

The utilization and degradation of d-ribose in archaea, the third domain of life, have not been analyzed so far. We show that Haloarcula species utilize d-ribose, which is degraded to α-ketoglutarate via a novel oxidative pathway. Evidence is presented that the oxidative degradation of d-ribose involves novel promiscuous enzymes, pentose dehydrogenase and pentonolactonase, and a novel sugar acid dehydratase highly specific for ribonate. This is the first report of an oxidative degradation pathway of d-ribose in archaea, which differs from the canonical nonoxidative pathway of d-ribose degradation reported for most bacteria. The data contribute to our understanding of the unusual sugar degradation pathways and enzymes in archaea.
Keywords: Haloarcula marismortui, d-ribose, d-xylose, l-arabinose, archaea, oxidative pentose degradation, ribonate dehydratase, UlaG protein family
ABSTRACT
The Haloarcula species H. marismortui and H. hispanica were found to grow on d-ribose, d-xylose, and l-arabinose. Here, we report the discovery of a novel promiscuous oxidative pathway of pentose degradation based on genome analysis, identification and characterization of enzymes, transcriptional analysis, and growth experiments with knockout mutants. Together, the data indicate that in Haloarcula spp., d-ribose, d-xylose, and l-arabinose were degraded to α-ketoglutarate involving the following enzymes: (i) a promiscuous pentose dehydrogenase that catalyzed the oxidation of d-ribose, d-xylose, and l-arabinose; (ii) a promiscuous pentonolactonase that was involved in the hydrolysis of ribonolactone, xylonolactone, and arabinolactone; (iii) a highly specific dehydratase, ribonate dehydratase, which catalyzed the dehydration of ribonate, and a second enzyme, a promiscuous xylonate/gluconate dehydratase, which was involved in the conversion of xylonate, arabinonate, and gluconate. Phylogenetic analysis indicated that the highly specific ribonate dehydratase constitutes a novel sugar acid dehydratase family within the enolase superfamily; and (iv) finally, 2-keto-3-deoxypentanonate dehydratase and α-ketoglutarate semialdehyde dehydrogenase catalyzed the conversion of 2-keto-3-deoxypentanonate to α-ketoglutarate via α-ketoglutarate semialdehyde. We conclude that the expanded substrate specificities of the pentose dehydrogenase and pentonolactonase toward d-ribose and ribonolactone, respectively, and the presence of a highly specific ribonate dehydratase are prerequisites of the oxidative degradation of d-ribose in Haloarcula spp. This is the first characterization of an oxidative degradation pathway of d-ribose to α-ketoglutarate in archaea.
IMPORTANCE The utilization and degradation of d-ribose in archaea, the third domain of life, have not been analyzed so far. We show that Haloarcula species utilize d-ribose, which is degraded to α-ketoglutarate via a novel oxidative pathway. Evidence is presented that the oxidative degradation of d-ribose involves novel promiscuous enzymes, pentose dehydrogenase and pentonolactonase, and a novel sugar acid dehydratase highly specific for ribonate. This is the first report of an oxidative degradation pathway of d-ribose in archaea, which differs from the canonical nonoxidative pathway of d-ribose degradation reported for most bacteria. The data contribute to our understanding of the unusual sugar degradation pathways and enzymes in archaea.
INTRODUCTION
The pentoses d-ribose, d-xylose, l-arabinose are abundant sugars in nature. d-Ribose is part of ribonucleotides and various cofactors, and d-xylose and l-arabinose are major components of hemicellulose plant material. The degradation pathways of these pentoses are well studied in bacteria. Typically, d-ribose is degraded to the pentose phosphate cycle intermediate xylulose-5-phosphate by d-ribokinase, d-ribose-5-phosphate isomerase, and ribulose-5-phosphate epimerase; this pathway is well characterized, e.g., for Escherichia coli (1, 2). Also, d-xylose and l-arabinose are degraded to xylulose-5-phosphate in most bacteria involving different isomerases, epimerases, and kinases (3). In a few bacteria, e.g., Azospirillum brasilense, Caulobacter crescentus, and Pseudomonas species, pentoses are metabolized via an alternative oxidative pathway to α-ketoglutarate (4–7).
In the domain of archaea, pentose degradation was first studied in the haloarchaeon Haloarcula marismortui growing on d-xylose. In this archaeon, enzymes of the classical degradation pathway of most bacteria are absent; instead, it was shown that the initial step of d-xylose degradation involves an inducible xylose dehydrogenase, suggesting an oxidative degradation pathway similar to the pentose degradation route in a few bacteria (8). Subsequently, the complete oxidative pathway of pentose degradation has been described in detail in Haloferax volcanii and in Sulfolobus species. In Sulfolobus solfataricus and Sulfolobus acidocaldarius, the pentoses d-arabinose, d-xylose, and l-arabinose are oxidatively degraded to α-ketoglutarate and/or l-malate (9–11). In H. volcanii, d-xylose and l-arabinose are oxidatively degraded to α-ketoglutarate (Fig. 1). Accordingly, the initial oxidation reactions of the pentoses are catalyzed by two distinct dehydrogenases (12, 13), generating the corresponding pentonolactones that are hydrolyzed by a lactonase (14). Further degradation of both xylonate and arabinonate proceeds via xylonate dehydratase, 2-keto-3-deoxyxylonate dehydratase, and α-ketoglutarate semialdehyde dehydrogenase generating α-ketoglutarate (12, 13). The genes encoding the enzymes of this pathway form a cluster and are transcriptionally activated by XacR, an IclR protein family-like transcriptional regulator of bacteria (15).
FIG 1.
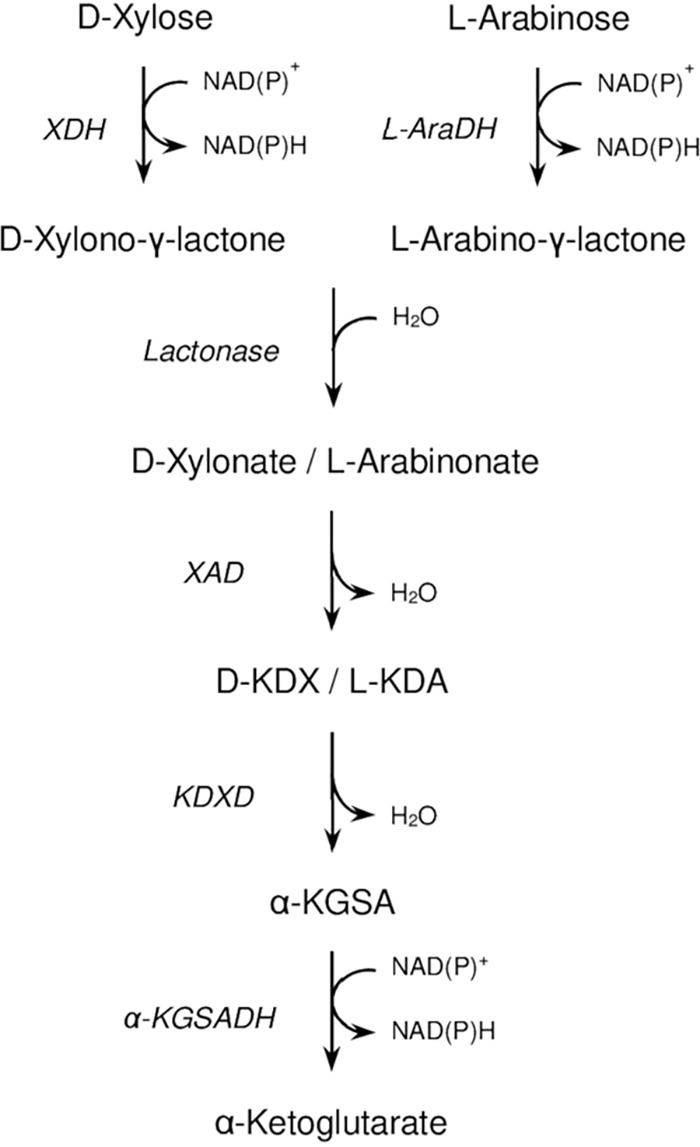
Proposed pathway of d-xylose and l-arabinose degradation in Haloferax volcanii. l-KDA, l-2-keto-3-deoxyarabinonate; α-KGSA, α-ketoglutarate semialdehyde; d-KDX, d-2-keto-3-deoxyxylonate; XDH, d-xylose dehydrogenase; l-AraDH, l-arabinose dehydrogenase; XAD, xylonate dehydratase; KDXD, KDX dehydratase; α-KGSADH, α-KGSA dehydrogenase.
So far, the degradation of d-ribose has not been reported in the domain of archaea. Here, we show that in contrast to H. volcanii, the Haloarcula species H. marismortui and H. hispanica grew efficiently on d-ribose in addition to d-xylose and l-arabinose. We report a detailed analysis of the degradation of d-ribose, d-xylose, and l-arabinose in Haloarcula species indicating the operation of an oxidative degradation pathway.
RESULTS AND DISCUSSION
The degradation of the pentoses d-xylose and l-arabinose in haloarchaea has been described in detail only for Haloferax volcanii. These pentoses were oxidized to α-ketoglutarate involving two dehydrogenases, a lactonase, and two dehydratases (12–14). d-Ribose as the substrate was not utilized by H. volcanii (13). Here, we report that H. marismortui utilizes d-ribose in addition to d-xylose and l-arabinose as the substrates. It is shown that these pentoses were degraded to α-ketoglutarate via a novel pathway involving promiscuous pentose dehydrogenase and pentonolactonase and a novel type of sugar acid dehydratase specific for ribonate.
Haloarcula species grow on d-ribose, l-arabinose, and d-xylose.
H. marismortui was grown on medium containing 25 mM d-ribose, d-xylose, or l-arabinose up to optical densities at 600 nm of about 4.0; during growth on d-ribose, the substrate was almost completely consumed (Fig. 2; see also Fig. S2 in the supplemental material). Without added sugars, cells grew on complex constituents up to an optical density of 2.5. Haloarcula hispanica DF60 also grew on a medium containing d-ribose or d-xylose but in contrast to H. marismortui did not show significant growth on l-arabinose (Fig. S3).
FIG 2.

Growth of H. marismortui on d-ribose. Cells were grown at 37°C on 25 mM d-ribose in the presence of 0.25% yeast extract and 0.5% Casamino Acids (circles), and the consumption of d-ribose (triangles) was monitored over time. Growth without the addition of d-ribose is indicated by squares.
Haloarcula species contain a pentose dehydrogenase promiscuous for d-ribose, d-xylose, and l-arabinose.
Cell extracts of H. marismortui grown on d-ribose, d-xylose, or l-arabinose contained dehydrogenase activities for d-ribose (224 mU/mg), d-xylose (161 mU/mg), and l-arabinose (200 mU/mg), respectively. The activity levels were 2- to 9-fold higher than those of glucose-grown cells indicating the induction of dehydrogenase activities by the three pentoses. In a previous study, d-xylose dehydrogenase activity from H. marismortui was purified, and the encoding gene was identified as rrnAC3034 (8). To find out whether separate enzymes or a promiscuous enzyme catalyze the dehydrogenase reactions of d-ribose and of l-arabinose, we purified the activities and identified the encoding genes.
d-Ribose dehydrogenase was purified 260-fold to a specific activity of 62 U/mg (Fig. S4). Based on the N-terminal amino acid sequence determined from the subunit of enzyme, MNVDALTGGFDRRDWQE, the encoding gene was identified in the genome of H. marismortui as rrnAC3034. This gene is identical to the gene encoding d-xylose dehydrogenase characterized previously (8) and thus codes for a promiscuous enzyme.
l-Arabinose dehydrogenase activity was purified starting with hydrophobic interaction chromatography as the first step, yielding two distinct activity peaks. Each of them was further purified, yielding a 37-kDa protein and a 52-kDa protein on SDS-PAGE, and by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) analysis, rrnAC0771 and rrnAC3034, respectively, were identified as the encoding genes. Both genes were overexpressed in H. volcanii H1209. The recombinant protein encoded by rrnAC3034 showed a high level of l-arabinose dehydrogenase activity, at 114 U/mg. These data indicate that rrnAC3034 encodes a promiscuous dehydrogenase for the three pentoses d-xylose, d-ribose, and l-arabinose. The catalytic efficiencies with d-ribose and d-xylose were almost identical, whereas the efficiency with l-arabinose was 20-fold lower due to a significant higher Km value for l-arabinose (Table 1). In contrast, the recombinant protein encoded by rrnAC0771, annotated as putative 3-hydroxybutyryl coenzyme A (3-hydroxybutyryl-CoA) dehydrogenase, showed low specific activity with l-arabinose (<0.1 U/mg at 20 mM l-arabinose), suggesting a minor role in the oxidation of l-arabinose in H. marismortui; the physiological role of this enzyme remains to be demonstrated.
TABLE 1.
Kinetic parameters of recombinant pentose dehydrogenase from Haloarcula marismortui
| Substance | Vmax (U/mg) | kcat (s−1) | Km (mM) | kcat/Km (s−1 M−1) |
|---|---|---|---|---|
| d-Xylose | 83.37 ± 8.46 | 2.50 × 102 ± 25.3 | 2.32 ± 0.26 | 10.77 × 104 |
| d-Ribose | 116.95 ± 2.66 | 3.51 × 102 ± 8.0 | 3.89 ± 0.22 | 9.01 × 104 |
| l-Arabinose | 114.25 ± 21.62 | 3.42 × 102 ± 64.8 | 63.93 ± 18.78 | 0.5 × 104 |
Transcriptional analysis of rrnAC3034 was performed by Northern blotting with RNA from H. marismortui cells grown on d-xylose, d-ribose, or l-arabinose compared to glucose. A specific transcript signal at 1,200 nucleotides could be detected only in pentose-grown cells but not in glucose-grown cells (Fig. 3A). The pentose-specific signal matches the length of rrnAC3034 (1,083 nucleotides). The data indicate that rrnAC3034 was induced transcriptionally by the three pentoses.
FIG 3.

Genes involved in d-ribose, d-xylose, and l-arabinose degradation to α-ketoglutarate in Haloarcula species and analysis of transcription. (A) Northern blotting of target genes from H. marismortui was performed with RNA from cells grown on glucose (G), d-xylose (X), d-ribose (R), and l-arabinose (A) using probes against genes of the cluster. 16S rRNA served as a loading control. (B) Genes involved in pentose degradation encode putative xylonate/gluconate dehydratase (96%), 2-keto-3-deoxypentanonate dehydratase (KDPD; 94%), ribonate dehydratase (91%), pentonolactonase (99%), pentose dehydrogenase (91%), α-ketoglutarate semialdehyde dehydrogenase (87%), and the putative transcriptional regulator of the pentose degradation pathway (96%). Sequence identities of homologous enzymes are given in parentheses. The bar indicates a length of 1,000 bp.
The gene rrnAC3034 in H. marismortui is part of a gene cluster that includes genes encoding putative lactonase (rrnAC3033), dehydratase (rrnAC3032), and α-ketoglutarate semialdehyde dehydrogenase (rrnAC3036). A homologous gene cluster was also identified in Haloarcula hispanica (Fig. 3B). Since a genetic system is established to generate gene deletion mutants for H. hispanica strain DF60 (16), we used this strain to prove the functional involvement of putative enzymes of pentose degradation in Haloarcula species. A deletion mutant of HAH_0291 encoding pentose dehydrogenase was generated, and growth was analyzed on d-xylose, d-ribose, and glucose. The mutant did not grow on d-ribose (Fig. 4), whereas growth on glucose was not affected (not shown). This indicates a functional involvement of the dehydrogenase in d-ribose degradation. Unexpectedly, the ΔHAH_0291 mutant grew on d-xylose as shown for the wild type; this might be explained by the presence of other dehydrogenases that replace the d-xylose dehydrogenase activity in the mutant. In accordance, d-xylose dehydrogenase activity of about 0.066 U/mg was detected in cell extracts of the ΔHAH_0291 mutant.
FIG 4.

Analysis of a deletion mutant of pentose dehydrogenase from Haloarcula hispanica. Shown is the growth of the ΔHAH_0291 mutant of H. hispanica DF60 on d-ribose (filled circles) compared to the wild type (filled squares) and the consumption of d-ribose of the ΔHAH_0291 mutant (open circles) and of the wild type (open squares).
Together, the data indicate that the pentose dehydrogenase from both Haloarcula species is promiscuous for the utilization of the three pentoses d-xylose, d-ribose, and l-arabinose. In contrast, H. volcanii contains two distinct dehydrogenases for the oxidation of d-xylose and l-arabinose, both of which do not utilize d-ribose (12, 13). The promiscuous pentose dehydrogenase from H. marismortui and specific d-xylose dehydrogenase from H. volcanii are both members of the GFO/IDH/MocA family (Pfam02894), in which the dehydrogenase of Haloarcula species evolved a novel substrate specificity toward d-ribose and l-arabinose.
Haloarcula species contain a pentonolactonase promiscuous for ribonolactone, xylonolactone, and arabinolactone.
Adjacent to each of the pentose dehydrogenase genes (Fig. 3B) is a putative lactonase encoded by rrnAC3033 in H. marismortui and HAH_0290 in H. hispanica. To analyze the catalytic properties of the lactonase, we expressed HAH_0290 in H. volcanii H1209, followed by purification and characterization. The recombinant lactonase had a molecular weight of 105 kDa and was composed of subunits with a calculated molecular mass of 32.4 kDa, indicating a homotrimeric structure of the enzyme (Fig. S5A). The enzyme catalyzed the hydrolysis of d-xylono-γ-lactone with high catalytic efficiency (Table 2). With d-ribono-γ-lactone (Fig. S5B) and l-arabino-γ-lactone, the enzyme showed 5.4- and 1.9-fold lower catalytic efficiencies. The enzyme hydrolyzed l-rhamnono-1,4-lactone with low activity (20 U/mg); d-galactono-γ-lactone, d-glucono-1,5-γ-lactone, and d-mannono-1,4-γ-lactone were not used as substrates. The lactonase activity was dependent on divalent cations being stimulated by Mn2+, with a Km value of 0.049 mM; Mn2+ could be replaced by Co2+ (41%, at 1 mM) rather than by Mg2+, Cu2+, Ca2+, Zn2+, or Ni2+. The data indicate that HAH_0290 encodes a metal-dependent lactonase that is specific for pentonolactones.
TABLE 2.
Kinetic parameters of recombinant pentonolactonase from Haloarcula hispanica
| Substance | Vmax (U/mg) | kcat (s−1) | Km (mM) | kcat/Km (s−1 M−1) |
|---|---|---|---|---|
| d-Xylono-γ-lactone | 211.6 ± 13.06 | 370.3 ± 22.86 | 3.54 ± 0.44 | 1.046 × 105 |
| d-Ribono-γ-lactone | 93.4 ± 7.12 | 163.5 ± 12.46 | 8.48 ± 0.38 | 0.193 × 105 |
| l-Arabino-γ-lactone | 139.6 ± 7.48 | 244.3 ± 13.09 | 4.49 ± 0.47 | 0.544 × 105 |
The involvement of pentonolactonase in the degradation of the three pentoses was analyzed by Northern blotting with RNA from H. marismortui grown on d-xylose, d-ribose, l-arabinose, and glucose. Using a probe against rrnAC3033 of H. marismortui, a specific transcriptional signal of 1,100 nucleotides was detectable in cells grown on d-xylose, d-ribose, and l-arabinose that matches well to the gene length of 861 nucleotides; no transcript could be detected in glucose-grown cells (Fig. 3A). The data indicate a specific function of the pentonolactonase in the degradation of the three pentoses.
The pentonolactonase from Haloarcula species differs from the recently characterized pentonolactonase from H. volcanii in various aspects. (i) In contrast to Haloarcula, the Haloferax enzyme did not utilize ribonolactone and showed a broad specificity toward hexonolactones (14). (ii) The Haloferax lactonase belongs to the senescence marker protein (SMP)-30/gluconolactonase/luciferin-regenerating enzyme-like (SGL) protein family (PF08450) (17), whereas the Haloarcula enzyme is a member of the metallo-β-lactamase (MBL) superfamily. This superfamily also comprises the lactonase UlaG of E. coli (18) that catalyzes the hydrolysis of l-ascorbate 6-phosphate to 3-keto-l-gulonate 6-phosphate as part of the ascorbate degradation pathway. Figure 5 shows a sequence alignment of the pentonolactonase of Haloarcula hispanica, the l-ascorbate 6-phosphate lactonase (UlaG) from E. coli, and selected homologous sequences from bacteria. The predicted secondary structure of pentonolactonase from H. hispanica matches well to the secondary structure elements concluded from the crystal structure of UlaG from E. coli. In addition, the residues involved in the binding of a divalent cation are conserved in the haloarchaeal enzyme (18, 19) (Fig. 5).
FIG 5.

Multiple-sequence alignment (calculated with ClustalX [28]) of pentonolactonase from Haloarcula hispanica, l-ascorbate 6-phosphate lactonase (UlaG) from E. coli, and UlaG homologs from other bacteria. Structure-based secondary structure elements of UlaG from E. coli and the predicted secondary structure (29) of pentonolactonase from H. hispanica are displayed using ESPript 3.0 (30). Conserved amino acid residues are indicated in red. Residues that contribute to the Mn2+ binding site of UlaG from E. coli are indicated by asterisks. Eco_2WYM (PDB identifier 2WYM), E. coli l-ascorbate 6-phosphate lactonase; CLL_A3267, Clostridium botulinum B strain Eklund 17B (NCBI accession no. YP_001887453); SpyM3_0142, Streptococcus pyogenes MGAS315 (NCBI accession no. NP_663946); Acear_0951, Acetohalobium arabaticum DSM 5501 (NCBI accession no. YP_003827546.1); HAH_0290, H. hispanica, pentonolactonase; and HAH_1228, H. hispanica, a paralog of pentonolactonase.
Further, UlaG from E. coli forms a hexameric structure that is unique within the MBL superfamily (18). This oligomeric structure is particularly stabilized by the UlaG-specific N-terminal motif of 35 to 40 amino acid residues (18). As shown in Fig. 5, this extreme N-terminal stretch is absent in the pentonolactonase sequence of H. hispanica and in other haloarchaeal homologs. It has been speculated that the lack of this N-terminal stretch in haloarchaeal UlaG homologs might prevent assembly of the protein as a hexamer (19); in accordance, we characterized the pentonolactonase of H. hispanica as a homotrimeric enzyme.
The pentonolactonase of H. hispanica represents the first characterized UlaG homolog in the archaeal domain. The finding that UlaG homologs are only present in haloarchaea rather than in any other archaeal species suggests that haloarchaea acquired the pentonolactonase from halophilic bacteria via lateral gene transfer events likely occurring in halophilic environments (19).
Haloarcula species contain a specific d-ribonate dehydratase and a promiscuous xylonate/gluconate dehydratase.
Downstream of rrnAC3033, which encodes pentonolactonase, rrnAC3032 is annotated as a gene encoding a putative sugar acid dehydratase of the enolase superfamily. We identified the catalytic function of the recombinant protein following the expression of rrnAC3032 in H. volcanii H1209 and subsequent purification.
The enzyme had a molecular mass of 320 ± 30 kDa; the calculated molecular mass of the subunit is 44.2 kDa, indicating a homo-octameric structure. The enzyme catalyzed the dehydration of ribonate at a rate of 0.25 U/mg, with a Km value of 1.64 mM. Activity was dependent on Mg2+, showing a Km value of 0.33 mM. The enzyme was highly specific for ribonate; other sugar acids, arabinonate, xylonate, gluconate, and galactonate (<1%), were not utilized.
Since ribonate dehydratase is highly specific for ribonate not utilizing xylonate, we identified the encoding gene of xylonate dehydratase following purification of xylonate dehydratase activity from H. marismortui cells grown on d-xylose. Xylonate dehydratase activity was purified 250-fold up to a specific activity of 0.060 U/mg (Fig. S6). By MALDI-TOF MS analysis, rrnAC0575 was identified as the encoding gene. rrnAC0575 was expressed in H. volcanii H1209, and the recombinant enzyme was purified as a 328-kDa protein showing a specific activity of 0.028 U/mg with xylonate (at 10 mM). With gluconate as the substrate, a significantly higher activity, 6.6 U/mg, was obtained; the Km value of gluconate was 7.85 mM. No activity could be detected with ribonate, arabinonate, and galactonate. The high activity with gluconate suggests that the enzyme has also a function as gluconate dehydratase in the glucose catabolism of H. marismortui. To test this hypothesis, we purified gluconate dehydratase activity from glucose-grown H. marismortui cells. By MALDI-TOF MS analysis of the subunit, rrnAC0575 was identified as the encoding gene. Thus, rrnAC0575 encodes a bifunctional dehydratase, catalyzing both the C-5 sugar acid xylonate and the C-6 sugar acid gluconate, and it is thus functionally involved in both pentose and hexose catabolism of H. marismortui.
Transcriptional analysis of xylonate/gluconate dehydratase and of ribonate dehydratase.
The transcription of rrnAC0575, encoding xylonate/gluconate dehydratase, was followed by Northern blot analysis using RNA from cells grown on d-xylose, glucose, d-ribose, and l-arabinose. As indicated in Fig. 3A, a strong transcriptional signal at 1,300 nucleotides was detected in cells grown on the three pentoses as well as on glucose; the length matches well to the length of 1,251 nucleotides of rrnAC0575.
The upregulation of rrnAC0575 in glucose- and pentose-grown cells indicates that the encoded dehydratase is promiscuous and is involved in both glucose and pentose catabolism.
Northern blotting using a probe against rrnAC3032 (gene length, 1,176 nucleotides), encoding ribonate dehydratase, revealed a transcriptional signal at 1,200 nucleotides in cells grown on the three pentoses rather than on glucose (Fig. 3A). Specific upregulation by pentoses was also observed (see above) for rrnAC3034, encoding the pentose dehydrogenase, and for rrnAC3033, encoding the pentonolactonase, indicating that these three genes are activated by a common transcriptional regulator.
Deletion mutant analysis of sugar acid dehydratases.
The functional involvement of ribonate dehydratase in d-ribose degradation in Haloarcula species was tested with a H. hispanica HAH_0289 deletion mutant. Growth of this mutant on d-ribose was impaired compared to the wild type, whereas growth on d-xylose and glucose was not affected (Fig. 6A), indicating the essential function of ribonate dehydratase in d-ribose degradation.
FIG 6.

Growth of the ΔHAH_0289 (A) and ΔHAH_1276 (B) mutants of H. hispanica on d-ribose, d-xylose, and glucose. Growth of the mutants (squares) compared to the wild type (circles) was analyzed on 20 mM each d-ribose (blue), d-xylose (red), and glucose (green). All experiments were performed with 0.25 g per liter yeast extract. Without added sugar, cells grew up to an optical density of about 0.3.
To analyze the function of xylonate/gluconate dehydratase in pentose and glucose catabolism, we deleted HAH_1276 of H. hispanica and performed growth experiments on glucose, d-xylose, and d-ribose (Fig. 6B). As expected, the ΔHAH_1276 mutant showed impaired growth on both d-xylose and glucose, whereas growth on d-ribose was not affected, supporting the role of the dehydratase in both d-xylose and glucose degradation.
The function of the mutants in l-arabinose degradation could not be tested since the H. hispanica wild type did not show significant growth on this pentose (Fig. S3). Therefore, we used a different approach to find out whether the xylonate/gluconate dehydratase also functions as arabinonate dehydratase. We performed growth experiments with a HVO_B0038A deletion mutant of H. volcanii that, in contrast to the wild type, is not able to grow on l-arabinose and d-xylose (12, 13). HVO_B0038A encodes a dehydratase that is promiscuous for xylonate and arabinonate, and thus, the ΔHVO_B0038A mutant can be used to identify the catalytic function of xylonate/gluconate dehydratase of H. marismortui as arabinonate dehydratase by complementation. As shown in Fig. S7, complementation of the ΔHVO_B0038A mutant with rrnAC0575 from H. marismortui effectively restored growth on both l-arabinose and d-xylose. This indicates that the xylonate/gluconate dehydratase from H. marismortui functions as arabinonate dehydratase as well as xylonate dehydratase. The latter result is in accordance with the impaired growth of the ΔHAH_1276 mutant on d-xylose (Fig. 6B). Complementation with rrnAC3032 encoding ribonate dehydratase did not restore growth of the ΔHVO_B0038A mutant on l-arabinose and d-xylose, which is in accordance with the high specificity of ribonate dehydratase for ribonate (Fig. S7).
Together, the data indicate that Haloarcula species contain two distinct sugar acid dehydratases, a ribonate dehydratase that is highly specific for ribonate and a xylonate/gluconate dehydratase that is promiscuous for the C-5 sugar acids xylonate and arabinonate and the C-6 sugar acid gluconate.
Phylogenetic analysis of sugar acid dehydratases.
The xylonate/gluconate dehydratase and ribonate dehydratase of Haloarcula species are members of the enolase-like superfamily. Xylonate/gluconate dehydratase shows a high degree of identity to xylonate dehydratase (XAD) (87%) from H. volcanii, involved in d-xylose/l-arabinose catabolism, and to gluconate dehydratase (GAD) (96%), involved in glucose catabolism (12, 13, 20). These dehydratases and putative homologs from other haloarchaea constitute a distinct family within the enolase superfamily. In contrast, gluconate dehydratases from the thermophilic archaea Thermoproteus tenax (21), Sulfolobus solfataricus (22), and Picrophilus torridus (23), which are part of modified Entner-Doudoroff (ED) pathways of glucose degradation, form a cluster distinct from the haloarchaeal XAD/GAD cluster (Fig. 7).
FIG 7.

Phylogenetic relationship of ribonate dehydratase and xylonate/gluconate dehydratase from Haloarcula spp. and related families of the enolase superfamily. The tree is based upon a multiple-amino-acid sequence alignment that was generated with ClustalX (28). Numbers at the nodes are bootstrapping values according to neighbor joining (NJ) (generated by using the NJ options of ClustalX). UniProt accession numbers are as follows: for ribonate D, Advenella mimigardefordensis, W0P9I6; Superficieibacter electus, A0A2P5GKV8; Caballeronia hypogeia, A0A157ZKC6; Paraburkholderia fungorum, A0A420FUX3; Salinigranum rubrum 1, A0A2I8VEK2; Salinigranum rubrum 2, A0A2I8VH23; Haloferax denitrificans, M0JBW3; Haloarcula marismortui, Q5UY97; Haloarcula hispanica, G0HRT2; Halopiger xanaduensis (H. xana.), F8DD77; Haloterrigena turkmenica, D2RXH3; for GAD/XAD, subcluster A, gluconate dehydratase of Haloferax volcanii, D4GYD4; xylonate/gluconate dehydratases of Haloarcula marismortui and Haloarcula hispanica, Q5V4G3 and G0HPX2, respectively; Halopiger xanaduensis 1 (H. xana 1), F8D4X4; Halopiger xanaduensis 2 (H. xana. 2), F8D4H7; Halorubrum lacusprofundi (H. lacus.), B9LNF4; subcluster B, XAD from Haloferax volcanii, D4GP40; Halorubrum lacusprofundi (H. lacus.), B9LRU5; Halopiger xanaduensis (H. xana.), F8DCB2. Accession numbers of enolases, rhamnonate dehydratases, fuconate dehydratases, and gluconate dehydratases can be found in references 20 and 25.
Ribonate dehydratase of Haloarcula spp. showed high sequence identity (70% to 80%) to homologs found in only a few other haloarchaea, e.g., Salinigranum rubrum, Haloterrigena turkmenica, Halopiger xanaduensis, and Haloferax denitrificans. Putative ribonate dehydratase homologs with lower sequence identity (45% to 58%) were also found in a few bacteria, e.g., Advenella mimigardefordensis, Superficieibacter electus, Caballeronia hypogeia, and Paraburkholderia fungorum. Together, the ribonate dehydratase of Haloarcula spp. and homologous sequences constitute a novel family within the enolase superfamily (Fig. 7). This superfamily also includes rhamnonate dehydratases, fuconate dehydratases, and enolases, which all form distinct clusters in accordance with their specific catalytic function.
It should be noted that homologous sequences of ribonate dehydratase, pentose dehydrogenase, and pentonolactonase are not present in Haloferax volcanii, which might explain the inability of this organism to grow on d-ribose.
Identification of 2-keto-3-deoxypentanonate dehydratase and of α-ketoglutarate semialdehyde dehydrogenase in Haloarcula species.
In H. marismortui and H. hispanica, genes (rrnAC1339 and HAH_1926) have been identified that are homologous to 2-keto-3-deoxyxylonate dehydratase (KDXD), the second dehydratase involved in the oxidative pentose degradation pathway of H. volcanii (12).
Transcriptional analysis was performed using a probe against rrnAC1339 and RNA from H. marismortui cells grown on pentoses and glucose. A specific transcript signal at 1,100 nucleotides was observed in cells grown on d-ribose-, d-xylose-, and l-arabinose rather than in glucose-grown cells (Fig. 3A). The upregulation of the KDXD homolog by the three pentoses indicates that the encoded protein is involved in the oxidative degradation of pentoses in H. marismortui. Downstream of rrnAC1339, a putative transcriptional regulator, rrnAC1338, is located that belongs to the IclR family (Fig. 3B). This IclR-like regulator shows high sequence identity (51%) to XacR of H. volcanii, which has been characterized as a transcriptional activator of xac genes involved in d-xylose and l-arabinose degradation (15). We propose that rrnAC1338 of H. marismortui and the homolog HAH_1925 of H. hispanica function as transcriptional regulators of pentose degradation in Haloarcula species.
The final step in oxidative degradation of pentoses in H. volcanii is catalyzed by α-ketoglutarate semialdehyde dehydrogenase (KGSADH) (Fig. 1). In cell extracts of H. marismortui grown on d-xylose, d-ribose, and l-arabinose, α-ketoglutarate semialdehyde dehydrogenase activities of 101 mU/mg, 93 mU/mg, and 39 mU/mg, respectively, could be detected. The activities were 2- to 6-fold higher than those of cell extract of glucose-grown cells (18 mU/mg), indicating induction by the three pentoses. Since several homologs of KGSADH from H. volcanii were identified in the genome of H. marismortui, we purified the enzyme from d-xylose-grown cells and identified the encoding gene. The enzyme was 40-fold enriched as a 209-kDa protein showing a specific activity of 2.2 U/mg (Fig. S8). By MALDI-TOF MS analysis of the subunit, rrnAC3036 was identified as the gene encoding α-ketoglutarate semialdehyde dehydrogenase with a subunit size of 50 kDa. This gene is located adjacent to genes coding for pentose dehydrogenase, pentonolactonase, and ribonate dehydratase (Fig. 3B).
The gene rrnAC3036 was cloned and expressed in H. volcanii H1209, and the recombinant enzyme was purified and characterized. The homotetrameric (203 kDa) α-ketoglutarate semialdehyde dehydrogenase showed a specific activity of 11 U/mg, with apparent Km values for glutardialdehyde and NADP+ of 0.15 mM and 0.02 mM, respectively. With NAD+, Vmax and Km values of 58.7 U/mg and 0.8 mM, respectively, were measured, indicating a 7.5-fold higher catalytic efficiency of the enzyme for NADP+. The specific activity with succinate semialdehyde (Km, 0.46 mM) was 0.139 U/mg, and that with α-ketoglutarate semialdehyde was 0.1 U/mg.
Northern blot analysis was performed with RNA from d-ribose-, d-xylose-, and l-arabinose-grown cells in comparison to glucose-grown cells. Using a probe against rrnAC3036, a strong transcriptional signal at 1,500 nucleotides was observed on the three pentoses, whereas on glucose, the signal had a significantly weaker intensity (Fig. 3A). These data correspond with activity measurements in cell extracts grown on the respective sugars (see above) and indicate that α-ketoglutarate semialdehyde dehydrogenase represents the final enzyme in the degradation of d-ribose, d-xylose, and l-arabinose to α-ketoglutarate.
Together, the data indicate that the KDXD homolog, the α-ketoglutarate semialdehyde dehydrogenase, and the putative IclR-like transcriptional regulator have the same function in oxidative pentose degradation in Haloarcula spp. as that reported for H. volcanii.
Conclusion.
We here report a novel oxidative pathway of pentose degradation to α-ketoglutarate in Haloarcula species that allows the utilization of d-ribose in addition to d-xylose and l-arabinose (Fig. 8). Evidence is presented that the oxidative degradation of d-ribose is due to an expanded substrate promiscuity of the pentose dehydrogenase and pentonolactonase and to an innovation of a novel enzyme, ribonate dehydratase, which is highly specific for ribonate. These enzymes are not present in Haloferax volcanii, which might explain the inability of this haloarchaeon to utilize d-ribose (Fig. 8). This is the first report of an oxidative degradation pathway of d-ribose to α-ketoglutarate in archaea.
FIG 8.

Oxidative degradation pathways for d-ribose, d-xylose, and l-arabinose to the citric acid cycle intermediate α-ketoglutarate in haloarchaea. The oxidative degradation of d-ribose is restricted to Haloarcula species involving promiscuous pentose dehydrogenase and pentonolactonase and the innovation of a specific ribonate dehydratase. Enzymes that belong to the same protein family are indicated by the same color, as follows: Gfo/Idh/MocA oxidoreductase family, red; short-chain dehydrogenase/reductase family, purple; metallo-β-lactamase superfamily, orange; senescence marker protein (SMP)-30/gluconolactonase/luciferin-regenerating enzyme-like (SGL) protein family, brown; enolase superfamily, light green and green; fumarylacetoacetate hydrolase superfamily, yellow; and aldehyde dehydrogenase family, blue. Specific ribonate dehydratase (green) and promiscuous xylonate/gluconate dehydratase (light green) of Haloarcula species together with xylonate/arabinonate dehydratase (light green) of Haloferax volcanii are members of the enolase superfamily. KDR/X, 2-keto-4(S),5-dihydroxy pentanoic acid; KDA, 2-keto-(R),5-dihydroxy pentanoic acid; α-KGSA, α-ketoglutarate semialdehyde.
MATERIALS AND METHODS
Strains, media, and growth conditions.
Haloarcula marismortui (DSM 3752) was grown aerobically at 37°C on a medium containing 25 mM d-ribose, d-xylose, l-arabinose, or d-glucose, 0.25% yeast extract, and 0.5% Casamino Acids; during growth, samples were taken to analyze the consumption of the pentoses over time using the orcinol assay (8). Haloarcula hispanica DF60 and the ΔHAH_0289, ΔHAH_0291, and ΔHAH_1276 knockout mutants were grown aerobically at 37°C on medium containing 25 mM d-ribose, d-xylose, l-arabinose, or d-glucose and yeast extract (0.1% or 0.025%) (16). The ΔHVO_B0038A mutant of Haloferax volcanii and cells that were transformed with the plasmid pTA963-rrnAC0575 or pTA963-rrnAC3032 were grown aerobically at 42°C in synthetic medium containing 25 mM d-xylose or l-arabinose (20). Growth was followed by measuring the optical density at 600 nm, and cell extracts were prepared from late-log-phase-grown cells by sonication and subsequent centrifugation.
Purification of enzymes from H. marismortui.
(i) Purification of d-ribose dehydrogenase activity. Cell extracts were prepared from d-ribose-grown cells in buffer A (100 mM Tris-HCl [pH 8.8] containing 2 M ammonium sulfate and 20 mM MgCl2) using a French press, followed by centrifugation. The supernatant was applied on a Sepharose CL4B column (GE Healthcare, Germany) that was equilibrated in buffer A; elution of protein was performed with a decreasing gradient of ammonium sulfate. Fractions containing d-ribose dehydrogenase activity were applied on a phenyl-Sepharose column (GE Healthcare) in buffer A, and protein was eluted by decreasing the concentration of ammonium sulfate. Further purification was performed on a Superdex 200 HiLoad 16/60 gel filtration column (GE Healthcare) in 50 mM Tris-HCl (pH 8.5) containing 150 mM NaCl, 20 mM MgCl2, and 10% glycerol.
(ii) Purification of l-arabinose dehydrogenase activity. Cell extracts were prepared from l-arabinose-grown cells in 100 mM Tris-HCl (pH 8.0) containing 2 M ammonium sulfate (buffer B). After application on a phenyl-Sepharose column, protein was eluted by decreasing the concentration of ammonium sulfate. Fractions containing l-arabinose dehydrogenase activity were pooled and purified by size exclusion chromatography using a Superdex 200 column and 50 mM Tris-HCl (pH 7.5) containing 2 M KCl. Further purification was performed using a butyl-Sepharose HP column (GE Healthcare) in buffer B; protein was eluted by decreasing the concentration of ammonium sulfate, and the buffer was exchanged using 50 mM Tris-HCl (pH 8.8) containing 150 mM NaCl and 50 mM MgCl2. This protein solution was applied onto a Q-Sepharose column (Bio-Rad, Germany), and elution of protein was performed by increasing the concentration of NaCl. By this procedure, two different enzymes with l-arabinose dehydrogenase activity were obtained.
(iii) Purification of ribonate dehydratase and xylonate/gluconate dehydratase. Cell extracts were prepared from d-xylose-grown cells in buffer B containing 50 mM MgCl2 and were applied on a phenyl-Sepharose column; protein was eluted by decreasing the concentration of ammonium sulfate. Fractions containing both xylonate and ribonate dehydratase activity were then applied on a Superdex 200 column, and protein was eluted in 50 mM Tris-HCl (pH 7.5) containing 1.5 M KCl (buffer C). This procedure resulted in a separation of partially purified ribonate and xylonate/gluconate dehydratase. Gluconate dehydratase was purified from glucose-grown cells by the same procedure.
(iv) Purification of KGSADH.
Cell extracts were prepared from d-xylose-grown cells in 100 mM Tris-HCl (pH 8.0) containing 2 M ammonium sulfate and 50 mM MgCl2 and were applied on a phenyl-Sepharose column; protein was eluted by decreasing the concentration of ammonium sulfate. Further purification of the enzyme was performed on a Superdex 200 column in buffer C.
In all cases, protein purity was assessed by SDS-PAGE, and protein concentration was determined using the Bradford method. The encoding genes of the purified enzymes were identified by either N-terminal amino acid sequencing or by MALDI-TOF MS analysis.
Determination of native molecular masses of enzymes.
Size exclusion chromatography was carried out with a flow rate of 1 ml per min on a Superdex 200 HiLoad column (1.6 by 60 cm). Calibration of the column was performed with HMW and LMW kits (GE Healthcare), as specified by the manufacturer.
Cloning and expression of target genes in Haloferax volcanii H1209.
Target genes were amplified from genomic DNA of H. hispanica or H. marismortui. DNA fragments were each ligated into the plasmid pTA963, and H. volcanii H1209 (24) was transformed with the respective plasmids. The expression of target genes in H. volcanii H1209 was induced by the addition of 2 mM l-tryptophan, followed by further growth for 16 h at 42°C (25).
Purification of recombinant enzymes.
(i) Pentose dehydrogenase (rrnAC3034), pentonolactonase (HAH_0290), and KGSADH (rrnAC3036). H. volcanii H1209 cell pellets (see above) were suspended in 50 mM Tris-HCl (pH 8.2), containing 1.5 M KCl, 50 mM MgCl2, and 5 mM imidazole, and disruption of cells was performed by passing through a French press cell, followed by a centrifugation step. The supernatants were applied on a nickel-nitrilotriacetic acid (Ni-NTA) column (GE Healthcare), and elution of recombinant proteins was performed with 100 mM imidazole. Recombinant proteins were further purified with a Superdex 200 column in 50 mM Tris-HCl (pH 7.5) containing 2 M KCl.
Recombinant rrnAC0771 protein was purified by affinity chromatography using a Ni-NTA column and 50 mM Tris-HCl (pH 8.2) containing 1.5 M KCl. Elution of protein was performed with 200 mM imidazole.
(ii) Xylonate/gluconate dehydratase (rrnAC0575) and ribonate dehydratase (rrnAC3032). Cell extracts were prepared in 50 mM Tris-HCl (pH 8.0) containing 5 mM imidazole and 2 M KCl and were applied on a Ni-NTA column. Elution of recombinant proteins was performed with 100 mM imidazole. Recombinant proteins were further purified using a Superdex 200 column and 100 mM Tris-HCl (pH 8.0) containing 2 M KCl.
Enzyme assays.
All dehydrogenases were measured by following the reduction of NADP+ at 340 nm. Enzymes from H. marismortui were measured at 37°C, and the pentonolactonase from H. hispanica was measured at 42°C. Sugar acid dehydratases were measured in a discontinuous thiobarbituric acid (TBA) assay (20). Protein concentrations were determined using the Bradford method.
Pentose dehydrogenase (rrnAC3034) from H. marismortui was measured in 20 mM Tris-HCl (pH 8.3) containing 1.5 M KCl, 1 mM NADP+, and 10 mM d-ribose, d-xylose, or l-arabinose. The enzyme encoded by rrnAC0771 of H. marismortui was measured in 50 mM HEPES-KOH (pH 8.0) containing 2 M KCl, 20 mM l-arabinose, and 1 mM NADP+.
Pentonolactonase (HAH_0290) from H. hispanica was measured by monitoring the pH change caused by acid formation from lactones using the pH indicator p-nitrophenol (14). The assays were performed in 10 mM Bis-Tris (pH 7.5) containing 1.5 M KCl, 1 mM MnCl2, and 10 mM lactone. The Km value of MnCl2 was measured in the presence of 15 mM xylonolactone.
Xylonate/gluconate dehydratase (rrnAC0575) from H. marismortui was measured in 10 mM xylonate, 25 mM MgCl2, and 1 M KCl in 0.1 M Tris-HCl (pH 8.5). Substrate specificity was tested by replacing xylonate with gluconate, arabinonate, ribonate, and galactonate (10 mM each).
Ribonate dehydratase (rrnAC3032) from H. marismortui was measured in 0.1 M Tris-HCl (pH 8.0) containing 25 mM MgCl2 and 5 mM ribonate. Arabinonate, xylonate, gluconate, and galactonate were tested as alternative substrates.
KGSADH (rrnAC3036) from H. marismortui was measured in an assay mixture containing 1 mM NADP+, 10 mM glutardialdehyde, and 1 M KCl in 0.1 M HEPES-KOH (pH 8.5). KGSADH was also measured with α-ketoglutarate semialdehyde (11) and succinate semialdehyde.
Transcriptional analysis.
RNA from exponentially grown cells of H. marismortui (optical density at 600 nm of about 3.0) was isolated as described previously (13). Northern blot analysis was performed with 8 and 15 μg RNA (26). Probes were generated by PCR using the PCR digoxigenin (DIG) probe synthesis kit (Roche Diagnostics, Mannheim, Germany); the primers used are summarized in Table S1. The sizes of the transcripts were calculated using the RiboRuler high-range RNA ladder (Thermo Fisher Scientific, Schwerte, Germany).
Generation of knockout mutants of H. hispanica DF60.
Deletion mutants were constructed using the pop-in/pop-out strategy (16, 27). The upstream and downstream regions of each target gene were amplified and fused by PCR (see Table S1 in the supplemental material). The fused PCR products were cloned into the suicide vector pHAR (16). For deletion of HAH_1276, the suicide vector pTA131 (27) was used in which the pyrE2 gene (plus promoter) of Haloferax volcanii was replaced by the pyrF gene of H. hispanica as a selection marker. H. hispanica DF60, the pyrF deletion strain, was transformed with the plasmids, and pop-in cells were cultivated in uracil-free medium with 1% Casamino Acids. Pop-out clones were selected by plating cells on medium containing uracil (50 μg/ml) and 5-fluoroorotic acid (150 μg/ml), followed by passaging the pop-out cells in liquid medium (16). Deletion of the target genes was identified by PCR, followed by DNA sequencing or Southern blot analysis (Fig. S1).
Supplementary Material
ACKNOWLEDGMENTS
We thank Roland Schmid (Osnabrück, Germany) for performing the N-terminal amino acid sequencing, Michael Bott (Jülich, Germany) for MALDI-TOF MS analysis, and Marius Orthjohann (Kiel, Germany) for replacing the pyrE2 selection marker of pTA131 with pyrF from Haloarcula hispanica.
This work was supported by the Deutsche Forschungsgemeinschaft (grant SCHO 316/11-1).
P.S. and U.J. initiated the project; J.-M.S., U.J., A.R., and A.P. performed the experiments; R.W. and H.X. generated the ΔHAH_0289 and ΔHAH_0291 mutants in Haloarcula hispanica DF60; and U.J., A.R., and P.S. were involved in writing the manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Orencio-Trejo M, Utrilla J, Fernández-Sandoval MT, Huerta-Beristain G, Gosset G, Martinez A. 2010. Engineering the Escherichia coli fermentative metabolism. Adv Biochem Eng Biotechnol 121:71–107. doi: 10.1007/10_2009_61. [DOI] [PubMed] [Google Scholar]
- 2.David J, Wiesmeyer H. 1970. Regulation of ribose metabolism in Escherichia coli. I. The ribose catabolic pathway. Biochim Biophys Acta 208:45–55. doi: 10.1016/0304-4165(70)90047-4. [DOI] [PubMed] [Google Scholar]
- 3.van de Werken HJ, Brouns SJ, van der Oost J. 2008. Pentose metabolism in archaea, p 71–94. In Blum P. (ed), Archaea: new models for prokaryotic biology. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 4.Watanabe S, Kodaki T, Kodak T, Makino K. 2006. Cloning, expression, and characterization of bacterial l-arabinose 1-dehydrogenase involved in an alternative pathway of l-arabinose metabolism. J Biol Chem 281:2612–2623. doi: 10.1074/jbc.M506477200. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe S, Shimada N, Tajima K, Kodaki T, Makino K. 2006. Identification and characterization of l-arabonate dehydratase, l-2-keto-3-deoxyarabonate dehydratase, and l-arabinolactonase involved in an alternative pathway of l-arabinose metabolism. Novel evolutionary insight into sugar metabolism. J Biol Chem 281:33521–33536. doi: 10.1074/jbc.M606727200. [DOI] [PubMed] [Google Scholar]
- 6.Stephens C, Christen B, Fuchs T, Sundaram V, Watanabe K, Jenal U. 2007. Genetic analysis of a novel pathway for d-xylose metabolism in Caulobacter crescentus. J Bacteriol 189:2181–2185. doi: 10.1128/JB.01438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weimberg R. 1961. Pentose oxidation by Pseudomonas fragi. J Biol Chem 236:629–635. [PubMed] [Google Scholar]
- 8.Johnsen U, Schönheit P. 2004. Novel xylose dehydrogenase in the halophilic archaeon Haloarcula marismortui. J Bacteriol 186:6198–6207. doi: 10.1128/JB.186.18.6198-6207.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nunn CE, Johnsen U, Schönheit P, Fuhrer T, Sauer U, Hough DW, Danson MJ. 2010. Metabolism of pentose sugars in the hyperthermophilic archaea Sulfolobus solfataricus and Sulfolobus acidocaldarius. J Biol Chem 285:33701–33709. doi: 10.1074/jbc.M110.146332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner M, Shen L, Albersmeier A, van der Kolk N, Kim S, Cha J, Bräsen C, Kalinowski J, Siebers B, Albers SV. 2018. Sulfolobus acidocaldarius transports pentoses via a carbohydrate uptake transporter 2 (CUT2)-type ABC transporter and metabolizes them through the aldolase-independent Weimberg pathway. Appl Environ Microbiol 84:e01273-17. doi: 10.1128/AEM.01273-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brouns SJ, Walther J, Snijders AP, van de Werken HJ, Willemen HL, Worm P, de Vos MG, Andersson A, Lundgren M, Mazon HF, van den Heuvel RH, Nilsson P, Salmon L, de Vos WM, Wright PC, Bernander R, van der Oost J. 2006. Identification of the missing links in prokaryotic pentose oxidation pathways: evidence for enzyme recruitment. J Biol Chem 281:27378–27388. doi: 10.1074/jbc.M605549200. [DOI] [PubMed] [Google Scholar]
- 12.Johnsen U, Dambeck M, Zaiss H, Fuhrer T, Soppa J, Sauer U, Schönheit P. 2009. d-Xylose degradation pathway in the halophilic archaeon Haloferax volcanii. J Biol Chem 284:27290–27303. doi: 10.1074/jbc.M109.003814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnsen U, Sutter J-M, Zaiß H, Schönheit P. 2013. l-Arabinose degradation pathway in the haloarchaeon Haloferax volcanii involves a novel type of l-arabinose dehydrogenase. Extremophiles 17:897–909. doi: 10.1007/s00792-013-0572-2. [DOI] [PubMed] [Google Scholar]
- 14.Sutter JM, Johnsen U, Schönheit P. 2017. Characterization of a pentonolactonase involved in d-xylose and l-arabinose catabolism in the haloarchaeon Haloferax volcanii. FEMS Microbiol Lett 364:fnx140. doi: 10.1093/femsle/fnx140. [DOI] [PubMed] [Google Scholar]
- 15.Johnsen U, Sutter JM, Schulz AC, Tästensen JB, Schönheit P. 2015. XacR–a novel transcriptional regulator of d-xylose and l-arabinose catabolism in the haloarchaeon Haloferax volcanii. Environ Microbiol 17:1663–1676. doi: 10.1111/1462-2920.12603. [DOI] [PubMed] [Google Scholar]
- 16.Liu H, Han J, Liu X, Zhou J, Xiang H. 2011. Development of pyrF-based gene knockout systems for genome-wide manipulation of the archaea Haloferax mediterranei and Haloarcula hispanica. J Genet Genomics 38:261–269. doi: 10.1016/j.jgg.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A. 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garces F, Fernández FJ, Montellá C, Penya-Soler E, Prohens R, Aguilar J, Baldomá L, Coll M, Badia J, Vega MC. 2010. Molecular architecture of the Mn2+-dependent lactonase UlaG reveals an RNase-like metallo-β-lactamase fold and a novel quaternary structure. J Mol Biol 398:715–729. doi: 10.1016/j.jmb.2010.03.041. [DOI] [PubMed] [Google Scholar]
- 19.Fernandez FJ, Garces F, Lopez-Estepa M, Aguilar J, Baldoma L, Coll M, Badia J, Vega MC. 2011. The UlaG protein family defines novel structural and functional motifs grafted on an ancient RNase fold. BMC Evol Biol 11:273. doi: 10.1186/1471-2148-11-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sutter JM, Tästensen JB, Johnsen U, Soppa J, Schönheit P. 2016. Key enzymes of the semiphosphorylative Entner-Doudoroff pathway in the haloarchaeon Haloferax volcanii: characterization of glucose dehydrogenase, gluconate dehydratase, and 2-keto-3-deoxy-6-phosphogluconate aldolase. J Bacteriol 198:2251–2262. doi: 10.1128/JB.00286-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed H, Ettema TJ, Tjaden B, Geerling AC, van der Oost J, Siebers B. 2005. The semi-phosphorylative Entner-Doudoroff pathway in hyperthermophilic archaea: a re-evaluation. Biochem J 390: 529–540. doi: 10.1042/BJ20041711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamble HJ, Milburn CC, Taylor GL, Hough DW, Danson MJ. 2004. Gluconate dehydratase from the promiscuous Entner-Doudoroff pathway in Sulfolobus solfataricus. FEBS Lett 576:133–136. doi: 10.1016/j.febslet.2004.08.074. [DOI] [PubMed] [Google Scholar]
- 23.Reher M, Fuhrer T, Bott M, Schönheit P. 2010. The nonphosphorylative Entner-Doudoroff pathway in the thermoacidophilic euryarchaeon Picrophilus torridus involves a novel 2-keto-3-deoxygluconate-specific aldolase. J Bacteriol 192:964–974. doi: 10.1128/JB.01281-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allers T, Barak S, Liddell S, Wardell K, Mevarech M. 2010. Improved strains and plasmid vectors for conditional overexpression of His-tagged proteins in Haloferax volcanii. Appl Environ Microbiol 76:1759–1769. doi: 10.1128/AEM.02670-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reinhardt A, Johnsen U, Schönheit P. 2019. l-Rhamnose catabolism in archaea. Mol Microbiol 111:1093–1108. doi: 10.1111/mmi.14213. [DOI] [PubMed] [Google Scholar]
- 26.Pickl A, Johnsen U, Schönheit P. 2012. Fructose degradation in the haloarchaeon Haloferax volcanii involves a bacterial type phosphoenolpyruvate-dependent phosphotransferase system, fructose-1-phosphate kinase, and class II fructose-1,6-bisphosphate aldolase. J Bacteriol 194:3088–3097. doi: 10.1128/JB.00200-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allers T, Mevarech M. 2005. Archaeal genetics–the third way. Nat Rev Genet 6:58–73. doi: 10.1038/nrg1504. [DOI] [PubMed] [Google Scholar]
- 28.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 29.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 30.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.