 mm long; ChromXP) and desalted before separation by gradient elution into a ChromXP C18 separation column (75 μm [i.d.] × 15 cm long). Column media consisted of 3-μm particles with 120-Å pores. The elution mobile phase consisted of two linear gradients using solvent A (98% water, 2% acetonitrile, 0.1% formic acid) and solvent B (95% acetonitrile, 5% water, 0.1% formic acid): 10–40% B for 70 min and then 40–80% for 10 min. Eluate was ionized with a NanoSpray III ion source (Sciex), and MS1 and MS2 fragments were obtained in Independent Data Acquisition (DDA) mode using a Sciex TripleTOF 5600 System, as described previously.15
Peptide sequences were obtained from fragment spectra using PEAKS 8.0 (Bioinformatics Solutions) at a precursor ion tolerance of 50 ppm and a product ion tolerance of 0.05 Da. For the H-2q produced in HeLa cells, the UniProt database with Homo sapiens taxonomy, iRT peptides, and the PyMT protein were used as a reference library for fragments. In the case of the mouse tumor samples, the database search was the UniProt Mus musculus taxonomy proteome, internal Retention Time (iRT, Biognosys) peptides, and the PyMT protein. Variable post-translational modifications analyzed included acetylation, deamination, pyroglutamate formation, oxidation, sodium adducts, phosphorylation, and cysteinylation. The identified peptides were synthesized with >95% purity (Atlantic Peptides) and analyzed with the same LC/MS method. Synthetic and eluted peptides were matched based on retention times, precursor ion m/z, b/y fragment ions, and normalized (%) signal intensity in the software PeakView (Sciex).
Peptide identification from MMTV-PyMT tumors
Anti-H-2q hybridoma and antibody purification
Anti-H-2q (28-14-8S and 34-1-2S, ATCC) hybridomas were grown in serum free media and purified with Protein G Sepharose 4 Fast Flow columns (GE Healthcare, Sweden). Immunoaffinity columns were generated by coupling the purified antibodies to CNBR-activated Sepharose 4 Fast Flow (GE Healthcare, Sweden). Column affinity for H2 was tested with soluble H-2Dq (28-14-8S column) or soluble H-2Kb (34-1-2S column) monomers, kindly provided by the NIH Tetramer Core (Emory University, Atlanta, GA).
Peptide extraction and 2-dimensional LC/MS identification
MHC-peptide complexes were extracted from tumors based on a previously published protocol.16 Whole tumors were flash frozen in liquid nitrogen, cryogenically milled (MM400, RETSCH), and suspended in lysis buffer containing octylphenoxy poly(ethyleneoxy)ethanol (IGEPAL) (Sigma) and cOmplete EDTA-free protease inhibitor cocktails (Roche). Lysates were rocked at 4ºC for 1 hour and clarified by ultracentrifugation at 100,000xg for 90 minutes. Filtered supernatant was passed twice over a protein A (Sigma) pre-column then sequential H-2Dq and -Kq columns. Columns were washed sequentially with buffers at pH 8.0: lysis buffer containing 5mM EDTA, 50mM Tris 150mM NaCl, 50mM Tris 450mM NaCl, and 50mM Tris in HPLC-grade water (JT Baker). After column washes, peptides were eluted in 0.2 N acid, resuspended to 10% acetic acid, and heated to 76ºC for 10ʹ. Peptides were collected in 1ʹ fractions and analyzed by LC/MS using DDA for de novo peptide identification in the manner described above. H-2q extraction was confirmed by tryptic digest and LC/MS analysis.
For high sensitivity PyMT peptide discovery of the H-2Dq peptides, Data Independent Acquisition(DIA)/SWATH spectra were collected on the LC/MS. The SWATH method cycle consisted of 1 250msec MS1 scan (300-1500m/z) followed by 25, 120msec SWATH scans with windows of 20m/z, overlapping by 1m/z spanning the range of 350-850m/z, giving at total cycle time of 3.0 sec. A fragment spectra library generated with DDA spectra of synthetic PyMT peptides, eluted H-2Dq ligands, and iRT peptides was built with Skyline 3.6 (MacCoss Lab Software) for peptide queries of SWATH spectra.
Peptides
A PyMT peptide library of 15mers (each overlapping by 12 amino acids) was synthesized (ThermoFisher, Waltham, MA) and resuspended in DMSO as 20mg/mL stocks. Peptides were grouped into 14 pools of 10, such that the final concentration of each peptide was 5ug/mL. Specific 15mer peptides within each pool were delineated with a second round of ELISPOTs at 20ug/mL per peptide. PyMT peptides (MT19-27:LPRQLWGDF, MT107-115:MPLTCLVNV, MT241-250: LPSLLSNPTY, MT288-296: YPRTPPELL, MT326-335:LPGEQVPQLI) were synthesized by Atlantic Peptides (Lewisburg, PA) at 95% purity and used at a concentration of 10ug/mL for restimulation assays and to generate tetramers. The synthetic PyMT peptides used to build the SWATH ligand library were synthesized by ThermoFisher (Waltham, MA) at >60% purity.
Vaccine plasmids
The intracellular domain of PyMT (corresponding to amino acids 1–394) and mGM-CSF were cloned into separate pVax1 plasmids (Life Technologies) using restriction digest by EcoRV and HindIII (New England Biolabs)and ligation with T4 DNA ligase (NEB). Plasmid constructs werevalidated by DNA sequencing. pVax1-PyMT and -mGM-CSF plasmids were produced in Top10 cells (Life Technologies) and purified using the PureYield MaxiPrep System (Promega). To confirm protein expression of pVax1-PyMT and pVax1-mGM-CSF in mammalian cells, 293T cells (ATCC) were transiently transfected with Lipofectamine 2000 (Life Technologies) and cell lysates were analyzed by Western blot 24 hours later. Antibodies used include: anti-PyMT (Abcam, ab15085), anti-mGM-CSF (R&D systems, MAB415), and HRP goat anti-rat (Jackson Immunoresearch). Cells were lysed with RIPA buffer containing protease inhibitor cocktails (Roche) then pelleted for protein quantification. Since mGM-CSF is a secreted protein, transfected 293T cells were treated ± the cellular transport inhibitor GolgiPlug (BD Biosciences) for 4 hours prior to analysis.
Animal husbandry
Female 5-8-week-old FVB/NJ (stock #1800) and MMTV-PyMT mice (stock # 002374) were purchased from Jackson Laboratories. Animal husbandry was in accordance with institutional guidelines at the University of Utah, and all studies were approved by the University of Utah IACUC committee.
Vaccination and syngeneic tumor transplants
Six-to-eight-week-old female FVB/NJ mice (n = 10 per group for epitope mapping, n = 5 per group for tumor studies) were vaccinated with pVax1-PyMT (70ug) plus adjuvant pVax1-mGM-CSF (20ug). As controls, a molar equivalent of pVax1 empty vector (50ug) and pVax1-mGM-CSF (20ug) were administered. Plasmids were suspended in HBSS in 100uL of volume, and 50uL was administered via bilateral intramuscular injections. Primary vaccination and 2 boosters were each administered 2 weeks apart. For anti-tumor vaccine experiments, 250,000 MMTV-PyMT tumor cells were implanted into cleared mammary fat pads of PyMT or control vaccinated mice two weeks after the final vaccine booster. Tumors were measured weekly and size was calculated using length and width caliper measurements with an ellipsoid formula (Tumor volume = 1/2(length × width2)). Mice were euthanized when tumors reached a maximum of 2 cm3. Whole blood was harvested by cardiac puncture of euthanized mice into 10:1 anticoagulant citrate dextrose solution and centrifuged for 5ʹ at 400xg. The plasma was isolated and frozen at −20ºC for antibody quantification. Splenocytes were collected by homogenizing between frosted glass slides (VWR) and passing through 100µm cell strainers (Corning). Red blood cells were lysed with tris-buffered ammonium chloride buffer pH 7.4 (Sigma) for 3 minutes, and splenocytes were then washed and resuspended in RPMI +10% FBS. CD8 + T cells were negatively sorted with magnetic beads conjugated with antibodies against CD4, CD11b, CD11c, CD19, CD45R (B220), CD49b (DX5), CD105, MHC-class II, Ter-119 and TCRγ/δ (Miltenyi Biotec). Sorted T cells were titrated back into CD8-negative flow-through sample for ELISPOT experiments.
PyMT protein purification
PyMT plasmids were a gift from Dr. Bryan Welm (University of Utah). The PyMT DNA was cloned using Gateway Cloning (ThermoFisher Scientific) technology into a pDEST-10 expression vector containing a poly-His tag. The protein was produced in E. Coli and affinity purified with a nickel column (ThermoFisher Scientific). Finally, PyMT protein purity was confirmed with Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis and Coomassie staining.
Anti-PyMT antibody ELISA
Ninety-six well plates (Cat#: L55XA, Meso Scale Discovery (MSD)) were coated with 20 ng/mL of PyMT protein overnight at 4ºC, then blocked the next day in 5% blocking buffer A at room temperature for 1 hour on a shaker. Plates were washed with Tris-buffered saline 0.05% Tween-20 (TBST) (Sigma) then incubated with Goat Anti-Mouse Sulfo Tag (Cat#: R32AC, MSD) combined with vaccinated mouse plasma (diluted 1:100). Following a 2 hour incubation, the plates were washed again with TBST and a reading buffer was added. Plates were read with the QuickPlex Instrument (MSD).
ELISPOT assays
Mouse IFNγ ELISPOT assays were performed using the Ready-SET-Go! kit (eBioscience) on Multiscreen HTSIP Filter Plates (EMD Millipore). Briefly, plates were coated with anti-IFNγ capture antibody overnight and then blocked for 2 hours at 37°C with RPMI in 20% FBS. Fifty thousand negatively sorted CD8 + T cells were combined with 150,000 CD8- cells (flow-through) from vaccinated mice. Cells were incubated at 37°C for 24 hours with 5ug/mL of pooled peptides or 10 ug/mL of a single peptide. Positive control wells stimulated with phorbol 12-myristate 13-acetate (PMA) (25ng/mL) and Ionomycin (500ug/mL) (Sigma) and negative control DMSO-only (Sigma) wells were included on each plate. After overnight stimulation, the wells were decanted and incubated with biotinylated anti-mouse IFNγ primary antibody for 2 hours at 37°C, followed by Streptavidin-HRP antibody for 45 minutes. Spots were developed using 3-Amino-9-ethylcarbazole chromogen (Sigma) and washed extensively with tap water. After drying, plates were imaged and counted with the ImmunoSpot Analyzer (CTL, Cleveland, OH).
Tetramer staining
PyMT peptides (MT107-115, MT241-250, MT288-296) were folded into H-2Dq tetramers with human β2m and conjugated to PE. Tetramers were kindly provided by the NIH Tetramer Core Facility (Emory University, Atlanta, GA). Splenocytes from PyMT-vaccinated mice or MMTV-PyMT tumor bearing mice were harvested as described above. One million cells were stained with fixable viability dye (eF780, eBiosciences) in PBS for 30 minutes at 4ºC. Cells were washed and incubated with tetramers (1:100 dilution) for 30ʹ at room temperature, then incubated for 30ʹ at 4ºC with the following antibodies (eBiosciences): anti-mouse Fc block, anti-CD8α (eF450, clone 53–6.7), CD3ϵ (FITC, clone 145-2C11), CD62L (APC, clone MEL-14), CD44 (PE-Cy7, clone IM7). Additionally, for compensation in the tetramer+ PE channel, anti-CD8α PE was used. Flow cytometry data was acquired with a 3-laser S1200Ex flow cytometer (Stratedigm). Each experiment included single-stained compensation controls and fluorescence-minus one (FMO) staining for the PE channel. Compensation and data analysis were conducted with FlowJo v10 (Tree Star, Inc.).
Results
Class I MHC peptide prediction identifies potential PyMT peptides
The Polyomavirus Middle T Antigen (PyMT or MT) drives tumor initiation and metastasis in the MMTV-PyMT tumor model.8 Deriving from the murine Polyomavirus, the MT antigen is essential to tumor development during Polyomavirus infection. In this infectious setting, antigen-specific T cells that recognize the MT antigen promoted tumor resistance in vivo, identifying PyMT as an important immunogenic target that can activate T cells.17 Antigen-specific T cell recognition of MMTV-PyMT tumors, more specifically the PyMT oncogene, has not been reported. This has, in large part, been due to poor characterization of the MHC H-2q haplotype, limiting the ability to study antigen-specific responses in the FVB/NJ strain on which the MMTV-PyMT mouse was developed and maintained. We recently characterized the FVB MHC class I molecules and generated a tool for predicting class I MHC epitopes.13 Here, we used this tool (NetH2pan) to predict PyMT peptides presented by the FVB class I MHC (H-2Dq), and proceeded to validate these epitopes with complementary approaches in vitro and in vivo. Table 1 shows the top 25 predicted H-2Dq PyMT epitopes sorted by % rank (a surrogate reading for binding affinity). As expected, these peptides contain a proline (P2) and a hydrophobic or aromatic anchor (P9) consistent with the H-2Dq motif.
Table 1.
NetH2pan PyMT peptide predictions. The PyMT protein amino acid sequence was entered into NetH2pan with the following search parameters: Peptide Length: 8-11mer peptides, Species/loci: Mouse (H-2), Allele: H-2Dq, and sorted by predicted affinity. Bold: eluted PyMT peptides from soluble MHC/tumors.
| Rank | Peptide | % Rank |
|---|---|---|
| 1 | VPQLIPPPI | 0.0148 |
| 2 | YPRTPPELL | 0.0420 |
| 3 | LPRQLWGDF | 0.0472 |
| 4 | LPSLLSNPTY | 0.0557 |
| 5 | MPMEDLYLDI | 0.1372 |
| 6 | VPQLIPPPII | 0.2031 |
| 7 | IPRAGLSPW | 0.2755 |
| 8 | TPTRDVLNL | 0.3715 |
| 9 | RLPSLLSNPTY | 0.4070 |
| 10 | LPSLLSNPT | 0.4477 |
| 11 | YPRTPPEL | 0.4783 |
| 12 | QVPQLIPPPI | 0.4825 |
| 13 | HPHILLEEDEI | 0.5053 |
| 14 | LPSLLSNPTYS | 0.5601 |
| 15 | LGRTLLLVTF | 0.6029 |
| 16 | MPLTCLVNV | 0.6511 |
| 17 | LAALLGICL | 0.6614 |
| 18 | YMPMEDLYLDI | 0.6718 |
| 19 | FIASMPIDW | 0.7104 |
| 20 | YPRTPPELLY | 0.7917 |
| 21 | HPDKGGSHAL | 0.7939 |
| 22 | LKLPRQLWGDF | 0.8135 |
| 23 | MPMEDLYLDIL | 0.8690 |
| 24 | LPGEQVPQLI | 0.8777 |
| 25 | IPPPIIPRAGL | 0.8827 |
Direct PyMT peptide elution from soluble H-2Dq uncovers two PyMT-specific peptides
To validate the PyMT predictions, we adopted an approach to directly elute PyMT-specific peptides using an in vitro sMHC transfection system. sMHC provides a powerful way to identify intracellular viral peptides and tumor neoantigens in vitro.15,18–21 Previous data showed that sMHC-bound peptides correlated extremely well with cell surface-presented MHCI peptides, making sMHC a robust system for PyMT neoantigen discovery.22 Engineered plasmids encoding the FVB/NJ sMHCI allele (H-2Dq) and FLAG-tagged PyMT were expressed in HeLa cells. Because murine MHCI α-chain binds human β-2-microglobulin more stably than murine β-2-microglobulin, a human cell line for murine sMHC production was chosen.23 Single-cell clones of high producing HeLa-H-2Dq-PyMT cells were identified by a sMHC ELISA of cell supernatant and fluorescently sorted for PyMT protein expression (Figure 1a,b). Following expansion of the double-transfected cells, sMHC:peptide complexes were immunoaffinity purified, and peptides were isolated using acetic acid and heat. LC/MS analysis of peptides identified 8,424 H-2Dq peptides with characteristic peptide lengths and binding motifs (Figure 1c,d). Most peptides were sampled 1–2 times from a single source protein (Figure 1e). Source protein analysis of these peptides revealed that high-turnover proteins involved in metabolic and cellular processes were more highly favored for MHCI presentation (Supplemental Figure 1).
Figure 1.
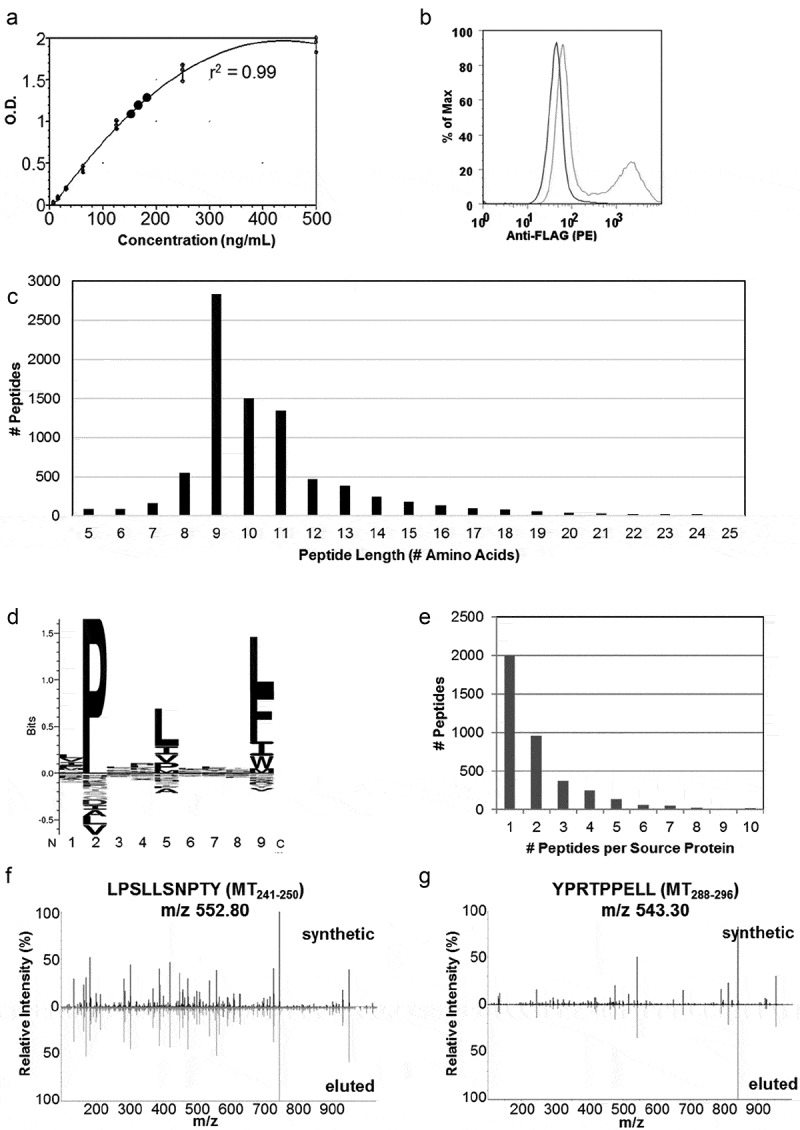
Soluble MHC elution matches H-2Dq profile and identifies H-2Dq PyMT-peptides . a) ELISA results of sMHC production from HeLa cells plotted against a standard curve of purified VLDL-tagged sMHC. b) Evaluation of PyMT expression using flow cytometry in HeLa-H-2Dq cells. An N-terminal flag-tag was engineered onto PyMT and an anti-FLAG antibody was used for staining. c) Peptide length distribution, d) Binding motif generated with Seq2Logo, e) Source protein distribution of eluted peptides, c-e) represent data from 8424 peptides eluted from soluble H-2Dq and PyMT co-transfected HeLa cells, f,g) MS2 fragment spectra from DDA acquisition containing b/y ions from synthetic MT241-250 and MT288-296 and sMHC-eluted peptides.
Peptide extraction from soluble H-2Dq yielded two PyMT-derived peptides (MT241-250 LPSLLSNPTY and MT 288–296 YPRTPPELL). The PyMT peptides were identified using de novo peptide discovery then searched against a Uniprot protein sequence database in PEAKS 8.0. Synthetic peptides matched the retention times and b/y ions of the eluted peptides, confirming that the solved PyMT peptide sequences were correct (Figure 1f,g, Supplemental Table 1). Based on the peptide elution data (Figure 1e), it was expected that only 1–2 peptides were chosen for presentation from each source protein, as was the case for PyMT. The PyMT peptides identified by sMHC elution ranked second and fourth in the NetH2pan prediction list and exhibited strong binding affinities (Table 1). This finding agreed with our previous observation that NetH2pan successfully predicted presented peptides within the top 38 theoretical binders.13 Importantly, the strongest predicted binder was not always presented by a cell since MHC class I peptide presentation is the culmination of proteosomal cleavage, peptide trimming, and trafficking to the cell surface. Variations at any point of peptide processing influences the peptides presented, emphasizing the importance of direct peptide elution from tissues for validation.
The identification of two putative MHCI PyMT peptides in vitro strongly indicated that these same PyMT peptides would also be present in complex with MHCI of MMTV-PyMT tumors in vivo. While many reports of MHCI downregulation exist, we previously conducted flow cytometric analysis of EPCAM+ MMTV-PyMT tumor cells and established comparable MHCI expression on tumors and naïve splenocytes.13 Therefore, we proceeded to elute MHCI:peptide complexes directly from tumors for the identification of PyMT tumor antigens.
MHC class I molecules on MMTV-PyMT tumor cells present PyMT peptides
Pooled spontaneous MMTV-PyMT tumors provided sufficient tissue for class I MHC pull-down followed by peptide extraction and proteomic identification. Cryogenically-milled tumors were lysed, clarified by centrifugation, and peptide:MHC complexes were immunoaffinity purified with two anti-H-2q antibodies (Figure 2a). Tryptic digest and MS/MS of the heavy chain-containing HPLC fractions confirmed successful purification of H-2Dq (raw .wiff files available upon request). Peptides identified from the primary tumors by 2D-LC/MS yielded four PyMT-specific peptides MT19-27, MT107-115 MT288-296, and MT326-335 (Figure 2b). This included one of the peptides, MT288-296, previously identified using the sMHC approach. The sequences of each PyMT-specific peptide were validated by matching the LC-retention time and MS-fragmentation patterns to their corresponding synthetic peptides (Figure 2b, Supplemental Table 1).
Figure 2.
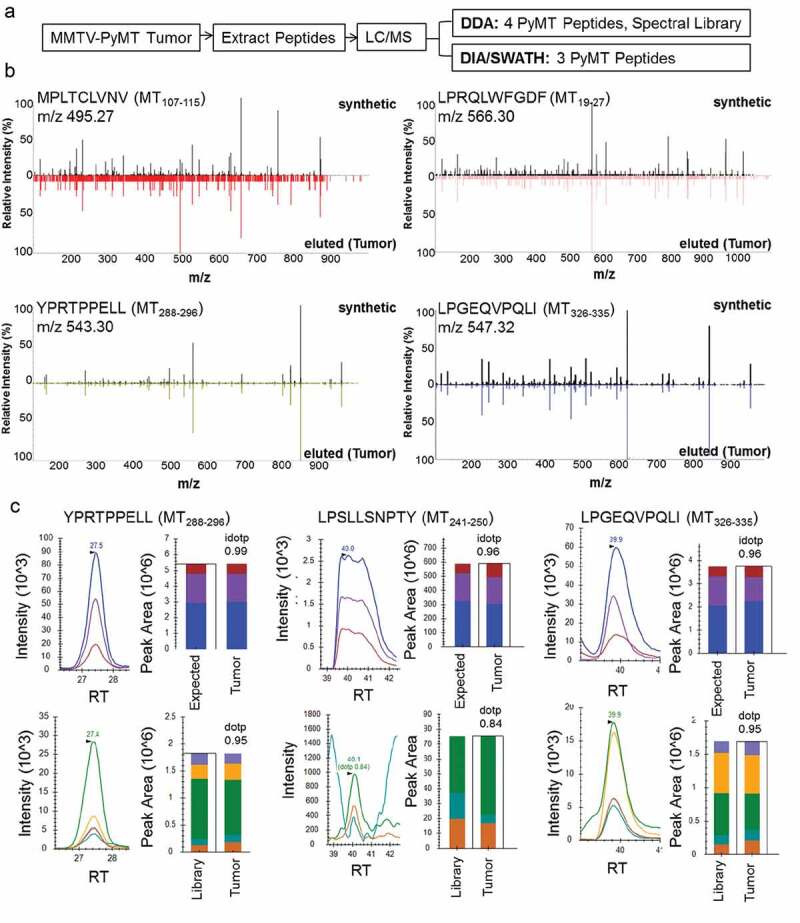
MMTV-PyMT tumors present PyMT-specific class I MHC peptides. a) Experimental approach to identify peptides directly from tumors. Peptides were extracted and identified using two types of data acquisition. DDA = Data Dependent Acquisition, DIA/SWATH = Data Independent Acquisition. b) MS2 spectra from DDA acquisition: b/y ions of PyMT peptides eluted from tumors vs. high-purity synthetic peptides. c) SWATH acquisition data matched to PyMT synthetics using Skyline Software. Peaks were matched by retention times and peak area. Top panel: precursor, [M + 1], [M + 2] compared to theoretical isotopic distribution, bottom panel: b/y ions compared to library. Analysis of idotp/dotp show close agreement to expected/library values. RT = Retention Time.
One PyMT peptide discovered on sMHC was not readily detected on tumors using the MHC pull-down:DDA proteomics approach (MT241-250), prompting us to explore a more sensitive method for epitope discovery. We utilized a Data Independent Acquisition (DIA) method, known as SWATH (Figure 2a). DIA or SWATH acquisition allows all precursors to be fragmented within preset retention time and mass-to-charge windows, eliminating bias for highly abundant peptides.24 While this approach is sensitive, it requires the use of a fragment spectrum library built from DDA fragment spectra of reference peptides. Here, we generated a synthetic fragment spectra library of the top 25 predicted PyMT peptides for interrogation of SWATH data (Table 1). The successful matching of a SWATH-acquired peptide to its library spectra is denoted in the “idot product” or “dot product”, which results from comparing the MS1 isotopes (precursor [M + 2H], [M + 1 + 2H], [M + 2 + 2H]) and MS2 (b/y fragment ion) relative peak intensities of the observed spectra with the library reference.
The application of SWATH proteomic data acquisition identified an additional PyMT peptide (MT241-250), along with MT288-296 and MT326-335, both of which had initially been identified using sMHC and tumor-eluted DDA data (Figure 2c). Notably, MT241-250 had a co-eluting isobaric peptide that caused ion suppression of this PyMT peptide, as evidenced by the saturated high-intensity MS1 peaks and the low intensity of the MS2 peaks (Figure 2c). Based on this observation, identification of MT241-250 using DDA would have proven challenging due to the presence of this additional peptide. With SWATH, MT241-250 could indeed be detected on the tumor surface. Although we focused on PyMT peptide identification, the SWATH dataset is available and can be revisited once new peptides of interest arise.24 In total, the methods of targeted neoantigen discovery shown here provide five potential T cell targets of PyMT.
T cells specific for PyMT peptides (MT19-27, MT107-115, MT241-250 and MT280-296) are confirmed using a PyMT DNA vaccine
Predictive algorithms and direct peptide discovery can provide many targets of interest, but the ability for a TCR to recognize a peptide on the MHC and become activated remains the primary bottleneck of neoantigen discovery. The unique ability of an individual’s T cells to perceive an MHCI peptide as foreign and mount a subsequent CTL response is subject to rigorous selection within the developing thymus and varies between even inbred mice. As such, validation of epitopes requires screening using vaccines or immunotherapy in vivo. We opted to generate antigen-specific T cells by vaccinating mice with a truncated PyMT DNA sequence followed by immunogenicity testing of the eluted PyMT peptides. Limiting the PyMT DNA sequence to the cytoplasmic domain minimized the theoretical risk of oncogenic transformation with full-length PyMT DNA introduction. Several iterations of vaccines and adjuvants were evaluated for their ability to generate a T cell response and inhibit tumor growth. Based on our preliminary experiments, we chose a plasmid DNA vaccine encoding PyMT and a separate plasmid encoding granulocyte-monocyte-colony stimulating factor (GM-CSF) as an adjuvant. GM-CSF is known to promote type 1 helper T cells (TH1) responses and is currently utilized as an adjuvant in HER2 breast vaccines.5 Transient transfection of the vaccine plasmids into mammalian 293T cells confirmed PyMT protein expression and GM-CSF production and secretion (Figure 3a). Plasmid vaccination of mice was performed, and splenocytes were collected two weeks after the final booster (Figure 3b).
Figure 4.
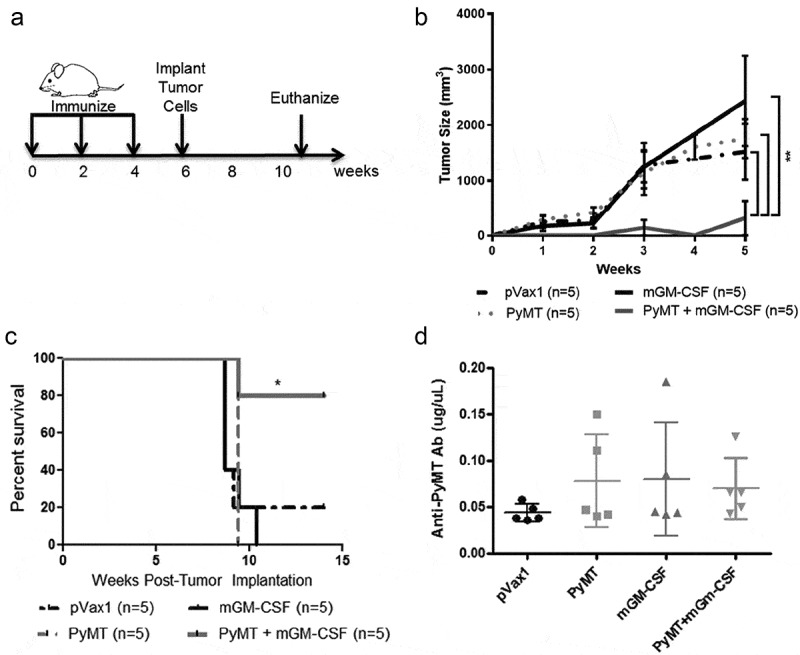
PyMT DNA vaccination prevented MMTV-PyMT tumor growth.
a) Prime/boost and tumor implantation timeline: mice were immunized with PyMT+GM-CSF (or control plasmid + GM-CSF) and boosted every 2 weeks, 250,000 MMTV-PyMT tumor cells were implanted at 6 weeks, and animals euthanized at 10 weeks or when tumors reached 2cm3, b) Tumor size measurements (0 = time of implantation) reveal PyMT+GM-CSF vaccine prevents tumor growth (**p < .005, linear regression analysis), c) Survival curve shows vaccine promotes survival up to 15 weeks (*p < .05, log-rank test), d) ELISA for PyMT-specific antibodies in the plasma of vaccinated mice one day after the 1st booster (n = 5).
As a complementary method to identify any immunogenic regions of the PyMT protein omitted by NetH2pan or elution approaches, we synthesized overlapping 15mers that spanned the entire length of the PyMT protein (Figure 3c, Supplemental Data). Epitope mapping with pools of ten peptides allowed rapid screening for immunogenic regions of PyMT. Anti-IFNγ ELISPOT cultures were enriched for CD8 + T cells by negative magnetic sorting, and re-combined with CD8-negative flow through splenocytes. Overlapping 15mers contained the entire PyMT protein sequence within discrete pools. In the first round of testing, splenocytes were re-stimulated ex vivo with pools of PyMT 15mers, revealing T cell responses to four pools (Figure 3d). The degree of response in each pool correlated to that obtained with the synthetic minimal epitopes (Figure 3e, Supplemental Figure 2) confirming that direct peptide elution from sMHC and tumors accurately identified PyMT-specific T cell epitopes. Surprisingly, although MT326-335 was eluted from tumors, it failed to elicit a positive ELISPOT response with overlapping 15mers or the minimal epitope. Thus, the minimal epitopes shown only include the first four PyMT peptides. Compared to the other PyMT peptides, MT107-115 induced a higher quantity response, evidenced in the greater number of spot-producing T cells and total responders per group (Figure 3e). A cumulative list of PyMT peptides and the method(s) used for their identification are listed in Table 2.
Table 2.
Summary of PyMT peptides: ELISPOT responses and complementary approaches used for each peptide identification; NetH2pan rank corresponds to “Rank%” from NetH2pan H-2Dq output.
 |
DNA vaccination with PyMT and GM-CSF prevents tumor growth
The complementary approaches to discover immunogenic PyMT peptides using sMHC, overlapping 15mer peptide pools, and direct elution from tumors converged upon five putative antigenic epitopes for MMTV-PyMT tumors. We hypothesized that PyMT vaccination would prime the host immune system to relevant immunogenic epitopes on MMTV-PyMT tumors and induce tumor rejection when challenged in vivo, validating PyMT-specific T cell responses. To test this, PyMT DNA was used in a clinically relevant setting as an anti-tumor vaccine. Mice received intramuscular vaccines and boosters, then underwent implantation of syngeneic MMTV-PyMT tumor cells into the mammary fat pad (Figure 4a). DNA vaccination prevented tumor growth only in the PyMT + GM-CSF vaccinated group (Figure 4b,c). Humoral IgG responses were assessed using an anti-PyMT antibody ELISA with a commercial mouse anti-PyMT antibody as a standard curve. Despite marked protection from tumor growth, the IgG responses against PyMT following DNA vaccination did not increase significantly from baseline (Figure 4d). These data show that vaccination effectively protected animals from PyMT-induced mammary tumorigenesis, despite the inability of a DNA vaccine to induce elevated antibody responses in this model. Importantly, the lack of a PyMT-specific antibody response does not preclude humoral involvement in anti-tumor immunity. However, given the immunogenicity of PyMT Class I peptides,we posit that vaccine-based protection from tumors in this model is through cellular immunity.
Figure 3.
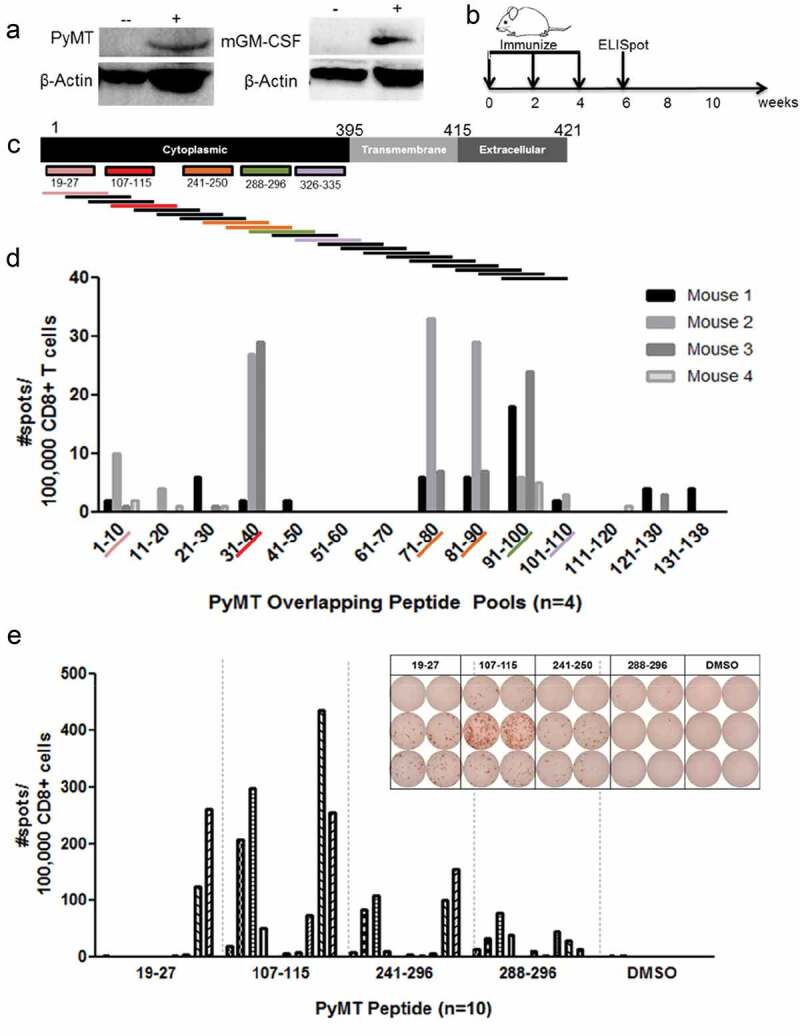
DNA vaccination confirms H-2Dq PyMT epitope immunogenicity: a) PyMT and GM-CSF western blot following transient transfection of 293T cells with DNA plasmid vaccine: (-) untransfected 293T, (+) transfected 293T. GM-CSF transfected cells were treated with cellular transport inhibitor for 4 hours prior to analysis, b) DNA vaccine timeline, c) Overlapping 15mer design. MT19–27: pool 1 (#1-10), MT107–115: pool 4 (#31-40), MT241–250: pool 8 (#71-80) and 9 (#81-90), MT288–296: pool 10 (#91-100), MT326–335: pool 11(#101-110), d) Anti-IFNγ ELISPOT following 24 hour overlapping peptide restimulation of splenocytes from vaccinated mice, e) Anti-IFNγ responses with representative ELISPOT picture to minimal epitopes of PyMT H-2Dq peptides following 24 hour peptide stimulation of splenocytes from vaccinated mice. Each row represents one mouse; columns represent duplicates of peptide or DMSO-only restimulation.
PyMT DNA vaccine produces antigen-specific T cells
Having established that PyMT DNA vaccination successfully blocks tumor growth, we designed PyMT-H-2Dq tetramers to evaluate antigen specific CD8 + T cells following vaccination. Three top-responding PyMT peptides (MT107-115, MT241-250, MT288-296) were folded into H-2Dq tetramers. Tetramer staining of splenocytes (gating strategy in Supplemental Figure 3) shows 3-5% of CD8 + T cells in vaccinated mice recognize each antigen (Figure 5a,b). MMTV-PyMT tumor-bearing FVB mice had sub-detectable antigen-specific CD8 + T cells despite clear evidence of PyMT antigen presentation in pull-down data, a finding that is consistent with global immunosuppression seen in cancer (Figure 5a,b).
Figure 5.
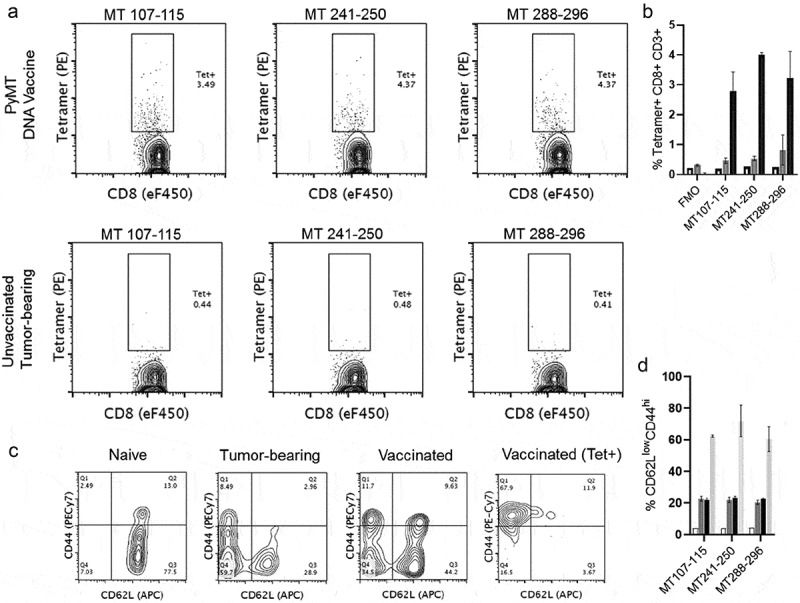
PyMT-Dq tetramers label antigen-specific CD8 + T cells after vaccination. a) Top Row) Naïve FVB mice were vaccinated and boosted twice with PyMT + mGM-CSF DNA plasmids. Two weeks after the final booster, splenocytes were harvested and analyzed with flow cytometry. Bottom Row) Naïve FVB mice received mammary fat pad transplants of 250,000 MMTV-PyMT tumor cells. Once tumors reached 2cm3, mice were euthanized and splenocytes were stained with tetramers for PyMT-specific T cells. Left-to-Right) H-2Dq tetramers in separate staining panels: MT107-115, MT241-250, and MT288-296. These data have previously been gated on viability/CD3+/CD8α+. b) Quantification of tetramer staining, c) Phenotype of CD8 + T cells in naïve, tumor-bearing, vaccinated, or antigen-specific (vaccinated) settings. Each of the panels were gated on viability/CD3+/CD8 α +, with an additional gate for tetramer+ staining. d) Quantification of CD62LlowCD44hi populations. Panels B&D groups (n = 2/group): Naïve Spleen (clear), Unvaccinated Tumor-bearing (dark gray), PyMT Vaccinated (black), PyMT Vaccinated>Tetramer+ (light gray).
Approximately two weeks after the antigenic stimulus, activated effector CTL populations will have undergone retraction. We next asked whether the tetramer-positive T cells at this timepoint would exhibit general markers of effector or memory T cells. T cell phenotypes can be interrogated by staining for surface markers CD44 and CD62L, particularly effector memory T cells (TEM CD62LlowCD44hi) and central memory T cells (TCM CD62LhiCD44hi). CD44 is an adhesion molecule that is upregulated upon activation. CD62L, L-selectin, is shed from T cells after activation, a process that enables T cell trafficking away from central lymphoid organs to the periphery. The combination of CD62LlowCD44hi can also mark a shift from naïve T cells to acute effector T cells. CD8 + T cells from vaccinated mice were stained with these markers and compared with CD8 + T cells from naïve and tumor-bearing mice (Figure 5c,d). PyMT DNA vaccination and elevated tumor burdens favored a shift to CD62LlowCD44hi cells (~10%) when compared with naïve unvaccinated mice. The tetramer-positive cells were enriched in the CD62LlowCD44hi quadrant, with ~60% of the cells exhibiting this phenotype. While further subset characterization of the antigen-specific T cells is necessary, the presence of these cell types is a promising start to identifying relevant T cell subsets and their role in tumor cell immunoediting during the elimination stage of cancer. Together, these data illustrate that PyMT DNA vaccination generated antigen-specific CD8 + T cells, prevented PyMT tumor growth, and enriched for activated, non-naïve memory T cells. These data also illustrate that our pipeline for tumor antigen discovery in a metastatic breast cancer model in FVB mice identified bona fide immunogenic targets in vivo.
Utility of PyMT epitopes extends to metastatic and therapeutic settings
Of paramount importance is the generalizability of PyMT-specific responses to settings beyond proof-of-concept prophylactic DNA vaccination. Prophylactic PyMT vaccination provided unambiguous identification of immunogenic peptides, but vaccination prior to tumor inception fails to encompass the complexity of immune tolerance that occurs during tumor growth. For these reasons, we deployed our PyMT-specific tools to settings where the host immune response has been potentially attenuated by the presence of primary PyMT tumors or metastases.
First, we applied the PyMT DNA vaccination as mono-therapy for tumor elimination after autologous transplant of tumor cells (Supplemental Figure 4A). Two rounds of therapeutic DNA vaccination were completed, but the tumors grew too rapidly for completion of a final vaccine booster (Supplemental Figure 4B-C). We found no difference in primary tumor growth between vaccinated and control groups (Supplemental Figure 4B-C). Furthermore, we found that PyMT-specific responses were undetectable in both syngeneic transplant settings and MMTV-PyMT transgenic settings (Figure 5, Supplemental Figure 4D). These data show that responses to validated immunodominant peptides become ineffective in the presence of established tumors, consistent with the well known phenomenon of suppressed tumor-specific immune responses in the established tumor setting. These negative findings, while disappointing, are consistent with previous cancer vaccine trials in breast cancer patients. Therapeutic vaccination alone for breast cancer immunotherapy has traditionally been met with mixed results, and most clinical trials are currently targetting multiple tumor-associated antigens or neoantigens in combination with other adjuvant chemotherapies.25 In this experimental model, the detection of T cell responses in the MMTV-PyMT transgenic mice, where T cells have been educated during development, would require significant immune stimuli to overcome tolerance to self. Therefore, we adopted a different form of immunotherapy to assess PyMT-specific T cell responses in the established tumor setting.
We employed intratumoral injection of N-dihydrogalactochitosan (GC), a novel water-soluble polysaccharide immunoadjuvant, as a method of augmenting immune responses against PyMT-tumor antigens. GC, especially in combination with photothermal therapy or ultrasound ablation, has been shown to stimulate broad host immune responses, likely to multiple tumor antigens, and eliminate epithelial-to-mesenchymal transition and other pro-invasive properties of breast cancer cells.26,27 Here, intratumoral GC provided a method of promoting multiple immune responses against tumor-associated antigens and neoantigens. MMTV-PyMT tumors were injected with GC or control solution and subsequently allowed to grow until an ethical endpoint. GC-treated tumors exhibited delayed tumor growth, prolonged survival, and splenocytes from these mice displayed positive ELISPOT responses to PyMT-peptides (Supplemental Figure 5A-E). Other non-PyMT tumor-associated antigens were also tested in this setting, but failed to elicit any immunogenicity (Supplemental File 4, Supplemental Figure 5E).13
As a final validation of PyMT-specific responses in the MMTV-PyMT mouse model, we evaluated PyMT tetramer staining in metastatic settings. Given that PyMT protein is the main oncogenic driver and is expressed within metastatic lesions, we expected conservation of its antigen presentation on metastases. We conducted tail-vein injections to seed tumor cells in a metastatic niche and subsequently conducted flow cytometric analysis of T cells in the spleen (Supplemental Figure 6A). Much like the previous data, PyMT-specific T cells were undetectable in tumor settings without another form of immunotherapy (Supplemental Figure 6B) and depletion of CD4 T cells greatly enhanced detection of antigen-specific CTLs using tetramers (Supplemental Figure 6A-B). These antigen-specific T cells also exhibited TEM-like (CD62LlowCD44hi) staining, consistent with populations detected following prophylactic PyMT DNA vaccination (Supplemental Figure 6C). Together, these findings demonstrate the utility of PyMT-specific tools for tracking T cell responses in the setting of established primary tumors and lung metastases, particularly in the presence of immune-activating therapies.
Discussion
We present a comprehensive approach to identifying T cell tumor antigen targets presented by class I MHC on tumors in FVB/NJ mice. The antigen of interest here is the PyMT protein as it is the driver oncogene responsible for tumorigenesis in the MMTV-PyMT metastatic breast cancer model. Direct peptide elution from sMHC and from cell-surface MHC of primary tumors identified five candidate PyMT neoantigen peptides. In the setting of DNA vaccination, epitope immunogenicity for four of the five tumor antigen peptides was confirmed using overlapping 15mers and synthetic minimal epitopes. Notably, the immunogenicity of these PyMT peptides differed among the mice receiving the vaccination. Despite being tested on congenic strains with identical genetic backgrounds, certain epitopes promoted T-cell responses in 20% of the recipients, while other epitopes generated responses in 40% of the mice. We have empirically observed similar variability to vaccination in both human and murine cohorts. Because of this, only the three most immunogenic PyMT peptides were chosen for tetramer synthesis and flow cytometric analysis. These immunogenic PyMT neoepitopes were accurately predicted by NetH2pan (rank order #2, 3, 4, 16, and 24), the online prediction tool trained for murine class I MHC peptide prediction. This demonstrates that not only does NetH2pan predict likely candidates for class I MHC presentation, but it also predicts likely targets for T cell recognition, a difficult hurdle to overcome for MHCI prediction algorithms. These data provide the first targeted proteomics approach to tumor-specific antigen identification from MMTV-PyMT tumors.
The work presented here provides a discovery tool that synergizes with in silico genomic approaches for neoantigen discovery. Recently, personalized cancer vaccine therapies have set a precedence for interrogation of whole genome sequencing (WGS) or whole exome sequencing (WES) data for the discovery of single-nucleotide variants (SNVs) in tumors.15 These approaches involve aligning sequencing data from tumors alongside that of healthy tissues, identifying SNVs unique to the tumor, interrogating these SNVs using HLA-binding prediction tools, and ultimately testing their immunogenicity with peptide vaccination and/or tetramer staining.28,29 Data generated from genomic sequencing ultimately converges upon HLA-peptide prediction tools, most of which are only validated for a handful of well-studied HLA-haplotypes.15,30 Such prediction tools can result in false-positive peptide predictions, requiring screening and proteomic confirmation of expression. In one study, of 962 identified mutations from WGS data, only 5% of SNVs emerged as likely candidates for peptide presentation, with one-third of these peptides actually generating an immune response.31 Therefore, in vivo validation of improved multi-epitope MHCI prediction tools such as NetH2pan provides a gateway for expanding Next-Generation-Sequencing(NGS)-neoantigen discovery to other murine models of cancer.
Traditionally, antigen-specific T cell responses have been difficult to study in breast cancer because the antigens are poorly characterized in available mouse models with high penetrance of breast cancer (FVB/NJ strain). Tumors are highly heterogeneous and considerable variation is present among different cells within a tumor.32 Furthermore, peptides presented by a tumor’s class I MHC undergo immunoediting as the tumor progresses, potentially downregulating the presentation of certain antigens.33,34 Therefore, a successful cancer vaccine must target driver mutations and essential oncogenes whose functions are crucial to tumor survival. The lack of tetramer staining in the tumor-bearing mice illustrated that the presence of a tumor is not sufficient to generate detectable antigen-specific T cells despite expression of PyMT peptides on cell-surface MHC. The requirement of a tumor-antigen vaccine was necessary to help educate CTLs against the tumor. In this way, the PyMT oncogene was an ideal candidate for such a proof-of-principle anti-tumor vaccination. The presence of four tumor-specific driver epitopes should facilitate antigen-specific tracking even if several of the epitopes are downregulated during tumorigenesis. Furthermore, residues NPTY of PyMT, contained C-terminally in the peptide LPSLLSNPTY, are required for association to Shc1, a chaperone important for Ras-MAPK signal transduction pathways in cell cycle progression and cell division.35 Since this PyMT peptide localized to a region required for cell transformation and cannot be eliminated, this peptide will serve as an excellent target for T cell recognition.
To date, only one H-2Dq tetramer has been reported and this is in the MMTV-Neu/FVB mouse.36,37 While NetH2pan prediction of H-2Dq peptides facilitated additional tumor neoantigen discovery in the MMTV-Neu model, several caveats are present in these primary mammary tumors. MMTV-Neu tumors take roughly 205 days to arise and require longer for metastatic disease, whereas MMTV-PyMT tumors are highly penetrant, exhibit metastasis, and are a ubiquitous clinical model for breast cancer.9,38 Therefore the development of tetramers for the MMTV-PyMT mouse provide invaluable tools for breast cancer research. Understandably, artifacts may arise since PyMT is a viral oncogene rather than a highly expressed self protein as in the case of MMTV-Neu or MMTV-Myc models. Still, an unambiguous tumor-specific antigen was required for preliminary studies and NetH2pan validation. Similarly, foreign antigens such as chicken ovalbumin or LCMV-NP or GP peptides are commonly employed as a means for T cell tracking, and we anticipate PyMT peptides to become as commonplace for those conducting immunology research in breast cancer.39–41
A major caveat of this study is that the MMTV-PyMT spontaneous breast cancer model remains, at its best, a murine model of breast cancer. The MMTV-PyMT tumor model is an excellent model of spontaneous tumor development and specifically mirrors human luminal B breast cancer.8,42,43 PyMT tumors reflect the cellular heterogeneity and histological stages of human luminal B breast cancer.43,44 Additionally, these tumors grow in an immune competent mouse and spontaneously metastasize to the lungs. Each of these important characteristics make the MMTV-PyMT mouse model a very useful tool for evaluating the stochastic dialogue between tumors and their microenvironment. Still, several limitations are present in this model compared to human disease. Although the PyMT antigen activates signaling pathways that are well known drivers in breast cancer, such as PI3K and MAPK pathways, PyMT itself is not present in the human disease.45,46 Another limitation of PyMT tumors is that they do not display the diverse breast cancer subtypes that, for example, can be captured by patient-derived xenograft models or primary human breast cancer tissue. Furthermore, the complexity of host MHC diversity is also minimal as PyMT tumors arise from inbred murine strains with identical MHC haplotypes. With these caveats in mind, we have previously shown that human breast cancers exhibit high levels of antigen presentation and that the methods used here for identification of PyMT epitopes can also be used to identify targets for human breast cancer.21,47 Therefore, the neoantigen approach described here is relevant to human breast cancer and extremely useful for studying one of the most commonly used murine models of the disease.
The central tenets of cancer immunoediting: elimination, equilibrium, and escape, have remained integral to the dogma of cancer immunotherapy development.33 As immunotherapy innovations occur, the evaluation of early stages of cancer immunoediting or metastatic burden poses particular challenges. Technological limitations succumb to small tumor sizes and sub-detectable T cell responses; yet these early stages of cancer immunoediting and metastatic cell seeding are where the therapeutic window of success may be the widest. Because of this, more sensitive tools for evaluating the immune response are crucial as cancer immunotherapy is now moving from end-stage, “hail-mary” treatments to precision-based, prophylactic ones. Provided here is a method that allows cancer biologists to study the early stages of anti-tumor immunity in a cancer that is particularly resilient to immunotherapy. We presented a method to generate and track antigen-specific T cells against a driver oncogene that is theoretically present from the very inception of the tumor.
DNA vaccination successfully prevented MMTV-PyMT tumor development in FVB/NJ mice, completely abrogating tumor growth. This effective vaccine response to newly identified PyMT tumor antigens represents a considerable milestone in the MMTV-PyMT/FVB metastatic model of breast cancer, allowing the integration of a suite of immunologic tools to identify key neoantigen targets. These tools can be leveraged to address a cluster of key issues: the phenotype of CD8 + T cells that confer protection, their status as TCM/TEM/TRM, the immunodominance hierarchy of PyMT neoepitopes, the number of CD8 + T cells that must be adoptively transferred to eliminate tumors, how these T cells become exhausted or tolerant to the tumor, the role of CTLs in metastasis, and other poorly defined factors. For example, our preliminary tetramer staining and flow cytometry analysis suggest that antigen-specific T cells exhibit a TEM phenotype (CD62LlowCD44hi), a finding that coincides with the observation that TEM cells confer anti-tumor immunity.48,49 Additionally, the stochastic toggling of TCM to TEM to TRM in the setting of tumor immunity can be studied. One can also envision the use of additional cell surface markers and high-throughput single cell RNA-sequencing to characterize how PyMT-restricted T cell subsets confer protection from primary tumors and metastases. Armed with these tools in a spontaneous, metastatic cancer model, cancer biologists will be finally able to identify which particular T cells, be they tissue-resident, central or peripheral, memory or effector, are responsible for sustained elimination of tumor cells.
Funding Statement
This work was supported by the National Institute of Allergy and Infectious Diseases [T32AI007633];National Institutes of Health [CA205348];Oklahoma Center for the Advancement of Science and Technology [HR16-085];Susan G. Komen [SAC160078];U.S. Department of Defense [W81XWH-12-1-0499].
Acknowledgments
We would like to thank Dr. Lauren Zenewicz and Alisha Chitrakar for technical assistance with the tetramer staining, Sean Osborn and Steven Cate for providing sequencing services, and the Flow Cytometry Core facilities at OUHSC and University of Utah. This work was supported by funding from a Department of Defense Breast Cancer Research Program Innovator and Scholar Concept Award (W81XWH-12-1-0499 to ALW), a Susan G. Komen Leadership Award (SAC160078 to ALW), a NIH NRSA NIAID Training Grant (T32AI007633 to CID), a NIH R01 Grant (CA205348 to WRC),and the Oklahoma Center for Advancement of Science and Technology Grant (HR16-085 to WRC).
Declaration of Interest
W.H. Hildebrand is chief scientist at, has ownership interest in, and is a consultant/advisory board member for Pure MHC. W.R. Chen has ownership interest in and is a member of the Board of Directors for Immunophotonics, Inc. No potential conflicts of interest were disclosed by the other authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A. Breast cancer statistics, 2015: convergence of incidence rates between black and white women. CA Cancer J Clin. 2016;66(1):31–15. doi: 10.3322/caac.21320. [DOI] [PubMed] [Google Scholar]
- 2.Russnes HG, Navin N, Hicks J, Borresen-Dale A-L. Insight into the heterogeneity of breast cancer through next-generation sequencing. J Clin Invest. 2011;121(10):3810–3818. doi: 10.1172/JCI57088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher R, Pusztai L, Swanton C.. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013;108(3):479–485. doi: 10.1038/bjc.2012.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Disis ML, Gooley TA, Rinn K, Davis D, Piepkorn M, Cheever MA, Knutson KL, Schiffman K. Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide-based vaccines. J Clin Oncol. 2002;20(11):2624–2632. doi: 10.1200/JCO.2002.06.171. [DOI] [PubMed] [Google Scholar]
- 5.Mittendorf EA, Clifton GT, Holmes JP, Clive KS, Patil R, Benavides LC, Gates JD, Sears AK, Stojadinovic A, Ponniah S, et al. Clinical trial results of the HER-2/neu (E75) vaccine to prevent breast cancer recurrence in high-risk patients. Cancer. 2012;118(10):2594–2602. doi: 10.1002/cncr.26574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12(3):954–961. doi: 10.1128/MCB.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54(1):105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 10.Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49(4):465–475. doi: 10.1016/0092-8674(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 11.Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell. 1984;38(3):627–637. doi: 10.1016/0092-8674(84)90257-5. [DOI] [PubMed] [Google Scholar]
- 12.Davie SA, Maglione JE, Manner CK, Young D, Cardiff RD, MacLeod CL, Ellies LG. Effects of FVB/NJ and C57Bl/6J strain backgrounds on mammary tumor phenotype in inducible nitric oxide synthase deficient mice. Transgenic Res. 2007;16(2):193–201. doi: 10.1007/s11248-006-9056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeVette CI, Andreatta M, Bardet W, Cate SJ, Jurtz VI, Jackson KW, Welm AL, Nielsen M, Hildebrand WH. NetH2pan: A computational tool to guide MHC peptide prediction on murine tumors. Cancer Immunol Res. 2018;6(6):636–644. doi: 10.1158/2326-6066.CIR-17-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trolle T, McMurtrey CP, Sidney J, Bardet W, Osborn SC, Kaever T, Sette A, Hildebrand WH, Nielsen M, Peters B. The length distribution of class I-restricted T cell epitopes is determined by both peptide supply and MHC allele-specific binding preference. J Immunol. 2016;196:1480–1487. doi: 10.4049/jimmunol.1501721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purcell AW. Isolation and characterization of naturally processed MHC-bound peptides from the surface of antigen-presenting cells. In: Aguilar M-I, editor. HPLC of peptides and proteins: methods and protocols. Totowa (NJ): Springer New York; 2004. p. 291–306. [DOI] [PubMed] [Google Scholar]
- 17.Lukacher AE, Moser JM, Hadley A, Altman JD. Visualization of polyoma virus-specific CD8+ T cells in vivo during infection and tumor rejection. J Immunol. 1999;163(6):3369–3378. [PubMed] [Google Scholar]
- 18.Yaciuk JC, Skaley M, Bardet W, Schafer F, Mojsilovic D, Cate S, Stewart CJ, McMurtrey C, Jackson KW, Buchli R, et al. Direct interrogation of viral peptides presented by the class I HLA of HIV-Infected T cells. J Virol. 2014;88(22):12992–13004. doi: 10.1128/JVI.01914-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McMurtrey CP, Lelic A, Piazza P, Chakrabarti AK, Yablonsky EJ, Wahl A, Bardet W, Eckerd A, Cook RL, Hess R, et al. Epitope discovery in West Nile virus infection: identification and immune recognition of viral epitopes. Proc Natl Acad Sci U S A. 2008;105(8):2981–2986. doi: 10.1073/pnas.0711874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wahl A, Schafer F, Bardet W, Buchli R, Air GM, Hildebrand WH. HLA class I molecules consistently present internal influenza epitopes. Proc Natl Acad Sci. 2009;106(2):540–545. doi: 10.1073/pnas.0811271106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hawkins OE, VanGundy RS, Eckerd AM, Bardet W, Buchli R, Weidanz JA, Hildebrand WH. Identification of breast cancer peptide epitopes presented by HLA-A*0201. J Proteome Res. 2008;7(4):1445–1457. doi: 10.1021/pr700761w. [DOI] [PubMed] [Google Scholar]
- 22.Scull KE, Dudek NL, Corbett AJ, Ramarathinam SH, Gorasia DG, Williamson NA, Purcell AW. Secreted HLA recapitulates the immunopeptidome and allows in-depth coverage of HLA A*02:01 ligands. Mol Immunol. 2012;51(2):136–142. doi: 10.1016/j.molimm.2012.02.117. [DOI] [PubMed] [Google Scholar]
- 23.Pedersen LO, Stryhn A, Holtet TL, Etzerodt M, Gerwien J, Nissen MH, Thøgersen HC, Buus S. The interaction of beta 2-microglobulin (beta 2m) with mouse class I major histocompatibility antigens and its ability to support peptide binding. A comparison of human and mouse beta 2m. Eur J Immunol. 1995;25(6):1609–1616. doi: 10.1002/(ISSN)1521-4141. [DOI] [PubMed] [Google Scholar]
- 24.Rost HL, Rosenberger G, Navarro P, Gillet L, Miladinović SM, Schubert OT, Wolski W, Collins BC, Malmström J, Malmström L, et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32(3):219–223. doi: 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- 25.Lopes A, Vandermeulen G, Préat V. Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J Exp Clin Cancer Res. 2019;38(1):146. doi: 10.1186/s13046-019-1154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y-L, Wang C-Y, Yang F-Y, Wang B-S, Chen JY, Lin L-T, Leu J-D, Chiu S-J, Chen F-D, Lee Y-J, et al. Synergistic effects of glycated chitosan with high-intensity focused ultrasound on suppression of metastases in a syngeneic breast tumor model. Cell Death Dis. 2014;5:e1178. doi: 10.1038/cddis.2014.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Ferrel GL, Guerra MC, Hode T, Lunn JA, Adalsteinsson O, Nordquist RE, Liu H, Chen WR. Preliminary safety and efficacy results of laser immunotherapy for the treatment of metastatic breast cancer patients. Photochem Photobiol Sci. 2011;10(5):817–821. doi: 10.1039/c0pp00306a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ER, Griffith M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016;8(1):11. doi: 10.1186/s13073-016-0264-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen Y-S, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lundegaard C, Lund O, Nielsen M. Prediction of epitopes using neural network based methods. J Immunol Methods. 2011;374(1–2):26–34. doi: 10.1016/j.jim.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castle JC, Kreiter S, Diekmann J, Löwer M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72(5):1081–1091. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 32.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 34.Marty R, Kaabinejadian S, Rossell D, Slifker MJ, van de Haar J, Engin HB, de Prisco N, Ideker T, Hildebrand WH, Font-Burgada J, et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell. 2017;171(6):1272–1283.e15. doi: 10.1016/j.cell.2017.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell KS, Ogris E, Burke B, Su W, Auger KR, Druker BJ, Schaffhausen BS, Roberts TM, Pallas DC. Polyoma middle tumor antigen interacts with SHC protein via the NPTY (Asn-Pro-Thr-Tyr) motif in middle tumor antigen. Proc Natl Acad Sci. 1994;91(14):6344–6348. doi: 10.1073/pnas.91.14.6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh R, Dominiecki ME, Jaffee EM, Paterson Y. Fusion to listeriolysin O and Delivery by Listeria monocytogenes Enhances the Immunogenicity of HER-2/neu and reveals subdominant epitopes in the FVB/N mouse. J Immunol. 2005;175(6):3663–3673. doi: 10.4049/jimmunol.175.6.3663. [DOI] [PubMed] [Google Scholar]
- 37.Ercolini AM, Ladle BH, Manning EA, Pfannenstiel LW, Armstrong TD, Machiels JPH, Bieler JG, Emens LA, Reilly RT, Jaffee EM, et al. Recruitment of latent pools of high-avidity CD8 + T cells to the antitumor immune response. J Exp Med. 2005;201(10):1591–1602. doi: 10.1084/jem.20042167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A. 1992;89(22):10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen H, Slifka MK, Matloubian M, Jensen ER, Ahmed R, Miller JF. Recombinant Listeria monocytogenes as a live vaccine vehicle for the induction of protective anti-viral cell-mediated immunity. Proc Natl Acad Sci. 1995;92(9):3987–3991. doi: 10.1073/pnas.92.9.3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S-K, Cornberg M, Wang XZ, Chen HD, Selin LK, Welsh RM. Private specificities of CD8 T cell responses control patterns of heterologous immunity. J Exp Med. 2005;201(4):523–533. doi: 10.1084/jem.20041337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schüler T, Blankenstein T. Cutting edge: CD8+ effector T cells reject tumors by direct antigen recognition but indirect action on host cells. J Immunol. 2003;170(9):4427–4431. doi: 10.4049/jimmunol.170.9.4427. [DOI] [PubMed] [Google Scholar]
- 42.Maglione J, Moghanaki D, Young LJ, Manner CK, Ellies LG, Joseph SO, Nicholson B, Cardiff RD, MacLeod CL. Transgenic polyoma middle-T mice model premalignant mammary disease. Cancer Res. 2001;61:8298–8305. [PubMed] [Google Scholar]
- 43.Drobysheva D, Smith BA, McDowell M, Guillen KP, Ekiz HA, Welm BE. Transformation of enriched mammary cell populations with polyomavirus middle T antigen influences tumor subtype and metastatic potential. Breast Cancer Res. 2015;17(1):132. doi: 10.1186/s13058-015-0641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim E, Wu D, Pal B, Bouras T, Asselin-Labat M-L, Vaillant F, Yagita H, Lindeman GJ, Smyth GK, Visvader JE, et al. Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res. 2010;12:R21. doi: 10.1186/bcr2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raptis L, Marcellus R, Corbley MJ, Krook A, Whitfield J, Anderson SK, Haliotis T. Cellular ras gene activity is required for full neoplastic transformation by polyomavirus. J Virol. 1991;65:5203–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Webster M, Hutchinson JN, Rauh MJ, Muthuswamy SK, Anton M, Tortorice CG, Cardiff RD, Graham FL, Hassell JA, Muller WJ, et al. Requirement for both Shc and phosphatidylinositol 3′ kinase signaling pathways in polyomavirus middle T-mediated mammary tumorigenesis. Mol Cell Biol. 1998;18:2344–2359. doi: 10.1128/MCB.18.4.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hawkins O, Verma B, Lightfoot S, Jain R, Rawat A, McNair S, Caseltine S, Mojsilovic A, Gupta P, Neethling F, et al. An HLA-presented fragment of macrophage migration inhibitory factor is a therapeutic target for invasive breast cancer. J Immunol. 2011;186(11):6607–6616. doi: 10.4049/jimmunol.1003995. [DOI] [PubMed] [Google Scholar]
- 48.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102(27):9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, McBain N, Wagner T, Edwards J, McConville R, et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature. 2019;565(7739):366–371. doi: 10.1038/s41586-018-0812-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.