

ABSTRACT
Vibrio mimicus is a foodborne pathogen, which is widely distributed in the aquatic environment. Moreover, it is often involved in aquatic animal diseases. In recent years, V. mimicus is an emerging pathogen in some species of Siluriformes. The strain SCCF01 was isolated from yellow catfish (Pelteobagrus fulvidraco). In this study, we aimed to perform genomic analysis of V. mimicus strain SCCF01 to identify genetic features and evolutionary relationships. Information on gene function and classification was obtained by functional annotation, and circular graph of strain SCCF01 genome, which was created by Circos v0.64. Information on virulence genes (adhesion, flagellum system, exotoxin, and secretory system, etc.) was obtained by virulence genes annotation. Genome element prediction showed that most of the mobile elements were distributed in chromosome I. Therefore, chromosome I of SCCF01 genome has more plasticity than chromosome II and might be larger in size. Genomic linear relationship between the strain of V. mimicus and strain SCCF01 was analyzed by linear pairwise comparison but was unable to determine the relationship. Gene family analysis predicted that the evolutionary direction of strain SCCF01 was: clinical strain → environmental strain → SCCF01 strain. Phylogenetic analysis showed that the strain SCCF01 was more closely related to environmental strains. According to gene family analysis and phylogenetic analysis, we speculated that strain SCCF01 has probably diverged from environmental strains.
KEYWORDS: Vibrio mimicus, virulence factors, genome element, genomic comparison
Introduction
Vibrio mimicus (V. mimicus) was initially considered as an atypical V. cholerae,[1] which is closely related to Vibrio cholerae. V. mimicus is a widely distributed aquatic bacterium that can cause disease in humans and massive death of aquatic animals. It is a foodborne pathogen [2], which can cause gastroenteritis, diarrhea and food poisoning [3–5]. V. mimicus infecting was also common in aquaculture (can infect shrimp, crab, and fish) [6–11]. In recent years, V. mimicus is an emerging pathogen in some species of Siluriformes. The epidemiological features of V. mimicus showed short disease duration and high mortality rate, which eventually leads to substantial economic loss in Siluriformes farmhouses [9,10]. Since 2011, large-scale V. mimicus infectious outbreaks have occurred continuously in Siluriformes farms in China. V. mimicus strain SCCF01, which is a highly virulent strain isolated from yellow catfish (Pelteobagrus fulvidraco) in China, causes almost 100% mortality in yellow catfish [9,12].
At present, there are three complete genome sequences and eight draft genome sequences of V. mimicus available in Genbank genome database (five strains were isolated from human, five strains were isolated from the environment and only one strain SCCF01 was isolated from fish) (Supplementary Table S1). Genome sequence of strain SCCF01 was the only complete genome sequence of V. mimicus from infected aquatic animals. Previous studies have shown that V. mimicus strain SCCF01 natural infection can cause high mortality rate in fishes [9], and is also a highly virulent in the artificial infection experiment [12,13]. However, the genetic features and evolutionary strategies of V. mimicus from fish remain unknown.
Materials and methods
Sources of strain and genome sequences
V. mimicus strain SCCF01, which was isolated from diseased yellow catfish (Pelteobagrus fulvidraco) at a commercial aquaculture site in Southwest China [9]. Challenges showed that bath immersion of strain SCCF01 (10 [6] CFU·mL−1) caused 100% mortality of yellow catfish. The whole-genome sequence of V. mimicus was obtained by single-molecule real-time (SMRT) sequencing using platform PacBio RS II [12]. The complete genomic sequences of SCCF01 have been deposited in GenBank under the accession numbers CP016383 (chromosome I) and CP016384 (chromosome II). The other genomic sequences which were used for comparative genome analysis were downloaded from the National Center for Biotechnology Information (NCBI) (Supplementary Table S1).
Analysis of genome plasticity
General genomic features: The genome of V. mimicus strain SCCF01 was annotated automatically using the GeneMarkS+ based on the NCBI Prokaryotic Genome Annotation Pipeline (PGAAP) [14]. RNA prediction: The rRNA identification was performed with RNAmmer 1.2 software [15], and the tRNA genes were predicted by tRNAscan-SE v2.0 [16]. COG classification: The predicted Open Reading Frame (ORF) sequences for the V. mimicus strain SCCF01 were translated into protein sequences and subsequently aligned against the COG database (http://www.ncbi.nlm.nih.gov/COG/). Accordingly, the predicted genes were divided into COG classes. Circular layouts were generated using Circos v0.64 (http://circos.ca/) [17]. Virulence factor analysis: Virulence factor database (VFDB) [18] was used to assess gene essentiality of ORFs predicted in SCCF01 with BLAST search. Prediction of repeat elements: Interspersed Repeat Sequences (IRS) and Tandem Repeat Sequences (TRS) in V. mimicus strain SCCF01 genome were screened by online program RepeatMasker 4.0.7 (http://www.repeatmasker.org/) using default parameters [19]. Prediction of genomic islands: IslandViewer 4 (http://www.pathogenomics.sfu.ca/islandviewer/browse/) [20] was applied to predict genomic islands in V. mimicus strain SCCF01 with default settings. Prophages prediction: Prophage sequences were predicted using Phage Search Tool (http://phast.wishartlab.com/) [21].
Comparative genome analysis
Collinearity analysis: Global collinearity was identified using Mummer v3.23 by genome-wide sequence comparisons (http://mummer.sourceforge.net/) [22], and LASTZ v1.03.54 (http://www.bx.psu.edu) [23] provides genome-local sequence comparisons to determine the detailed collinearity (Translocation/Trans, Inversion/Inv and Trans+Inv) between two sequences. Gene family analysis: The pairwise alignments of the genome using BLAST [24] were performed to filter out untrustworthy results. Meanwhile, a gene family clustering table was constructed based on the results of alignment similarity by Hcluster-sg v 0.5.1 [25]. Phylogenetic analysis: The two phylogenetic gene trees (Vibrio mimicus species and Vibrio genus) were constructed based on locally collinear block searching in the sample and reference strains as previously reported [26]. The output file in HomBlocks alignments was input into RAxML [27] to construct the phylogenetic trees using the GTR+I + G model with a bootstrap value of 1,000. The phylogenetic trees were displayed and customized using Evolview (http://www.evolgenius.info/evolview/) [28]. Structural variation (SV) identification: The global alignments using Mummer v3.23 [22] were performed between SCCF01 and each reference strain, then Structural variation (SV) was identified using the LASTZ v1.03.54 [23] by pairwise alignment.
Results and discussion
General genomic features of V. mimicus strain SCCF01
The genome of V. mimicus strain SCCF01 was sequenced using PacBio RS II with the P6-C4 Reagent Kit, which resulted in 35,089 pair polymerase reads with Read N50 of 12,135-bp and their characteristics are summarized in Table 1. After filtering, all reads were assembled into two circular chromosomes of 3,213,040 bp for Chromosome I and 1,272,975 bp for Chromosome II, with a G + C content of 46.61% and 45.88%, respectively (Supplementary Table S2). A total of 4,160 genes (4,018 CDSs and 140 RNA genes) were predicted in the SCCF01 genome by PGAAP (Table 1). The strain SCCF01 genome encodes 18 rRNAs and 69 tRNAs. The predicted ORFs are further classified into COGs functional groups (Figure 1) which summarized in Supplementary Table S3.
Table 1.
The results of sequencing and assembly.
| Attribute | Value | |
|---|---|---|
| PacBio statistic data | Polished Contigs | 2 |
| Adapter Dimers (0–10bp) | 0.0% | |
| Short Inserts (11-100bp) | 0.0% | |
| Number of Bases | 306,431,068 | |
| Number of Reads | 35,089 | |
| N50 Read Length | 12,135 | |
| Mean Read Length | 8,732 | |
| Mean Read Score | 0.84 | |
| Mapped Reads | 33,254 | |
| Mapped Read Length of Insert | 7,032 | |
| Average Reference Length | 1,500,987 | |
| Average Reference Bases Called | 100.0% | |
| Average Reference Consensus Concordance | 100.0% | |
| Average Reference Coverage | 56.73 | |
| Characteristics of the genome of Vibrio mimicus strain SCCF01 genome | genes (total) | 4,160 |
| CDS | 4,018 | |
| RNA | 142 | |
| rRNAs | 11, 10, 10 (5S, 16S, 23S) | |
| complete rRNAs | 11, 10, 10 (5S, 16S, 23S) | |
| tRNAs | 105 | |
| ncRNAs | 6 |
Figure 1.
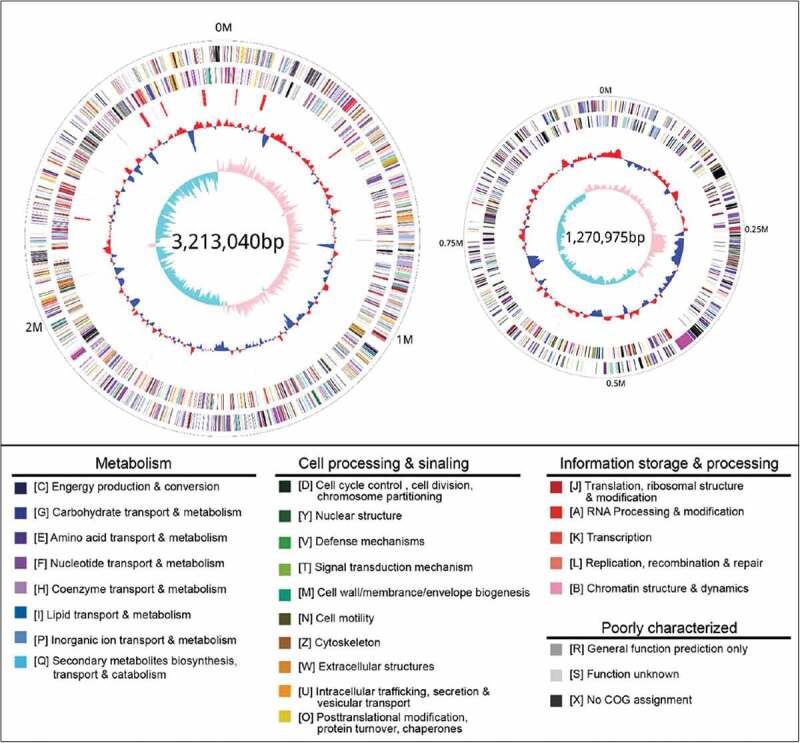
Circular graph of V. mimicus strain SCCF01 genome. Rings from the outermost to the center: (1) scale marks of the genome; (2) protein-coding genes on the forward strand; (3) protein-coding genes on the reverse strand; (4) tRNA (black) and rRNA (red) genes on the forward strand; (5) tRNA (black) and rRNA (red) genes on the reverse strand; (6) GC content; (7) GC skew. Protein-coding genes are color-coded according to their COG categories.
Virulence factors
Based on the VFDB database [18], we scanned the V. mimicus SCCF01 genomes for virulence-related features. In total, we identified 107 putative orthologs involved in the production of any of the above virulence factors (summarized in Table 2). This reported dataset should be applied to the development of gene attenuated vaccine and can serve as the basis for future studies concerning interactions of V. mimicus strain SCCF01 and diseases. Here, we also compared the genomic virulence genes of V. mimicus SCCF01 with the clinical strain (ATCC33655) and environmental strain (ATCC33654). The three genomes analyzed shared 539 virulence genes in the core genome and the Specific virulence genes were 74 (SCCF01), 73 (ATCC33655) and 52 (ATCC33654) respectively (Supplementary Figure 2). Characteristics of common and specific virulence genes were summarized in Supplementary Table. The results showed that various virulence genes related to intestinal infections exists only in clinical strain (ATCC33655), such as cholera enterotoxin (ctx), accessory cholera enterotoxin (ace), zona occludens toxin(zot) and toxin co-regulated pilus (tcp). Interestingly, the other type VI secretion system (T6SS) gene cluster (VM_12775 ~ VM_12835) which is different with Vibrio exists in strain SCCF01. In addition, various virulence genes that are from other species exist only in strain SCCF01. Whether these extraneous virulence factors cause Siluriformes diseases need to be investigated further.
Table 2.
Virulence factors of V. mimicus strain SCCF01.
| Annotation | Virulence factor | Virulence factor | |
|---|---|---|---|
| Adherence | Accessory colonization factor | - | - |
| Mannose-sensitive hemagglutinin | mshA~mshQ | VM13460 ~ VM13540 | |
| Outer Membrane Protein U | OmpU | VM12035 | |
| Toxin-coregulated pilus | - | - | |
| Type IV pilus | pilD, pilC, pilB, pilA | VM02850 ~ VM02865 | |
| Flagellum system | Capsular polysaccharide | flaA and flaC | VM03930 ~ VM03935 |
| flaE, flaD, flab, flaG | VM04235 ~ VM04250 | ||
| flhB | VM04355 | ||
| fliD, flaI, fliS, flrA, flrB, flrC | VM04255 ~ VM04280 | ||
| flgB ~ flgL | VM03875 ~ VM03925 | ||
| flgT, flgO, flgP, flgN, flgM, flgA | VM03835 ~ VM03860 | ||
| Flagella motor protein | motA and motB | VM10690 ~ VM10685 | |
| motY and motX | VM10115, VM01930 | ||
| Flagella | flgA~flgN | VM19705 ~ VM19770 | |
| fliE~fliR | VM04285 ~ VM04350 | ||
| Chemotaxis protein | cheY, cheZ, cheA, cheB, cheW | VM04615 ~ VM04645 | |
| cheV and cheR | VM03865 ~ VM03870 | ||
| Toxin | Hemolysin | VMH, TLH | VM17590, VM17595 |
| Enterotoxin | Enterotoxin | VM17705 | |
| Proteases | Metalloproteases, Neuraminidase | VM20315, VM15685 | |
| Secretion system | type III secretion system | - | - |
| EPS type II secretion system | epsC ~ epsN | VM00880 ~ VM00935 | |
| type VI secretion system | vgrG-3, vasa ~ vasL, vipA~B | VM18140 ~ VM18220 | |
| Iron uptake | Heme receptors | HutA | VM16145 |
| Siderophore receptors | FhuA | VM14565 | |
| Siderophores (Enterobactin, Vibriobactin and Aerobactin) | vctA, irgA | VM17540, VM17545 | |
| viuA, viuB | - | ||
| iutA | VM09630 | ||
| TonB1 system | ExbB1, ExbD1, TonB1 | VM20125 ~ VM20135 | |
| TonB2 system | ExbB2, ExbD2, TonB2 | VM07220 ~ VM07230 | |
| Transport of iron complexes | hmuV and hutC | VM08650 ~ VM08655 | |
| FhuC, FhuD, FhuB | VM14550 ~ VM14560 | ||
| vctC, vctG, vctD, vctP | VM17550 ~ VM17565 | ||
| Feo system | FeoA and FeoB | VM04550 ~ VM04555 | |
| Fbp system | FbpA, FbpB, FbpC | VM12160 ~ VM12150 |
Genome element prediction
Mobile elements including repetitive elements, genomic islands, and phages within genomes have driven bacterial horizontal gene transfer and evolution [29–31].
Repetitive elements (also known as repeated sequences) are repetitive multiple copies of DNA sequences that do not have transcriptional activity. According to their structure, repetitive elements can be divided into Interspersed Repeat Sequences (IRS) and Tandem Repeats Sequence (TRS). Repetitive elements within genomes play an important role in the evolutionary process [32]. SCCF01 genome was screened by online program RepeatMasker, and the results (Table 3) showed that the total IRS percentage of the strain SCCF01 genome in Chromosome I and Chromosome II was 21.79% and 13.75%, respectively. Similarly, the total TRS percentage of the strain SCCF01 genome in Chromosome I and Chromosome II was 7.30% and 8.30%, respectively. The percentage of total IRS in Chromosome I was greater than Chromosome II. However, the percentage of total TRS in Chromosome II was greater than Chromosome I. The IRS is derived from transposable elements (TEs), that are largely responsible for horizontal gene transfer [33]. The TRS can exhibit high-mutation rates [34]. Therefore, we speculated that chromosome I is responsible for structural variation and chromosome II is responsible for single nucleotide change in the evolutionary process of strain SCCF01.
Table 3.
Repeat elements prediction in V. mimicus strain SCCF01.
| Chromosome I |
Chromosome II |
|||||
|---|---|---|---|---|---|---|
| number | length | percentage | number | length | percentage | |
| SINE | 26 | 1700 bp | 5.29% | 1 | 148 bp | 1.16% |
| LINE | 51 | 4310 bp | 13.41% | 15 | 1046 bp | 8.23% |
| LTR | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| DNA element | 14 | 991 bp | 3.08% | 7 | 553bp | 4.35% |
| Unclassified | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| Total IRS | 81 | 7001 bp | 21.79% | 23 | 1747 bp | 13.75% |
| Satellites | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| Simple repeats | 44 | 1870 bp | 5.82% | 16 | 836 bp | 6.58% |
| Low complexity | 9 | 475 bp | 1.48% | 5 | 218 bp | 1.72% |
| Total TRS | 53 | 0 | 7.30% | 21 | 0 | 8.30% |
Genomic islands (GI) are large genomic regions that mediated horizontal gene transfer in bacteria [31]. There are 16 GIs predicted in the genome of the strain SCCF01 by IslandViewer 4 and localization of predicted GIs as shown in Figure 2. Interestingly, 15 predicted GIs were located in the Chromosome I, in this case only one predicted GI was located in Chromosome II. The results of genomic islands prediction indicated that Chromosome I was more likely than Chromosome II to acquire genes via horizontal gene transfer.
Figure 2.
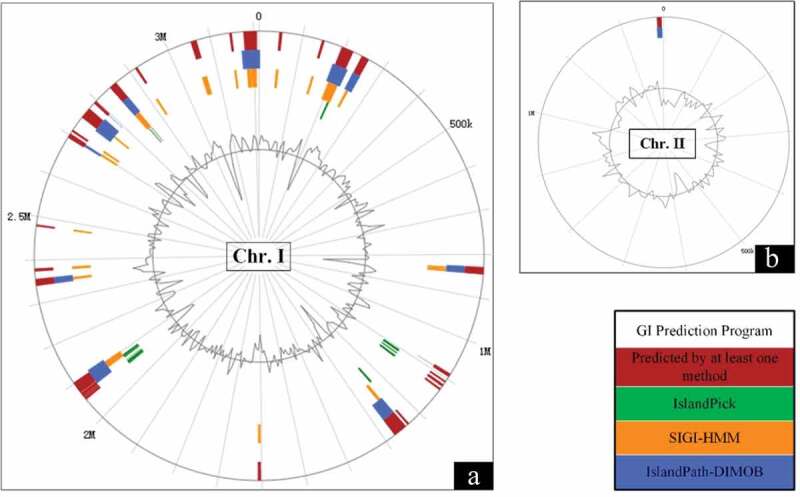
Genomic islands (GI) prediction in V. mimicus SCCF01 by IslandViewer; A: Chromosome I (Chr.I), Chromosome II (Chr.II).
Prophage sequences of 11 V. mimicus strains were predicted by PHAST and the features are listed in Supplementary Table S4. Prophages prediction showed that the strains (SCCF01, ATCC33654, ATCC33655, SX-4, and VM573) contained intact prophage sequences and other strains contained incomplete or questionable prophage sequences. V. mimicus strain SCCF01 uniquely harbored two integrated prophages in the large chromosome (chromosome I) of strain SCCF01, and its CDSs sharing greater identity to the Vibrio phage 12B12 [GenBank: NC_021070.1] (Figure 3). However, no prophage sequences could be detected in chromosome II of strain SCCF01. Recently, many researches have shown that prophage can mediate horizontal gene transfer [35,36]. The results of prophage prediction indicated that chromosome I was capable of horizontal gene transferring by a prophage.
Figure 3.
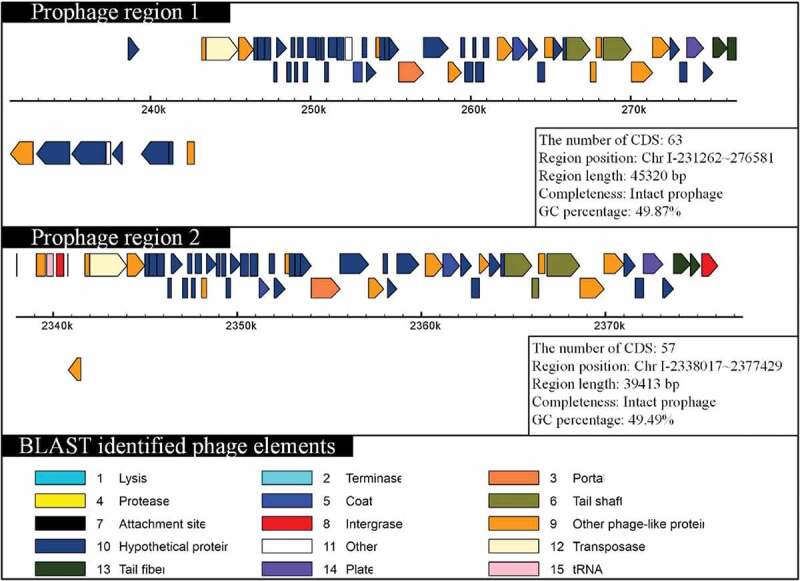
Schematic diagram of prophage organization.
In general, the majority of mobile elements (repetitive elements, genomic islands, and prophage) were detected in chromosome I. These mobile elements are connected closely to horizontal gene transfer contribution to acquire genes [33,35–37]. Therefore, chromosome I of the SCCF01 genome has more plasticity than chromosome II and chromosome I might be enlarged in size.
Collinearity analysis
Herein, Mummer and LASTZ program were applied for a genome-wide collinearity analysis between the SCCF01 genome and standard strains (ATCC33654 and ATCC33655). Our analysis of the V. mimicus SCCF01 genome, suggested that the evolution of the strain SCCF01 genome structure is marked by interchromosomal rearrangements (Figure 4). Structural variation (translocation, inversion, deletion, insertion, and complexindel) is shown in Supplementary Figure 1. Genome-wide collinearity relation and detailed structural variation can be visualized. However, it is difficult to confirm evolutionary relationships.
Figure 4.
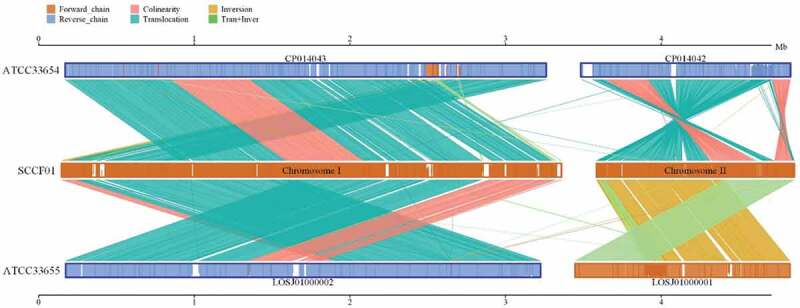
Linear pairwise comparison of the SCCF01 strain genome.
Note: the upper axis and lower axis are standard strain genome, the medial axis is SCCF01 strain genome, sense strand is shown in the yellow box, antisense strand is shown in blue line, color depth in the box shows the similarity of alignment, full-fit indicates 100% similarity.
Gene family analysis
A gene family is a set of several similar genes, formed by duplication of a single original gene. The statistics of gene family numbers were obtained according to the cluster of orthologous group based on protein sequences of SCCF01, ATCC33654, ATCC33655, VM223, and MB-451. Gene family analysis showed that the number (genes number, genes in families, unclustered genes, family number, and unique families) of strain SCCF01 were calculated more than other strains (Table 4). In addition, the number (genes number, genes in families, unclustered genes, family number, and unique families) of environmental strains (ATCC33654 and VM223) were computed more than clinical strain (ATCC33655 and MB-451). The gene gain/loss events might have occurred during the evolution of the genus Vibrio, and the gene gain events in the evolutionary process of V. mimicus and V. cholerae were more frequent than the gene loss events [38]. Therefore, gene family analysis predicted that the evolutionary direction of strain SCCF01 was clinical strain → environmental strain → SCCF01 strain.
Table 4.
The result of gene family analysis.
| Strain | SCCF01 | ATCC33654 | ATCC33655 | VM223 | MB-451 |
|---|---|---|---|---|---|
| Genes number | 4,031 | 3,874 | 3,791 | 3,902 | 3,841 |
| Genes in families | 3,786 | 3,638 | 3,608 | 3,698 | 3,674 |
| Unclustered genes | 245 | 236 | 183 | 204 | 167 |
| Family number | 2,846 | 2,748 | 2,787 | 2,823 | 2,821 |
| Unique families | 46 | 19 | 13 | 24 | 6 |
Note (From left to right): 1. Genes number: Total number of genes, 2. Genes in families:Total number of genes in families, 3. Unclustered genes: The number of, 4. Family number: The gene family numbers, 5. Unique families: The unique numbers.
Phylogenetic analysis
In order to determine the phylogenetic relationship of the SCCF01, genome tree analysis was performed from V. mimicus species (Figure 5) and Vibrio genus (Supplementary Figure 2) based on locally collinear block searching. The phylogenetic tree from V. mimicus species (Figure 5) showed that the isolates were roughly divided into two clusters: clinical V. mimicus (ATCC33655, SX-4, and MB-451) and environmental V. mimicus (CAIM1882, CAIM1883, ATCC33654, SCCF01, and VM223). The evolutionary relationships inferred by this tree suggest that SCCF01 is more closely related to the environmental isolate.
Figure 5.
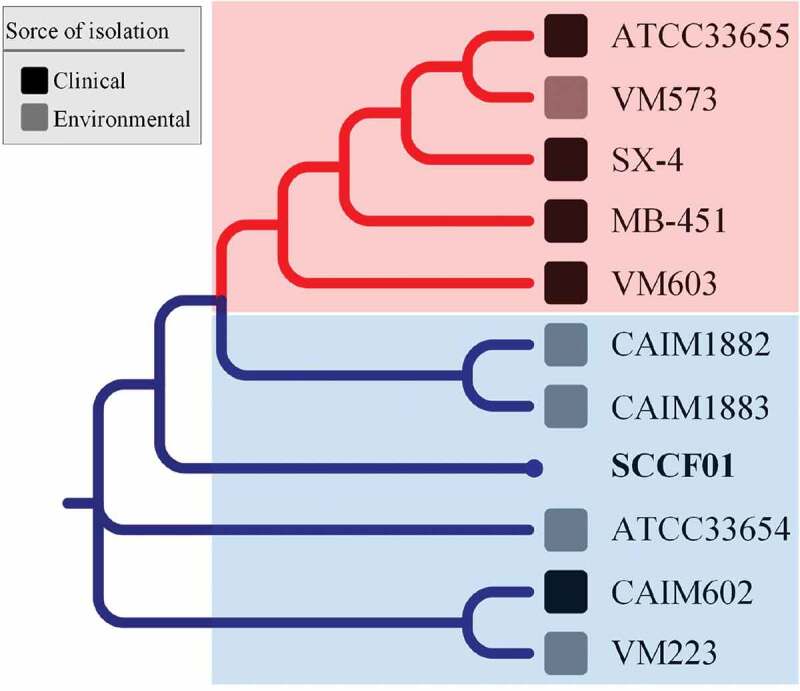
Phylogenetic tree of Vibrio mimicus based on locally collinear block searching.
The phylogenetic tree from Vibrio genus (Supplementary Figure 3) showed that the strain SCCF01 was classified into the V. mimicus cluster and further proved that the strain SCCF01 was determined to be V. mimicus on the genome level.
Conclusions
First, Genome analysis of V. mimicus strain SCCF01 revealed common basic features. The information of virulence (adhesion, flagellum system, exotoxin, and secretory system) was obtained by virulence genes annotation and will be useful for the development of gene attenuated vaccine and pathogenesis study for this pathogen. Secondly, chromosome I of the SCCF01 genome has more plasticity than chromosome II and might be larger in size. Finally, we speculate that the strain SCCF01 has probably diverged from environmental strains based on gene family analysis and phylogenetic analysis.
Funding Statement
This work was supported by the Sichuan Innovation Team Project of Agricultural Industry Technology System [2017SICAD002]; Key research and development program of Sichuan province (No. 2018NZ0007).
Acknowledgments
We thank Dr. Mujibur Rahman (Sichuan Agricultural University) for providing editorial comments on an earlier draft of our manuscript and Mr. Madan Mohan (National University of Singapore) for proofreading the language of our manuscript. This work was supported by Key research and development program of Sichuan province (No. 2018NZ0007), and Sichuan Innovation Team Project of Agricultural Industry Technology System (No.2017SICAD002).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Davis BR, Fanning GR, Madden JM, et al. Characterization of biochemically atypical Vibrio cholerae strains and designation of a new pathogenic species, Vibrio mimicus. J Clin Microbiol. 1981;14:631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vieira VV, Teixeira LFM, Vicente ACP, et al. Differentiation of environmental and clinical isolates of Vibrio mimicus from Vibrio cholerae by multilocus enzyme electrophoresis. Appl Environ Microbiol. 2001;67:2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Muller S, Chau H, Boudreaux K.. Vibrio Mimicus as the rare cause of acute diarrheal illness. J Louisiana State Med Soc Official Organ Louisiana State Med Soc. 2016;168:192. [PubMed] [Google Scholar]
- [4].Terceroalburo JJ, Gonzálezmárquez H, Bonillagonzález E, et al. Identification of capsule, biofilm, lateral flagellum, and type IV pili in Vibrio mimicus strains. Microb Pathog. 2014;76:77–83. [DOI] [PubMed] [Google Scholar]
- [5].Shandera WX, Johnston JM, Davis BR, et al. Disease from infection with Vibrio mimicus, a newly recognized Vibrio species. Clinical characteristics and edipemiology. Ann Intern Med. 1983;99:169–171. [PubMed] [Google Scholar]
- [6].Raja RA, Panigrahi A, De D, et al. Investigations on white spot disease outbreak in Penaeus monodon (Fabricius, 1798) in association with Vibrio mimicus infection in the Sunderbans, West Bengal, India. Indian J Fish. 2017;64:56. [Google Scholar]
- [7].Kay MK, Cartwright EJ, Maceachern D, et al. Vibrio mimicus infection associated with crayfish consumption, Spokane, Washington, 2010. J Food Prot. 2012;75:762. [DOI] [PubMed] [Google Scholar]
- [8].Li YY, Li JN, Yu WY. Selection of optimal producing condition of the exotoxin of Vibrio mimicus from Eriocheir sinensis. J Fish China. 2003;27:468–473. [Google Scholar]
- [9].Geng Y, Liu D, Han S, et al. Outbreaks of vibriosis associated with Vibrio mimicus in freshwater catfish in China. Aquaculture. 2014;433:82–84. [Google Scholar]
- [10].Zhang X, Li YW, Mo ZQ, et al. Outbreak of a novel disease associated with Vibrio mimicus infection in fresh water cultured yellow catfish, Pelteobagrus fulvidraco. Aquaculture. 2014;432:119–124. [Google Scholar]
- [11].Austin B. Vibrios as causal agents of zoonoses. Vet Microbiol. 2010;140:310–317. [DOI] [PubMed] [Google Scholar]
- [12].Yu Z, G Y, W K, et al. Complete genome sequence of Vibrio mimicus strain SCCF01 with potential application in fish vaccine development. Virulence. 2017;8:1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen C, Geng Y, Yu ZH, et al. Dynamic distribution of Vibrio mimicus in infected yellow catfish (Pelteobagrus fulvidraco) and its hispathological changes. South China Fish Sci. 2017;13:10–18. [Google Scholar]
- [14].Angiuoli SV, Gussman A, Klimke W, et al. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS. 2008;12:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lagesen K, Hallin P, Rødland EA, et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lowe TM, Chan PP. tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–W7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Krzywinski M, Schein J, Birol İ, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen L, Zheng D, Bo L, et al. VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016;44:D694–D7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tempel S. Using and understanding repeatmasker. Methods Mol Biol. 2012;859:29. [DOI] [PubMed] [Google Scholar]
- [20].Bertelli C, Laird MR, Williams KP, et al. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017;45:W30–W5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhou Y, Liang Y, Lynch KH, et al. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Harris RS. Improved pairwise alignment of genomic DNA. [Dissertations & Theses – Gradworks]; 2007. [Google Scholar]
- [24].Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. [DOI] [PubMed] [Google Scholar]
- [25].Ruan J, Li H, Chen Z, et al. TreeFam: 2008 update. Nucleic Acids Res. 2008;36:D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bi G, Mao Y, Xing Q, et al. HomBlocks: A multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 2017;110:18–22. [DOI] [PubMed] [Google Scholar]
- [27].Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Oxford University Press; 2006:2688–2890. [DOI] [PubMed] [Google Scholar]
- [28].He Z, Zhang H, Gao S, et al. Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016;44:W236–W41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jr KH. Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. [DOI] [PubMed] [Google Scholar]
- [30].Brownjaque M, Calerocáceres W, Muniesa M. Transfer of antibiotic-resistance genes via phage-related mobile elements. Plasmid. 2015;79:1–7. [DOI] [PubMed] [Google Scholar]
- [31].Juhas M, Meer JRVD, Gaillard M, et al. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev. 2009;33:376–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bourque G. Transposable elements in gene regulation and in the evolution of vertebrate genomes. Curr Opin Genet Dev. 2009;19:607–612. [DOI] [PubMed] [Google Scholar]
- [33].Ullastres A, Merenciano M, Guio L, González J . Transposable Elements: A Toolkit for Stress and Environmental Adaptation in Bacteria. Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria. US: Wiley; 2016. p.137–145. [Google Scholar]
- [34].Gelfand Y, Rodriguez A, Benson G. TRDB – the tandem repeats database. Nucleic Acids Res. 2006;35:D80–7. Advance Access Published December. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Eggers CH, Gray CM, Preisig AM, et al. Phage-mediated horizontal gene transfer of both prophage and heterologous DNA by φBB-1, a bacteriophage of Borrelia burgdorferi. Pathog Dis. 2016;74:107. [DOI] [PubMed] [Google Scholar]
- [36].Lemire S, Figueroabossi N, Bossi L. Horizontal gene transfer in the evolution of pathogenesis: prophage contribution to salmonella virulence and diversity. UK: Cambridge University Press; 2008. [Google Scholar]
- [37].Hacker J, Kaper JB. Pathogenicity islands and the evolution of microbes. Germany: Springer; 2002. [DOI] [PubMed] [Google Scholar]
- [38].Lin H, Yu M, Wang X, et al. Comparative genomic analysis reveals the evolution and environmental adaptation strategies of vibrios. Bmc Genomics. 2018;19:135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.