

Abstract
Polo-like kinase 1 (PLK1) is a key regulator of mitosis and a recognized drug target for cancer therapy. Inhibiting the polo-box domain of PLK1 offers potential advantages of increased selectivity and subsequently reduced toxicity compared with targeting the kinase domain. However, many if not all existing polo-box domain inhibitors have been shown to be unsuitable for further development. In this paper, we describe a novel compound series, which inhibits the protein–protein interactions of PLK1 via the polo-box domain. We combine high throughput screening with molecular modeling and computer-aided design, synthetic chemistry, and cell biology to address some of the common problems with protein–protein interaction inhibitors, such as solubility and potency. We use molecular modeling to improve the solubility of a hit series with initially poor physicochemical properties, enabling biophysical and biochemical characterization. We isolate and characterize enantiomers to improve potency and demonstrate on-target activity in both cell-free and cell-based assays, entirely consistent with the proposed binding model. The resulting compound series represents a promising starting point for further progression along the drug discovery pipeline and a new tool compound to study kinase-independent PLK functions.
Introduction
Polo-like kinase (PLK1) is a key regulator of cell cycle progression and is a member of a family of closely related multifunctional kinases comprising PLK1, PLK2, PLK3, and PLK4. During mitosis, PLK1 has many crucial functions, from timely mitotic entry to successful cytokinesis and is critical for bipolar spindle formation and for correct chromosome segregation. In clinical terms, PLK1 is dysregulated in a wide range of human tumors, and its level of expression correlates with a poor prognosis.1 In addition, numerous cell-line studies demonstrate that PLK1 overexpression induces a transformed phenotype in noncancer cell lines, and inhibition of PLK1 can selectively kill tumor cells.2−6 Drug discovery approaches to targeting protein kinases have traditionally focussed around the kinase active site, and such inhibitors of PLK1 show some promise in the clinic.7,8 However, accompanying toxicity and the potential for the emergence of resistance are likely to limit the use of such agents. The development of protein–protein interaction (PPI) inhibitors provides an alternative and potentially more selective approach and one, which lends itself well to targeting PLK1 due to the modular nature of the enzyme. PLK1 is composed of two main structural elements: a kinase domain, which phosphorylates a great many proteins during mitosis, and a polo-box domain (PBD), comprising two polo-box units, which provides spatio-temporal regulation through interaction with its substrates.9,10 These two domains are joined by a linker of approximately 50 residues. The kinase domain has been crystallized in the presence and absence of bound inhibitors and shows similarity to many other kinases.11 The PBD has been crystallized in the presence and absence of bound phosphopeptides, and, in contrast, it has a unique structure, shared only with other members of the PLK family.12 The PBD offers both the phosphopeptide binding groove and the more recently described tyrosine pocket10,13−15 as possible sites for small-molecule inhibition and potentially a means to overcome some of the problems of kinase inhibitors. In the last few years, studies on a number of small-molecule inhibitors of the PBD of PLK1 have been published.16−21 However, recent work has raised questions over the mechanism of action for some of these inhibitors, casting doubt over the suitability for further development.22 Here, we describe a novel drug-like series for the inhibition of the PLK1 PBD. We characterize key members of the series using a panel of cell-free, biophysical, and cell-based assays to show on-target activity, alongside the use of molecular modeling to inform synthetic chemistry and improve physicochemical properties.
Results and Discussion
Fluorescence Polarization Screen Identifies Inhibitors of the PLK1 Polo-box Domain
A fluorescence polarization (FP) assay was developed to identify compounds that inhibit the binding of a consensus phosphopeptide to the PBD domain of PLK1. The assay was then used to screen a diverse library of approximately 86 000 compounds as described previously.23 Some of the primary hits were not reproducible or were identified as pan assay interference compounds (PAINS)24 (Table S1). In some cases, highly fluorescent compounds (Table S1) also generated false positives. These compounds were excluded from further development. After removing these spurious results, the screen yielded a hit rate of 0.06% leaving 53 remaining compounds as robust primary hits. The primary hits contained a number of promising chemical scaffolds including the recently reported Poloppins.23 In this paper, we focus on the series of compounds derived from compound 1 (Table 1).
Table 1. FP Results for the Primary Hit Compound 1 and a Set of Its Commercially Available Analoguesa.

All values represent the mean of three independent experiments ± standard deviation. ND = Not detectable.
Following confirmation of activity by repeat screening in the FP assay, a number of commercially available analogues were purchased for all primary hits, to generate early structure–activity relationship (SAR) data. We used the clusters generated in the library design stage to randomly select analogues of each hit molecule. These analogues were then tested by FP (Table 1 and Figure S1). The LHSpT peptide, a minimal phosphopeptide recognizing the PLK1 PBD,25 was included as a positive control. All of the compounds in Table 1, with the exception of compound 5, showed dose-dependent inhibition of phosphopeptide binding in the FP assay (Figure S1). Further, additional compound 1 analogues were purchased and tested in the FP assay; however, at a concentration of 50 μM, none showed activity above 10% inhibition in the assay and the inherent poor aqueous solubility of these compounds prevented testing at higher concentrations (Table S2). Compound 1 remained the most potent of the compounds tested. Compounds 2 and 5 indicate the importance of the terminal phenyl ring, while compounds 3 and 4 highlight the methyl substituent and the sulfur atom in the benzothiazinone, respectively (Table 1).
Molecular Modeling Informs Synthetic Chemistry To Improve Solubility
Despite the promising results from FP screening, we were unable to further characterize the compounds in biophysical or cell-based assays due to the observed poor solubility. To remedy this problem, we attempted to improve the physicochemical properties of compound 1 to generate compounds that retained activity while showing increased aqueous solubility. To aid in this process, we generated binding modes for compound 1 using molecular docking with Glide. The predicted pose can be seen in Figure 1.
Figure 1.
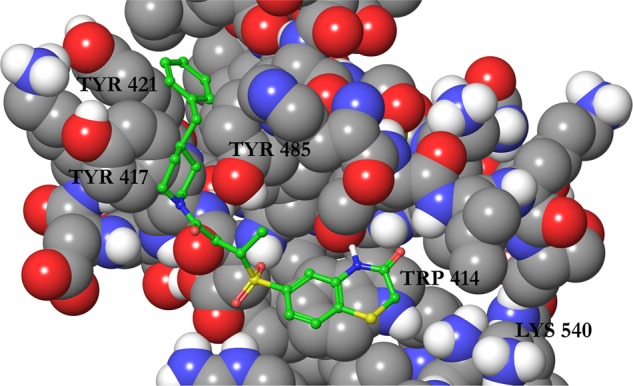
Binding mode of compound 1 to the polo-box domain (PBD) of PLK1 (PDBID 3BZI) predicted by Glide docking. The protein is displayed as atom colored space filling with gray carbons and the ligand is displayed as atom colored balls and sticks with green carbons. Only polar hydrogen atoms are displayed, and a selection of residues are labeled.
Compound 1 was docked to two PLK1 PBD structures (PBDID 3BZI and PDBID 3P37). Interestingly, the compound is not predicted to bind to the His 538/Lys 540 pincer, which binds the phosphate group of the phosphopeptide binding partners.9,26 This is in line with the difficulty in disrupting the strongly bound network of water molecules in this region.10 The predicted binding modes for the two structures show an excellent overlap, and in both cases, the molecule is predicted to lie in the phosphopeptide binding groove by stacking on the residue Trp 414 and extends into a cryptic hydrophobic pocket lined by Tyr 417, Tyr 421, and Tyr 485. This pocket has previously been implicated in the binding of artificial peptide analogues14 and more recently a subset of natural PLK1 substrates.15 The high Glide XP Scores of −11.7 and −12.1 kcal/mol alongside the MM-GBSA scores of −95.3 and −92.9 kcal/mol were encouraging, and the predicted binding mode was in line with the SAR derived from the compounds in Table 1, increasing its credibility. In particular, the terminal phenyl ring fills the cryptic hydrophobic pocket, the methyl group is positioned in a small cavity between Leu 119, Val 415, and Tyr 485, and the sulfur atom lies on the hydrophobic surface formed from the sidechain of Trp 414. Using this model, we designed and synthesized a series of close analogues of compound 1 that were predicted to show improved solubility without having a deleterious effect on the activity. Notably, changes around the terminal phenyl ring (one edge of which is solvent-exposed in the putative binding pose) were predicted to be tolerated. The docking mode suggests that changes to the ortho, meta, and para positions should all be allowed (see Figure 1). For ease of synthesis, we focused on changes to this end of the molecule by amide coupling a number of commercially available amines to a commercially available acid. Compounds were selected with high similarity to compound 1 but with an increased polar atom count. No changes were made to the sulfone group, alkyl chain, or amide groups, which link the two ends of the molecule together. These orient the molecule, and the sulfone is predicted to make a hydrogen bond with the protein backbone. The solubility of these analogues is shown in Table 2, where the turbidity of compounds in aqueous solution is used to indicate solubility.
Table 2. Solubility of a Set of Compound 1 Analogues Designed to Increase Aqueous Solubilitya.

Turbidity was experimentally determined by measuring absorbance at 625 nm at 250 μM, and values are relative to dimethyl sulfoxide (DMSO). All turbidity values are the mean of three independent experiments ± standard deviation.
Compounds 6 and 9 showed a substantial improvement in solubility through increased polarity around the terminal phenyl ring, Crucially, this increase in solubility enabled further characterization of compound 9 (Figure 2A).
Figure 2.

Optimized soluble analogue, compound 9, is active in FP (A) and isothermal titration calorimetry (ITC) (B) assays. (C) Activity of the isolated enantiomers of compound 9 in the FP assay. For FP assays, values are the mean of three independent experiments ± SD. A representative experiment is shown for ITC. The experiment was carried out in duplicate.
Compound 9 was more active in the FP assay with an IC50 of 36 μM. The improved aqueous solubility for compound 9 also allowed us to determine a Kd of 20 μM using isothermal titration calorimetry (ITC) (Figure 2B), which is consistent with the results of the FP assay. As a part of an ongoing campaign to test the predicted binding mode in Figure 1 and improve the binding affinity, we designed and tested a number of further analogues, and among them, compounds (−)–9 and (+)–9, the pair of enantiomers comprising compound 9, are a racemic mix (Table 3).
Table 3. FP Results for a Set of Compound 1 Analogues Designed to Test the Predicted Binding Mode and Improve the Binding Affinitya.

All IC50 values were measured by FP, and values represent the mean of three independent experiments ± standard deviation. ND = not determinable. * Stereochemistries were assigned arbitrarily.
In the FP assay, (−)–9 showed an increase in potency over the parental compound (24.4 μM), while (+)–9 was considerably less potent, showing only mild activity (Figure 2C). The predicted binding mode can accommodate both enantiomers, consistent with some activity for both compounds in the assay, but in one case, the methyl group is solvent-exposed and in the other, it contacts the protein. This data is thus commensurate with the predicted binding mode. Additional analogues added further confidence to the predicted binding mode (Table 3). For example, the para position of the phenyl ring can accommodate a number of substitutions (compounds 12 and 13), consistent with its lack of protein contacts in the predicted binding mode. The importance of the 3-thiomorpholinone heterocycle is underlined by a lack of measurable affinity for compound 16 and this is also in line with the predicted binding mode, where the amide hydrogen and carbonyl oxygen in the ring make hydrogen bonds with the backbone of residue Trp 414. When compound 15 is docked, the binding mode is maintained, but the bond between the sulfone group and the thiomorpholinone ring is rotated such that it still stacks on Trp 414 but no longer makes hydrogen-bonding contacts with the backbone. This is in line with its slightly reduced activity (111.5 vs 73.1 μM). Compound 16 loses the stacking as well as the hydrogen-bonding contacts and is inactive. Conversely, converting the 3-thiomorpholinone ring to a 3-morpholinone ring (compound 10) retains activity, consistent with its lack of protein contacts. Although compound 11 appeared to have improved potency, neither of the isolated enantiomers, (−)–11 and (+)–11, matched this improvement. The reason for this observation was not clear, so this compound was not taken further.
Improved Soluble Analogue Shows on-Target Activity in Cell-Based Assays
With excellent solubility and in vitro evidence for target engagement from the FP and ITC assays (Figure 2), compound 9 was tested for cell-based activity alongside the isolated enantiomer pair, (−)–9 and (+)–9. The compounds were tested in a high-content mitotic index assay designed to identify molecules, which are able to cause a mitotic arrest (Figure 3A).
Figure 3.

Dose-dependent increase in mitotic arrest with chromosome congression defects is caused upon treatment with Compound 9 and its enantiomers. (A) Cells were treated with Compound 9 and its enantiomers, compounds (−)–9 and (+)–9 (at 200, 100, 50, 25, 12.5, 6.25, and 0 μM) and corresponding DMSO controls for 12 h. Mitotic cells scored as phospho-histone H3-stained cells per 2000 Hoechst 33342-stained nuclei in a high-content screening platform as described earlier.27 Each bar is a mean of three replicates ± standard error of the mean (SEM). The data presented is representative of two independent experiments. (B) HeLa cells were treated with compound (−)–9/DMSO for 12 h, as shown in A. The cells were fixed and stained for DNA and β-tubulin. Mitotic cells were identified by microscopy and scored under three categories: (a) misaligned chromosomes and monopolar spindles, (b) misaligned chromosomes and bipolar spindles, and (c) aligned chromosomes and bipolar spindles. The quantification of the cellular phenotype is shown in the histogram. (C) Representative maximal-intensity projection images of cells in each category (as in B) showing DNA in blue and spindle microtubules in red. The scale bar is 3 μm.
Small-molecule inhibition of PLK1 by either kinase inhibitors28−31 or inhibitors of the polo-box domain23 has been shown to inhibit timely progression through mitosis, giving rise to an increased mitotic index. Compound 9 and (−)–9 both showed a robust increase in mitotic arrest (4-fold), while (+)–9 gave a much milder effect, confirming that (−)–9 is the active enantiomer as predicted by the FP assay results (Figure 2C). In addition, examination of the images from high-content analysis revealed cells arrested in prometaphase with misaligned chromosomes, a phenotype characteristic of inhibition of PLK1 via the PBD (Figure S2). Closer examination and quantitation of the mitotic phenotype by confocal microscopy confirmed that the predominant phenotype was arrested in prometaphase with a bipolar spindle and noncongressed chromosomes (Figure 3B,C). This phenotype has been consistently reported as a direct result of inhibition of the PBD, and it is distinct from the monopolar phenotype seen when PLK1 kinase activity is directly inhibited either by small molecules or by depletion of the protein using RNA interference.26,32 Thus, these observations provide strong support that compound (−)–9 is an inhibitor of the PLK1 PBD both in a cell-free setting and within the cellular milieu.
We sought further evidence that the action of compound (−)–9 was through inhibition of the PBD of PLK1 by examining localization of the PLK1 protein in compound-treated cells. Using a previously described HeLa cell line with a doxycycline-inducible overexpression of GFP-tagged PLK1,15 we looked at localization of the fluorescently tagged protein in mitosis. Mutations of the PBD at residues His 538 and Lys 540 within the phosphopeptide binding groove or at residues Tyr 421, Leu 478, and Tyr 481 within the tyrosine pocket have been shown to disrupt the interaction of PLK1 with critical mitotic substrates and result in loss of GFP-PLK1 from the kinetochores.15 This phenotype was recapitulated upon treatment of cells with compound (−)–9, showing a clear decrease in kinetochore localization of GFP-PLK1 (Figure 4).
Figure 4.

Treatment with compound (−)–9 causes mislocalization of PLK1 from kinetochores in mitotic cells. (A) Cells expressing GFP-PLK1 were treated with compound (−)–9 or DMSO for 9.5 h after double thymidine release. The cells were fixed, stained for DNA (Hoechst) and kinetochores (CREST antisera), and analyzed by immunofluorescence microscopy. Prometaphase mitotic cells were identified based on DNA morphology, and 1 μm confocal Z-stacks were taken for each cell. Quantification of GFP-PLK1 intensity on CREST-stained kinetochores was carried out using ImageJ. The data is represented as the Whisker maximum to minimum plot with a horizontal bar indicating mean GFP-PLK1 intensity on kinetochores. Statistical analysis was done using a nonparametric, Mann–Whitney two-tailed test with a 95% confidence interval, **p = 0.0037. 8 cells were analyzed for DMSO and 7 for compound (−)–9. (B) Representative maximal-intensity projection images of cells showing CREST-stained kinetochores (red), GFP-PLK1 (green), and DNA (blue) used for quantification of GFP-PLK1 intensity in Figure 4A. The scale bar is 5 μm.
While this provides strong evidence that this compound can inhibit the PBD of PLK1 in cells, we cannot exclude the possibility that these compounds may also inhibit the PBD of other PLK family members. Indeed, a sequence alignment of the PBD of the PLK family suggests that achieving selectivity for PLK1 will be challenging but possible. The overall sequence identity with the PLK1 PBD is 38.67% for PLK2 and 39.01% for PLK3, but the key contact residues have a high similarity (Figure 5).
Figure 5.

Sequence alignment of the PBDs of the PLK family. Sequences of human PLK1 residues 410–603, PLK2 residues 503–685, and PLK3 residues 463–646 were taken from UniProt. PLK4 and PLK5 were excluded from the analysis as they have markedly different structures and functions. The alignment was performed by Clustal Omega using default parameters. Residues in the phosphopeptide binding groove and cryptic hydrophobic pocket are highlighted in bold. Residues in the phosphopeptide binding groove and cryptic hydrophobic pocket with complete conservation across the PLK family are in red and those which differ are in blue.
In particular, selectivity may be achievable by targeting the Thr 477/Leu 478 (TL) pair at the end of the cryptic hydrophobic pocket, which are TV in PLK2 and GI in PLK3. In addition, the pair of residues Arg516/Phe535 are lysine and tyrosine in PLK2 and PLK3. Measuring the selectivity of this series across the PLK family should be the focus of future work.
Conclusions
This paper describes screening for PPI inhibitors and subsequent hit identification and optimization of a promising new compound series for inhibition of the PBD of PLK1. Protein–protein interactions have long been considered challenging targets for drug discovery, and to address this, we started with a PPI-focused library of approximately 86 000 commercially available compounds. Many of the primary hits from FP screening were false positives, highlighting the need for thorough validation and awareness of common liabilities. Due to the nature of PPIs, poor compound solubility can be a problem as many small-molecule inhibitors have significant hydrophobic character and derive their binding affinity from nonpolar interactions.33 In this case, poor solubility of the primary hit precluded the initial advancement of the series, but we were able to use molecular modeling to generate a predicted binding pose, which guided the introduction of small structural changes designed to improve solubility. It is worth noting that such improvements may be easier to achieve in PPIs than traditional targets, due to the inherent solvent exposure of the inhibitors.34 Sequence analysis suggests that achieving selectivity for PLK1 over PLK2 and PLK3 may be challenging but should be achievable by targeting specific residues that differ between these domains.
Thus, we have combined computational techniques, synthetic chemistry, and a robust FP assay for the design, synthesis, and characterization of analogues, with small structural changes, to successfully remedy the solubility problems and facilitate further development of the compound series. This initial campaign has resulted in a cell-permeable, small-molecule series with low molecular weight and evidence of target engagement in both cell-free and cell-based assays. Further work will be required to identify whether this is a PLK1-specific or pan-PLK inhibitor. However, we propose that this series may be a good candidate for further elaboration towards chemical probe development and/or a cancer drug discovery program.
Methods
Library Design
The small-molecule library for high throughput screening (HTS) was design and assembled in-house from commercial vendors (Table 4).
Table 4. Chemical Vendors Used to Source the HTS Library.
| supplier | compounds available | compounds selected |
|---|---|---|
| Asinex | 364 407 | 9999 |
| Chembridge | 442 051 | 23 086 |
| ChemDiv | 789 603 | 20 000 |
| Enamine | 1 116 406 | 23 200 |
| Life Chemicals | 327 211 | 10 016 |
| total | 3 039 678 | 86 301 |
The complete set of approximately 3 million compounds was filtered aggressively using the FILTER program (OpenEye). We employed the REOS substructure filters,35 OpenEye’s FILTER filters, and a set of physicochemical property filters developed for PPI inhibitors (Table 5).
Table 5. Limits on the Physicochemical Properties Used to Filter the HTS Library.
| property | minimum | maximum |
|---|---|---|
| molecular weight | 250 | 650 |
| number of heavy atoms | 0 | 50 |
| total charge | –2 | 2 |
| hydrogen bond acceptors | 0 | 10 |
| hydrogen bond donors | 0 | 6 |
| clog P | –5.0 | 6.0 |
| rotatable bonds | 0 | 10 |
| chiral centers | 0 | 4 |
These criteria are wider than those typically used to generate screening libraries,36 and these are based on recent studies.37,38 This filtering step removed approximately 50% of the compounds from consideration. The next stage was then selection of a diverse subset of the remaining compounds. This process was performed using directed sphere exclusion39 with Canvas (Schrödinger, LLC). Each compound was assigned radial fingerprints40 with daylight atom types and a radius of 4. The compound similarity was assessed using the Tanimoto metric.41 Compounds were then selected iteratively, with the requirement that no selected compound had a Tanimoto similarity greater than 0.8 to any previously selected compound. This process yielded a set of approximately 43 000 compounds. Each of these compounds was defined as the center of a cluster, and another molecule with a Tanimoto similarity greater than 0.8 to the cluster center was randomly selected for each cluster. This ensures that some basic SAR will be present in the primary screening data. This led to the selection of approximately 86 000 compounds. An additional 12 000 peptide-mimic compounds were also tested,42 bringing the screening library to 98 000 compounds.
Protein Preparation
A region of the human PLK1 cDNA sequence (residues 345–603) was amplified by polymerase chain reaction (PCR) and cloned into a pGEX-6P1 vector (Invitrogen, Carlsbad, CA) to generate a recombinant protein in C41 strain Escherichia coli.43 Bacteria were disrupted using an Emulsiflex c5 homogenizer (Avestin), and lysates were passed onto a glutathione S-transferase (GST)-Sepharose column in the presence of 50 mM HEPES pH 7.5, 200 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 1 mM DTT. Bound fractions were cleaved on the column at 4 °C overnight with PreScission protease (GE healthcare). Purified fractions were polished by gel chromatography using a Sephadex G25 (GE healthcare) column, collected and concentrated. Fractions were tested by Western blotting using the PLK1 antibody (Invitrogen 33–1700) to validate protein purification.
Fluorescence Polarization Assay
The HTS was conducted using a fluorescence polarization (FP) assay, as described in Narvaez et al.23 All peptides and phosphopeptides were synthesized using standard chemistry (Designer Bioscience Ltd., Cambridge, U.K.). The fluorescently labeled probe was the phosphopeptide sequence MAGPMQSpTPLNGAKK with N-terminal TAMRA. FP measurements were carried out in 384-well, low-volume, black, flat-bottom polystyrene N-bromosuccinimide (NBS) microplates (Corning 3820) using a PHERAstar Plus plate reader (BMGLabtech). The final assay volume of 45 μL contained a 10 nM labeled probe peptide, 84 nM PBD, and varying concentrations of the competitor. Assays were carried out in phosphate-buffered saline (PBS) (pH 7.4) plus 0.03% tween. FP values were obtained in millipolarization units at an excitation wavelength of 540 nm and an emission wavelength of 590 nm. Compounds were screened at an average concentration of 125 μM. All of the hits identified in the primary screen were then reanalyzed at a range of concentrations to generate dosimetry curves. DMSO controls were run alongside all experimental compounds, and % inhibition was normalized to these controls. The unlabeled LHSpT peptide was used as a positive control. % inhibition values were calculated using GraphPad Prism.
Isothermal Titration Calorimetry
ITC measurements were performed in duplicate, as described in Narvaez et al.23 In all titrations, protein PBD345–603 was used at 25 μM and buffered in HEPES (50 mM, pH 7.4), NaCl (200 mM), and DTT (1 mM). Protein was diluted into a buffer with DMSO at 5% (v/v). Compounds were prepared by diluting from DMSO stock into the same buffer containing DMSO at 5% (v/v). Great care was taken to match the concentration of DMSO in the ligand and protein samples as closely as possible. In a typical experiment, protein (25 μM) was loaded in the sample cell, and a total of 20 injections of 8 μL were made at 2 min intervals from a 200 μL syringe rotating at 1000 rpm and loaded with the ligand solution (0.5 mM). In all titrations, an initial injection of 2 μL ligand was made, and these data were discarded during data analysis. The thermodynamic parameters were obtained by fitting the data to a single-site binding model with a stoichiometry of 1. ITC experiments were performed at 20 °C with a MicroCal ITC200 instrument, and all data were analyzed with the software implemented in Origin (version 7).
Turbidity Assay
50 mM compound stock solutions in DMSO were diluted to 250 μM in dH2O. 50 μL of the diluted compounds was aliquoted into flat-bottom 96-well plates and incubated at room temperature for 20 min before absorbance at 625 nM and it was read using a Tecan Infinite 200 Pro plate reader. Absorbance readings were normalized to DMSO controls.
Binding Pose Prediction
Before analysis, all structures were prepared using Schrödinger’s Preparation Wizard44 using the default settings to check all protonation states as well as the orientations of asparagine, glutamine, and histidine residues. All water molecules were deleted prior to the analysis. Molecular docking was performed using Schrödinger’s Glide 4.045 with the PDB structures 3BZI(43) and 3P37.13 These structures have the PLK1 PBD cryptic pocket in an open state, as we believed that this hydrophobic pocket would be important in binding small-molecule inhibitors. For the grid generation and ligand docking procedures, the default Glide settings were used. All structures were docked and scored using the Glide extra-precision (XP)46 and rescored using an MM-GBSA method.47
Cell Culture
HeLa FlpIn T-REx cells (a kind gift from Professor Steve Taylor, University of Manchester) were cultured at 37 °C under 5% CO2 in DMEM with GlutaMAX (Life Technologies), supplemented with 10% fetal calf serum, Zeocin (Invitrogen) at 0.05 mg/mL, and Blasticidin (InvivoGen) at 4 μg/mL. All GFP-PLK1 HeLa FlpIn T-REx cell lines were generated and maintained as described in Sharma et al.15 Doxycycline (Sigma-Aldrich) at 0.1 mg/mL was used for induction of the GFP-fusion protein.
Mitotic Index (MI) Assay
HeLa FlpIn T-REx cells were arrested in early S-phase in growth media containing 2 mM thymidine (Sigma) for 16 h. Cells were then released for 8 h in thymidine-free media, and these were then arrested again with thymidine for 16 h followed by the release in thymidine-free media. Cells were treated for 12 h with DMSO or compounds, and then these were fixed in 3.7% formaldehyde (Agar Scientific). Cells were permeabilized with 0.1% Triton X-100, and then these were incubated with anti-phospho-histone H3 (Ser10) antibody (Abcam ab5176). The cells were washed with PBS, and then these were incubated with Alexa Fluor 488-labeled goat anti-rabbit IgG (Invitrogen A11034) in the presence of 4 μg/mL Hoechst 33342 (Invitrogen H3570). Cells were washed in PBS, and then these were imaged on an Arrayscan VTi HCS instrument (Thermo Fisher) using the Target Activation V4 BioApplication. Mitotic cells were scored as phospho-histone H3-stained cells per 2000 Hoechst 33342-stained nuclei, as described in Ibbeson et al.27
Immunofluorescence (IF) and Image Analysis
Cells were fixed on a coverslip with 4% formaldehyde (Agar Scientific) for 10 min. Cells were permeabilized with 0.1% Triton-100 (Fisher), 0.1% Tween-20 (NBS Biologicals) in 1× PBS (PBS-Triton-Tween) for 10 min and blocked with 1% BSA (Fisher Scientific) in PBS-Triton-Tween for 30 min. CREST antisera (Europa FZ90C-CS1058) was diluted 1:1000 in the blocking solution, and cells were incubated in humidified chambers for 1 h at room temperature. The cells were then washed with blocking solution, and the cells were incubated with Alexa Fluor-conjugated secondary antibodies (Life Technologies, 1:500) for 30 min. Cells were washed thrice in the blocking solution and mounted in the 4,6-diamidino-2-phenylindole (DAPI) containing medium (Vectashield). The samples were stored in dark at 4 °C before microscopy. The fixed cell images were captured using a Leica SP5 confocal microscope using a 63× or 100× 1.4 NA/oil objective with Z-stacks of confocal slices taken at 1 μm intervals. Pixel intensities were never saturated; laser exposure and detector settings were identical between samples across an experiment. The ImageJ software was used for image analysis. CREST staining was used as a mask to determine the average GFP-PLK1 staining intensity in CREST-stained kinetochores.
Acknowledgments
Work in D.J.H.’s laboratory is supported by the Medical Research Council under grant ML/L007266/1. Work in D.R.S.’s laboratory is supported by the European Union, Engineering and Physical Sciences Research Council, Biotechnology and Biological Sciences Research Council, Medical Research Council, Cancer Research U.K., and the Wellcome Trust. Work in A.R.V.’s laboratory is supported by the Wellcome Trust and Medical Research Council program grants MC_UU_12022/1 and MC_UU_12022/8. All calculations were performed using the Darwin Supercomputer of the University of Cambridge High Performance Computing Service (http://www.hpc.cam.ac.uk/) provided by Dell Inc. using Strategic Research Infrastructure Funding from the Higher Education Funding Council for England, and these were funded by the EPSRC under grants EP/F032773/1 and EP/J017639/1. D.W.W., R.B., and R.G.B. are employees and shareholders of Sentinel Oncology Ltd. G.J.M. is an employee and shareholder of PhoreMost Ltd. A.R.V. is a founder, shareholder, and scientific advisor of Sentinel Oncology Ltd. and Phoremost Ltd. David Huggins is a founder and shareholder of Integrated Biomedical Solutions Ltd.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03626.
Primary hits removed due to liabilities, FP results for a set of compound 1 analogues, FP curves, representative images used for mitotic index determination, and detailed synthesis methods (PDF)
Author Present Address
¶ Discovery Sciences, Innovative Medicines and Early Development Biotech Unit, AstraZeneca, Darwin Building, Cambridge CB4 0WG, United Kingdom (A.J.N.).
Author Present Address
◆ PhoreMost Ltd., Babraham Research Campus, Cambridge CB22 3AT, United Kingdom (R.B. and G.J.M.).
Author Present Address
○ College of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou 310014, Zhejiang, China (F.Z.).
Author Present Address
∇ Department of Chemistry, Technical University of Denmark, Kemitorvet 207, 2800 Kgs. Lyngby, Denmark (L.L.).
Author Present Address
# Cancer Research UK-AstraZeneca Antibody Alliance Laboratory The Portway Building, Granta Park, Great Abington, Cambridge, CB21 6GS, United Kingdom (P.S.).
Author Present Address
⊥ Tri-Institutional Therapeutics Discovery Institute, Belfer Research Building, 413 East 69th Street, New York, New York 10021, United States (D.J.H.).
The authors declare the following competing financial interest(s): David Walker, Richard Boyce and Robert G. Boyle are employees and shareholders of Sentinel Oncology Ltd. Grahame McKenzie is an employee and shareholder of PhoreMost Ltd. Ashok R. Venkitaraman is a founder, shareholder and scientific advisor of Sentinel Oncology Ltd. and Phoremost Ltd. David Huggins is a founder and shareholder of Integrated Biomedical Solutions Ltd.
Supplementary Material
References
- Eckerdt F.; Yuan J.; Strebhardt K. Polo-like kinases and oncogenesis. Oncogene 2005, 24, 267–276. 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- Strebhardt K.; Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321–330. 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- Liu X.; Lei M.; Erikson R. L. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol. Cell. Biol. 2006, 26, 2093–2108. 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner A. G.; Dos Santos C.; Recher C.; Bailly C.; Créancier L.; Kruczynski A.; Payrastre B.; Manenti S. Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood 2009, 114, 659–662. 10.1182/blood-2008-12-195867. [DOI] [PubMed] [Google Scholar]
- Lee K. S.; Burke T. R. Jr.; Park J.-E.; Bang J. K.; Lee E. Recent advances and new strategies in targeting Plk1 for anticancer therapy. Trends Pharmacol. Sci. 2015, 36, 858–877. 10.1016/j.tips.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutteridge R. E. A.; Ndiaye M. A.; Liu X.; Ahmad N. Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. 10.1158/1535-7163.MCT-15-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjertsen B. T.; Schöffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015, 29, 11–19. 10.1038/leu.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.-E.; Hymel D.; Burke T. R. Jr.; Lee K. S. Current progress and future perspectives in the development of anti-polo-like kinase 1 therapeutic agents. F1000Research 2017, 6, 1024. 10.12688/f1000research.11398.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia A. E. H.; Rellos P.; Haire L. F.; Chao J. W.; Ivins F. J.; Hoepker K.; Mohammad D.; Cantley L. C.; Smerdon S. J.; Yaffe M. B. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 2003, 115, 83–95. 10.1016/S0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- Huggins D. J.; McKenzie G.; Robinson D.; Narváez A.; Hardwick B.; Roberts-Thomson M.; Venkitaraman A.; Grant G.; Payne M. Computational Analysis of Phosphopeptide Binding to the Polo-Box Domain of the Mitotic Kinase PLK1 Using Molecular Dynamics Simulation. PLoS Comput. Biol. 2010, 6, e1000880 10.1371/journal.pcbi.1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothe M.; Kohls D.; Low S.; Coli R.; Cheng A. C.; Jacques S. L.; Johnson T. L.; Lewis C.; Loh C.; Nonomiya J.; Sheils A. L.; Verdries K. A.; Wynn T. A.; Kuhn C.; Ding Y. H. Structure of the catalytic domain of human polo-like kinase 1. Biochemistry 2007, 46, 5960–5971. 10.1021/bi602474j. [DOI] [PubMed] [Google Scholar]
- Cheng K. Y.; Lowe E. D.; Sinclair J.; Nigg E. A.; Johnson L. N. The crystal structure of the human polo-like kinase-1 polo box domain and its phospho-peptide complex. EMBO J. 2003, 22, 5757–5768. 10.1093/emboj/cdg558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Śledź P.; Stubbs C. J.; Lang S.; Yang Y. Q.; McKenzie G. J.; Venkitaraman A. R.; Hyvönen M.; Abell C. From Crystal Packing to Molecular Recognition: Prediction and Discovery of a Binding Site on the Surface of Polo-Like Kinase 1. Angew. Chem., Int. Ed. 2011, 50, 4003–4006. 10.1002/anie.201008019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Park J.-E.; Qian W.-J.; Lim D.; Gräber M.; Berg T.; Yaffe M. B.; Lee K. S.; Burke T. R. Jr. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat. Chem. Biol. 2011, 7, 595–601. 10.1038/nchembio.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P.; Mahen R.; Rossmann M.; Stokes J. E.; Hardwick B.; Huggins D. J.; Emery A.; Kunciw D. L.; Hyvönen M.; Spring D. R.; McKenzie G. J.; Venkitaraman A. R. A cryptic hydrophobic pocket in the polo-box domain of the polo-like kinase PLK1 regulates substrate recognition and mitotic chromosome segregation. Sci. Rep. 2019, 9, 15930 10.1038/s41598-019-50702-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M.; Sanhaji M.; Pietsch L.; Béquignon I.; Herbrand A. K.; Süß E.; Gande S. L.; Caspar B.; Kudlinzki D.; Saxena K.; Sreeramulu S.; Schwalbe H.; Strebhardt K.; Biondi R. M. Modulation of the allosteric communication between the polo-box domain and the catalytic domain in Plk1 by small compounds. ACS Chem. Biol. 2018, 13, 1921–1931. 10.1021/acschembio.7b01078. [DOI] [PubMed] [Google Scholar]
- Berg A.; Berg T. Inhibitors of the Polo-Box Domain of Polo-Like Kinase 1. ChemBioChem 2016, 17, 650–656. 10.1002/cbic.201500580. [DOI] [PubMed] [Google Scholar]
- Watanabe N.; Sekine T.; Takagi M.; Iwasaki J.; Imamoto N.; Kawasaki H.; Osada H. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J. Biol. Chem. 2009, 284, 2344–2353. 10.1074/jbc.M805308200. [DOI] [PubMed] [Google Scholar]
- Liao C.; Park J.-E.; Bang J. K.; Nicklaus M. C.; Lee K. S. Probing Binding Modes of Small Molecule Inhibitors to the Polo-Box Domain of Human Polo-like Kinase 1. ACS Med. Chem. Lett. 2010, 1, 110–114. 10.1021/ml100020e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reindl W.; Yuan J.; Kramer A.; Strebhardt K.; Berg T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem. Biol. 2008, 15, 459–466. 10.1016/j.chembiol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- Reindl W.; Yuan J.; Kramer A.; Strebhardt K.; Berg T. A pan-specific inhibitor of the polo-box domains of polo-like kinases arrests cancer cells in mitosis. ChemBioChem 2009, 10, 1145–1148. 10.1002/cbic.200900059. [DOI] [PubMed] [Google Scholar]
- Archambault V.; Normandin K. Several inhibitors of the Plk1 Polo-Box Domain turn out to be non-specific protein alkylators. Cell Cycle 2017, 16, 1220–1224. 10.1080/15384101.2017.1325043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narvaez A. J.; Ber S.; Crooks A.; Emery A.; Hardwick B.; Almeida E. G.; Huggins D. J.; Perera D.; Roberts-Thomson M.; Azzarelli R.; et al. Modulating Protein-Protein Interactions of the Mitotic Polo-like Kinases to Target Mutant KRAS. Cell Chem. Biol. 2017, 24, 1017–1028.e7. 10.1016/j.chembiol.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Yun S. M.; Moulaei T.; Lim D.; Bang J. K.; Park J. E.; Shenoy S. R.; Liu F.; Kang Y. H.; Liao C.; Soung N. K.; Lee S.; Yoon D. Y.; Lim Y.; Lee D. H.; Otaka A.; Appella E.; McMahon J. B.; Nicklaus M. C.; Burke T. R. Jr.; Yaffe M. B.; Wlodawer A.; Lee K. S. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat. Struct. Mol. Biol. 2009, 16, 876–882. 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch A.; Wehner A.; Nigg E. A.; Sillje H. H. Different Plk1 functions show distinct dependencies on Polo-Box domain-mediated targeting. Mol. Biol. Cell 2006, 17, 448–459. 10.1091/mbc.e05-08-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibbeson B. M.; Laraia L.; Alza E.; O’Connor C. J.; Tan Y. S.; Davies H. M.; McKenzie G.; Venkitaraman A. R.; Spring D. R. Diversity-oriented synthesis as a tool for identifying new modulators of mitosis. Nat. Commun. 2014, 5, 3155 10.1038/ncomms4155. [DOI] [PubMed] [Google Scholar]
- Steegmaier M.; Hoffmann M.; Baum A.; Lenart P.; Petronczki M.; Krssak M.; Gurtler U.; Garin-Chesa P.; Lieb S.; Quant J.; Grauert M.; Adolf G. R.; Kraut N.; Peters J. M.; Rettig W. J. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Santamaria A.; Neef R.; Eberspächer U.; Eis K.; Husemann M.; Mumberg D.; Prechtl S.; Schulze V.; Siemeister G.; Wortmann L.; et al. Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate functions of Plk1 in early and late stages of mitosis. Mol. Biol. Cell 2007, 18, 4024–4036. 10.1091/mbc.e07-05-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleixner K. V.; Ferenc V.; Peter B.; Gruze A.; Meyer R. A.; Hadzijusufovic E.; Cerny-Reiterer S.; Mayerhofer M.; Pickl W. F.; Sillaber C.; Valent P. Polo-like kinase 1 (Plk1) as a novel drug target in chronic myeloid leukemia: overriding imatinib resistance with the Plk1 inhibitor BI 2536. Cancer Res. 2010, 70, 1513–1523. 10.1158/0008-5472.CAN-09-2181. [DOI] [PubMed] [Google Scholar]
- Degenhardt Y.; Greshock J.; Laquerre S.; Gilmartin A. G.; Jing J.; Richter M.; Zhang X.; Bleam M.; Halsey W.; Hughes A.; et al. Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability. Mol. Cancer Ther. 2010, 9, 2079–2089. 10.1158/1535-7163.MCT-10-0095. [DOI] [PubMed] [Google Scholar]
- Seong Y. S.; Kamijo K.; Lee J. S.; Fernandez E.; Kuriyama R.; Miki T.; Lee K. S. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of plk1 in u-2 OS cells. J. Biol. Chem. 2002, 277, 32282–32293. 10.1074/jbc.M202602200. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Laraia L.; McKenzie G.; Spring D. R.; Venkitaraman A. R.; Huggins D. J. Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein-protein interactions. Chem. Biol. 2015, 22, 689–703. 10.1016/j.chembiol.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters W. P.; Stahl M. T.; Murcko M. A. Virtual screening - an overview. Drug Discovery Today 1998, 3, 160–178. 10.1016/S1359-6446(97)01163-X. [DOI] [Google Scholar]
- Huggins D. J.; Venkitaraman A. R.; Spring D. R. Rational methods for the selection of diverse screening compounds. ACS Chem. Biol. 2011, 6, 208–217. 10.1021/cb100420r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé C. M.; Laconde G.; Kuenemann M. A.; Villoutreix B. O.; Sperandio O. iPPI-DB: A manually curated and interactive database of small non-peptide inhibitors of protein–protein interactions. Drug Discovery Today 2013, 18, 958–968. 10.1016/j.drudis.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Higueruelo A. P.; Schreyer A.; Bickerton G. R. J.; Pitt W. R.; Groom C. R.; Blundell T. L. Atomic Interactions and Profile of Small Molecules Disrupting Protein-Protein Interfaces: the TIMBAL Database. Chem. Biol. Drug Des. 2009, 74, 457–467. 10.1111/j.1747-0285.2009.00889.x. [DOI] [PubMed] [Google Scholar]
- Gobbi A.; Lee M. L. DISE: Directed Sphere Exclusion. J. Chem. Inf. Comput. Sci. 2003, 43, 317–323. 10.1021/ci025554v. [DOI] [PubMed] [Google Scholar]
- Bender A.; Jenkins J. L.; Scheiber J.; Sukuru S. C. K.; Glick M.; Davies J. W. How Similar Are Similarity Searching Methods? A Principal Component Analysis of Molecular Descriptor Space. J. Chem. Inf. Model. 2009, 49, 108–119. 10.1021/ci800249s. [DOI] [PubMed] [Google Scholar]
- Willett P. Similarity-based virtual screening using 2D fingerprints. Drug Discovery Today 2006, 11, 1046–1053. 10.1016/j.drudis.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Janeček M.; Rossmann M.; Sharma P.; Emery A.; Huggins D. J.; Stockwell S. R.; Stokes J. E.; Tan Y. S.; Almeida E. G.; Hardwick B.; et al. Allosteric modulation of AURKA kinase activity by a small-molecule inhibitor of its protein-protein interaction with TPX2. Sci. Rep. 2016, 6, 28528 10.1038/srep28528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Alvarez B.; de Carcer G.; Ibanez S.; Bragado-Nilsson E.; Montoya G. Molecular and structural basis of polo-like kinase 1 substrate recognition: Implications in centrosomal localization. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 3107–3112. 10.1073/pnas.0609131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Lyne P. D.; Lamb M. L.; Saeh J. C. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 2006, 49, 4805–4808. 10.1021/jm060522a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.