

replying to A. Liston et al. Nature Genetics 10.1038/s41588-019-0543-3 (2019)
The origin of octoploid strawberry has been the focus of several phylogenetic studies over the past decade (for example, refs. 1–3). Our previous study, using the octoploid genome and transcriptomes of every extant diploid Fragaria species, provided support for four species (Fragaria vesca, Fragaria iinumae, Fragaria viridis and Fragaria nipponica) as the closest extant relatives of the diploids that contributed to the origin of octoploid strawberry4. In a response paper5, Liston et al. stated “that only two extand diploids were progenitors” with one subgenome being contributed by F. vesca and three by F. iinumae–like ancestors. Our reanalysis of the transcriptome data and comparative genomic analyses of a chromosome-scale F. iinumae genome support our previous model for the origin of octoploid strawberry4.
Liston et al.5 raised a concern regarding one of the steps in the phylogenetic analysis of the subgenome tree-searching algorithm (PhyDS) tool we developed to identify extant relatives of diploid progenitors of allopolyploids. Specifically, they argue that we may have incorrectly identified F. viridis and F. nipponica as extant relatives because in-paralogs were excluded from our previous phylogenetic analysis4. Our reanalysis of the data using PhyDS, now including in-paralogs, yielded results consistent with those presented in our previous study (Fig. 1; Supplementary Information and Supplementary Dataset 1). Furthermore, their alternative model for the origin of octoploid strawberry (1× F. vesca–like and 3× F. iinumae–like subgenomes) is not supported by comparative genomic analyses of a new chromosome-scale F. iinumae genome (Fig. 2).
Fig. 1. Phylogenetic analyses.

a, Number of genes from species identified as being sister to a homoeolog from the octoploid genome, by using PhyDS with bootstrap support value (BSV) cutoffs. Based on previous results4. b, Reanalysis of the data, including in-paralogs and BSV50 cutoff, identified the same progenitor species. The prevalence and biased patterns of homoeologous exchanges between subgenomes resulted in the dominant F. vesca subgenome replacing a greater number of corresponding regions in each of the recessive subgenomes4. Thus, a greater number of genes from the dominant F. vesca subgenome were identified, with the F. iinumae–like subgenome being second.
Fig. 2. Divergence of Ks rates among subgenomes.

a, Synonymous substitution divergence for all syntenic genes between the F. iinumae and Fragaria × ananassa genomes4. The median Ks divergence values for the seven chromosomes previously assigned to each progenitor species are plotted. The F. iinumae and F. vesca subgenomes exhibit the lowest and highest Ks divergence, respectively. b–d, Ks analysis of F. iinumae (b), F. viridis (c) and F. nipponica (d) transcriptomes against the phylogenetically supported homoeolog in the octoploid genome. The Ks distributions of F. viridis and F. nipponica transcriptomes are both unique and distinct from that of F. iinumae.
Phylogenetic analysis of the subgenome tree-searching algorithm searched a set of gene trees to identify sequences most closely related to a set of user-provided paralogs (or homoeologs in polyploids). Homoeologs are orthologous genes that were brought back into the same nucleus by allopolyploidization6. For our analyses, we used syntenic (that is, positionally conserved) homoeologs that were present on all subgenomes in octoploid strawberry. Gene trees were estimated using RAxML7 based on orthologs identified using established orthogrouping approaches8 applied to de novo assembled transcriptomes for each diploid Fragaria species4. PhyDS performs a relatively simple and straightforward analysis of gene trees. First, it identifies the user-provided paralog present in a gene tree and then moves to the direct ancestral node of the paralog. Second, PhyDS then returns to the user the direct descendants (that is, sequence identities including the paralog) of that ancestral node with its bootstrap support value (Fig. 1).
We have two major concerns regarding the methods used in refs. 2,5. First, phylogenetic analyses aimed at estimation of species relationships are reliant first on correct identification of orthologs9. These authors used a sequence similarity-based approach to identify putative orthologs that has relatively high error rates10. Furthermore, pangenome studies have shown that up to one-half of gene content exhibits presence–absence variation at the species level in plants11. In other words, many genes are individual- or population-specific. Thus, many of the putative ortholog predictions in their studies may be inaccurate. Second, Liston et al.5 performed analyses of 100-kb windows across each of the seven base chromosomes. This could be problematic because chromosomal regions from one parental species can be replaced with chromosomal regions from the other parental species during meiosis in polyploids (referred to as homoeologous exchanges12). Homoeologous exchanges can range in size from large megabase-sized regions to single genes (see a recent review on its impact on subgenome assignment in ref. 13). We identifed extensive homoeologous exchanges throughout the octoploid strawberry genome4. Thus, the 100-kb windows Liston et al. used consist of genes with different evolutionary histories reflecting each of the different progenitor species. This could result in inaccurate estimates of species relationships.
Here we present a chromosome-scale genome of F. iinumae with a scaffold minimum scaffold length needed to cover 50% of the genome of 33.98 Mb and 23,665 protein-coding genes (see Supplementary Information). This genome was used to calculate the synonymous substitution (Ks) divergence between F. iinumae to each of the four subgenomes (Fig. 2a). This revealed that only one of the subgenomes of octoploid strawberry is F. iinumae–like, which does not support the model presented by Liston et al.5 that the origin of octoploid strawberry involved three F. iinumae–like and one F. vesca–like progenitor species. Instead, these results are consistent with our phylogenetic estimates supporting more than two diploid progenitors (Fig. 2b–d). The F. viridis (Fig. 2c) and F. nipponica (Fig. 2d) subgenomes are not F. iinumae–like.
Our new phylogenetic analyses support four distinct progenitor species, which is consistent with our previous results4 and that of other groups3. The conflicting results obtained by Liston et al.5 are probably due to differences in methodology. As pointed out above, establishing gene orthology is crucial for molecular phylogenetics. Our pipeline started by identifying high-confidence syntenic 1:1 homoeologs present on each of the subgenomes. This step alone filtered out 82.1% of genes from the octoploid strawberry genome4. The number of genes analyzed in our study was further reduced due to absence across transcriptome data, stringent orthogroup filtering and bootstrap value filtering. In short, more data are not always better if one introduces ‘phylogenetic noise’. It is unclear to us how Liston et al.5 obtained high unique mapping rates (~89% alignment) across the F. vesca genome, which consists of ~31% transposable elements and hundreds of duplicate genes. Furthermore, many genes are species-specific based on previous pangenome studies.
As pointed out by Liston et al.5, incomplete lineage sorting can impact phylogenetic inferences. However, that is far more likely to impact within-species than between-species estimates. This is exactly what was observed in our study. Other F. vesca subspecies were identified as contributors but were present at notably lower levels than F. viridis and F. nipponica (Fig. 1a). These patterns provide further support for F. viridis and F. nipponica as extant relatives of the progenitors that contributed to the origin of the intermediate hexaploid ancestor. Lastly, we did state that F. moschata may be an extant relative of the intermediate hexaploid ancestor. Given the high frequency of polyploid formation in Fragaria14 and birth–death dynamics of polyploids15, we agree it is possible that the hexaploid ancestor may be extinct. This remains to be properly evaluated using robust phylogenetic approaches and datasets.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41588-019-0544-2.
Supplementary information
Supplementary Note and Tables 1–4
Results from PhyDS analysis.
Acknowledgements
We thank J. Lei and L. Xue for sample preparation of F. iinumae. This work was supported by Michigan State University AgBioResearch (to P.P.E.), USDA-NIFA HATCH (no. 1009804 to P.P.E.), USDA-NIFA (no. SCRI 2014-51181-22378) and NSF-DEB (no. 1737898) to P.P.E., USDA-NIFA (no. SCRI 2017-51181-26833 to S.J.K.), the California Strawberry Commission (to S.J.K.), the University of California (to S.J.K.) and the National Natural Science Foundation of China (nos. 31770408 to T.Z. and 31760082 to Q.Q.).
Extended data
Extended Data Fig. 1. Anchoring genome sequence to the genetic map.
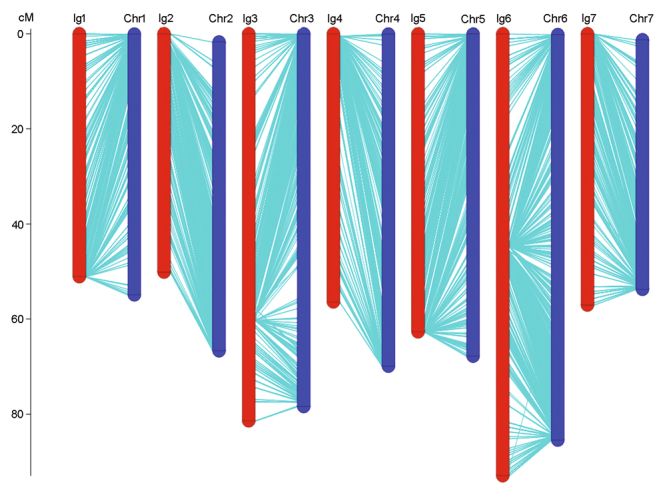
Previously a high-density linkage map of F. iinumae was constructed by 4173 markers, with 3280 from the Array and 893 from genotyping by sequencing7. Here we anchored the contigs to this genetic map to obtain a chromosome-scale genome of F. iinumae.
Author contributions
P.P.E., M.R.M., A.E.Y., S.J.K., Q.Q. and T.Z. perfomed research and/or analyzed data. P.P.E. and M.R.M. drafted the manuscript. P.P.E., M.R.M., A.E.Y., S.J.K., Q.Q. and T.Z. reviewed and edited the manuscript.
Data availability
The phylogenetic trees and alignments are available on Dryad (10.5061/dryad.b2c58pc). The genome assembly and annotation files are available on the Genome Database for Rosaceae (https://www.rosaceae.org/) and NCBI GenBank under BioProjects PRJNA544784 and PRJNA508389. The raw sequence data are available in the Sequence Read Archive under the same NCBI BioProject numbers, PRJNA544784 and PRJNA508389.
Code availability
Custom software for running PhyDS phylogenetic analyses is available on GitHub (https://github.com/mrmckain/PhyDS/).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Patrick P. Edger, Michael R. McKain.
Contributor Information
Patrick P. Edger, Email: edgerpat@msu.edu
Qin Qiao, Email: qiaoqin@ynu.edu.cn.
Ticao Zhang, Email: zhangticao@mail.kib.ac.cn.
Extended data
is available for this paper at 10.1038/s41588-019-0544-2.
Supplementary information
is available for this paper at 10.1038/s41588-019-0544-2.
References
- 1.Rousseau-Gueutin M, et al. Tracking the evolutionary history of polyploidy in Fragaria L. (strawberry): new insights from phylogenetic analyses of low-copy nuclear genes. Mol. Phylogenet. Evol. 2009;51:515–530. doi: 10.1016/j.ympev.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 2.Tennessen JA, Govindarajulu R, Ashman T-L, Liston A. Evolutionary origins and dynamics of octoploid strawberry subgenomes revealed by dense targeted capture linkage maps. Genome Biol. Evol. 2014;6:3295–3313. doi: 10.1093/gbe/evu261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y, Davis TM. A new perspective on polyploid Fragaria (strawberry) genome composition based on large-scale, multi-locus phylogenetic. Analysis. Genome Biol. Evol. 2017;9:3433–3448. doi: 10.1093/gbe/evx214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edger PP, et al. Origin and evolution of the octoploid strawberry genome. Nat. Genet. 2019;51:541–547. doi: 10.1038/s41588-019-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liston Aaron, Wei Na, Tennessen Jacob A., Li Junmin, Dong Ming, Ashman Tia-Lynn. Revisiting the origin of octoploid strawberry. Nature Genetics. 2019;52(1):2–4. doi: 10.1038/s41588-019-0543-3. [DOI] [PubMed] [Google Scholar]
- 6.Glover NM, Redestig H, Dessimoz C. Homoeologs: what are they and how do we infer them? Trends Plant Sci. 2016;21:609–621. doi: 10.1016/j.tplants.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duarte JM, et al. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 2010;10:61. doi: 10.1186/1471-2148-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nichio BTL, Marchaukoski JN, Raittz RT. New tools in orthology analysis: a brief review of promising perspectives. Front. Genet. 2017;8:165. doi: 10.3389/fgene.2017.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon SP, et al. Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure. Nat. Commun. 2017;8:2184. doi: 10.1038/s41467-017-02292-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong Z, Gaeta RT, Pires JC. Homoeologous shuffling and chromosome compensation maintain genome balance in resynthesized allopolyploid Brassica napus. Proc. Natl Acad. Sci. USA. 2011;108:7908–7913. doi: 10.1073/pnas.1014138108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edger PP, McKain MR, Bird KA, VanBuren R. Subgenome assignment in allopolyploids: challenges and future directions. Curr. Opin. Plant Biol. 2018;42:76–80. doi: 10.1016/j.pbi.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Hummer K. The discovery and naming of the cascade strawberry (Fragaria cascadensis) Kalmiopsis. 2015;21:26–31. [Google Scholar]
- 15.Mayrose I, et al. Recently formed polyploid plants diversify at lower rates. Science. 2011;333:1257. doi: 10.1126/science.1207205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Note and Tables 1–4
Results from PhyDS analysis.
Data Availability Statement
The phylogenetic trees and alignments are available on Dryad (10.5061/dryad.b2c58pc). The genome assembly and annotation files are available on the Genome Database for Rosaceae (https://www.rosaceae.org/) and NCBI GenBank under BioProjects PRJNA544784 and PRJNA508389. The raw sequence data are available in the Sequence Read Archive under the same NCBI BioProject numbers, PRJNA544784 and PRJNA508389.
Custom software for running PhyDS phylogenetic analyses is available on GitHub (https://github.com/mrmckain/PhyDS/).