

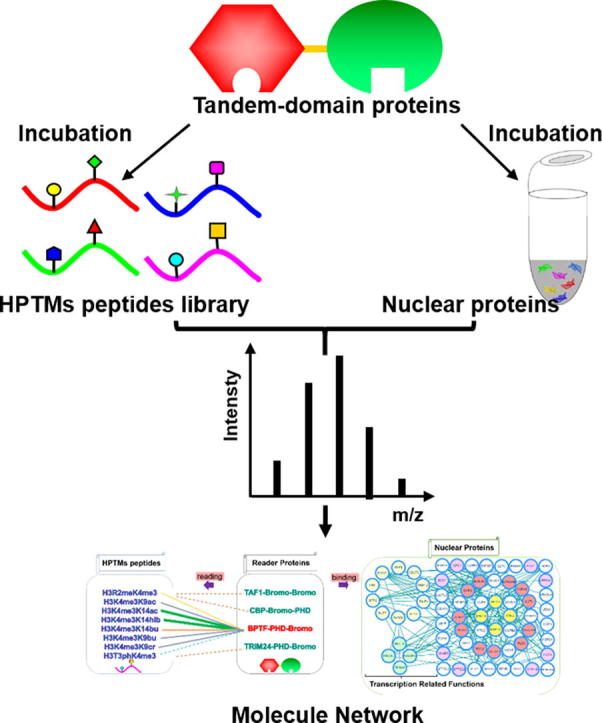
Graphical abstract
Keywords: Posttranslational modification, Histone, Protein-protein interaction, Reader proteins, Binding proteins
Highlights
-
•
The interaction between combinatorial histone modifications and tandem-domain reader proteins was identified. Four tandem-domain proteins (BPTF-PB, CBP-BP, TRIM24-PB, TAF1-BB) could read the peptides with dual-modifications.
-
•
The binding affinities were detected by isothermal titration calorimetry. The interaction between BPTF-PB and peptides with PTMs is the strongest.
-
•
The binding proteins to the tandem-domains were quantified. 78 enriched proteins were further characterized.
-
•
The molecule network of “histone modification-reader-binding proteins” was analyzed.
Abstract
Histone posttranslational modifications (HPTMs) play important roles in eukaryotic transcriptional regulation. Recently, it has been suggested that combinatorial modification codes that comprise two or more HPTMs can recruit readers of HPTMs, performing complex regulation of gene expression. However, the characterization of the multiplex interactions remains challenging, especially for the molecular network of histone PTMs, readers and binding complexes. Here, we developed an integrated method that combines a peptide library, affinity enrichment, mass spectrometry (MS) and bioinformatics analysis for the identification of the interaction between HPTMs and their binding proteins. Five tandem-domain-reader proteins (BPTF, CBP, TAF1, TRIM24 and TRIM33) were designed and prepared as the enriched probes, and a group of histone peptides with multiple PTMs were synthesized as the target peptide library. First, the domain probes were used to pull down the PTM peptides from the library, and then the resulting product was characterized by MS. The binding interactions between PTM peptides and domains were further validated and measured by isothermal titration calorimetry analysis (ITC). Meanwhile, the binding proteins were enriched by domain probes and identified by HPLC-MS/MS. The interaction network of histone PTMs-readers-binding complexes was finally analyzed via informatics tools. Our results showed that the integrated approach combining MS analysis with ITC assay enables us to understand the interaction between the combinatorial HPTMs and reading domains. The identified network of “HPTMs-reader proteins-binding complexes” provided potential clues to reveal HPTM functions and their regulatory mechanisms.
Introduction
Recently, a number of histone posttranslational modifications (HPTMs) were reported (e.g., acetylation (ac), methylation (me), phosphorylation (ph), butyrylation (bu) [1], crotonylation (cr) [2], succinylation (su) [3], 2-hydroxybutyrylation (hib) [4] or lactylation [5]). Meanwhile, it has been suggested that two or more histone modifications may form combinatorial HPTMs, which act as a recognition platform to recruit reader proteins and further regulate gene transcription [6], [7], [8], [9]. For example, the methylation of lysine H3 and its adjacent phosphorylation (H3S10ph) can modulate the binding of hetero-chromatin protein 1 with histone H3 and thus alter chromosome alignment and segregation [10]. Therefore, the readers that display distinct binding abilities to different HPTMs play critical roles in translating the complex PTM codes to certain meaningful biological functions [8], [9], [11], including transcription, cell cycle progression, cell growth and differentiation, and apoptosis [12]. The majority of reader proteins can recognize combinatorial HPTMs by their multiple domains. For example, a trans-histone PTM platform that was formed by tri-methylation of lysine 4 on histone H3 (H3K4me3) and acetylation of lysine 16 on histone H4 (H4K16ac) to coordinately interact with the PHD finger and Bromodomain of BPTF [13]. Recently, a dual histone methyl-lysine binding module of SHORT LIFE was reported to recognize both tri-methylation on H3K4 and H3K27 via its BAH and PHD domains, respectively [14]. Moreover, tandem-domain-reader proteins not only lead to multivalent binding of the combinatorial histone modifications but also interact with nuclear proteins to form large multiprotein complexes, which are involved in many chromatin-dependent functions [6], [12], [15], [16], [17]. A further complication is the fact that the “HPTMs-reader proteins-binding complexes” coordinately interact with each other to reveal epigenetic codes [8], [18]. Previous reports have indicated that the network of “HPTMs-reader proteins-binding complexes” is related to numerous diseases [19].
Currently, research on the interactions between HPTMs and proteins has developed rapidly, and most studies have been performed on the peptide level, such as peptide microarrays. However, the interactions are usually transient and characterized by modest tens-to-hundreds micromolar affinity [19], [20]. Peptide microarrays are largely limited to peptides containing individual PTMs, and the integrity and spatial orientation of the peptides also have some nonspecific influence on the interaction [21]. Another popular technology, chromatin immunoprecipitation (ChIP)-based methods [22], has enabled the mapping and understanding of histone modifications at the genomic level. However, ChIP has also been limited by the weakness of the antibodies, which exhibit cross-reactivity and epitope closure [23], [24]. Recently high throughput strategy based on the semi-synthesis of DNA-barcoded nucleosome libraries had been developed for screening the recruitment and modulation of HPTM binders, as well as known combinatorial HPTM crosstalks [25], [26]. The method is very attractive, however the construction of analysis tool still has a high requirement in histone protein synthesis and the reassemble nucleosome. To date, more and more methods, such as surface plasmon resonance imaging technique [27] and NMR spectroscopy [28] have been implemented to profile readers of combinatorial HPTMs or to reveal their interactions. Notably, MS, as a more detailed, high-throughput, and unbiased method [29], [30], plays important roles in the study of HPTMs and has promising potential to elucidate the communication of “HPTMs-reader proteins-binding complexes”.
In this work, we focused on several tandem-domain-reader proteins and analyzed their readout of HPTMs peptides and binding nucleoproteins to profile the complicated interactions between combinatorial HPTMs and tandem domains. The interactions were identified by MS, and then the binding abilities were quantified by isothermal titration calorimetry (ITC). In addition, tandem-domain proteins were used as probes to pull down the nucleoproteins. The interaction network of the histone PTMs-readers-binding complexes was analyzed by bioinformatics analysis. A schematic view of the work is provided in the supplemental information (Fig. S1). The results showed that four transcription-associated proteins, such as MBB1A, HELLS, PRKDC and TRRAP, could modify the histones alone or as a component of the complex. Finally, we constructed the network of “HPTMs-reader proteins-binding complexes” and revealed the molecular mechanism of the effects of tandem-domain protein-mediated histone crosstalk on epigenetic regulation.
Material and methods
Protein expression and purification
The chromatin-associating domains from human BPTF (PHD-Bromo 8326–8832), human CBP (Bromo-PHD 4038–4745), human TAF1 (Bromo-Bromo 4033–4908), human TRIM24 (PHD-Bromo 2658–3341), and human TRIM33 (PHD-Bromo 2789–3511) were N-terminally fused to GST. All proteins were expressed in E. coli Rosetta and induced overnight by 1 mM isopropyl b-D-thiogalactoside at 16℃ in LB medium supplemented with 50 mM ZnCl2 [13]. After cell lysis and centrifugation, the supernatant was applied to Glutatahione Sepharose 4B agarose (GE Healthcare, Pittsburgh, USA). The resultant plasmid sequences (BPTF-PB, CBP-BP, TRIM24-PB, TRIM33-PB, and TAF1-BB) are provided in the supplemental information (Figs. S8–S12).
Peptide pull-down
All histone peptides bearing combinational modifications were purchased from SciLight Biotechnology (China) and derived by chemical synthesis. The combinatorial HPTMs peptides were designed based on previous reports [1], [2], [3], [4], [31], [32], [33], [34]. The peptide mixture was prepared by mixing equivalent amounts of different peptides. GST-tagged proteins were first attached to Glutatahione Sepharose 4B beads (GE Healthcare, Pittsburgh, USA). After washing the beads five times (wash buffer, 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.05% NP40), the peptide mixture was incubated with the beads. During the peptide pull-down, the peptide mixture (100 mg/ml, 5 μl) was incubated with the GST-tagged proteins (10 mg/ml, 20 μl). After incubation, all the beads were washed by wash buffer I two times (wash buffer I, 50 mM Tris-HCl (pH 8.0), 350 mM NaCl, 0.5% NP40), and by wash buffer II three times (wash buffer II, 50 mM Tris-HCl (pH 8.0), 350 mM NaCl, 0.05% NP40) to remove false positive binding peptides. Finally, 30% acetic acid was added, and the enriched peptides were eluted and detected by MALDI-TOF MS.
Matrix-assisted laser desorption ionization time-of-flight analysis (MALDI-TOF)
Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) analysis was performed using Autoflex III TOF/TOF mass spectrometer (Bruker Daltonics, Leipzig, Germany) (mass tolerance: ≤4 ppm). The measurements were conducted in reflex positive-ion mode with delayed ion extraction. Prior to analysis, the instrument was externally calibrated with a mixture of peptide standards. 2,5-Dihydroxybenzoic acid (DHB) was used as the matrix for the analysis of peptides. Sample aliquots of 1.0 µl were placed onto the MALDI plate. Then, 1.0 µl of the DHB matrix was added and dried at room temperature. MS data were analyzed using Flexanalysis software (3.3.65.0) for spectral processing and peak detection.
Isothermal titration calorimetry (ITC)
For ITC measurement, synthetic histone peptides (SciLight Biotechnology, Beijing, China) and proteins were extensively dialyzed against the ITC buffer: PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4) (pH 6.5). During the ITC assay procedure, four GST tagged proteins (75 μM, 350 μl) were titrated with each peptide (1.5 mM, 80 μl), separately. The titration experiment was monitored using a MicroCal iTC200 system (GE Healthcare, Pittsburgh, USA) at 25 °C. Each ITC titration comprised 18 successive injections. Each peptide was titrated into different proteins and tested by ITC. The resultant ITC curves were processed using Origin (v.8.0) software (OriginLab) in accordance with the ‘‘One Set of Sites’’ fitting model.
Protein pull-down experiment
All GST-tagged proteins (BPTF-PB, CBP-BP, TRIM24-PB, and TAF1-BB) were first incubated with Glutatahione Sepharose 4B beads. After washing the beads five times (wash buffer, 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.05% NP40), 1 mg HEK293T nuclear extract was added to the beads which were previously bound to the tandem domain proteins and incubated overnight at 4 °C. The nuclear proteins enriched by the tandem-domain-protein probes (BPTF-PB, CBP-BP, TRIM24-PB, and TAF1-BB) served as sample groups. The beads incubated only with 1 mg nuclear extracts served as the negative control group. After incubation, all the beads were washed with wash buffer I two times (wash buffer I, 50 mM Tris-HCl (pH 8.0), 350 mM NaCl, 0.5% NP40) and with wash buffer II three times (wash buffer II, 50 mM Tris-HCl (pH 8.0), 350 mM NaCl, 0.05% NP40) to remove false positive binding peptides. Finally, 5 × loading buffer was added to the beads, and the mixture was boiled at 95℃ for 5 min. Then, the enriched proteins were separated by a 10–12% gradient PAGE gel. The gel was dealt with silver staining and subjected to LC-MS/MS analysis.
LC-MS/MS analysis
All proteins were first subject to in-gel trypsin digestion. Then, each sample of peptides was reconstituted in 7 µl HPLC buffer A (0.1% (v/v) formic acid in water), and 5 µl was injected into a Nano-LC system (EASY-nLC 1000, Thermo Fisher Scientific, Waltham, USA). We used C18 columns (50-μm inner diameter × 15 cm, 2 μm C18) to separate each sample via an 85-minute HPLC-gradient (linear gradient from 2 to 35% HPLC buffer B and 0.1% formic acid in acetonitrile for 75 min and then to 90% buffer B in 10 min). The HPLC elution was electro-sprayed to an Orbit rap Q-Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, USA). The source was operated at 1.8 kV. We carried out mass spectrometric analysis in a data-dependent mode with an automatic switch between a full MS scan and an MS/MS scan in the orbit rap. The automatic gain control (AGC) target was 3e6, and the scan range was from 400 to 1350 with a resolution of 70,000 in the full MS survey scan. We selected the 10 most intense peaks with a charge state of 2 and above for fragmentation by higher-energy collision dissociation (HCD) with a normalized collision energy of 27%. The MS2 spectra were acquired with 17,500 resolutions. Finally, we searched the MS/MS data results against the UniProt database using MaxQuant software (v1.5.2.8) with a less than 1% overall false discovery rate (FDR) for peptides. The mass tolerance of LC-MS/MS is 0.05 Da. The peptide sequences were searched using trypsin specificity with a maximum of two missed cleavages. We performed three replicate experiments to evaluate experimental reproducibility. The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE [35] partner repository with dataset identifier PXD014909.
Label-free analysis
The nuclear proteins enriched by the GST-tagged tandem domain probes (BPTF-PB, CBP-BP, TRIM24-PB, and TAF1-BB) served as the sample group, while nuclear proteins enriched only by the Glutatahione Sepharose 4B beads (GE Healthcare, Pittsburgh, USA) served as the control group. After SDS-PAGE, the proteins of sample group and control group were digested in gel and identified by LC-MS/MS. Every identified protein has an LFQ intensity after searching with MaxQuant software (v1.5.2.8). The ratio was obtained when the experimental LFQ intensity divided by the control LFQ intensity of each identified protein. A ratio greater than 2.0-fold (ratio > 2.0) was defined as indicative of “enriched” proteins. Then, bioinformatics analysis of enriched proteins was further carried out with the DAVID and KEGG database.
Bioinformatics analysis
Categorical annotation was performed using Gene Ontology (GO). Database for Annotation Visualization and Integrated Discovery (DAVID) was used to analyze the biological process (BP), molecular function (MF) and cellular component (CC) of the proteins. The distribution of the different proteins in the metabolic pathways was demonstrated by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Results and discussion
The structures of tandem-domain proteins and readout of combinatorial HPTMs by these proteins
The proteins BPTF [13], [36], TAF1 [37], CBP [38], TRIM24 [39] and TRIM33 [40] all have tandem PHD fingers and Bromodomain, except TAF1, which contains tandem Bromodomain. The plant homeodomain (PHD) finger recognizes methylated lysine [13], [36], [41], [42], [43], while Bromodomain recognizes acetylated lysine, which is involved in the suppression or activation of transcription [19], [44], [45]. Located in tandem with other reader domains, these domains often perform dual recognition of HPTMs, suggesting a potential cross-talk among readers [46]. PHD fingers and Bromodomain are conservative in different proteins or species [47], [48]. However, the interactions or orientation in the Bromodomain-PHD or PHD-Bromodomain tandem modules are different, as shown in Fig. 1A. This finding implies that the unique function of a single domain or the whole function of tandem domains may be distinguished. It has been reported that these tandem domain proteins can read combinatorial HPTMs peptides, namely, BPTF-PHD-Bromo (BPTF-PB) reads H3K4me3/H4K16ac [15], CBP-Bromo-PHD (CBP-BP) reads H4K12acK16ac [48], TRIM24-PHD-Bromo (TRIM24-PB) reads H3K4me3K23ac [39], TRIM33-PHD-Bromo (TRIM33-PB) reads H3K9me3K18ac [40] and TAF1-Bromo-Bromo (TAF1-BB) reads H4K5ac/K8ac [37]. Here, we present schematics of the binding models of the five proteins (Fig. 1B).
Fig. 1.

The protein structures and readout of combinatorial HPTMs peptides. (A). The BPTF [13], [36], TAF1 [37], CBP [38], TRIM24 [39] and TRIM33 [40], [54] proteins have conservative tandem domains (BPTF PDB code 2F6J, CBP PDB code 4N4F, TAF1 PDB code 6FIC, TRIM24 PDB code 3O33, and TRIM33 PDB code 3U5M); (B). These proteins can also read combinatorial HPTMs peptides, i.e., BPTF-PHD-Bromo (BPTF-PB) reads H3K4me3/H4K16ac [15], CBP-Bromo-PHD (CBP-PB) reads H4K12acK16ac [48], TRIM24-PHD-Bromo (TRIM24-PB) reads H3K4me3K23ac [39], TRIM33-PHD-Bromo (TRIM33-PB) reads H3K9me3K18ac [37], [40] and TAF1-Bromo-Bromo (TAF1-BB) reads H4K5ac/K8ac [37].
To explain the relationship of the “HPTMs-reader proteins-binding complexes”, we carried out the experiments in two parts (Fig. S1). First, to profile the complicated interactions between combinatorial HPTMs and reader proteins, we incubated the peptide library with tandem-domain proteins using a new integrated method that screened and quantified the interactions by MALDI-TOF MS and ITC. Then, we used the tandem domain proteins as probes to enrich the interactive nucleoproteins and identified these proteins by HPLC-MS/MS analysis.
The MALDI-TOF MS screening of the peptides recognized by tandem-domain proteins
To reveal the transient and slight interactions between combinatorial HPTMs peptides and tandem-domain proteins, we used an integrated method that combines MS and ITC analysis. The peptide library and highly sensitive MS were used to measure the interaction between the HPTMs peptides and the tandem-domain proteins. Then, ITC was used to quantitatively verify the interactions.
We incubated the proteins with a peptide library (Table 1) that contains combinatorial modifications on different amino acid sites. The identification of tandem-domain proteins and combinatorial HPTMs peptides by MS is provided in the supplemental information (Fig. S2, Figs. S3 and S7). To exclude nonspecific binding peptides, we incubated peptides only with GST beads as the negative control for comparison with peptides incubated with BPTF-PB (Fig. S4). Then, the enriched interactive peptides were identified by MALDI-TOF MS (Fig. 2). The results showed that the peptides H3K4me3, H3K9ac, H3R2meK4me3, H3K9bu, H3K9cr, H3K9hib, H3K4me3K9ac, H3K4me3K9bu, H3K4me3K9cr and H3K4me3K9hib could be enriched by the four tandem-domain proteins, but the intensities of these peptides were different. This finding is consistent with previous reports that the domain-domain interactions or orientation between PHD fingers and Bromodomain affected the binding properties of these peptides [20]. However, we did not detect any peptides readout by TAIM33-PB.
Table 1.
The list of HPTM peptides.
| Peptide | Name | Sequence and Modification |
|---|---|---|
| H3(1–17) | Control | ARTKQTARKSTGGKAPR |
| H3(1–17) | H3K4me3 | ARTK(me3)QTARKSTGGKAPR |
| H3(1–17) | H3K9ac | ARTKQTARK(ac)STGGKAPR |
| H3(1–17) | H3K9cr | ARTKQTARK(cr)STGGKAPR |
| H3(1–17) | H3K9bu | ARTKQTARK(bu)STGGKAPR |
| H3(1–17) | H3K9hib | ARTKQTARK(hib)STGGKAPR |
| H3(1–17) | H3R2meK4me3 | AR(me)TK(me3)QTARKSTGGKAPR |
| H3(1–17) | H3T3phK4me3 | ART(ph)K(me3)QTARKSTGGKAPR |
| H3(1–17) | H3K4me3K9ac | ARTK(me3)QTARK(ac)STGGKAPR |
| H3(1–17) | H3K4me3K9bu | ARTK(me3)QTARK(bu)STGGKAPR |
| H3(1–17) | H3K4me3K9cr | ARTK(me3)QTARK(cr)STGGKAPR |
| H3(1–17) | H3K4me3K9hib | ARTK(me3)QTARK(hib)STGGKAPR |
aAbbreviation of acylation modifications: methylation (me), tri-methylation (me3), acetylation (ac), phosphorylation (ph), butyrylation (bu), crotonylation (cr), succinylation (su), and 2-hydroxybutyrylation (hib).
Fig. 2.

Identification of combinatorial HPTM peptides by MALDI-TOF MS screening. (A). The peptides interacted with TRIM24-PB; (B). The peptides interacted with TAF1-BB; (C). The peptides interacted with CBP-BP; (D). The peptides interacted with BPTF-PB; (a. H3K4me3; b. H3K9ac; c. H3R2meK4me3; d. H3K9bu; e. H3K9cr; f. H3K9hib; g. H3K4me3K9ac; h. H3K4me3K9bu; i. H3K4me3K9cr; j. H3K4me3K9hib).
The HPTMs readout patterns of the tandem-domain proteins revealed different readout properties. For example, the distribution of H3T3ph is opposite to that of H3K4me3, which reveals a Tph-mediated binary switch mechanism in active genes [49]. The diverse outcomes of the histone combinatorial readout by tandem-domain-reader proteins indicates a complex regulation based on multiple histone modifications. Using the integrated approach in this research, we could understand the interaction between combinational HPTMs and reading proteins by combining the sensitive MS technique with ITC analysis.
The quantitative analysis of the binding ability of combinatorial HPTM peptides with tandem-domain proteins by ITC
To quantify the binding affinities, we carried out ITC assays between the peptides containing combinatorial modifications and tandem-domain proteins based on the MS screening results (Fig. 3). The tandem-domain proteins used in ITC were digested by thrombin proteases to remove the GST tag and the digestion was verified by SDS-PAGE (Fig. S5). We found that only BPTF-PB could efficiently bind different combinatorial HPTM peptides. No apparent heat change was detected in the other three proteins during ITC. KD and other thermodynamic parameters are reported in the supplemental information (Table S4).
Fig. 3.


Quantitative analysis of the binding ability was determined by isothermal titration calorimetry (ITC). Each tandem-domain protein was titrated with HPTM peptides. The titration in the same group was conducted under the same experimental parameters. (A). TRIM24-PB titrated with H3K4me3K9ac; (B) TRIM24-PB titrated with H3K4me3K9bu; (C). TRIM24-PB titrated with H3K4me3K9cr; (D) TRIM24-PB titrated with H3K4me3K9hib; (E). TAF1-BB titrated with H3K4me3K9ac; (F). TAF1-BB titrated with H3K4me3K9bu; (G). TAF1-BB titrated with H3K4me3K9cr; (H) TAF1-BB titrated with H3K4me3K9hib; (I). CBP-PB titrated with H3R2meK4me3; (J). CBP-PB titrated with H3K4me3K9ac; (K) CBP-PB titrated with H3K4me3K9bu; (L). CBP-PB titrated with H3K4me3K9cr; (R) CBP-PB titrated with H3K4me3K9hib; (M). BPTF-PB titrated with H3R2meK4me3; (N). BPTF-PB titrated with H3K4me3K9ac; (O) BPTF-PB titrated with H3K4me3K9bu; (P). BPTF-PB titrated with H3K4me3K9cr; (Q) BPTF-PB titrated with H3K4me3K9hib.
BPTF-PB could interact with several combinatorial HPTM peptides in our peptide library: H3R2meK4me3 (KD 13 μM), H3K4me3K9ac (KD 15.8 μM), H3K4me3K9bu (KD 27.6 μM), H3K4me3K9cr (KD 17.5 μM) and H3K4me3K9hib (KD 14.1 μM) (Fig. 3 M−Q). The measured interactions between BPTF-PB and H3R2meK4me3, H3K4me3K9ac or H3K4me3K9cr are consist with the previously reported interaction [13], [33]. The other peptides (H3K4me3K9bu and H3K4me3K9hib) were found to interact with BPTF-PB for the first time. As known, the BPTF-PB readout of combinatorial HPTM peptides mainly depended on the PHD finger readout of H3K4me3 [36], and the Bromodomain of BPTF-PB could read acetylated lysine, which exhibits a synergistic effect [36]. These acetylation modifications (butyrylation, crotonylation, or 2-hydroxybutyrylation) are similar to lysine acetylation but exhibit different hydrocarbon chain lengths and hydrophobicity or charges [33], [34]. Thus, the modifications might have the potential to stretch into the binding pocket of Bromodomain and ultimately lead to different binding abilities of BPTF-PB. Thus, given the strong interaction between PHD and H3K4me3, the peptides with combined modifications (H3K4me3K9bu, H3K4me3K9cr and H3K4me3K9hib) also interacted with BPTF-PB. In contrast, the other three proteins CBP-BP, TRIM24-PB, and TAF1-BB did not exhibit binding ability when titrated with the peptides in ITC. We hypothesized that these proteins lack of the molecular basis of BPTF-PB for which the interaction mainly relied on the PHD finger readout of H3K4me3 [36]. The extended hydrocarbon chains of butyrylation, crotonylation and hydroxybutyrylation on lysine increased the hydrophobicity and the bulk of the modified lysine residues in histones compared with lysine acetylation [33]. Thus, the interaction between these proteins and peptides was weak and transient, and it was difficult to detect. In general, compared with ITC, MALDI-TOF MS is a more sensitive technology to detect weak and transient interactions, and this integrated approach enabled us to detect the signal of the interactions between the combinatorial HPTMs and reading domains, which could not be easily observed in vivo.
The qualitative and quantitative analysis of differential enriched-proteins of these tandem-domain proteins by label-free analysis
To explore the nuclear proteins enriched by the tandem-domain proteins, we identified and analyzed these nucleoproteins by HPLC-MS/MS. Here, we used four different proteins, BPTF-PB, TAF1-BB, CBP-BP and TRIM24-PB, as probes that were incubated with HEK293T cell nuclear extracts.
Nucleoproteins enriched by tandem-domain-protein probes (BPTF-PB, CBP-BP, TRIM24-PB and TAF1-BB) served as the sample group, while nuclear proteins enriched only by the beads served as the control group (Fig. S6). The ratio was obtained when the experimental LFQ intensity divided by the control LFQ intensity of each identified protein. The proteins which ratio > 2.0 were defined as enriched proteins and we found that almost all the differential proteins belonged to enriched proteins (Fig. 4A) [40]. All these differential nucleoproteins could be separately enriched by BPTF-PB, TAF1-BB, CBP-BP or TRIM24-PB (Tab S1 and Tab S2), and these differential proteins were further analyzed by bioinformatics (Fig. 4B). Among the enriched proteins, HELLS [50], PRKDC [51], PLK1 [52], and PSIP1 [53] have been reported to interact with BPTF, while TRIP12 [54], BPTF [55], and PARP1 [56] interact with CBP.
Fig. 4.

Quantitative and qualitative analyses of differential proteins using label-free. (A). Enriched proteins screened by label-free quantitative analysis; (B). Quantitative analysis of proteins enriched by tandem-domain proteins; (C). The biological process of the proteins enriched by BPTF-PB, TAF1-BB, CBP-BP and TRIM24-PB were analyzed by DAVID; (D). The molecular function of the proteins enriched by BPTF-PB, TAF1-BB, CBP-BP and TRIM24-PB were analyzed by DAVID.
To annotate and assess the biological roles of these proteins, we used Database for Annotation Visualization and Integrated Discovery (DAVID) to analyze the biological process, molecular function and cellular components of the proteins (Fig. 4C and 4D). The distribution map of cluster analysis reveals that the molecular functions of these 78 enriched proteins were associated with various binding processes, such as ATP binding or RNA binding (Fig. 4D). GO biological process analysis results showed that these proteins were significantly enriched in many cellular gene expression and transcription processes (Fig. 4C). In particular, 14 of these 78 enriched proteins could bind with DNA, RNA, histone or chromatin, which directly participated in transcription-related epigenetics processes (Fig. 7). The distribution of different proteins in metabolic pathways was analyzed by Kyoto Encyclopedia of Proteins and Genomes (KEGG) pathways. These enriched proteins were mainly involved in five different metabolic pathways, particularly cell cycle and spliceosome (Tab S3). Collectively, these data revealed a high-probability role of these enriched proteins in the regulation of cellular transcription.
Fig. 7.

The molecular network of “HPTMs-reader proteins-binding proteins”. From the network, the tandem-domain-reader proteins could read different combinatorial HPTMs peptides and interact with nucleoproteins. The interactive nucleoproteins were separated into two parts based on function, namely, transcription and other functions. Among the 78 proteins, 14 proteins (HELLS, MBB1A, PRKDC, LBR, SFPQ, PSIP1, GTF2I, HLTF, TRIPC, RAD50, CDC73, DDX21, and PHF8) have functions related to transcription, and in particular, and 4 of these proteins (HELLS, MBB1A, PRKDC, and TRRAP) modified histones directly.
The cluster analysis of these enriched proteins
To discover the regulation patterns revealed by the similarity of enriched proteins, we carried out cluster analysis. We used Perseus software (v1.5.6) to cluster these 78 enriched proteins and to identify the relationship among BPTF-PB, TAF1-BB, CBP-BP and TRIM24-PB. The result reveals that the intensities of these proteins was relatively similar, even they were enriched by different tandem-domain proteins (Fig. 5). In addition, all these nuclear proteins have functions related to multiple cellular processes, such as transcription regulation and chromatin remodeling [12]. In general, the cluster analysis of these enriched proteins is of great significance in establishing the molecular network.
Fig. 5.

Cluster analysis of the enriched proteins of BPTF-PB, CBP-BP, TRIM24-PB and TAF1-BB by Perseus software. The proteins enriched by BPTF-PB, CBP-BP, TRIM24-PB and TAF1-BB exhibited some common features. All these proteins can interact with these four tandem domain proteins.
The special binding proteins in the network of “HPTMs-reader proteins-binding complexes”
To further reveal the relationship of the “binding complexes” and “HPTMs”, we tried to screen and identify the proteins associated with both of them using the EpiFactors database. From the EpiFactors database, four nucleoproteins (MBB1A, HELLS, PRKDC and TRRAP) were identified that directly participated in epigenetic transcriptional regulation. These four proteins were perfectly identified by HPLC/MS-MS. The specific peptides of each protein are presented in Fig. 6.
Fig. 6.

Identification of the special peptides of the screened binding proteins by MS. Four proteins were screened and identified in the EpiFactors database. (A). Myb-binding protein 1A (MBB1A); (B) DNA-dependent protein kinase catalytic subunit (PRKDC); (C) transformation/transcription domain-associated protein (TRRAP); (D) Lymphoid-specific helicase (HELLS, also known as LSH).
Myb-binding protein 1A (MBB1A) [57] interacts with sequence-specific DNA binding proteins to activate or inhibit transcription. MBB1A is one of the components of the histone phosphorylation-modifying complex and chromatin-remodeling complex that can phosphorylate H2AXY142. DNA-dependent protein kinase catalytic subunit (PRKDC) [58] is a serine/threonine protein kinase. As a molecular sensor of DNA damage, PRKDC participates in double-strand breaks and repairs. PRKDC is a HPTM writer that can phosphorylate histones, especially H2AXS139 and H2AFXS139. Transformation/transcription domain-associated protein (TRRAP) [59] is a histone acetylation writer cofactor and provides specific tags for transcriptional activation. Lymphoid-specific helicase (HELLS, also as LSH) [60], [61], [62] participates in the formation of heterochromatin and chromatin remodeling. HELLS could also maintain DNA methylation and modify histones. Thus, all of these four proteins that can interact with reader proteins can also modify the histones. Finally, direct links of the network “HPTMs-reader proteins-binding complexes” have been established by these four proteins.
The molecular network of “HPTMs-reader proteins-binding complexes”
Finally, the two functions of reader proteins, which read combinatorial histone modifications and interact with nucleoproteins, were linked together to explain their relationship. As an important linker, the tandem-domain-reader proteins connect the HPTMs with binding complexes to form the network (Fig. 7). From the network, the tandem-domain-reader proteins could read different combinatorial HPTMs peptides and interact with nucleoproteins. The interactive nucleoproteins were separated into two parts based on function, namely, transcription and other functions.
These reader proteins often exist in protein complexes associated with chromatin [12], [47]. In addition to HPTM-dependent histone bindings, reader proteins could also mediate other protein-protein or protein-DNA interactions, ultimately contributing to the overall chromatin interaction [46], [63]. Given the presence of multiple proteins in the complex, their multivalent interaction increases the affinity and specificity and leads to different biological outcomes [15], [16]. The knowledge of this network is beneficial for interpreting the mechanism and for translating this molecular network language into downstream functions given their significant roles in gene activation.
Conclusions
In the study, we established an integrated method to identify the molecular network of the “HPTMs-reader proteins-binding complexes” for understanding the molecular mechanism of HPTM. Our results showed that the method was beneficial for the comprehensive analysis of combinatorial HPTMs crosstalk. Focusing on the interaction between tandem-domain proteins and peptides containing combinatorial modifications, we found that all the tandem-domain proteins (BPTF-PB, CBP-BP, TRIM24-PB, TRIM33-PB, and TAF1-BB) have the potential to read dual-modification peptides, especially BPTF-PB, which could interact strongly with H3R2meK4me3, H3K4me3K9ac, H3K4me3K9bu, H3K4me3K9cr and H3K4me3K9hib. The results also indicated the effect of the new modifications (such as butyrylation, crotonylation, or 2-hydroxybutyrylation) on such interactions. Then, we analyzed the relationship between the tandem-domain proteins and nuclear proteins. Using GST pull-down and HPLC-MS/MS identification, we identified 78 enriched proteins and screened out differential enrichment of the reading domains. Together, we identified the interaction network of “HPTMs-reader proteins-binding complexes”, which will be helpful to understand the crosstalk between combinatorial HPTMs, to interpret the binding mechanisms of tandem domain histone readers, and to dissect the regulatory mechanism of dual HPTMs and the downstream activities regulated by histone codes.
Compliance with ethics requirements
This article does not contain any studies with human or animal subjects
Declaration of Competing Interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 21874100 and 21904097), Tianjin Municipal Science and Technology Commission (Nos. 19JCZDJC35000 and 19JCQNJC08900), and Talent Excellence Program from Tianjin Medical University.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2019.11.003.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Chen Y., Sprung R., Tang Y., Ball H., Sangras B., Kim S.C. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteom. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan M.J., Luo H., Lee S., Jin F.L., Yang J.S., Montellier E. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1015–1027. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park J., Chen Y., Tishkoff D.X., Peng C., Tan M., Dai L. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. 2013;50:919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dai L.Z., Peng C., Montellier E., Lu Z.K., Chen Y., Ishii H. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat Chem Biol. 2014;10:365-U73. doi: 10.1038/nchembio.1497. [DOI] [PubMed] [Google Scholar]
- 5.Zhang D., Tang Z., Huang H., Zhou G., Cui C., Weng Y. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–580. doi: 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Strahl B.D., Allis C.D. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 8.Jenuwein T., Allis C.D. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y., Li M., Fan M., Song Y., Yu H., Zhi X. Chromodomain Y-like protein-mediated histone crotonylation regulates stress-induced depressive behaviors. Biol Psych. 2019;85:635–649. doi: 10.1016/j.biopsych.2018.11.025. [DOI] [PubMed] [Google Scholar]
- 10.Flanagan J.F., Mi L.Z., Chruszcz M., Cymborowski M., Clines K.L., Kim Y.C. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 11.Berger S.L. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 12.Musselman C.A., Lalonde M.E., Cote J., Kutateladze T.G. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H.T., Ilin S., Wang W.K., Duncan E.M., Wysocka J., Allis C.D. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian S.M., Lv X.C., Scheid R.N., Lu L., Yang Z.L., Chen W. Dual recognition of H3K4me3 and H3K27me3 by a plant histone reader SHL. Nat Commun. 2018;9:2425. doi: 10.1038/s41467-018-04836-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruthenburg A.J., Li H., Patel D.J., Allis C.D. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taverna S.D., Li H., Ruthenburg A.J., Allis C.D., Patel D.J. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yap K.L., Zhou M.M. Keeping it in the family: diverse histone recognition by conserved structural folds. Crit Rev Biochem Mol Biol. 2010;45:488–505. doi: 10.3109/10409238.2010.512001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Longbotham J.E., Chio C.M., Dharmarajan V., Trnka M.J., Torres I.O., Goswami D. Histone H3 binding to the PHD1 domain of histone demethylase KDM5A enables active site remodeling. Nat Commun. 2019;10:94. doi: 10.1038/s41467-018-07829-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez R., Zhou M.M. The role of human bromodomains in chromatin biology and gene transcription. Curr Opin Drug Discov Devel. 2009;12:659–665. [PMC free article] [PubMed] [Google Scholar]
- 20.Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.P., Barsyte-Lovejoy D. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilkinson A.W., Gozani O. Histone-binding domains: strategies for discovery and characterization. Biochim Biophys Acta. 2014;1839:669–675. doi: 10.1016/j.bbagrm.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egelhofer T.A., Minoda A., Klugman S., Lee K., Kolasinska-Zwierz P., Alekseyenko A.A. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 2011;18:91–93. doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuchs S.M., Krajewski K., Baker R.W., Miller V.L., Strahl B.D. Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr Biol. 2011;21:53–58. doi: 10.1016/j.cub.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dann G.P., Liszczak G.P., Bagert J.D., Muller M.M., Nguyen U.T.T., Wojcik F. ISWI chromatin remodellers sense nucleosome modifications to determine substrate preference. Nature. 2017;548:607–611. doi: 10.1038/nature23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liszczak G., Muir T.W. Nucleic acid-barcoding technologies: converting DNA sequencing into a broad-spectrum molecular counter. Angew Chem Int Ed. 2019;58:4144–4162. doi: 10.1002/anie.201808956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao S., Zhang B., Yang M., Zhu J., Li H. Systematic profiling of histone readers in arabidopsis thaliana. Cell Rep. 2018;22:1090–1102. doi: 10.1016/j.celrep.2017.12.099. [DOI] [PubMed] [Google Scholar]
- 28.Gatchalian J., Wang X.D., Ikebe J., Cox K.L., Tencer A.H., Zhang Y. Accessibility of the histone H3 tail in the nucleosome for binding of paired readers. Nat Commun. 2017;8:1489. doi: 10.1038/s41467-017-01598-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan Z.F., Arnaudo A.M., Garcia B.A. Mass spectrometric analysis of histone proteoforms. Annu Rev Anal Chem (Palo Alto Calif) 2014;7:113–128. doi: 10.1146/annurev-anchem-071213-015959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang H., Lin S., Garcia B.A., Zhao Y. Quantitative proteomic analysis of histone modifications. Chem Rev. 2015;115:2376–2418. doi: 10.1021/cr500491u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali M., Yan K., Lalonde M.E., Degerny C., Rothbart S.B., Strahl B.D. Tandem PHD fingers of MORF/MOZ acetyltransferases display selectivity for acetylated histone H3 and are required for the association with chromatin. J Mol Biol. 2012;424:328–338. doi: 10.1016/j.jmb.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minguez P., Letunic I., Parca L., Bork P. PTMcode: a database of known and predicted functional associations between post-translational modifications in proteins. Nucleic Acids Res. 2013;41:D306–D311. doi: 10.1093/nar/gks1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu Y., Xu Q., Liu Y., Yu Y., Cheng Z.Y., Zhao Y. Dynamics and functional interplay of histone lysine butyrylation, crotonylation, and acetylation in rice under starvation and submergence. Genome Biol. 2018;19:144. doi: 10.1186/s13059-018-1533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rousseaux S., Khochbin S. Histone acylation beyond acetylation: terra incognita in chromatin biology. Cell J. 2015;17:1–6. doi: 10.22074/cellj.2015.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruthenburg A.J., Li H.T., Milne T.A., Dewell S., McGinty R.K., Yuen M. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell. 2011;145:692–706. doi: 10.1016/j.cell.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobson R.H., Ladurner A.G., King D.S., Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- 38.Kremer EA, N Gaur, MA Lee, O Engmann, J Bohacek, IM Mansuy. Interplay between TETs and microRNAs in the adult brain for memory formation. Scientific Reports. 2018;8:1678. [DOI] [PMC free article] [PubMed]
- 39.Tsai W.W., Wang Z., Yiu T.T., Akdemir K.C., Xia W., Winter S. TRIM24 links a non-canonical histone signature to breast cancer. Nature. 2010;468:927–932. doi: 10.1038/nature09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xi Q., Wang Z., Zaromytidou A.I., Zhang X.H., Chow-Tsang L.F., Liu J.X. A poised chromatin platform for TGF-beta access to master regulators. Cell. 2011;147:1511–1524. doi: 10.1016/j.cell.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pascual J., Martinez-Yamout M., Dyson H.J., Wright P.E. Structure of the PHD zinc finger from human Williams-Beuren syndrome transcription factor. J Mol Biol. 2000;304:723–729. doi: 10.1006/jmbi.2000.4308. [DOI] [PubMed] [Google Scholar]
- 42.Shi X., Hong T., Walter K.L., Ewalt M., Michishita E., Hung T. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wysocka J., Swigut T., Xiao H., Milne T.A., Kwon S.Y., Landry J. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 44.Dhalluin C., Carlson J.E., Zeng L., He C., Aggarwal A.K., Zhou M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 45.Sartor G.C., Malvezzi A.M., Kumar A., Andrade N.S., Wiedner H.J., Vilca S.J. Enhancement of BDNF expression and memory by HDAC inhibition requires BET bromodomain reader proteins. J Neurosci. 2019;39:612–626. doi: 10.1523/JNEUROSCI.1604-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Z., Patel D.J. Combinatorial readout of dual histone modifications by paired chromatin-associated modules. J Biol Chem. 2011;286:18363–18368. doi: 10.1074/jbc.R111.219139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yun M., Wu J., Workman J.L., Li B. Readers of histone modifications. Cell Res. 2011;21:564–578. doi: 10.1038/cr.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plotnikov A.N., Yang S., Zhou T.J., Rusinova E., Frasca A., Zhou M.M. Structural insights into acetylated-histone H4 recognition by the bromodomain-PHD finger module of human transcriptional coactivator CBP. Structure. 2014;22:353–360. doi: 10.1016/j.str.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bai X., Bi W.J., Dong H.Y., Chen P., Tian S.S., Zhai G.J. An integrated approach based on a DNA Self-assembly technique for characterization of crosstalk among combinatorial histone modifications. Anal Chem. 2018;90:3692–3696. doi: 10.1021/acs.analchem.7b05174. [DOI] [PubMed] [Google Scholar]
- 50.Kimura M., Morinaka Y., Imai K., Kose S., Horton P., Imamoto N. Extensive cargo identification reveals distinct biological roles of the 12 importin pathways. Elife. 2017:6. doi: 10.7554/eLife.21184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frey W.D., Chaudhry A., Slepicka P.F., Ouellette A.M., Kirberger S.E., Pomerantz W.C.K. BPTF maintains chromatin accessibility and the self-renewal capacity of mammary gland stem cells. Stem Cell Rep. 2017;9:23–31. doi: 10.1016/j.stemcr.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galdeano C., Ciulli A. Selectivity on-target of bromodomain chemical probes by structure-guided medicinal chemistry and chemical biology. Future Med Chem. 2016;8:1655–1680. doi: 10.4155/fmc-2016-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bochynska A., Luscher-Firzlaff J., Luscher B. Modes of interaction of KMT2 histone H3 lysine 4 methyltransferase/COMPASS complexes with chromatin. Cells. 2018;7:E17. doi: 10.3390/cells7030017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park H.Y., Lee S.B., Yoo H.Y., Kim S.J., Kim W.S., Kim J.I. Whole-exome and transcriptome sequencing of refractory diffuse large B-cell lymphoma. Oncotarget. 2016;7:86433–86445. doi: 10.18632/oncotarget.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim R.N., Moon H.G., Han W., Noh D.Y. Perspective insight into future potential fusion gene transcript biomarker candidates in breast cancer. Int J Mol Sci. 2018;19:E502. doi: 10.3390/ijms19020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perniola R. Twenty years of AIRE. Front Immunol. 2018;9:98. doi: 10.3389/fimmu.2018.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith R.S., Meyers D.A., Peters S.P., Moore W.C., Wenzel S.A., Bleecker E.R. Sequence analysis of HSPA1A and HSPA1B in a multi-ethnic study population. DNA Seq. 2007;18:47–53. doi: 10.1080/10425170601060707. [DOI] [PubMed] [Google Scholar]
- 58.Chen M.H., Tan K.T., Cheng J.H., Fang W.L., Yeh Y.C., Yeh C.N. A new candidate for checkpoint blockade immunotherapy? J Clin Oncol. 2017;35:3022. [Google Scholar]
- 59.Tapias A., Zhou Z.W., Shi Y., Chong Z., Wang P., Groth M. Trrap-dependent histone acetylation specifically regulates cell-cycle gene transcription to control neural progenitor fate decisions. Cell Stem Cell. 2014;14:632–643. doi: 10.1016/j.stem.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 60.Geiman T.M., Durum S.K., Muegge K. Characterization of gene expression, genomic structure, and chromosomal localization of Hells (Lsh) Genomics. 1998;54:477–483. doi: 10.1006/geno.1998.5557. [DOI] [PubMed] [Google Scholar]
- 61.Han Y.X., Ren J.K., Lee E., Xu X.P., Yu W.S., Muegge K. Lsh/HELLS regulates self-renewal/proliferation of neural stem/progenitor cells. Sci Rep. 2017;7:1136. doi: 10.1038/s41598-017-00804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Litwin I., Bakowski T., Maciaszczyk-Dziubinska E., Wysocki R. The LSH/HELLS homolog Irc5 contributes to cohesin association with chromatin in yeast. Nucleic Acids Res. 2017;45:6404–6416. doi: 10.1093/nar/gkx240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Su Z., Denu J.M. Reading the Combinatorial Histone Language. ACS Chem Biol. 2016;11:564–574. doi: 10.1021/acschembio.5b00864. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.