

Abstract
IL-17A is an important cytokine in intestinal inflammation. However, anti-IL-17A therapy does not improve clinical outcomes in patients with Crohn’s disease. We aimed to evaluate the role of RORγt+ innate lymphoid cells (ILCs) in murine colitis models in the absence of IL-17A. An acute colitis model was induced with either dextran sulfate sodium (DSS) or trinitrobenzenesulfonic acid (TNBS) and a chronic colitis model was induced by CD4+CD45RBhi T cell transfer from either wild-type C57BL/6 or Il17a−/− mice. An anti-IL-17A antibody, secukinumab, was also used to inhibit IL-17A function in the colitis model. Flow cytometry was performed to analyze the population of RORγt+ ILCs in the colonic lamina propria of mice with chronic colitis. Acute intestinal inflammation due to DSS and TNBS was attenuated in IL-17A knockout mice, whereas chronic colitis was not relieved by T cell transfer from Il17a−/− mice (% of original body weight: wild-type mice vs. Il17a−/− mice, 81.9% vs. 82.2%; P = 0.922). However, the mean proportion of Lin-RORγt+ lymphocytes was higher after T cell transfer from Il17a−/− mice than that after T cell transfer from wild-type mice (28.8% vs. 18.5%). The proportion of Lin-RORγt+ was also increased in Rag2−/− mice that received T cell transfer from wild-type mice when anti-IL-17A antibody was administered (31.7%). Additionally, Il6 and Il22 tended to be highly expressed after T cell transfer from Il17a−/− mice. In conclusion, RORγt+ ILCs may have an important role in the pathogenesis of chronic colitis in the absence of IL-17A. Blocking the function of IL-17A may upregulate Il6 and recruit RORγt+ ILCs in chronic colitis, thereby upregulating IL-22 and worsening the clinical outcomes of patients with Crohn’s disease.
Subject terms: Innate immunity, Crohn's disease
Introduction
In patients with Crohn’s disease, IL-17A-producing cells are highly prevalent in the intestinal mucosa, and intestinal mucosal cells exhibit high transcript expression levels of IL-17A1,2. Fecal IL-17A has also been observed in patients with active Crohn’s disease. Based on these findings, anti-IL-17A therapy would be expected to be effective in treating patients with Crohn’s disease3. However, initial trials of anti-IL-17A therapy for Crohn’s disease has yielded disappointing results. In a clinical trial of anti-IL-17A antibody, secukinumab, for Crohn’s disease did not improve symptoms3. Moreover, severe adverse events, including cases of Crohn’s disease worsening further, occurred in the secukinumab group. A phase II trial of brodalumab, an IL-17R blocker, also showed no benefit for Crohn’s disease compared to placebo4. In the context of the successful outcomes of secukinumab treatment for psoriasis and rheumatoid arthritis5–7, the lack of efficacy for secukinumab in treating Crohn’s disease was highly disappointing. Unlike the negative results of anti-IL-17A therapy, treatment with IL-12/IL-23 antagonists or selective IL-23 antagonists, including ustekinumab, risankizumab, and brazikumab, which block the upstream pathway of IL-17, are effective against Crohn’s disease8–10.
In the phase 3 trials of secukinumab in plaque psoriasis and psoriatic arthritis, two patients developed Crohn’s disease after treatment with secukinumab. Moreover, a case of rapid onset of fulminant inflammatory bowel disease after a single secukinumab infusion has been reported11,12. We hypothesized that innate immunity influences the response to anti-IL-17A therapy for Crohn’s disease, since RORγt+ ILCs are primarily distributed in the intestine rather than the skin or joints13,14.
Several hypotheses have been proposed to explain the lack of efficacy of anti-IL-17A therapy in treating Crohn’s disease; these hypotheses have included the complexity of Th17 biology, the role of IL-17A in yeast immunity, and the action of IL-17A on intestinal epithelium to promote barrier function15–17. However, Th17 cells’ complex polarity and epithelial barrier integrity may not be the only possible explanation for the failure of anti-IL-17A therapy in Crohn’s disease, because RORγt+ innate lymphoid cells (ILCs) also produce IL-17A18. Moreover, RORγt+ ILCs are almost exclusively found in the intestinal lamina propria and have been suggested to play a crucial role in the pathogenesis of Crohn’s disease13. If anti-IL-17A therapies influence the development of RORγt+ ILCs, these therapies might have unintended outcomes in patients with chronic colitis that are distinct from those in patients with psoriasis.
Therefore, we aimed to evaluate the impact of blocking IL-17A function in acute and chronic colitis mouse models; to this end, we utilized Il17a−/− knockout mice. Subsequently, acute and chronic colitis mouse models were validated using the IL-17A inhibitor secukinumab. We also assessed whether RORγt+ ILCs are recruited in the absence of IL-17A in our chronic colitis mouse model.
Results
Role of IL-17A in the acute colitis model
To investigate the role of IL-17A in acute intestinal inflammation, 3.7 mg of trinitrobenzenesulfonic acid (TNBS) was administered to wild-type (WT) and Il17a−/− mice (Fig. 1). After TNBS treatment, body weight decreased on the first and second days in both groups. The mean body weight of WT and Il17a−/− mice that received TNBS were lower than those that did not (WT, P < 0.001; Il17a−/−, P < 0.001; Fig. 1A). However, among the mice that received TNBS, Il17a−/− mice tended to be heavier than WT mice (P = 0.020). In addition, the mean total colon length of Il17a−/− mice was longer than that of WT mice (Fig. 1B). Although macroscopic inflammation scores increased after TNBS administration in both WT and Il17a−/− mice (WT, P < 0.001; Il17a−/−, P < 0.001), the post-TNBS intestinal inflammation scores of the Il17a−/− mice were lower than those of the WT mice (P = 0.001; Fig. 1C).
Figure 1.

TNBS-induced colitis model. Bodyweight changes (A), representative gross photos of the colorectal area (B), inflammation scores (C), and representative microscopic images of histopathological examination (x200) (D) of wild-type and Il17a−/− mice (wild-type mice without TNBS: n = 4; wild-type mice with TNBS: n = 8; Il17a−/− mice without TNBS: n = 5; Il17a−/− mice with TNBS: n = 10). Bars represent standard errors. * P < 0.05, ** P < 0.01. TNBS, 2,4,6-trinitrobenzenesulfonic acid.
Next, we compared colonic inflammation between WT and Il17a−/− mice in the dextran sodium sulfate (DSS)-induced colitis model (Fig. 2). On the 8th day after DSS treatment, the body weight of both WT and Il17a−/− mice began to decrease. However, Il17a−/− mice tended to lose less weight than WT mice (P = 0.442; Fig. 2A). The total colon length of WT and Il17a−/− mice was similar (Fig. 2B). Microscopic examination of H&E-stained colonic tissue specimens revealed that the intestinal inflammation scores of the Il17a−/− mice after DSS administration tended to be lower than those in the WT mice after DSS administration (P = 0.074; Fig. 2C). The validation study using anti-IL-17A antibody showed similar results to the DSS-induced colitis model in Il17a−/− mice (Fig. 2E,F). WT mice that received anti-IL-17A antibody tended to lose less weight than those who did not (Fig. 2E).
Figure 2.

DSS-induced colitis model. Bodyweight changes (A), representative gross photos of the colorectal area (B), inflammation scores (C), and representative microscopic images of histopathological examination (x200) (D) after administration of DSS for wild-type and Il17a−/− mice (wild-type mice without DSS: n = 5; wild-type mice with DSS: n = 8; Il17a−/− mice without DSS: n = 7; Il17a−/− mice with DSS: n = 9). Bodyweight changes (E), representative gross photos of the colorectal area (F), and representative microscopic images of histopathological examination (x200) (G) according to administration of anti-IL-17A antibody (wild-type mice without DSS: n = 3; wild-type mice with DSS: n = 3; wild-type mice with anti-IL-17A antibody and DSS: n = 4). In the anti-IL-17A antibody group, 100 μL of secukinumab was injected intraperitoneally three times per week starting two weeks before DSS administration, continuing until the end of the experiment. Bars represent standard errors. * P < 0.05, ** P < 0.01. DSS, dextran sulfate sodium; NS, not significant.
Role of IL-17A in the chronic colitis model
To determine the effects of blocking the function of IL-17A in chronic colitis, we induced colonic inflammation in Rag2−/− mice via a transfer of CD4+CD45RBhi T cells isolated from WT or Il17a−/− mice. As shown in Fig. 3A, the body weight of Rag2−/− mice that received T cells from WT mice decreased starting the 2nd week after the transfer. In contrast, Rag2−/− mice that received T cells from Il17a−/− mice maintained their weight until the 4th week, although they also presented with lower body weight than control mice (Rag2−/− mice that did not receive T cells). Throughout the monitoring period (week 0 to week 9), the mean body weight of mice that received T cells from Il17a−/− mice was higher than that of mice that received T cells from WT mice (P = 0.012 by repeated measures analysis of variance (ANOVA)). Eventually, however, the body weight of Rag2−/− mice that received T cells from Il17a−/− mice decreased from the 6th week onward. Nine weeks after T cell transfer, body weight did not differ between mice that received T cells from WT mice and those that received T cells from Il17a−/− mice (% of original body weight: 81.9% vs. 82.2%; P = 0.922). The total colon length was also similar between mice that received T cells from WT mice and mice that received T cells from Il17a−/− mice (Fig. 3B). In addition, the intestinal inflammation scores of H&E-stained colonic tissue specimens from the two groups were not significantly different (P = 0.494; Fig. 3C).
Figure 3.

CD4+CD45RBhi T cell transfer-induced colitis model. Bodyweight changes (A), representative gross photos of the colorectal area (B), inflammation scores (C), and representative microscopic images of histopathological examination (x200) (D) after T cell transfer from wild-type or Il17a−/− mice (Rag2−/− mice without T cell transfer: n = 13; Rag2−/− mice with T cell transfer from wild-type mice: n = 8; Rag2−/− mice with T cell transfer from Il17a−/− mice: n = 8). Bodyweight changes (E), representative gross photos of the colorectal area (F), representative microscopic images of histopathological examination (x200) (G) after T cell transfer from wild-type mice according to administration of anti-IL-17A antibody (Rag2−/− mice without T cell transfer [control]: n = 3; Rag2−/− mice received T cell transfer from wild-type mice: n = 2; Rag2−/− mice received T cell transfer from wild-type mice and anti-IL-17A antibody: n = 2). In the anti-IL-17A antibody group, 100 μL of secukinumab was injected intraperitoneally three times per week from the time of T cell transfer to the end of the experiment. Bars represent standard errors. *P < 0.05, **P < 0.01. NS, not significant.
In the validation study using anti-IL-17A antibody, chronic colitis was induced in all the Rag2−/− mice that received T cells (Fig. 3E–G). Although statistical significance was not achieved in the comparison between the T cell transfer with anti-IL-17A antibody group and the group that did not receive anti-IL-17A antibody due to a small sample size, administration of anti-IL-17A antibody could not attenuate chronic colitis.
These data indicate that blocking IL-17A function did not prevent chronic colitis, although we noted that induction of colitis was delayed in mice that received T cells from Il17a−/− mice. To compare the difference between T cell transfer from WT mice and that from Il17a−/− mice, we analyzed the expression levels of various inflammatory cytokines including IL-12, IFN-γ, and IL-6 in colonic tissue samples from Rag2−/− mice via enzyme-linked immunosorbent assay (ELISA) (Fig. S1). Although none of the differences were statistically significant, all cytokines tended to show higher expression after T cell transfer (from both WT and Il17a−/− mice). The expression levels of IL-12 and IFN-γ, which are associated with Th1 and group 1 ILC differentiation, were similar between mice that received T cells from WT versus Il17a−/− mice (IL-12p40: 837.8 ± 183.3 pg/mL vs. 830.9 ± 216.4 pg/mL, P = 0.982; IFN-γ: 205.4 ± 34.4 pg/mL vs. 147.2 ± 57.7 pg/mL, P = 0.416). However, expression of IL-6, which is associated with Th17 and RORγt+ ILC differentiation, tended to be higher in mice that received T cells from Il17a−/− mice than in mice that received T cells from WT mice (T cell transfer from WT mice vs. T cell transfer from Il17a−/− mice: 14.7 ± 4.0 pg/mL vs. 28.9 ± 6.1 pg/mL, P = 0.134).
Innate lymphoid cells in the chronic colitis model
To evaluate ILC changes in the chronic colitis model, we analyzed colonic lamina propria cells from WT, Il17a−/−, and Rag2−/− mice with flow cytometry. As shown in Fig. 4, relatively small populations of RORγt+ cells were observed in WT and Il17a−/− mice (4.5% and 6.0%, respectively). Of particular note, the percentages of Lin-RORγt+ cells [which include group 3 ILCs (LTi cells and ILC3s)] were 4.5% and 5.9% in WT and Il17a−/− mice, respectively. In the absence of adaptive immunity (i.e. in Rag2−/− mice), the proportion of RORγt+ ILCs was higher than in WT or Il17a−/− mice (WT vs. Il17a−/− vs. Rag2−/− mice, 4.5% vs. 5.9% vs. 16.8%; Fig. 4C), which is consistent with a previous study19.
Figure 4.

Flow cytometry analysis of colonic lamina propria lymphocytes in wild-type C57BL/6 mice (A), Il17a−/− mice (B), and Rag2−/− mice (C). FSC, forward scatter; SSC, side scatter.
After transferring CD4+CD45RBhi T cells from WT mice, the proportion of Lin-RORγt+ lymphocytes was similar to that in Rag2−/− mice that did not receive T cells (T cell transfer from WT mice vs. without T cell transfer: 18.5% vs. 16.8%, Figs. 4C and 5A). The majority of RORγt+ ILCs expressed CD4 and did not express NKp46 (Fig. 5A). These results show that, in the absence of adaptive immunity, most RORγt+ ILCs were CD4+LTi cells, while CD4-LTi cells and ILC3s represented only a minor portion of ILCs.
Figure 5.

Flow cytometry analysis of colonic lamina propria lymphocytes in Rag2−/− mice that received T cell transfers from wild-type mice (A) or Il17a−/− mice. (B) Flow cytometry analysis was performed on the 9th week after the T cell transfer. FSC, forward scatter; SSC, side scatter.
In mice that received T cells from Il17a−/− mice, the proportion of Lin-RORγt+ lymphocytes increased to 28.8% (Fig. 5B). The majority of RORγt+ ILCs also expressed CD4 and did not express NKp46 (T cell transfer from WT mice vs. T cell transfer from Il17a−/− mice: CD4+RORγt+ ILCs, 21.7% vs. 26.7%; NKp46+RORγt+ ILCs, 0.02% vs. 0.25%). However, the proportion of CD4-RORγt+ ILCs was also slightly elevated compared to the proportion of mice that received T cells from WT mice (T cell transfer from WT mice vs. T cell transfer from Il17a−/− mice: 1.4% vs. 4.7%). In addition, the proportion of CD4-RORγt- ILCs, which included ILC1s, was increased in Rag2−/− mice that received T cells from Il17a−/− mice compared to mice that received T cells from WT mice (T cell transfer from WT mice vs. T cell transfer from Il17a−/− mice: CD4-RORγt- ILCs, 9.4% vs. 26.9%). Overall, RORγt+ ILCs and ILC1s were more prevalent in Rag2−/− mice that received T cells from Il17a−/− mice than in mice that received T cells from WT mice.
Findings were similar in experiment using anti-IL-17A antibody (Fig. 5C). RORγt+ ILCs were observed in large numbers in Rag2−/− mice that received T cells from WT mice when anti-IL-17A antibody was administered. Furthermore, the majority of RORγt+ ILCs expressed CD4 (39.8%), but they barely expressed NKp46 (0.1%).
Innate lymphoid cells in the spleens of mice with chronic colitis
To determine whether ILCs were exclusively enriched in the intestine, we used flow cytometry to analyze spleen cells from mice with T cell transfer-induced colitis. As shown in Fig. S2, the lymphocyte proportions of RORγt+ ILCs were similar among Rag2−/− mice that did not receive T cells, Rag2−/− mice that received T cells from WT mice, and Rag2−/− mice that received T cells from Il17a−/− mice (6.4% vs. 7.6% vs. 5.2%, respectively). Additionally, the lineage-negative lymphocyte proportions of CD4+RORγt+ ILCs were similar between Rag2−/− mice that received T cells from WT mice and those that received T cells from Il17a−/− mice (4.6% vs. 4.5%).
Innate lymphoid cells in the colonic lamina propria with acute colitis model
To determine whether the increase in the proportion of Lin-RORγt+ lymphocytes is a characteristic finding in the chronic colitis model, we analyzed colonic lamina propria cells from WT and Il17a−/− mice with and without DSS treatment. As shown in Fig. S3, only small populations of RORγt+ cells were observed in the acute colitis model (both WT and Il17a−/− mice [1.1% and 2.0%, respectively]).
Transcript expression in mice with chronic colitis
The transcript expression levels of various cytokines, including IL-12, IFN-γ, IL-6, Il-23, IL-17, and IL-22 were evaluated next via quantitative real-time polymerase chain reaction (Fig. 6). Although statistical significance was not reached due to a limited sample size, Il6 and Il22, which are involved in Th17 and RORγt+ ILC differentiation, tended to be highly expressed in mice that received T cells from Il17a−/− mice. Additionally, Il12p40 and Ifng also showed a trend of high expression in mice that received T cells from Il17a−/− mice, even though IL-12 and IFN-γ are more closely related to Th1 and ILC1 differentiation than Th17 and RORγt+ ILC differentiation. Since a subpopulation of RORγt+ ILCs may downregulate RORγt and acquire the capacity to produce IFNγ20, these findings imply that innate immunity (especially RORγt+ ILCs) is involved in the pathogenesis of T cell transfer-induced colitis.
Figure 6.
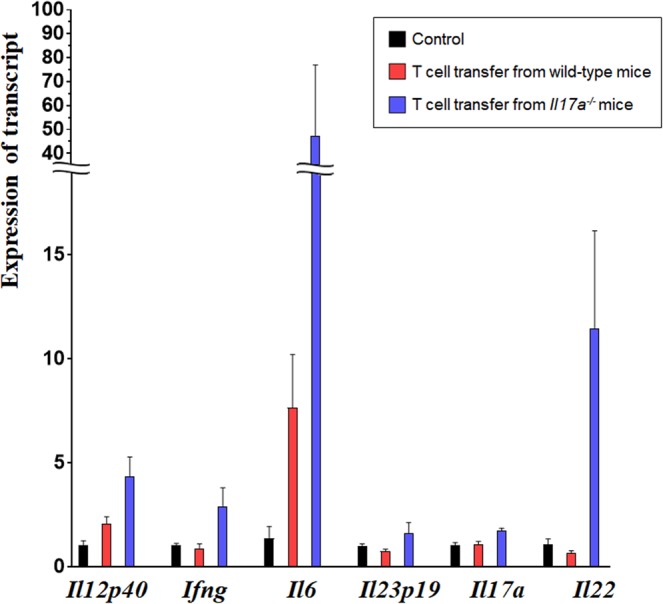
Expression of transcripts in colon tissue from mice with T cell-induced colitis. The relative expression of each transcript was measured by quantitative real-time polymerase chain reaction and calculated with the 2−Δ ΔCT method using β-actin levels for normalization (Rag2−/− mice without T cell transfer: n = 3; Rag2−/− mice with T cell transfer from wild-type mice: n = 3; Rag2−/− mice with T cell transfer from Il17a−/− mice: n = 3). There was no significant difference between any of the groups. Although statistical power was insufficient, Il6 and Il22 tended to be higher in Rag2−/− mice with T cell transfer from Il17a−/− mice than those with T cell transfer from wild-type mice. Bars represent standard errors. All reactions were performed in duplicate.
Discussion
We found that the absence of IL-17A tended to attenuate acute colitis. These results are consistent with previous studies of TNBS-induced and DSS-induced colitis models, both of which demonstrated that IL-17A plays a pathogenic role in acute colitis17,21. In contrast, the severity of chronic colitis was not affected by T cell transfer from Il17a−/− mice or anti-IL-17A therapy22,23. Our study also demonstrated that blocking IL-17A function did not attenuate chronic colitis. In addition, we found that weight loss in mice that received T cells from Il17a−/− mice occurred later than in mice that received T cells from WT mice.
One of the possible reasons for the difference in the outcome of anti-IL-17A therapy between acute and chronic colitis models may be the different effects of cytokines depending on the phase of colitis. It has been shown that the mucosal pattern of effector cytokines differs according to the different stages of Crohn’s disease24. For example, in the early stages of Crohn’s disease, macroscopically unaffected neoterminal ileum contained high levels of IFN-γ and IL-21, which are produced by Th1 cells. On the contrary, in the advanced lesions of Crohn’s disease, marked up-regulation of IL-17A and induction of IL-23 and IL-6 were observed.
In the current study, we hypothesize that the delay in colitis development between mice that received T cells from WT mice and those that received T cells from Il17a−/− mice was due to the differentiation of ILCs that are involved in chronic colitis in the absence of IL-17A. Anti-IL-17A therapy can attenuate acute colitis because IL-17A is a potent inducer of neutrophil-promoting cytokines. In cases of chronic colitis, however, disruption of epithelial barrier integrity and activation of ILC may worsen disease activity due to treatment with anti-IL-17A therapy. Although weight loss was similar at the 9th week between the two chronic colitis mouse models (T cell transfer from WT mice vs. T cell transfer from Il17a−/− mice), the proportions of ILCs in the intestinal lamina propria were different. In particular, the proportion of RORγt+ ILCs was increased in the absence of IL-17A. Additionally, the majority of RORγt+ ILCs were CD4+LTi cells. In the intestinal lamina propria, CD4+LTi cells may play a major role in the development of chronic colitis in the absence of IL-17A. In addition, Il6 and Il22, which are involved in LTi cell differentiation13, were highly expressed in mice that received T cells from Il17a−/− mice. The elevated expression of Il6 in the absence of IL-17A is particularly noteworthy because IL-6 is a critical cytokine that promotes IL-17A production in both humans and mice13,25. RORγt+ ILCs (which include LTi cells) respond to IL-6 and produce the effector cytokine IL-22. Thus, a lack of IL-17A may upregulate IL-6 and recruit RORγt+ ILCs via negative feedback mechanisms; thus, IL-22 can be upregulated and worsen the clinical outcomes of patients with Crohn’s disease13. In order to prevent this unintended response of ILC, simultaneous blocking of IL-17A and IL-6 may be considered as a potential strategy in the treatment of Crohn’s disease.
Although IFN-γ is an effector cytokine of ILC1s and not RORγt+ ILCs, we observed a trend of high expression of Ifng in the mice that received T cells from Il17a−/− mice. An increase of IFN-γ may be caused by the activation of pathogenic T cells; however, we proposed that IFN-γ would be increased by ILC1s in the absence of IL-17A. In our study, we found that the proportions of ILCs other than RORγt+ ILCs (e.g., ILC1s) were also increased in mice with chronic colitis in the absence of IL-17A. Although the majority of the elevated ILCs were CD4+LTi cells that belonged to the RORγt+ ILC group, a subpopulation of RORγt+ ILCs may downregulate RORγt and gain the ability to produce IFNγ20. Our flow cytometry analysis showed that Lin-RORγt- cells, which included ILC1s, were more abundant in Rag2−/− mice that received T cells from Il17a−/− mice than in those that received T cells from WT mice. Immune cell plasticity is a distinctive characteristic in adaptive immunity as well as innate immunity. Previous studies have demonstrated that Tregs can be transdifferentiated from Th17 cells by myeloid-derived suppressor cells26–28. Our results indicate that blocking IL-17A function may also increase the population of RORγt+ ILCs (which include LTi cells) via IL-6 production. Moreover, some RORγt+ ILCs may transform into ILC1s. RORγt+ ILCs and transformed ILC1s may eventually worsen intestinal inflammation via IL-22 and IFN-γ, respectively, in chronic colitis when IL-17A is absent. Therefore, additional targets besides IL-17A are required to control intestinal inflammation in chronic colitis. As mentioned above, we propose that IL-6 as a potential target for Crohn’s disease. A study reporting a pilot randomized trial of anti-IL-6R monoclonal antibodies suggested a clinical effect in active Crohn’s disease29, which supports our theory.
In addition, we demonstrated that the proportion of RORγt+ ILCs in spleen lymphocytes did not increase in mice with T cell transfer-induced colitis. Since ILCs are distributed in a tissue-specific manner, the effects of ILC differentiation may drive different outcomes according to the type of disease. These characteristics of ILCs may explain the different outcomes of anti-IL-17A therapy in the context of psoriasis, RA, and Crohn’s disease, each of which involves different organs.
Additionally, we identified that an increase in the proportion of RORγt+ ILCs was observed only in the chronic colitis model and not in the acute colitis model. Anti-IL-17A therapy in patients with chronic colitis seems to be unable to improve inflammation, perhaps because it may induce RORγt+ ILCs.
In addition to the potential causes mentioned above, changes in the gut microbiome may influence the deterioration of chronic colitis after anti-IL-17A therapy. In the previous study, a higher frequency of fungal infection was reported in patients with Crohn’s disease after treatment with secukinumab3. If anti-IL-17A therapy affects not only fungi but also the gut microbiome, this may increase disease activity of Crohn’s disease.
In this article, we outlined a potential mechanism by which the unexpected results of anti-IL-17A therapy in Crohn’s disease can be explained, and we suggested potential treatment targets. However, our study did have several limitations. First, we mainly evaluated the outcomes of anti-IL-17A therapy in Rag2−/− mice that received T cells from Il17a−/− mice. Although we validated the findings using additional experiments with an anti-IL-17A antibody, the sample size of the validation study was limited. However, overall findings were similar between the study with T cell transfer from Il17a−/− mice and that with anti-IL-17A antibody. Second, the results for the mRNA profile and flow cytometry were rather descriptive. Although we showed that blocking IL-17A function may be associated with upregulation of Il6 and recruits RORγt+ ILCs in chronic colitis models, the pathogenic relevance of these findings should be investigated further. Moreover, longitudinal measurements of the proportion of ILCs based on the clinical course of the disease after initiating anti-IL-17A therapy may be helpful for understanding ILC differentiation in the context of blocking IL-17A in Crohn’s disease.
Despite these limitations, our study provides a better understanding of the possible mechanisms underlying unresolved intestinal inflammation in the presence of anti-IL-17A therapy in Crohn’s disease. Blocking IL-17A function did not attenuate chronic colitis, although it did reduce intestinal inflammation in acute colitis and in the early phase of chronic colitis. This discrepancy may be caused by Th17 cells heterogeneity or Th17 polarization depending on the stage of intestinal inflammation. Besides, IL-17A blockage may increase the proportion of RORγt+ ILCs (which include CD4+LTi cells) and ILC1s, thereby eventually worsening chronic colitis. RORγt+ ILCs may have an important role in the pathogenesis of chronic colitis in the absence of IL-17A.
Materials and Methods
Mice
WT, Il17a−/−, and Rag2−/− C57BL/6 mice were bred and maintained under specific pathogen-free conditions at the accredited animal facilities at Hanyang University. All experiments were performed in accordance with appropriate guidelines for animal experimentation. The experimental protocol was approved by the Institutional Animal Care and Use Committee of Hanyang University.
Induction of acute colitis
Acute colitis was induced by administering either 3.7 mg of 2,4,6-TNBS in 50% ethanol or 2% DSS. The TNBS- and DSS-induced colitis models were analyzed in separate experiments.
In the TNBS-induced colitis model, animals were fasted overnight for 12 hours prior to hapten TNBS (Sigma-Aldrich, St. Louis, MO) administration. Next, mice were lightly anesthetized using a mixture of 20% v/v isoflurane in propylene glycol. Then, TNBS solution (3.7 mg in 50% ethanol) was administered via an 18 G 1.3 mm diameter IV catheter polyethylene tube. The IV catheter tube was advanced through the rectum into the colon until the tip was 4 cm proximal to the anus. To ensure retention of the haptenating agent within the colon, mice were held in a vertical position for 30 s after TNBS administration to the rectum. Control mice were administered 50% ethanol using the same technique. All mice were sacrificed by cervical dislocation on the 4th day of the experiment.
In the DSS-induced colitis model, DSS (molecular weight 36,000–50,000; MP Biochemicals, Solon, OH) was added to drinking water (2% final concentration) for 5 days. The control group was given the same drinking water without DSS. After 5 days, DSS was removed from the drinking water and the animals received normal drinking water. All mice were sacrificed by cervical dislocation on the 12th day of the experiment.
For the experiments using the acute colitis model, C57BL/6 WT and Il17a−/− mice were randomly assigned to either the control or TNBS (or DSS) group. Therefore, each mouse was allocated to one of the following four groups: WT control, WT TNBS (or DSS), Il17a−/− control, and Il17a−/− TNBS (or DSS).
To validate the results in the Il17a−/− mice, we performed additional experiments using the anti-IL-17A antibody secukinumab. In the validation study, WT mice were randomly assigned to one of the following three groups: control, DSS, or anti-IL-17A antibody with DSS. For the anti-IL-17A antibody with DSS group, 100 μL of secukinumab was injected intraperitoneally three times per week starting two weeks before the DSS administration until the end of the experiment.
Induction of chronic colitis
Chronic colitis was induced via a transfer of naïve CD4+CD45RBhi T cells. The naïve CD4+CD45RBhi T cells were enriched by nylon passage of spleen cells obtained from C57BL/6 WT or Il17a−/− mice and isolated using PE-conjugated anti-CD4 and FITC-conjugated anti-CD45RB monoclonal antibodies (eBioscience, CA) with an FACSAria III Cell Sorting System. The purity of the isolated CD4+CD45RBhi T cells was >99%. Purified CD4+CD45RBhi T cells (1 × 106 cells) from C57BL/6 WT or Il17a−/− mice were injected into Rag2−/− mice intraperitoneally. For histopathological analysis of colitis, mice were sacrificed at 9 weeks after the CD4+CD45RBhi T cell transfer. The body weight of each mouse was checked every two days during the monitoring period.
The validation study using anti-IL-17A antibody was also performed in the chronic colitis model. WT mice were randomly allocated to one of the following groups: control, T cell transfer, or T cell transfer with anti-IL-17A antibody. Intraperitoneal injection of 100 μL of secukinumab was carried out three times per week starting at the time of T cell transfer until the end of the experiment.
Assessment of intestinal inflammation
After sacrificing the mice, the colons were removed, and the entire length of each colon was measured. Then, the colon tissue was washed with ice-cold phosphate-buffered saline. The tissue was cut into several segments and then fixed with 10% phosphate-buffered formalin. Tissue segments were embedded in paraffin, stained with hematoxylin and eosin, and assessed under a light microscope.
In the TNBS-induced colitis model, colonic inflammation was assessed based on the following macroscopic scoring criteria for intestinal inflammation30: 0 points, no injury; 1 point, focal hyperemia or bowel wall edema with no hemorrhage; 2 points, focal hyperemia or bowel wall edema with hemorrhage at one site; 3 points, extended hyperemia or bowel wall edema with hemorrhage at >1 site; and 4 points, extended hyperemia or bowel wall edema with hemorrhage at >1 site and perforation. In the DSS-induced colitis model, intestinal inflammation was graded based on the following histological grading system of colitis, with a number of points being assigned for each category31: (1) inflammation (0–3 points), (2) extent (0–3 points), (3) regeneration (0–4 points), (4) crypt damage (0–4 points), and (5) percent involvement (1–4 points). The total score was the sum of the scores of each category and ranged from 1 to 18. In the CD4+CD45RBhi T cell transfer-induced colitis model, intestinal inflammation was scored based on previously reported criteria, as follows32: (1) degree of inflammation in the lamina propria (0–3 points), (2) goblet cell loss (0–2 points), (3) abnormal crypts (0–3 points), (4) presence of crypt abscesses (0–1 points), (5) mucosal erosion and ulceration (0–1 points), (6) submucosal spread to transmural involvement (0–3 points), and (7) number of neutrophils counted at ×40 magnification (0–4 points). The total score was calculated by adding the scores for each of the seven parameters, for a maximum score of 17.
ELISA
Colon segments (100–200 mg of tissue) were washed with cold phosphate-buffered saline (PBS) and shaken at room temperature in Roswell Park Memorial Institute medium (RPMI) containing 50 µg/mL gentamycin for 30 min at 280 rpm. The colonic tissue fragments were distributed (0.05 g per well) into 24-well flat-bottomed culture plates and incubated in RPMI 1640 medium with 5% fetal bovine serum and 50 µg/mL gentamycin for 24 h at 37 °C. After incubation, the supernatants were collected, spun by centrifugation at 842 g at 4 °C for 15 min, and stored at −70 °C until analysis. IL-12p40, IFN-γ, and IL-6 were measured in colonic culture supernatants by a commercially available ELISA (OptEIA kit, BD Biosciences, USA).
Isolation of ILC subpopulations and flow cytometry
Cell suspensions were prepared from the colonic lamina propria and spleen as previously described19,33. To isolate mononuclear cells from the intestinal lamina propria, gut fragments were cut open and washed three times with PBS by vigorous shaking. Washed gut pieces were subsequently cut into pieces 1–2 cm in length and incubated for 30 min at 37 °C in PBS containing 5% fetal bovine serum, 500 mM EDTA, 1 M HEPES, 100 mM sodium pyruvate, and 1X penicillin/streptomycin. Tissue pieces were washed three times in warm PBS by vigorous shaking and then incubated for 30 min with fresh medium containing collagenase D (1 mg/mL; Roche) and DNase I (1 mg/mL; Roche). The remaining intestinal fragments were filtered through the 100-μm mesh, and the cell suspensions were spun by centrifugation at 431 g for 8 min. After discarding the supernatant, the cell pellets were resuspended in 75% (wt/vol) Percoll (GE Healthcare). Next, 40% (wt/vol) Percoll was added to the suspension. After spinning by centrifugation for 20 min at 1,350 g, mononuclear cells were collected from the 75%/40% interphase. To obtain single spleen cells, the spleens were cut into pieces 5 mm in length and incubated for 30 min at 37 °C in RPMI containing 5% FBS and 1% penicillin/streptomycin. The pieces were then pressed through 100-μm mesh. All cells were first preincubated with monoclonal antibody 2.4G2 (anti-mouse CD16/CD32 mAb; BD Pharmingen) to block Fcγ receptors, after which the cells were washed and incubated for 40 min with the appropriate monoclonal antibody conjugates. Incubations were performed in a total volume of 100 μL PBS containing 2 mM EDTA and 2% (vol/vol) bovine serum. Cells were analyzed on an FACSCanto II instrument (BD Biosciences) with FlowJo software (TreeStar). Cells were sorted with a FACSAria III Cell Sorting System to a purity of 95–98%.
The following antibodies were used for flow cytometry: anti-mouse lineage antibodies conjugated to FITC, anti-RORγt antibodies conjugated to APC, anti-NKp46 antibodies conjugated to PE, and anti-CD4 antibodies conjugated to PE (eBioscience).
Quantitative real-time polymerase chain reaction
Colon tissues were obtained from Rag2−/− mice at 9 weeks after the CD4+CD45RBhi T cell transfer. Total RNA was extracted using QIAzol Lysis Reagent (QIAGEN, Valencia, CA), dissolved in diethylpyrocarbonate (DEPC)-treated water, and quantified using a Biospec-nano spectrophotometer (Life Science, Columbia, MD). A TOPscript™ cDNA synthesis kit (Enzynomics, Daejeon, Republic of Korea) was used for cDNA synthesis. Quantitative real-time PCR was performed with a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA) using TOPreal™ qPCR 2X PreMIX (Enzynomics) and a final volume of 25 μL reactions. All reactions were performed in duplicate in a 96-well plate using the following cycling conditions: 40 cycles of 95 °C for 30 s, 63 °C for 30 s, and 68 °C for 1 min. The relative expression of each transcript was calculated with the 2−Δ ΔCT method using β-actin levels for normalization. The sequences of the primers used for PCR were as follows: Il12p40 forward: CGCAAGAAAGAAAAGATGAAGGAG, Il12p40 reverse: TTGCATTGGACTTCGGTAGATG; Ifng forward: CTTCCTCATGGCTGTTTCTGG, Ifng reverse: ACGCTTATGTTGTTGCTGATGG; Il6 forward: ATGGATGCTACCAAACTGGAT, Il6 reverse: TGAAGGACTCTGGCTTTGTCT; Il23p19 forward: GTCACTAAGAACTAACAGGACTACCA, Il23p19 reverse: TGAAAAGTTCCCTTCCCACTT; Il17a forward: GGTCAACCTCAAAGTCTTTAACTC, Il17a reverse: TTAAAAATGCAAGTAAGTTTGCTG; and Il22 forward: TTGAGGTGTCCAACTTCCAGCA, Il22 reverse: AGCCGGACGTCTGTGTTGTTA. The housekeeping control gene β-actin was used as an internal control.
Statistical analyses
Continuous and ordinal variables, including body weight, intestinal inflammation score, mRNA expression level, and protein expression level, are expressed as means with standard errors. A two-tailed Student’s t-test was used to compare means between groups. For comparisons of longitudinal data such as body weight changes, repeated measures ANOVA was performed. A P-value of < 0.05 was considered to denote a significant difference. All statistical analyses were conducted using R statistical software (version 3.6.0; R Foundation for Statistical Computing, Vienna, Austria).
Supplementary information
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT)(No.2018R1A2B6004475).
Author contributions
Conception and design: D.S.H., Acquisition of data: A.L. and S.B.A., Analysis and interpretation of data: C.H.P., C.S.E. and D.S.H., Drafting of Manuscript: C.H.P., Review of manuscript: C.H.P., A.L., S.B.A., C.S.E. and D.S.H., Study Supervision: D.S.H.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-57233-w.
References
- 1.Fujino S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hölttä V, et al. IL-23/IL-17 immunity as a hallmark of Crohn’s disease. Inflamm. bowel Dis. 2008;14:1175–1184. doi: 10.1002/ibd.20475. [DOI] [PubMed] [Google Scholar]
- 3.Hueber W, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Targan SR, et al. A Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Safety, Tolerability, and Efficacy of AMG 827 in Subjects With Moderate to Severe Crohn’s Disease. Gastroenterology. 2012;143:e26. doi: 10.1053/j.gastro.2012.07.084. [DOI] [Google Scholar]
- 5.Xiong HZ, et al. Efficacy and safety of secukinumab in the treatment of moderate to severe plaque psoriasis: a meta-analysis of randomized controlled trials. Int. J. Clin. Exp. Med. 2015;8:3156–3172. [PMC free article] [PubMed] [Google Scholar]
- 6.Papp KA, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 2012;366:1181–1189. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- 7.Kunwar S, Dahal K, Sharma S. Anti-IL-17 therapy in treatment of rheumatoid arthritis: a systematic literature review and meta-analysis of randomized controlled trials. Rheumatol. Int. 2016;36:1065–1075. doi: 10.1007/s00296-016-3480-9. [DOI] [PubMed] [Google Scholar]
- 8.Feagan BG, et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016;375:1946–1960. doi: 10.1056/NEJMoa1602773. [DOI] [PubMed] [Google Scholar]
- 9.Feagan BG, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. 2017;389:1699–1709. doi: 10.1016/S0140-6736(17)30570-6. [DOI] [PubMed] [Google Scholar]
- 10.Sands BE, et al. Efficacy and Safety of MEDI2070, an Antibody Against Interleukin 23, in Patients With Moderate to Severe Crohn’s Disease: A Phase 2a Study. Gastroenterology. 2017;153:77–86 e76. doi: 10.1053/j.gastro.2017.03.049. [DOI] [PubMed] [Google Scholar]
- 11.Langley RG, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N. Engl. J. Med. 2014;371:326–338. doi: 10.1056/NEJMoa1314258. [DOI] [PubMed] [Google Scholar]
- 12.Mease PJ, et al. Secukinumab Inhibition of Interleukin-17A in Patients with Psoriatic Arthritis. N. Engl. J. Med. 2015;373:1329–1339. doi: 10.1056/NEJMoa1412679. [DOI] [PubMed] [Google Scholar]
- 13.Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med. 2015;21:698–708. doi: 10.1038/nm.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mielke LA, et al. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J. Exp. Med. 2013;210:1117–1124. doi: 10.1084/jem.20121588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaser A. Not all monoclonals are created equal - lessons from failed drug trials in Crohn’s disease. Best. Pract. Res. Clin. Gastroenterol. 2014;28:437–449. doi: 10.1016/j.bpg.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Colombel JF, Sendid B, Jouault T, Poulain D. Secukinumab failure in Crohn’s disease: the yeast connection? Gut. 2013;62:800–801. doi: 10.1136/gutjnl-2012-304154. [DOI] [PubMed] [Google Scholar]
- 17.Maxwell JR, et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity. 2015;43:739–750. doi: 10.1016/j.immuni.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 18.Giuffrida P, Corazza GR, Di Sabatino A. Old and New Lymphocyte Players in Inflammatory Bowel Disease. Dig. Dis. Sci. 2018;63:277–288. doi: 10.1007/s10620-017-4892-4. [DOI] [PubMed] [Google Scholar]
- 19.Sawa S, et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 2011;12:320–326. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- 20.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells–how did we miss them? Nat. Rev. Immunol. 2013;13:75–87. doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- 21.Song X, et al. Growth Factor FGF2 Cooperates with Interleukin-17 to Repair Intestinal Epithelial Damage. Immunity. 2015;43:488–501. doi: 10.1016/j.immuni.2015.06.024. [DOI] [PubMed] [Google Scholar]
- 22.Izcue A, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yen D, et al. IL-23 is essential for T cell–mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investigation. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zorzi F, et al. Distinct profiles of effector cytokines mark the different phases of Crohn’s disease. PLoS One. 2013;8:e54562. doi: 10.1371/journal.pone.0054562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Powell N, et al. Interleukin 6 Increases Production of Cytokines by Colonic Innate Lymphoid Cells in Mice and Patients With Chronic Intestinal Inflammation. Gastroenterology. 2015;149:456–467 e415. doi: 10.1053/j.gastro.2015.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji J, et al. Myeloid-derived suppressor cells contribute to systemic lupus erythaematosus by regulating differentiation of Th17 cells and Tregs. Clin. Sci. (Lond.) 2016;130:1453–1467. doi: 10.1042/CS20160311. [DOI] [PubMed] [Google Scholar]
- 27.Hoechst B, et al. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood. 2011;117:6532–6541. doi: 10.1182/blood-2010-11-317321. [DOI] [PubMed] [Google Scholar]
- 28.Centuori SM, et al. Myeloid-derived suppressor cells from tumor-bearing mice impair TGF-beta-induced differentiation of CD4+CD25+FoxP3+ Tregs from CD4+CD25-FoxP3- T cells. J. Leukoc. Biol. 2012;92:987–997. doi: 10.1189/jlb.0911465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ito Hiroaki, Takazoe Masakazu, Fukuda Yoshihiro, Hibi Toshifumi, Kusugami Kazuo, Andoh Akira, Matsumoto Takayuki, Yamamura Takehira, Azuma Junichi, Nishimoto Norihiro. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease☆. Gastroenterology. 2004;126(4):989–996. doi: 10.1053/j.gastro.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, et al. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm. Bowel Dis. 2006;12:382–388. doi: 10.1097/01.MIB.0000218764.06959.91. [DOI] [PubMed] [Google Scholar]
- 31.Dieleman LA, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 1998;114:385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostanin DV, et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G135–146. doi: 10.1152/ajpgi.90462.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uhlig HH, et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J. Immunol. 2006;177:5852–5860. doi: 10.4049/jimmunol.177.9.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.