

Abstract
In this study, we describe the correction of single-point mutations in mammalian cells by repair-polypurine reverse Hoogsteen hairpins (repair-PPRHs). These molecules consist of (1) a PPRH hairpin core that binds to a polypyrimidine target sequence in the double-stranded DNA (dsDNA), producing a triplex structure, and (2) an extension sequence homologous to the DNA sequence to be repaired but containing the wild-type nucleotide instead of the mutation and acting as a donor DNA to correct the mutation. We repaired different point mutations in the adenosyl phosphoribosyl transferase (aprt) gene contained in different aprt-deficient Chinese hamster ovary (CHO) cell lines. Because we had previously corrected mutations in the dihydrofolate reductase (dhfr) gene, in this study, we demonstrate the generality of action of the repair-PPRHs. Repaired cells were analyzed by DNA sequencing, mRNA expression, and enzymatic activity to confirm the correction of the mutation. Moreover, whole-genome sequencing analyses did not detect any off-target effect in the repaired genome. We also performed gel-shift assays to show the binding of the repair-PPRH to the target sequence and the formation of a displacement-loop (D-loop) structure that can trigger a homologous recombination event. Overall, we demonstrate that repair-PPRHs achieve the permanent correction of point mutations in the dsDNA at the endogenous level in mammalian cells without off-target activity.
Keywords: PPRH, gene-repair, Hoogsteen, mutation, recombination, gene-correction, aprt, Triplex, D-loop, gene-editing
Graphical Abstract
Introduction
Monogenic disorders present a global prevalence at birth of 10 out of 1,000 cases, thus affecting millions of people worldwide.1 These diseases are the result of single-point mutations in the DNA sequence of a specific gene that lead to the production of nonfunctional versions of the protein. In recent years, different gene-editing tools have been developed to correct mutations in the double-stranded DNA (dsDNA). On the one hand, several molecular tools such as zinc-finger nucleases (ZFNs),2, 3, 4, 5 transcription activator-like nucleases (TALENs),6, 7, 8, 9, 10 and CRISPR/Cas9 RNA-guided nucleases11, 12, 13, 14, 15 have been used to conduct gene correction therapies. These editing technologies rely on the usage of nucleases to generate locus-specific dsDNA breaks near the mutation and a donor DNA sequence that acts as a template for the correction. However, new CRISPR/Cas9 approaches such as base editing and prime editing that do not rely on dsDNA breaks to produce the correction have been also developed. Briefly, base editing is based on the deamination of the purine or pyrimidine base to eventually convert one base pair to another in the dsDNA.16, 17, 18 Prime editing technology can directly write new genetic information into a specific DNA site by a Cas9 endonuclease fused to a reverse transcriptase programmed with a guide RNA that both specifies the target and encodes the desired editing.19 In this case, it is necessary to generate a nick in one of the strands (protospacer-adjacent motif strand). Nevertheless, one of the main concerns upon using these nuclease-dependent technologies is the appearance of off-target effects in the genome of the host after the treatment.20,21 On the other hand, modified or non-modified single-stranded oligodeoxynucleotides (ssODNs) have also been developed to produce the correction of single-point mutations in the dsDNA. In some cases, the ssODN containing the corrected sequence binds to its target dsDNA in a sequence-specific manner, leading to a recombination event that incorporates the corrected nucleotide.22, 23, 24, 25 In other cases, modified molecules such as peptide nucleic acids (PNAs) and their derivatives (e.g., γPNAs) are presently used to bind to the dsDNA, creating a triplex helical structure that stimulates the recombination between a nearby sequence and a provided donor DNA that contains the corrected nucleotide.26, 27, 28, 29, 30
Polypurine reverse Hoogsteen hairpins (PPRHs) are single-stranded and non-modified oligodeoxynucleotides composed of two antiparallel polypurine mirror repeat domains linked by a five-thymidine loop. The intramolecular linkage consists of reverse-Hoogsteen bonds between the purines, allowing the formation of the hairpin structure. PPRHs bind in a sequence-specific manner to polypyrimidine stretches in the dsDNA via Watson-Crick bonds while maintaining the hairpin conformation, thus producing a triplex structure and displacing the fourth strand of the dsDNA.31,32 During the last decade, PPRHs have been used to silence genes involved in resistance to chemotherapeutic drugs,33 cancer progression,32,34, 35, 36, 37 and immunotherapy approaches.38, 39, 40 Recently, we performed a pharmacogenomic study showing the specificity of the PPRH toward its target sequence and the absence of off-target effects when using a negative DNA hairpin. We also demonstrated that PPRHs do not cause hepatotoxicity or nephrotoxicity in vitro.41
Repair-PPRHs are hairpins that bear an extension sequence at one end of the molecule (usually the 5′ end) that is homologous to the DNA sequence to be corrected but contains the wild-type nucleotide instead of the mutated one. A previous study performed in our laboratory demonstrated that repair-PPRHs were able to correct a single-point mutation in a plasmid containing a mutated version of the dihydrofolate reductase (dhfr) minigene. The correction was also achieved when the plasmid was stably transfected into a dhfr-deficient Chinese hamster ovary (CHO) cell line.42 Furthermore, we also corrected different types of point mutations (substitutions, insertions, and deletions) at the endogenous locus of the dhfr gene using repair-PPRHs in various dhfr mutant CHO cell lines.43
In this work, we show the generality of action of repair-PPRHs by correcting single-point mutations at the endogenous locus of the aprt gene in mammalian cells (Figure 1), and we assess the absence of off-target effects of this gene-editing technology. Moreover, we gain insight into the molecular mechanism involving homologous recombination that could be responsible for the gene correction event.
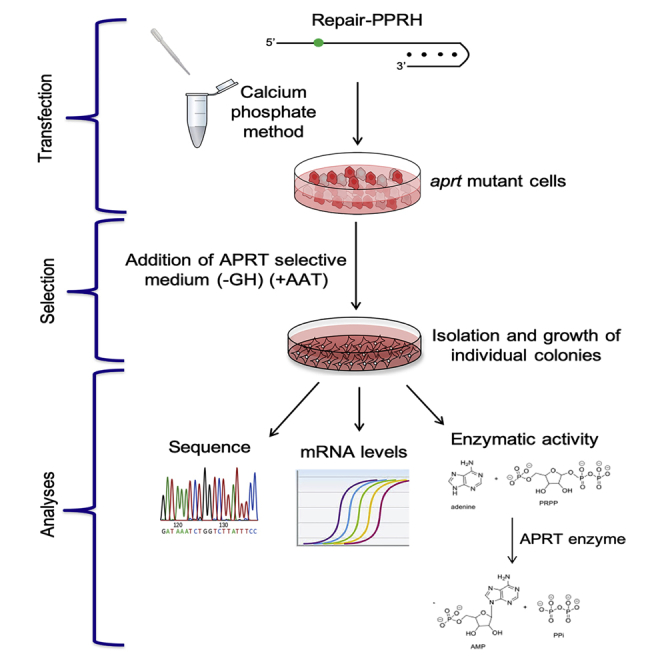
Figure 1.

Gene Correction Strategy Using Repair-PPRHs
(A) Structure of a repair-PPRH, consisting of a hairpin core linked to a repair domain. The core is formed by two polypurine domains linked by five thymidines, which are bound intramolecularly by reverse-Hoogsteen bonds that bind to its polypyrimidine target sequence in the dsDNA via Watson-Crick bonds. The repair domain is a sequence homologous to the DNA sequence to be repaired but containing the corrected nucleotide instead of the mutation. Mutation in the dsDNA is represented in red, whereas the correct nucleotide contained in the repair-PPRH is shown in green. (B) Scheme of a long-distance repair-PPRH containing a hairpin core that binds to a polypyrimidine target sequence away from the location of the mutation. This hairpin core is linked to a repair domain containing the wild-type nucleotide (green) by five additional thymidines (5T). Mutation in the DNA is represented in red. (C) General procedure for the targeted correction using repair-PPRHs of the adenosyl phosphoribosyl transferase (aprt) gene in a collection of aprt-deficient CHO cell lines. The approach involved transfection and selection (+AAT) of the repaired cells followed by sequencing analyses and determination of mRNA levels and enzymatic activity.
Results
Targeted Correction of Point Mutations at the Endogenous Locus of the aprt Gene
Our goal was to correct three aprt-deficient CHO cell lines presenting different point mutations in the aprt locus, leading to premature stop codons. For that reason, we designed different repair-PPRHs as described in the Materials and Methods and shown in Table S2. Each repair-PPRH contained a hairpin core that binds to a specific polypyrimidine sequence near the mutation to direct the repair domain.
The first cell line to be corrected was the S23 mutant, in which the substitution of a guanine for a thymidine in exon 1 led to a TAA stop codon (ochre) in situ. The repair-PPRH (HpS23E1rep) contained three pyrimidine interruptions in the hairpin core, and the repair domain contained a 51-nt sequence as an extension of the hairpin core. Upon transfection and selection, the analyzed colonies bore the corrected nucleotide, rescuing the wild-type triplet (GAA) encoding for a glutamic acid (Figure 2A).
Figure 2.

DNA Sequencing Results
(A–C) DNA sequences from mutants S23 (A), S62 (B), and S1 (C) and their repaired counterparts obtained after transfection with the corresponding repair-PPRH and subsequent selection. The underlined nucleotides represent the mutated/repaired codon, and the arrows indicate the specific nucleotide subjected to correction. Each experiment was conducted a minimum of three times, and a minimum of three different colonies were analyzed.
The next mutant, S62, contained a substitution of a guanine for a thymidine in exon 5 that led to a TGA stop codon (opal) in place. The repair-PPRH (HpS62E5rep) contained two pyrimidine interruptions in the hairpin core, and the length of the repair domain was 57 nt. We confirmed the restoration of the wild-type codon (glycine) in the analyzed colonies (Figure 2B).
Finally, the S1 mutant bore a substitution of a cytosine for a guanine in exon 2, producing a TAG premature stop codon (amber). The repair-PPRH (HpS1E2rep) contained one pyrimidine interruption in the hairpin core and a 57-nt repair domain, restoring the wild-type codon (tyrosine) (Figure 2C).
To test the requirement of the hairpin core to correct the mutation, the repair domains of the different repair-PPRHs were transfected alone in their respective mutant cell lines. No surviving colonies were observed after transfecting the RD-S23E1rep, RD-S62E5rep, and RD-S1E2rep in the S23, S62, and S1 mutant cell lines, respectively. As an additional negative control, we transfected a full repair-PPRH containing a scrambled polypurine hairpin attached to the specific repair domain of the S23 mutant (HpS23E1rep-Sc) into S23 cells, and we did not obtain any surviving cell colony.
All of the previous repair-PPRHs contained repair domains attached directly to the hairpin core, which was close to the mutation site. We wanted to explore whether we could repair a mutation using a hairpin core that was binding farther away from the mutation site and connected to the repair domain through an additional pentathymidine loop. Therefore, we designed a long-distance repair-PPRH (LD-HpS1E2rep) oligonucleotide in which the target sequence of the repair domain was located 24 nt upstream of the polypyrimidine target sequence of the hairpin core. This long-distance repair-PPRH containing a 52-nt repair domain was also able to correct the mutation, restoring the wild-type nucleotide.
APRT mRNA Levels Are Increased in the Repaired Cells
We assessed the restoration of APRT mRNA levels in the repaired cells in comparison with the mutant cell lines and the wild-type D422 cell line. In S23 repaired clones, APRT mRNA levels were increased between 1.25- and 2-fold when compared with those of the S23 mutant cell line (Figure 3A). Regarding S62 repaired clones, APRT mRNA levels were similar to those of the mutant, since the mutation is located in the last exon of the gene (exon 5) and, therefore, non-sense-mediated decay does not take place (Figure 3B). The increase in APRT mRNA levels was also observed in the case of S1 repaired colonies (Figure 3C). There were no significant differences between APRT mRNA levels from clones repaired by the HpS1E2rep repair-PPRH and by the LD-HpS1E2rep long-distance repair-PPRH (Figure 3C).
Figure 3.

APRT mRNA and Enzymatic Analyses
(A–C) APRT mRNA levels of mutants S23 (A), S62 (B), and S1 (C) and their repaired counterparts obtained after transfection with the corresponding repair-PPRHs and subsequent selection are shown. In (C), also represented are the aprt mRNA levels in three clones repaired using the long-distance repair-PPRH (LD-HpS1E2rep). APRT mRNA levels were determined by qRT-PCR and normalized with TBP mRNA. Data are plotted relative to the wild-type cell line D422. (D–F) APRT enzymatic activity was determined for mutants S23 (D), S62 (E), and S1 (F) and their repaired counterparts obtained after transfection with the corresponding repair-PPRHs and subsequent selection. In (F), also shown are the APRT enzymatic activity levels of three clones repaired with the long-distance repair-PPRH (LD-HpS1E2rep). The D422 wild-type cell line was included in the determination as the positive control. Data are represented as mU of APRT enzyme divided by the mg of total protein extract. Error bars represent the standard error of the mean of three experiments. Statistical analysis was performed comparing the mean value of each clone with the mean value of the mutant sample. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
APRT Enzymatic Activity Is Restored in Repaired Cells
We determined the enzymatic activity of APRT protein in the mutant cell lines and the repaired clones. S23 and S62 mutants did not show any APRT activity, whereas the activity levels in their respective repaired clones were similar to those of the parental cell line D422 (Figures 3D and 3E). The S1 mutant presented a very low activity in contrast with the repaired clones that showed a very similar activity to the D422 cell line (Figure 3F). There were no differences in APRT activity levels between the clones repaired by the HpS1E2rep repair-PPRH and by the LD-HpS1E2rep repair-PPRH (Figure 3F).
Gene Correction Frequency Increases in S Phase
The determination of the gene correction frequency was performed using the HpS23E1rep repair-PPRH in the S23 mutant cell line. The frequency of correction was 0.1% in the asynchronous condition. However, when cells were transfected in S phase the frequency was 0.25%, corresponding to a 2.5-fold increase compared with cells transfected in the asynchronous state (Figure 4).
Figure 4.

Gene Correction Frequency
Gene correction frequency values were calculated as the ratio between the number of surviving colonies and the total number of cells initially plated. Also shown is a representative image of the number of S23 repaired colonies obtained after the treatment with HpS23E1rep repair-PPRH in cells transfected either in asynchronous conditions or in S phase. After selection, surviving cell colonies were fixed with formaldehyde and stained with crystal violet. Error bars represent the standard error of the mean of three experiments. *p < 0.05.
Whole-Genome Sequencing Analyses Reveal No Off-Target Effects
The sequenced reads that aligned at position 960,367 in contig NW_003613583 confirmed that the sample S23 mutant had a T in that genomic position and that in S23 repaired cells the T was replaced with a G. To check whether there was any major difference in the S23 repaired cells in comparison with original mutant cells, we looked at (1) the number of total variants and (2) if there was any evidence of the insertion of the construct in other genomic locations. Regarding the total number of variants, we compared the number of variants (single-nucleotide variants, insertions, and deletions) in the genomic positions with enough coverage in both samples and we did not find any major discrepancy. Therefore, we did not see any clear evidence of a major increase in the number of variants in the S23 repaired sample (Table 1). To investigate the possible integration of HpS23E1rep in multiple genomic regions, reads with similarity to the construct were scrutinized. Under the assumption that if multiple insertions occur, the genomic fragments with this new insertion would have been sequenced but not mapped, because the sequence would not be found in the reference genome, we searched within all of the original reads (before mapping) for those with similarity with the construct using BLAST, as explained in “Whole-Genome Sequencing Analyses” in the Materials and Methods. Table 2 shows the reads with similarity to the construct in both samples. All the found reads were mapped in the target region in contig NW_003613583 (Table 2). No unmapped reads or mapped reads anywhere else with similarity were found.
Table 1.
Number of Variants per Sample
| S23 Mutant | S23 Repaired | All Variants | Filtered Variants | Filtered SNV | Filtered Del | Filtered Insert |
|---|---|---|---|---|---|---|
| 0/0 | 0/1 | 66,758 | 46,002 | 36,147 | 4,681 | 5,174 |
| 0/1 | 0/0 | 67,563 | 46,414 | 36,637 | 4,684 | 5,093 |
| 0/0 | 1/1 | 8,579 | 644 | 105 | 307 | 232 |
| 1/1 | 0/0 | 8,414 | 729 | 92 | 345 | 292 |
| 0/1 | 1/1 | 23,814 | 6,322 | 4,490 | 890 | 942 |
| 1/1 | 0/1 | 22,472 | 5,675 | 4,046 | 769 | 860 |
| 0/1 | 0/1 | 1,074,296 | 971,609 | 798,079 | 92,812 | 80,718 |
| 1/1 | 1/1 | 1,036,972 | 549,872 | 441,444 | 35,296 | 73,132 |
| Total | 2,308,868 | 1,627,267 | 1,321,040 | 139,784 | 166,443 | |
The genotype conventions were followed: “0/0” refers to homozygous reference position (no variant), “0/1” heterozygous, and “1/1” homozygous alternative. “Filtered” refers to the minimum coverage of 10 in both samples and a minimum of three for the alternative alleles, which reduces the number of variants by 30% but ensures that both samples have a good coverage. We can observe that the new variants in sample S23 repaired (0/0 0/1 or 0/0 1/1) and the new variants in sample S23 mutant (0/1 0/0 or 1/1 0/0) are very similar. Thus, we cannot see a big increase or decrease in the overall number of variants between these two samples. SNV, single nucleotide variant; Del, deletions; Insert, insertions.
Table 2.
Genomic Integration of the Repair-PPRH
| S23 Mutant | ||||||
|---|---|---|---|---|---|---|
| Read ID | Identity % | Align Length | Exp. Val. | In Region | CIGAR | mapQ |
| J00148:62:HMFTCBBXX:7:1205:28544:42583 | 98 | 50 | 4.99e−16 | yes | 151M | 60 |
| J00148:62:HMFTCBBXX:7:1228:19431:11143 | 98 | 50 | 4.99e−16 | yes | 151M | 60 |
| K00310:156:HMFGLBBXX:8:1222:4980:29677 | 98 | 50 | 5.47e−16 | yes | 1S150M | 30 |
| K00310:156:HMFGLBBXX:8:2107:11464:21500 | 98 | 50 | 5.47e−16 | yes | 151M | 30 |
| K00310:158:HMFF5BBXX:6:1115:14154:37800 | 98 | 50 | 5.47e−16 | yes | 151M | 60 |
| K00310:158:HMFF5BBXX:6:1208:21846:33475 | 98 | 50 | 5.47e−16 | yes | 151M | 60 |
| K00310:158:HMFF5BBXX:6:1215:8471:30450 | 98 | 50 | 5.47e−16 | yes | 151M | 60 |
| K00310:156:HMFGLBBXX:8:1120:27011:20762 | 98 | 49 | 1.88e−15 | yes | 151M | 60 |
| J00148:62:HMFTCBBXX:7:2208:23876:43779 | 96 | 49 | 2.32e−14 | yes | 1S147M3S | 60 |
| K00310:156:HMFGLBBXX:8:2218:24728:11425 | 95 | 42 | 1.90e−10 | yes | 151M | 60 |
| K00310:158:HMFF5BBXX:6:2204:26210:16770 | 98 | 50 | 5.47e−16 | yes | 74M77S | 0 |
| K00310:158:HMFF5BBXX:6:1115:20811:45010 | 96 | 47 | 3.29e−13 | yes | 31S115M5S | 0 |
| J00148:60:HLL3CBBXX:7:2222:31598:29624 | 86 | 44 | 5.15e−06 | yes | 101M50S | 0 |
| S23 Repaired | ||||||
| Read ID | Identity % | Align Length | Exp. Val. | In Region | CIGAR | mapQ |
| K00310:157:HMFGJBBXX:3:1104:7476:24085 | 100 | 51 | 1.08e−17 | yes | 151M | 60 |
| K00310:157:HMFGJBBXX:3:1227:21734:7468 | 100 | 51 | 1.08e−17 | yes | 151M | 60 |
| K00310:159:HMFFVBBXX:4:2125:23957:2527 | 100 | 51 | 1.10e−17 | yes | 151M | 60 |
| K00310:157:HMFGJBBXX:3:2228:25915:42812 | 100 | 51 | 1.08e−17 | yes | 41M110S | 0 |
| K00310:159:HMFFVBBXX:3:1101:10338:27567 | 100 | 51 | 1.10e−17 | yes | 43M108S | 0 |
| J00148:61:HMFGCBBXX:3:1218:21521:21869 | 100 | 20 | 5.08e−06 | yes | 55S96M | 0 |
| K00310:159:HMFFVBBXX:3:1117:27661:25474 | 100 | 27 | 2.41e−04 | yes | 151M | 60 |
Reads are similar to the construct HpS23E1rep found with BLAST before being mapped into the genome. “Identity %,” “Align Length” (alignment length), and “Exp. Val.” (experimental value) refer to the values obtained in the BLAST search. Accordingly, at the most, only the 51 bases that are identical to the complementary genome region were found in the reads. In the S23 repaired we can observe that the alignment length is 51 and the identity is 100% because it contains the replaced G. The columns “In Region,” “CIGAR,” and “mapQ” refer to the statistics of the reads when mapped into the genome using GEM3. In region means that the read has been mapped in the region near position 960,367 in contig NW_003613583. CIGAR refers to the alignment code, and mapQ refers to the mapping quality score. Most of the reads have a quality score of 60, but some have lower quality due to bad score qualities in the bases of the read. In any case, all reads were mapped uniquely to the expected genomic region. Therefore, we could not find any evidence of integration of the construct in the genome.
Repair-PPRHs Bind to Their Target Sequence
To test the binding of repair-PPRHs to their polypyrimidine target sequence in the aprt locus, we performed gel-shift assays using a 160-bp radioactive probe containing the mutation present in S23 cells and its adjacent sequences (dsDNA-S23). The binding of the HpS23E1-core to its polypyrimidine target sequence produced two shifted bands corresponding to two different molecular species (Figure 5A, lane 2). The shifted band with the highest mobility corresponded to a triplex structure in which the HpS23E1-core was binding only to the polypyrimidine target sequence located in one of the two DNA strands of the probe (Figure 5A, red panel). The shifted bands with the lowest mobility corresponded to the binding of the HpS23E1-core to its polypyrimidine target sequence together with the rest of the dsDNA probe that was still bound by intramolecular Watson-Crick bonds (Figure 5A, green panel). However, we did not observe any shifted band when incubating the probe only with the RD-S23E1rep repair domain (Figure 5A, lane 3). The incubation of the full HpS23E1rep repair-PPRH produced three shifted bands (Figure 5A, lane 4). The two highest mobility-shifted bands corresponded to the same molecular species shown in lane 2. The lowest mobility-shifted band corresponded to the binding of the HpS23E1rep repair-PPRH to its polypyrimidine sequence together with the repair domain bound to its complementary strand (Figure 5A, blue panel). As negative controls, we used a hairpin core, a repair domain, or a scrambled full repair-PPRH (Figure 5A, lanes 5–7). We only observed one shifted band with the incubation of the scramble hairpin core with the probe that corresponded to an unspecific and/or partial binding of the hairpin to the sequence, but with different mobility than that of the specific hairpin core (Figure 5A, lane 5).
Figure 5.

Binding of the Repair-PPRH to Its Target Sequence
Gel-shift assays using a 160-bp 32P-radiolabeled dsDNA probe (dsDNA-S23) containing the mutation present in the S23 mutant and its flanking regions. The unlabeled oligodeoxynucleotides present in each binding reaction are indicated. (A) Lane 1, dsDNA-S23 probe alone; lane 2, dsDNA-S23 plus HpS23E1-core (100 nM); lane 3, dsDNA-S23 plus RD-S23E1rep (100 nM); lane 4, dsDNA-S23 plus HpS23E1rep repair-PPRH (100 nM); lane 5, dsDNA-S23 plus Hp-core-Sc (100 nM); lane 6, dsDNA-S23 plus RD-Sc (100 nM); lane 7, dsDNA-S23 plus Hp-rep-Sc (100 nM). Color arrows indicated the different molecular species that are generated upon incubation with the different oligodeoxynucleotides. The color of the arrows matches those of the panels corresponding to the proposed structures shown on the right panel. (B) Competition assay. Lane 1, dsDNA-S23 probe alone; lane 2, dsDNA-S23 probe plus HpS23E1rep repair-PPRH (100 nM); lane 3, dsDNA-S23 probe plus HpS23E1rep (100 nM) competed with HpS23E1-core (2 μM). 20x indicates that the oligonucleotide was added in a concentration 20 times greater than x.
To confirm the nature of the lowest mobility-shifted band that appeared when incubating the full repair-PPRH with the probe (Figure 5A, lane 4), we performed a competition assay (Figure 5B). The amount of radiolabeled probe incubated with the HpS23E1rep repair-PPRH was competed with 20-fold of the HpS23E1rep-core hairpin core (Figure 5B, lane 3), which resulted in the decrease of the band that corresponded to the structure depicted in the blue panel, thus demonstrating that the full repair-PPRH is needed to produce this structure.
Binding of the PPRH to Its Target Sequence Produces a Displacement Loop
To study the molecular mechanism responsible for the repair event, we performed gel-shift assays incubating the HpS23E1-core of the repair-PPRH with the radioactive probe dsDNA-S23. To check whether this binding led to the formation of a displacement-loop (D-loop) structure, we designed three invading oligonucleotides of different lengths (O-16, O-40, and O-60) that were complementary to the displaced strand of the probe (Figure 6A). The hypothesis was that the binding of the HpS23E1-core to its polypyrimidine target sequence would form a D-loop structure, allowing the binding of the invading oligonucleotides to the displaced strand and thus producing different migration patterns in the gel shifts depending on the length of the oligonucleotide. As shown in Figure 6B, lane 2, when incubating the probe with the HpS23E1-core, two shifted bands that reproduced the same pattern as in Figure 5A, lane 2, were obtained. The incubation of each invading oligonucleotide, either O-16, O-40, or O-60, with the probe alone in the absence of the hairpin produced two shifted bands (Figure 6B, lanes 3, 5, and 7). The shifted band with the highest mobility corresponded to the binding of the invading oligonucleotide to its complementary strand present in the probe, whereas the one with the lowest mobility represented the sandwich structure between the invading oligonucleotide and the probe. Finally, when the probe was first incubated with the HpS23E1-core and then the different invading oligonucleotides were added, prominent shifted bands appeared (color arrows) with different mobilities depending on the length of the invading oligonucleotide (Figure 6B, lanes 4, 6, and 8). Therefore, the binding of the hairpin core to the probe provoked the formation of a D-loop structure of a determined length (Figure 6B, color panels). The invading oligonucleotides can be bound to the probe either completely (red panel, O-16) or partially (green and blue panels, O-40 and O-60, respectively) depending on their length. This would form structures in which the ends of the longest invading oligonucleotides would be overhanging the complex.
Figure 6.

Strand Displacement upon Binding of the Repair-PPRH to the Target Sequence
(A) Schematic representation of the strategy used to study the formation of a displacement-loop (D-loop) structure. The hairpin core (HpS23E1-core) of the repair-PPRH was incubated with the dsDNA-S23 probe, and then different oligonucleotides varying in length and complementary to the displaced strand were added to the binding reaction. (B) Gel-shift assays using a 160-bp 32P-radiolabeled dsDNA probe (dsDNA-S23) containing the mutation in S23 cells and its flanking regions. In all cases, an amount of 60 ng of each one of the unlabeled oligodeoxynucleotides was added to the binding reaction. Lane 1, dsDNA-S23 probe alone; lane 2, dsDNA-S23 plus HpS23E1-core; lane 3, dsDNA-S23 plus O-16; lane 4, dsDNA-S23 plus HpS23E1-core plus O-16; lane 5, dsDNA-S23 plus O-40; lane 6, dsDNA-S23 plus HpS23E1-core plus O-40; lane 7, dsDNA-S23 plus O-60; lane 8, dsDNA-S23 plus HpS23E1-core plus O-60. Color arrows indicate the different molecular species that are generated upon incubation with the different oligodeoxynucleotides. The colors of the arrows in the gel image match those of the panels corresponding to the proposed structures shown on the right of the figure.
Discussion
In this study, we demonstrate the generality of action of the repair-PPRHs technology that we previously used to correct six different point mutations (single substitutions, insertions, deletions, and a double substitution) in a different gene that codes for the DHFR. In that study, we used different polypurine hairpin cores against polypyrimidine target sequences ranging from 10 to 23 nt for the successful correction of the dhfr gene.43 In the present work, we carried out gene correction experiments in aprt-deficient cell lines that contained different single-point substitutions in the endogenous locus of the aprt gene, serving as a model of a disease mutation in CHO cells, because its deficiency in humans is an inherited condition that affects the kidneys and urinary tract.44,45 In this study, we designed polypurine hairpins containing between 19 and 22 nt to assure the specificity toward its polypyrimidine target sequence and reduce the possible off-target effects. In all of the cases we demonstrated the correction of the mutation not only at the genomic level, but also at the mRNA and enzymatic activity levels, showing that the translated protein was functional. One limitation of the repair-PPRHs is the requirement of a polypyrimidine tract near the mutation to be corrected. Although the frequency of these polypyrimidine stretches around the human genome is more abundant than that predicted by simple random models,46 it can be difficult to find an appropriate sequence adjacent to the location of the point mutation. We dealt with this situation by designing a long-distance repair-PPRH in which the target for the repair domain was located 24 nt upstream of the hairpin core. In this case, the repair domain was linked to the hairpin core by an additional pentathymidine loop. The long-distance repair-PPRH was able to correct its targeted mutation, indicating that adjacency between the repair domain and the hairpin core was not necessary to achieve the correction. This is in accordance with our previous data showing that a long-distance repair-PPRH containing a hairpin core binding 662 nt away from the mutation was able to produce the correction.43
The highest level of gene repair was achieved when cells were transfected just after release from S phase synchronization. We had already observed an increase in repair efficiency after synchronization when we corrected a point mutation in the dhfr gene using repair-PPRHs.42 This is also consistent with the work of Brachman and Kmiec47 that showed increased repair frequencies when using modified ssODNs by lengthening the S phase and stalling the replication fork, thus inducing the homologous recombination pathway. Other reports also determined gene correction frequencies among different cell cycle stages, confirming that the S phase stage was the most prone to achieve the correction of the mutation.48,49 We did not obtain any surviving cell colony when cells were transfected only with the repair domain of the repair-PPRH or when this domain was attached to the scrambled polypurine hairpin that did not show any binding to the target dsDNA, establishing the requirement of the specific hairpin core to induce the triplex structure and stimulate the gene repair event. This fact corroborates our previous observation that repair domains bearing hairpin cores bound by intramolecular Watson-Crick bonds (unable to bind to the target dsDNA) instead of Hoogsteen bonds did not produce the correction.42
Nowadays, most popular gene-editing technologies (CRISPR/Cas9, ZFN, and TALEN systems) rely on the activity of nucleases that create extrinsic breaks in the dsDNA to achieve the correction of the mutation. One of the main concerns with these gene-editing tools is the presence of off-target effects in the repaired genome such as small insertions, deletions, or substitutions,50, 51, 52, 53, 54 usually produced by unspecific cuts of the nuclease. Haapaniemi et al.55 showed that CRISPR/cas9 genome editing induced a p53-mediated DNA damage response and cell cycle arrest in human retinal pigment epithelial cells. On-target mutagenesis such as large deletions in the target site56 and unexpected chromosomal truncations57 have also been reported. In this regard, repair-PPRHs did not produce any off-target effects in the genome of the repaired cells. No random insertions or deletions caused by the repair-PPRH were detected, and the repair-PPRH itself was not inserted in any region of the repaired genome. Moreover, the presence of a preexisting effector T cell response directed toward Cas9 proteins in human beings has been described since Staphylococcus aureus and Staphylococcus pyogenes cause infections in the human population at high frequencies.58,59 In contrast, PPRHs are non-modified, economical, and non-immunogenic DNA molecules that do not activate the innate inflammatory response.60 Additionally, it has already been described that natural oligonucleotides,61,62 oligonucleotides including phosphorothioate bonds,63 morpholinos,64 and locked nucleic acids65 are not genotoxic.
One of the aims of this work was to get an insight into the molecular mechanism behind the repair event. On the one hand, we demonstrated the specific binding of the hairpin core of the repair-PPRH to the polypyrimidine target sequence in the dsDNA, thus producing the triplex structure. On the other hand, we explored the formation of a D-loop upon the incubation of the hairpin core of the repair-PPRH to the target sequence that could eventually stimulate the repair event. We designed different invading oligonucleotides varying in length and complementary to the displaced strand of the dsDNA upon formation of the triplex by the action of the hairpin core. We showed that after the binding of the hairpin core to the target sequence there was indeed a displacement of the complementary strand, which allowed the different invading oligonucleotides to bind, generating structures with retarded mobility that indicated the formation of a D-loop. In this regard, it is known that the potential of molecules such as triplex-forming oligonucleotides to stimulate recombination with donor DNAs depends on the homology-directed repair (HDR)66,67 and the nucleotide excision repair (NER) pathways.67, 68, 69 Concerning the HDR pathway, RAD51 is one of the main proteins involved in this process by promoting the homologous pairing of a single-stranded DNA to a duplex DNA in a structure similar to a D-loop,70, 71, 72 as the one shown in the present work. Therefore, this structure can stimulate the HDR pathway to finally achieve the correction of the mutation using the repair domain attached to the hairpin core of the repair-PPRH. Our previous data showed that the transfection of a repair-PPRH along with a Rad51 expression vector increased gene correction frequency by 10-fold,42 thus confirming that the HDR pathway is involved in the repair process triggered by the repair-PPRH. Regarding the NER pathway, it has been reported that noncanonical DNA structures such as triple helices can be identified by the XPA/RPA DNA damage recognition complex that recruits NER machinery to these distorted sites, leading to DNA repair activity that generates recombination intermediates.73 However, the entire mechanism by which these triplex structures stimulate recombination remains unclear.
In summary, this work demonstrates the generality of repair-PPRHs to specifically correct point mutations in their endogenous locus in mammalian cells without detecting off-target modification in the genome. We also determined the formation of a D-loop structure upon the binding of the PPRH to its target sequence that is involved in the HDR pathway, giving information about the mechanism by which the repair event may occur. Collectively, this study provides further knowledge for the usage of this technology. We envision repair-PPRHs as an alternative gene-editing tool to correct single-point mutations responsible for monogenic diseases in human cells. These molecules can be associated with advanced delivery systems such as new liposomes and gold/polymeric nanoparticles that could improve their delivery, thus allowing the implementation of this technology for in vivo approaches.
Materials and Methods
Cell Culture
Several aprt-deficient CHO cell lines were used for gene correction. All cell lines contained a single nucleotide substitution within the coding sequence that produced a premature stop codon (nonsense mutation), thus generating a truncated protein. The mutant cell lines were isolated using different mutagens from the parental cell line D422,74 which is a CHO cell line hemizygous for the aprt gene.75 The different cell lines and their corresponding mutations are described in Table S1. Cells were grown in Ham’s F12 medium containing 10% fetal bovine serum (both from Gibco, Madrid, Spain) at 37°C in a 5% CO2-controlled humidified atmosphere. Trypsinization of the cells was performed using 0.05% trypsin (Sigma-Aldrich, Madrid, Spain).
Oligodeoxynucleotides
To design the different PPRHs we used the Triplex-Forming Oligonucleotide Target Sequence Search software (http://utw10685.utweb.utexas.edu/tfo/, Austin, TX, USA), which searches for stretches of polypurines within the DNA sequence of interest. Repair-PPRHs were designed by attaching an extension sequence (repair domain) to the hairpin core that ultimately binds to the polypyrimidine target sequence (Figure 1A).
We also designed a long-distance repair-PPRH (LD-HpS1E2rep) in which the hairpin core was located 24 nt away from the repair domain. In this case, an additional pentathymidine loop between the hairpin core and the repair domain was included to provide flexibility to the repair domain (Figure 1B). All oligodeoxynucleotides were synthesized by Sigma-Aldrich (Haverhill, UK), dissolved at 10 μg/μL (stock solution) in a sterile RNase-free Tris-EDTA buffer (1 mM EDTA and 10 mM Tris, pH 8.0; both from Sigma-Aldrich, Madrid, Spain), and stored at −20°C until use.
As negative controls, different oligodeoxynucleotides that contained only the repair domain for each mutant, without the hairpin core, were used. In addition, a scrambled polypurine hairpin core attached to the repair domain of the S23 mutant was transfected into S23 mutant cells as an additional negative control. Sequences and abbreviations of all oligodeoxynucleotides are described in Table S2.
Transfection
Different numbers of cells ranging from 50,000 to 300,000 were plated the day before transfection. Transfections were carried out using the original calcium phosphate method.76 Briefly, 10 μg of the repair-PPRH was mixed with 100 μL of a 2.5 M CaCl2 solution and sterile Milli-Q H2O in a final volume of 500 μL. This solution was added dropwise to an equal volume of a sterile 2× HEPES-buffered saline (HBS; 280 mM NaCl, 50 mM HEPES, and 1.5 mM NaH2PO4, pH adjusted to 7.1) while bubbling with a 1-mL sterile glass pipette. The calcium phosphate-DNA precipitate was allowed to form for 30 min at room temperature without agitation. Then, 1-mL mixtures were added dropwise to 100-mm plates containing the recipient cells in 10 mL of culture medium. After 5 h of incubation at 37°C, culture medium was replaced with fresh medium and cells were incubated for an additional 48 h before selection (Figure 1C).
Selection of the Repaired Cells
APRT selection was applied to transfected cells using RPMI 1640 selective medium (Gibco, Madrid, Spain) lacking glycine and hypoxanthine (−GH medium), supplemented with 50 μM adenine, 5 μM aminopterin, and 10 μM thymidine (+AAT medium) (Sigma-Aldrich, Madrid, Spain) containing 7% dialyzed fetal bovine serum (Gibco, Madrid, Spain). Each experiment was performed a minimum of three times, and a minimum of three colonies from each replicate were analyzed (Figure 1C).
DNA Sequencing
Total genomic DNA was extracted using the Wizard genomic DNA purification kit (Promega, Madrid, Spain), following the manufacturer’s instructions. PCR was carried out to amplify the specific genomic region containing the mutated site. OneTaq DNA polymerase (New England Biolabs, Ipswich, MA, USA) was used following the standard PCR cycling conditions recommended by the manufacturer. The primer sequences for each amplification were as follows: 5′-TTACCCTTGTTCCCGGACTG-3′ and 5′-TGATCTCACCTAAACAGCAC-3′ for the S23 cell line, 5′-CAGGAACCATGTGCGCTGCCTGTGAGC-3′ and 5′-GGTAAGGCTGAGCCACTGTTCAACCG-3′ for the S62 cell line, and 5′-CTTGTTCCCAGGGATATCTCG-3′ and 5′-GGTTGAAGAAAGAAGGGATAGG-3′ for the S1 cell line. The PCR-amplified products were sequenced by Macrogen (Amsterdam, the Netherlands).
mRNA Analyses
Total RNA was extracted using TRI Reagent (Life Technologies, Barcelona, Spain) following the instructions provided by the manufacturer. RNA concentration was determined by measuring its absorbance at 260 nm using a Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Barcelona, Spain). cDNA was synthesized in a 20-μL reaction mixture containing 1 μg of total RNA, 125 ng of random hexamers (Roche, Barcelona, Spain), 0.5 mM of each deoxyribonucleotide triphosphate (dNTP; Bioline, London, UK), 2 μL of buffer (10×), 20 U of RNase inhibitor, and 200 U of Moloney murine leukemia virus reverse transcriptase (last three from Lucigen, Middleton, WI, USA). The reaction was incubated at 42°C for 1 h. 3 μL of the cDNA mixture was used for quantitative real-time PCR amplification.
A StepOnePlus real-time PCR system (Applied Biosystems, Barcelona, Spain) was used to perform the experiments. PCR cycling conditions were 10 min denaturation at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. An APRT mRNA TaqMan probe (assay ID: Cg04465038_g1) was used to determine the mRNA expression of the aprt gene. The relative mRNA amount of the target gene was calculated using the ΔΔCT method, where CT is the threshold cycle that corresponds to the cycle where the amount of amplified mRNA reaches the threshold of fluorescence. A TATA box-binding protein (TBP) mRNA TaqMan probe (assay ID: Cg04504571_m1) was used as an endogenous control. All TaqMan probes were purchased from Life Technologies (Barcelona, Spain). The final volume of the reaction was 20 μL containing 1× TaqMan Universal PCR Master Mix II, 1× TaqMan probe (both from Life Technologies, Barcelona, Spain), 3 μL of cDNA, and nuclease-free Milli-Q H2O.
APRT Enzymatic Activity Assay
The assay was adapted from the original paper of Johnson et al.77 with some modifications. Basically, this method is based on the change in absorbance of adenine at 260 nm due to its conversion to AMP by APRT. First, mutant or repaired cells (5 × 106) were lysed with 200 μL of 0.1% Triton X-100 in 100 mM Tris-HCl (pH 7.4). After vortexing, cell extracts were centrifuged at 10,000 × g for 10 min at 4°C and the supernatant was kept in ice until APRT activity determination. Protein concentration of the cell lysates was determined by the Bio-Rad protein assay following the instructions of the manufacturer. The 500-μL incubation mixture consisted of 0.25 mM adenine, 5 mM MgCl2, 1 mM phosphoribosyl pyrophosphate, and 50 mM Tris-HCl (pH 7.4) (all from Sigma-Aldrich, Madrid, Spain). To start the reaction, 190 μL of cell lysate was added to the incubation mixture at 37°C. After vortexing, 200 μL of sample was immediately removed and placed into a new tube containing 200 μL of LaCl3 (Sigma-Aldrich, Madrid, Spain), followed by rapid mixing to stop the reaction (t0). After 30 min of reaction, another sample was removed and processed as mentioned before (t30). Finally, 400 μL of 40 mM Na2HPO4 was added to each sample, mixed, and centrifuged at 10,000 × g for 5 min at 4°C. The absorbance of the clear supernatant was measured at a wavelength of 260 nm in a UV-1700 UV-Vis spectrophotometer (Shimadzu, Duisburg, Germany). Data were represented as enzyme milliunits (mU), corresponding to the amount of enzyme that catalyzes the conversion of 1 nmol of adenine per minute, divided by the mg of total protein extract.
A scheme representing the different assays (DNA sequencing, mRNA expression, and enzymatic activity) performed to confirm the functionality of aprt upon repair are shown in Figure 1C.
Gene Correction Frequency
Gene correction frequency was determined for the S23 mutant cell line using the HpS23E1rep repair-PPRH. Transfection was performed either in asynchronous cells or with cells in the S phase of the cell cycle. Synchronization in S phase was achieved following the protocol described by Chin et al.,26 which consisted basically in incubating the cells in medium supplemented with 0.1% serum for 72 h followed by incubation with 1.5 mM hydroxyurea for 15 h. After transfection and selection in +AAT medium, surviving cell colonies were fixed with 6% formaldehyde, stained with crystal violet (both from Sigma-Aldrich, Madrid, Spain), and counted.
Whole-Genome Sequencing Analyses
Total genomic DNA was extracted from both S23 mutant cells and S23 repaired cells using the method previously described in “DNA Sequencing” above. Both samples were sequenced with a whole-genome approach with an average target coverage of 26× in the facilities of the National Center for Genomic Analyses (CNAG), Barcelona, Spain. The CHO genome and annotation CHO-K1[ATCC]_RefSeq_2014 assembly was obtained from https://chogenome.org/ and used as a reference genome. The sequenced reads were mapped into the CHO-K1[ATCC]_RefSeq_2014 assembly using the GEM3 aligner78 with default parameters. The CHO-K1[ATCC]_RefSeq_2014 assembly was very fragmented with more than 100,000 contig fragments. To avoid mapping problems in small contigs that could lead to false-positive variant calls and to simplify the analysis, a set of 3,158 contigs longer than 100 kb covering around 90% of the genomic sequence (2.2 Gb) was used to analyze the number of variants and the genotype of both samples. For this analysis, we used the variants calls computed with HaplotypeCaller from the Genome Analysis Toolkit79 following their best practices guidelines. As the covered regions could be different among samples and genomic regions, we applied a filter to ensure that there was enough reliable information in both samples. A set of variants with enough support in the two samples was created. Thus, we required that the genomic position had a coverage of at least 10 reads in both samples, and if there was an alternative allele, it had to have a coverage of at least three reads with the alternative variant. The filter reduced the total number of reported variants around 30% but ensured that all calls were reliable and comparable between the two samples.
To check the possible off-target effects by insertion of the repair-PPRH construct in different regions of the genome, we performed similarity searches to identify genomic sequenced reads that could contain the repair-PPRH sequence. The alignment software BLAST80 was run with word size parameter set to 15 to speed up the search. With a word size of 15, BLAST required a seed of 15 bases from the query to be identical to the target sequence in the database to start the alignment. Only the sequences with an alignment with an expected p value lower than 1e−03 were taken for the analysis.
DNA Binding Assays
To prepare the probe for the binding assays, the target dsDNA sequence of the HpS23E1rep repair-PPRH was PCR amplified using genomic DNA from S23 mutant cells and the S23-Fw and S23-Rv primers. The dsDNA PCR product (200 ng) was 5′ end labeled with [γ-32P]ATP (3,000 Ci/mmol) (PerkinElmer, Madrid, Spain) using T4 polynucleotide kinase (New England Biolabs, USA) in a 10-μL reaction mixture, according to the manufacturer’s protocol. After incubation at 37°C for 1 h, 90 μL of Tris-EDTA buffer (1 mM EDTA and 10 mM Tris, pH 8.0; Sigma-Aldrich, Madrid, Spain) was added to the reaction mixture, which was subsequently filtered through a Sephadex G-50 (Sigma-Aldrich, Madrid, Spain) spin column to eliminate the unincorporated [γ-32P]ATP.
Binding experiments were carried out by incubating the radiolabeled DNA probe (20,000 cpm) with a length of 160 bp with different unlabeled oligodeoxynucleotides in a buffer containing 10 mM MgCl2, 100 mM NaCl, and 50 mM HEPES (pH 7.2). Binding reactions (20 μL) were incubated 5 min at 92°C followed by 25 min of cooling down until reaching room temperature. In the case of the D-loop formation gel-shift assays, binding reactions were incubated 5 min at 92°C and 5 min at 37°C (invading oligonucleotides were added to the mix at this point) and 20 min of cooling down until reaching room temperature. Unspecific DNA (poly(dI:dC)) was included in each condition at a 1:2 unspecific DNA/specific DNA ratio. Electrophoresis was performed on a nondenaturing 10% polyacrylamide gels containing 10 mM MgCl2, 5% glycerol, and 50 mM HEPES (pH 7.2). Gels were run at a fixed voltage of 220 V (4°C) using a running buffer containing 10 mM MgCl2 and 50 mM HEPES (pH 7.2). Finally, gels were dried at 80°C, exposed to photostimulable phosphor plates, and visualized on a Storm 840 Phosphorimager (Molecular Dynamics, GE Healthcare, Barcelona, Spain). ImageQuant software v5.2 was used to visualize the results.
Statistical Analyses
Data are represented as the mean ± SEM values of three experiments. Statistical analyses were performed using ordinary one-way ANOVA with Dunnett’s multiple comparison tests. Gene frequency data were analyzed with an unpaired Student’s t test. Analyses and representation of the data were carried out using GraphPad Prism version 6.0 software (GraphPad, La Jolla, CA, USA).
Author Contributions
A.J.F. designed and performed the experiments and wrote the manuscript. C.J.C. and V.N. conceived the study, participated in its design and supervision, and revised the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We would like to thank Dr. Lawrence Chasin (Columbia University) for providing the aprt-deficient CHO mutant cell lines. This work was supported by grant RTI2018-093901-B-I00 from Plan Nacional de Investigación Científica (Spain). Our group holds the Quality Mention from Generalitat de Catalunya 2017-SGR-94. A.J.F. is the recipient of a fellowship (FPU) from the Ministerio de Educación (Spain).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2019.12.015.
Supplemental Information
References
- 1.(1996). Control of hereditary diseases. Report of a WHO Scientific Group. World Health Organ. Tech. Rep. Ser. 865, 1–84. [PubMed]
- 2.Urnov F.D., Miller J.C., Lee Y.L., Beausejour C.M., Rock J.M., Augustus S., Jamieson A.C., Porteus M.H., Gregory P.D., Holmes M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 3.Yusa K., Rashid S.T., Strick-Marchand H., Varela I., Liu P.Q., Paschon D.E., Miranda E., Ordóñez A., Hannan N.R., Rouhani F.J. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen F., Pruett-Miller S.M., Huang Y., Gjoka M., Duda K., Taunton J., Collingwood T.N., Frodin M., Davis G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods. 2011;8:753–755. doi: 10.1038/nmeth.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mori T., Mori K., Tobimatsu T., Sera T. Sandwiched zinc-finger nucleases demonstrating higher homologous recombination rates than conventional zinc-finger nucleases in mammalian cells. Bioorg. Med. Chem. Lett. 2014;24:813–816. doi: 10.1016/j.bmcl.2013.12.096. [DOI] [PubMed] [Google Scholar]
- 6.Ding Q., Lee Y.K., Schaefer E.A.K., Peters D.T., Veres A., Kim K., Kuperwasser N., Motola D.L., Meissner T.B., Hendriks W.T. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosbach V., Poggi L., Viterbo D., Charpentier M., Richard G.F. TALEN-induced double-strand break repair of CTG trinucleotide repeats. Cell Rep. 2018;22:2146–2159. doi: 10.1016/j.celrep.2018.01.083. [DOI] [PubMed] [Google Scholar]
- 8.Yahata N., Matsumoto Y., Omi M., Yamamoto N., Hata R. TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m.13513G>A mutation. Sci. Rep. 2017;7:15557. doi: 10.1038/s41598-017-15871-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bedell V.M., Wang Y., Campbell J.M., Poshusta T.L., Starker C.G., Krug R.G., 2nd, Tan W., Penheiter S.G., Ma A.C., Leung A.Y. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–118. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Low B.E., Krebs M.P., Joung J.K., Tsai S.Q., Nishina P.M., Wiles M.V. Correction of the Crb1rd8 allele and retinal phenotype in C57BL/6N mice via TALEN-mediated homology-directed repair. Invest. Ophthalmol. Vis. Sci. 2014;55:387–395. doi: 10.1167/iovs.13-13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain A., Zode G., Kasetti R.B., Ran F.A., Yan W., Sharma T.P., Bugge K., Searby C.C., Fingert J.H., Zhang F. CRISPR-Cas9-based treatment of myocilin-associated glaucoma. Proc. Natl. Acad. Sci. USA. 2017;114:11199–11204. doi: 10.1073/pnas.1706193114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park H., Oh J., Shim G., Cho B., Chang Y., Kim S., Baek S., Kim H., Shin J., Choi H. In vivo neuronal gene editing via CRISPR-Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer’s disease. Nat. Neurosci. 2019;22:524–528. doi: 10.1038/s41593-019-0352-0. [DOI] [PubMed] [Google Scholar]
- 13.Amoasii L., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Bassel-Duby R., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ousterout D.G., Kabadi A.M., Thakore P.I., Majoros W.H., Reddy T.E., Gersbach C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruan J., Hirai H., Yang D., Ma L., Hou X., Jiang H., Wei H., Rajagopalan C., Mou H., Wang G. Efficient gene editing at major CFTR mutation loci. Mol. Ther. Nucleic Acids. 2019;16:73–81. doi: 10.1016/j.omtn.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komor A.C., Badran A.H., Liu D.R. Editing the genome without double-stranded DNA breaks. ACS Chem. Biol. 2018;13:383–388. doi: 10.1021/acschembio.7b00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X.H., Tee L.Y., Wang X.G., Huang Q.S., Yang S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids. 2015;4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin H.Y., Wang C., Lee H.K., Yoo K.H., Zeng X., Kuhns T., Yang C.M., Mohr T., Liu C., Hennighausen L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017;8:15464. doi: 10.1038/ncomms15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Ravesteyn T.W., Dekker M., Fish A., Sixma T.K., Wolters A., Dekker R.J., Te Riele H.P. LNA modification of single-stranded DNA oligonucleotides allows subtle gene modification in mismatch-repair-proficient cells. Proc. Natl. Acad. Sci. USA. 2016;113:4122–4127. doi: 10.1073/pnas.1513315113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Igoucheva O., Alexeev V., Yoon K. Targeted gene correction by small single-stranded oligonucleotides in mammalian cells. Gene Ther. 2001;8:391–399. doi: 10.1038/sj.gt.3301414. [DOI] [PubMed] [Google Scholar]
- 24.Ellis H.M., Yu D., DiTizio T., Court D.L. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. USA. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLachlan J., Fernandez S., Helleday T., Bryant H.E. Specific targeted gene repair using single-stranded DNA oligonucleotides at an endogenous locus in mammalian cells uses homologous recombination. DNA Repair (Amst.) 2009;8:1424–1433. doi: 10.1016/j.dnarep.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 26.Chin J.Y., Kuan J.Y., Lonkar P.S., Krause D.S., Seidman M.M., Peterson K.R., Nielsen P.E., Kole R., Glazer P.M. Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids. Proc. Natl. Acad. Sci. USA. 2008;105:13514–13519. doi: 10.1073/pnas.0711793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lonkar P., Kim K.H., Kuan J.Y., Chin J.Y., Rogers F.A., Knauert M.P., Kole R., Nielsen P.E., Glazer P.M. Targeted correction of a thalassemia-associated β-globin mutation induced by pseudo-complementary peptide nucleic acids. Nucleic Acids Res. 2009;37:3635–3644. doi: 10.1093/nar/gkp217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bahal R., Ali McNeer N., Quijano E., Liu Y., Sulkowski P., Turchick A., Lu Y.C., Bhunia D.C., Manna A., Greiner D.L. In vivo correction of anaemia in β-thalassemic mice by γPNA-mediated gene editing with nanoparticle delivery. Nat. Commun. 2016;7:13304. doi: 10.1038/ncomms13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ricciardi A.S., Bahal R., Farrelly J.S., Quijano E., Bianchi A.H., Luks V.L., Putman R., López-Giráldez F., Coşkun S., Song E. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun. 2018;9:2481. doi: 10.1038/s41467-018-04894-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McNeer N.A., Anandalingam K., Fields R.J., Caputo C., Kopic S., Gupta A., Quijano E., Polikoff L., Kong Y., Bahal R. Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium. Nat. Commun. 2015;6:6952. doi: 10.1038/ncomms7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Almagro M.C., Coma S., Noé V., Ciudad C.J. Polypurine hairpins directed against the template strand of DNA knock down the expression of mammalian genes. J. Biol. Chem. 2009;284:11579–11589. doi: 10.1074/jbc.M900981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ciudad C.J., Rodríguez L., Villalobos X., Félix A.J., Noé V. Polypurine reverse Hoogsteen hairpins as a gene silencing tool for cancer. Curr. Med. Chem. 2017;24:2809–2826. doi: 10.2174/0929867324666170301114127. [DOI] [PubMed] [Google Scholar]
- 33.Mencia N., Selga E., Noé V., Ciudad C.J. Underexpression of miR-224 in methotrexate resistant human colon cancer cells. Biochem. Pharmacol. 2011;82:1572–1582. doi: 10.1016/j.bcp.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 34.Rodríguez L., Villalobos X., Dakhel S., Padilla L., Hervas R., Hernández J.L., Ciudad C.J., Noé V. Polypurine reverse Hoogsteen hairpins as a gene therapy tool against survivin in human prostate cancer PC3 cells in vitro and in vivo. Biochem. Pharmacol. 2013;86:1541–1554. doi: 10.1016/j.bcp.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 35.Oleaga C., Welten S., Belloc A., Solé A., Rodriguez L., Mencia N., Selga E., Tapias A., Noé V., Ciudad C.J. Identification of novel Sp1 targets involved in proliferation and cancer by functional genomics. Biochem. Pharmacol. 2012;84:1581–1591. doi: 10.1016/j.bcp.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 36.Villalobos X., Rodríguez L., Solé A., Lliberós C., Mencia N., Ciudad C.J., Noé V. Effect of polypurine reverse Hoogsteen hairpins on relevant cancer target genes in different human cell lines. Nucleic Acid Ther. 2015;25:198–208. doi: 10.1089/nat.2015.0531. [DOI] [PubMed] [Google Scholar]
- 37.de Almagro M.C., Mencia N., Noé V., Ciudad C.J. Coding polypurine hairpins cause target-induced cell death in breast cancer cells. Hum. Gene Ther. 2011;22:451–463. doi: 10.1089/hum.2010.102. [DOI] [PubMed] [Google Scholar]
- 38.Bener G., J Félix A., Sánchez de Diego C., Pascual Fabregat I., Ciudad C.J., Noé V. Silencing of CD47 and SIRPα by polypurine reverse Hoogsteen hairpins to promote MCF-7 breast cancer cells death by PMA-differentiated THP-1 cells. BMC Immunol. 2016;17:32. doi: 10.1186/s12865-016-0170-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Medina Enríquez M.M., Félix A.J., Ciudad C.J., Noé V. Cancer immunotherapy using Polypurine reverse Hoogsteen hairpins targeting the PD-1/PD-L1 pathway in human tumor cells. PLoS One. 2018;13:e0206818. doi: 10.1371/journal.pone.0206818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciudad C.J., Medina Enriquez M.M., Félix A.J., Bener G., Noé V. Silencing PD-1 and PD-L1: the potential of PolyPurine Reverse Hoogsteen hairpins for the elimination of tumor cells. Immunotherapy. 2019;11:369–372. doi: 10.2217/imt-2018-0215. [DOI] [PubMed] [Google Scholar]
- 41.Félix A.J., Ciudad C.J., Noé V. Functional pharmacogenomics and toxicity of PolyPurine Reverse Hoogsteen hairpins directed against survivin in human cells. Biochem. Pharmacol. 2018;155:8–20. doi: 10.1016/j.bcp.2018.06.020. [DOI] [PubMed] [Google Scholar]
- 42.Solé A., Villalobos X., Ciudad C.J., Noé V. Repair of single-point mutations by polypurine reverse Hoogsteen hairpins. Hum. Gene Ther. Methods. 2014;25:288–302. doi: 10.1089/hgtb.2014.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Solé A., Ciudad C.J., Chasin L.A., Noé V. Correction of point mutations at the endogenous locus of the dihydrofolate reductase gene using repair-PolyPurine Reverse Hoogsteen hairpins in mammalian cells. Biochem. Pharmacol. 2016;110–111:16–24. doi: 10.1016/j.bcp.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 44.Edvardsson V.O., Sahota A., Palsson R. University of Washington; 2019. Adenine phosphoribosyltransferase deficiency. In GeneReviews.https://www.ncbi.nlm.nih.gov/books/NBK100238/ [PubMed] [Google Scholar]
- 45.Bollée G., Harambat J., Bensman A., Knebelmann B., Daudon M., Ceballos-Picot I. Adenine phosphoribosyltransferase deficiency. Clin. J. Am. Soc. Nephrol. 2012;7:1521–1527. doi: 10.2215/CJN.02320312. [DOI] [PubMed] [Google Scholar]
- 46.Goñi J.R., de la Cruz X., Orozco M. Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res. 2004;32:354–360. doi: 10.1093/nar/gkh188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brachman E.E., Kmiec E.B. Gene repair in mammalian cells is stimulated by the elongation of S phase and transient stalling of replication forks. DNA Repair (Amst.) 2005;4:445–457. doi: 10.1016/j.dnarep.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 48.Majumdar A., Puri N., Cuenoud B., Natt F., Martin P., Khorlin A., Dyatkina N., George A.J., Miller P.S., Seidman M.M. Cell cycle modulation of gene targeting by a triple helix-forming oligonucleotide. J. Biol. Chem. 2003;278:11072–11077. doi: 10.1074/jbc.M211837200. [DOI] [PubMed] [Google Scholar]
- 49.Olsen P.A., Randol M., Krauss S. Implications of cell cycle progression on functional sequence correction by short single-stranded DNA oligonucleotides. Gene Ther. 2005;12:546–551. doi: 10.1038/sj.gt.3302454. [DOI] [PubMed] [Google Scholar]
- 50.Cradick T.J., Fine E.J., Antico C.J., Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P., Páleníková P., Khodak A., Kiselev V., Kosicki M. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2018;37:64–82. doi: 10.1038/nbt.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson K.R., Haeussler M., Watanabe C., Janakiraman V., Lund J., Modrusan Z., Stinson J., Bei Q., Buechler A., Yu C. CRISPR off-target analysis in genetically engineered rats and mice. Nat. Methods. 2018;15:512–514. doi: 10.1038/s41592-018-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin Y., Cradick T.J., Brown M.T., Deshmukh H., Ranjan P., Sarode N., Wile B.M., Vertino P.M., Stewart F.J., Bao G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42:7473–7485. doi: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schaefer K.A., Wu W.H., Colgan D.F., Tsang S.H., Bassuk A.G., Mahajan V.B. Unexpected mutations after CRISPR-Cas9 editing in vivo. Nat. Methods. 2017;14:547–548. doi: 10.1038/nmeth.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 56.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cullot G., Boutin J., Toutain J., Prat F., Pennamen P., Rooryck C., Teichmann M., Rousseau E., Lamrissi-Garcia I., Guyonnet-Duperat V. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019;10:1136. doi: 10.1038/s41467-019-09006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wagner D.L., Amini L., Wendering D.J., Burkhardt L.M., Akyüz L., Reinke P., Volk H.D., Schmueck-Henneresse M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019;25:242–248. doi: 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
- 59.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Villalobos X., Rodríguez L., Prévot J., Oleaga C., Ciudad C.J., Noé V. Stability and immunogenicity properties of the gene-silencing polypurine reverse Hoogsteen hairpins. Mol. Pharm. 2014;11:254–264. doi: 10.1021/mp400431f. [DOI] [PubMed] [Google Scholar]
- 61.Berman C.L., Barros S.A., Galloway S.M., Kasper P., Oleson F.B., Priestley C.C., Sweder K.S., Schlosser M.J., Sobol Z. OSWG recommendations for genotoxicity testing of novel oligonucleotide-based therapeutics. Nucleic Acid Ther. 2016;26:73–85. doi: 10.1089/nat.2015.0534. [DOI] [PubMed] [Google Scholar]
- 62.European Medicines Agency . 2005. CHMP SWP reflection paper on the assessment of the genotoxic potential of antisense oligodeoxynucleotides.https://www.ema.europa.eu/en/documents/scientific-guideline/chmp-swp-reflection-paper-assessment-genotoxic-potential-antisense-oligodeoxynucleotides_en.pdf [Google Scholar]
- 63.Henry S.P., Monteith D.K., Matson J.E., Mathison B.H., Loveday K.S., Winegar R.A., Matson J.E., Lee P.S., Riccio E.S., Bakke J.P., Levin A.A. Assessment of the genotoxic potential of ISIS 2302: a phosphorothioate oligodeoxynucleotide. Mutagenesis. 2002;17:201–209. doi: 10.1093/mutage/17.3.201. [DOI] [PubMed] [Google Scholar]
- 64.Sazani P., Weller D.L., Shrewsbury S.B. Safety pharmacology and genotoxicity evaluation of AVI-4658. Int. J. Toxicol. 2010;29:143–156. doi: 10.1177/1091581809359206. [DOI] [PubMed] [Google Scholar]
- 65.Guérard M., Andreas Z., Erich K., Christine M., Martina M.B., Christian W., Franz S., Thomas S., Yann T. Locked nucleic acid (LNA): based single-stranded oligonucleotides are not genotoxic. Environ. Mol. Mutagen. 2017;58:112–121. doi: 10.1002/em.22076. [DOI] [PubMed] [Google Scholar]
- 66.Knauert M.P., Kalish J.M., Hegan D.C., Glazer P.M. Triplex-stimulated intermolecular recombination at a single-copy genomic target. Mol. Ther. 2006;14:392–400. doi: 10.1016/j.ymthe.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 67.Datta H.J., Chan P.P., Vasquez K.M., Gupta R.C., Glazer P.M. Triplex-induced recombination in human cell-free extracts. Dependence on XPA and HsRad51. J. Biol. Chem. 2001;276:18018–18023. doi: 10.1074/jbc.M011646200. [DOI] [PubMed] [Google Scholar]
- 68.Rogers F.A., Vasquez K.M., Egholm M., Glazer P.M. Site-directed recombination via bifunctional PNA-DNA conjugates. Proc. Natl. Acad. Sci. USA. 2002;99:16695–16700. doi: 10.1073/pnas.262556899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Faruqi A.F., Datta H.J., Carroll D., Seidman M.M., Glazer P.M. Triple-helix formation induces recombination in mammalian cells via a nucleotide excision repair-dependent pathway. Mol. Cell. Biol. 2000;20:990–1000. doi: 10.1128/mcb.20.3.990-1000.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Papaioannou I., Simons J.P., Owen J.S. Oligonucleotide-directed gene-editing technology: mechanisms and future prospects. Expert Opin. Biol. Ther. 2012;12:329–342. doi: 10.1517/14712598.2012.660522. [DOI] [PubMed] [Google Scholar]
- 71.Gupta R.C., Bazemore L.R., Golub E.I., Radding C.M. Activities of human recombination protein Rad51. Proc. Natl. Acad. Sci. USA. 1997;94:463–468. doi: 10.1073/pnas.94.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krejci L., Altmannova V., Spirek M., Zhao X. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40:5795–5818. doi: 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vasquez K.M., Christensen J., Li L., Finch R.A., Glazer P.M. Human XPA and RPA DNA repair proteins participate in specific recognition of triplex-induced helical distortions. Proc. Natl. Acad. Sci. USA. 2002;99:5848–5853. doi: 10.1073/pnas.082193799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Phear G., Armstrong W., Meuth M. Molecular basis of spontaneous mutation at the aprt locus of hamster cells. J. Mol. Biol. 1989;209:577–582. doi: 10.1016/0022-2836(89)90595-0. [DOI] [PubMed] [Google Scholar]
- 75.Simon A.E., Taylor M.W., Bradley W.E., Thompson L.H. Model involving gene inactivation in the generation of autosomal recessive mutants in mammalian cells in culture. Mol. Cell. Biol. 1982;2:1126–1133. doi: 10.1128/mcb.2.9.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wigler M., Pellicer A., Silverstein S., Axel R., Urlaub G., Chasin L. DNA-mediated transfer of the adenine phosphoribosyltransferase locus into mammalian cells. Proc. Natl. Acad. Sci. USA. 1979;76:1373–1376. doi: 10.1073/pnas.76.3.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johnson L.A., Gordon R.B., Emmerson B.T. Adenine phosphoribosyltransferase: a simple spectrophotometric assay and the incidence of mutation in the normal population. Biochem. Genet. 1977;15:265–272. doi: 10.1007/BF00484458. [DOI] [PubMed] [Google Scholar]
- 78.Marco-Sola S., Sammeth M., Guigó R., Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods. 2012;9:1185–1188. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- 79.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.