

Abstract
Multiple doses of tenofovir disoproxil fumarate (TDF) together with emtricitabine is effective for HIV preexposure prophylaxis (PrEP). TDF is converted to tenofovir (TFV) in circulation, which is subsequently cleared via tubular secretion by organic ion transporters (OATs; OAT1 and OAT3). Using in vitro kinetic parameters for TFV and the OAT1 and OAT3 inhibitor probenecid, a bottom‐up physiologically‐based pharmacokinetic model was successfully developed for the first time that accurately describes the probenecid–TFV interaction. This model predicted an increase in TFV plasma exposure by 60%, which was within 15% of the observed clinical pharmacokinetic data, and a threefold decrease in renal cells exposure following coadministration of a 600 mg TDF dose with 2 g probenecid. When compared with multiple‐dose regimens, a single‐dose probenecid‐boosted TDF regimen may be effective for HIV PrEP and improve adherence and safety by minimizing TFV‐induced nephrotoxicity by reducing TFV accumulation in renal cells.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Tenofovir disoproxil fumarate (TDF), in combination with emtricitabine, is used for HIV preexposure prophylaxis (PrEP). TDF is rapidly hydrolyzed to tenofovir (TFV) in plasma. TFV is a substrate for renal secretion via organic anion transporters (OATs; OAT1 and OAT3). Probenecid (PRO) is a potent inhibitor of OAT1 and OAT3.
WHAT QUESTION DID THE STUDY ADDRESS?
☑ This work developed a physiologically‐based pharmacokinetic (PBPK) model‐informed predictive tool for the transporter‐mediated drug–drug interaction (DDI) between TFV and PRO in plasma to be used for on‐demand HIV PrEP.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ An accurate PBPK model describing the transporter‐mediated DDI between TFV and PRO in plasma was developed and predicted TFV plasma maximum concentration (Cmax) and area under the curve from zero to infinity (AUC0−INF) within 15% of observed after a single dose of 600 mg TDF.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS?
☑ This model‐informed strategy serves as a useful predictive tool to inform clinical study design and accurately predict the impact of PRO on TFV pharmacokinetics to optimize TDF efficacy and safety.
Tenofovir disoproxil fumarate (TDF), in combination with emtricitabine, is an effective oral therapy for the prevention of new HIV infections. In 2016, a nondaily on‐demand therapy of TDF from the IPERGAY study showed promising effects for HIV preexposure prophylaxis (PrEP) comparable to daily administration.1 Although effective, this on‐demand regimen relies on adherence to intermittent drug administration both before and after sexual activity. Perhaps of more practical importance, sexually active individuals (> 2 sexual encounters per week) would require nearly daily drug administration to maintain protection. Poor adherence, especially in the setting of participants not currently in a monogamous relationship, has been suggested as a possible reason why some multiple day treatments may have failed in preventing HIV infections. As such, methods to improve adherence to PrEP regimens with oral antiretrovirals in HIV‐negative individuals would be of great value to the HIV community at large.2 Reported reasons for nonadherence from patients include pill burden, cost, and long‐term adverse events,3 so perhaps a single‐dose HIV PrEP regimen with TDF may be a solution.
TDF, a potent antiretroviral for PrEP, rapidly releases tenofovir (TFV) following oral administration that subsequently is eliminated from the body entirely by the kidneys. Part of this elimination process involves active secretion of TFV by uptake renal organic anion transporters (OATs; OAT1 and OAT3). TFV may also be distributed into cells by unknown mechanisms where it is available for phosphorylation to the active moiety tenofovir di‐phosphate, TFV‐DP. The polarity of TFV‐DP causes “ion‐trapping” inside cells and results in TFV‐DP accumulation and longer exposure compared with the moiety found in circulation, TFV. For example, with daily TDF administration, the accumulation of the TFV‐DP in the lymphocytes (peripheral blood mononuclear cells (PBMCs)) is greater than eightfold.4 Although this accumulation provides beneficial protection from HIV acquisition in the PBMCs, it may lead to nephrotoxicity in kidney cells. Probenecid (PRO) is a well‐tolerated oral drug that blocks renal organic anion transporters (OATs) located on the basolateral side of the renal cell. By doing so, taking PRO with TDF would be expected to reduce the renal elimination of TFV and thereby increase the length of time that TFV remains active in the body. Thus, the administration of TDF with PRO may allow less‐frequent dosing of TDF and reduce the harmful exposure of the drug to the kidneys.
Physiologically‐based pharmacokinetic (PBPK) models can be used as predictive tools for drug–drug interactions (DDIs), particularly for small molecules such as TFV. They have been used commonly in predicting both the impact of drug inhibition on substrate pharmacokinetics and informing clinical study designs.5, 6 In some cases, PBPK models are used in lieu of clinical pharmacokinetic data for dosing recommendations and product labeling.6 Traditionally, DDI PBPK models were developed for enzyme‐mediated interactions, but as our understanding of transporter‐mediated drug disposition advances, transporter‐mediated models are also being studied.5 In this proposal, a PBPK model was developed to predict the effects of the transporter‐mediated DDI between PRO and TFV for a single‐dose HIV PrEP regimen. In this model‐informed strategy for on‐demand HIV PrEP, a PRO‐boosted TFV PBPK model was used to prospectively optimize PRO dosing for a clinical study design based on in vitro TFV and PRO parameters. Then retrospectively, the simulated DDI pharmacokinetic parameters were compared with available observed data to assess model performance. The model used extravascular compartments including PBMCs and renal cells for the surrogate interpretations of TFV efficacy and nephrotoxicity, respectively.
The objectives of the current analyses were to (i) develop and verify PBPK models that accurately described TFV and TFV‐DP plasma and PBMC pharmacokinetic profiles, (ii) simulate prospective DDIs between PRO and TFV that may be applicable for use as an on‐demand PrEP regimen, (iii) retrospectively evaluate DDI predictions with observed data, and (iv) assess the impact of coadministered PRO on TFV renal exposure and toxicity.
Methods
The model flow for this article is shown in Figure 1.
Figure 1.
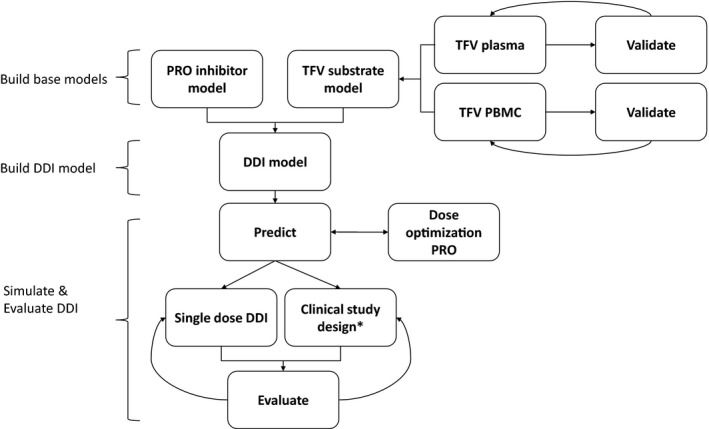
Proposed drug–drug interactions (DDI) physiologically‐based pharmacokinetic (PBPK) model workflow. *A randomized, cross‐over, two‐treatment clinical study conducted in healthy volunteers for the treatments predicted by DDI PBPK model were evaluated: (i) Test treatment: 600 mg tenofovir disoproxil fumarate (TDF) + 2 g probenecid (PRO) on day 1; (ii) control treatment: 600 mg TDF on day 1, 300 mg TDF day 2, 3. TFV, tenofovir.
Model development and verification
TFV substrate model
The base PBPK model in plasma was adapted from a previously published model of TFV exposure in pregnancy7 and modified with supporting literature.8, 9, 10, 11, 12, 13, 14, 15 The PBPK model was built using SimCYP (version 15; Certara, Inc., Princeton, NJ). Briefly, the published TFV base model was a first‐order absorption, full distribution, permeability‐limited kidney PBPK model. The B:P ratio was updated from more accurate information available for TFV alafenamide under the assumption that there would be no changes in this parameter for TFV.9 It was then enhanced by adding mechanistic description of OAT‐mediated TFV uptake in the kidney via both OAT1 and OAT3 and MRP4‐mediated efflux.13, 14, 15 Renal clearance in SimCYP is calculated using the following equation:
where Clr is renal clearance of drug for individual, Q r is renal blood flow, CLUsec,int is active secretion of drug, GFR is glomerular filtration rate for individual, and F re–abs is the fractional tubular reabsorption.16 The PBPK model input parameters are shown in Table 1. The clinically observed data for oral 300 mg TDF dosing in healthy volunteers reported by Wenning et al. 17 was retrieved by the GetData Graph Digitizer (version 2.26.0.20; GetData Pty Ltd., Kogarah, Australia) and used as a verification set for the TFV base model. The base model was further expanded to describe the active TFV‐DP concentrations in PBMCs using previously published kinetic parameters to describe the rate of uptake and efflux of TFV‐DP into these cells.11, 12 This was described as an additional organ compartment with parameters shown in Table 1. The model was able to adequately predict clinically observed TFV‐DP concentrations in PBMCs at steady state in healthy volunteers.11
Table 1.
SimCYP input parameters for TFV (substrate) and PRO (inhibitor)
| TFV parameter | Input |
|---|---|
| MW | 2877 |
| logP | 1.258 |
| B/P | 19 |
| pKa | 3.758 |
| f u | 0.938 |
| f a | 0.28 |
| k a | 1.03 1/h11 |
| Absorption | |
| Simulation | 1st order7 |
| LLC‐PK1 | 265 10−6 cm/second7 |
| Distribution | |
| Simulation | Full PBPK |
| V ss | 0.307 L/kg (method 2)10 |
| Transport (elimination) | |
| CLPD (hepatic) | 4 × 10‐5 mL/minute/10−6 cells6 |
| Sinusoidal uptake (Clint) (hepatic) | 3.125 mL/minute/10−6 cellsa |
| OAT1 transporter (Clint) (renal) | 5.8 µL/min/10−6 cells13, 14, 15 |
| OAT3 transporter (Clint) (renal) | 3.6 µL/minute/10−6 cells13, 14, 15 |
| MRP4 transporter (Clint) (renal) | 1 µL/minute/10−6 cells15 |
| PBMC (additional organ PD) | |
| Uptake J max | 10.44 pmol/minute/whole organ11, 12 |
| Uptake K m | 0.355 µM11, 12 |
| Efflux Clint,t | 1.288 µL/minute11, 12 |
| PRO parameterb | Input |
|---|---|
| OAT1 Transporter (K i) (renal) | 4.03 µM18, 19 |
| OAT3 Transporter (K i) (renal) | 1.3 µM18, 19 |
B/P, blood to plasma ratio; Clint, intrinsic clearance; CLPD, passive diffusion clearance; f a, fraction absorbed; f u, fraction unbound; J max, maximum transporter rate; k a, absorption rate constant; K i, inhibitor rate constant; K m, Michaelis‐Menten constant; logP, partition coefficient; MW, molecular weight; PBMC, peripheral blood mononuclear cells; PBPK, physiologically‐based pharmacokinetic; pKa, dissociation constant; PRO, probenecid; TFV, tenofovir; V ss, volume at steady‐state.
Optimized to fit observed data by automatic sensitivity analysis in SimCYP.
SimCYP library model was used for PRO with the additional modifications.
PRO inhibitor model
The “boosting” effects of PRO were evaluated using the verified SimCYP library PBPK model and incorporating modified inhibition potencies toward OAT1‐mediated and OAT3‐mediated renal uptake of TFV.18, 19 The modified inhibitory input parameters are shown in Table 1. This approach has been previously used to accurately predict the impact of PRO interactions with the antiinfective agents cidofovir, oseltamivir, and cefuroxime.20
DDI model predictions and evaluation
The initial models developed previously for TFV (substrate) and PRO (inhibitor) were combined in the DDI model and used for dynamic predictions of PRO inhibitory effects on TFV plasma and TFV‐DP PBMC pharmacokinetics. First, the model was used to optimize the dose of PRO for maximum boosting effects on single‐dose TFV for HIV PrEP. Next, trial designs and drug administration identical to observed (Figure S1 ) were simulated and compared retrospectively when information became available. The dose‐optimized PRO‐TFV PBPK model was used to predict pharmacokinetic parameter changes of a single 600 mg TDF dose in the presence and absence of 2 g PRO. Similarly, the PBPK model was then used to predict multiple doses of TDF seen in the IPERGAY study. Briefly, the IPERGAY study drug administration was a 3‐day medication course of 600 mg TDF on day 1, followed by doses of 300 mg TDF on days 2 and 3. Both predicted clinical scenarios were compared with observed data from healthy volunteers.21 Single‐dose TFV pharmacokinetic parameters in the presence and absence of 2 g PRO from the clinical study were recovered by noncompartmental analysis performed by Phoenix WinNonlin (version 6.4; Certara, Inc.). More information on the study design is provided in the Supplemental Information. The single‐dose DDI predictions were evaluated for accuracy through visual predictive checks and pharmacokinetic parameters: maximum concentration (Cmax) geometric mean ratio (GMR; GMR + PRO/−PRO), and area under the curve (AUC) GMR, and renal clearance (Clr). If the pharmacokinetic parameters were within twofold of the observed and the observed data overlay within the 95% confidence intervals of predicted TFV concentration‐time profiles, the model was considered accurate. For intracellular TFV‐DP PBMC concentrations and multiple‐dose TFV simulations, the models were evaluated by visual predictive checks only. Finally, the PBPK model was used to predict the effects of PRO on TFV exposure in renal cells. The mechanistic kidney model within the SimCYP simulator was selected to evaluate the different kidney processes (i.e., filtration and secretion) contributing to TFV renal clearance with or without PRO.
Trial design simulations
Initially, the TFV base model was developed in a single trial with 14 healthy volunteers. Next, trial simulations of 10 trials with 14 healthy volunteers for a total population size of 140 were simulated and selected for model performance (verification and evaluation data sets). The multiple trial predicted mean (and 95% confidence intervals) for plasma and PBMC concentration‐time profiles are shown in the figures.
Sensitivity analysis
Sensitivity analysis was performed by the automated sensitivity analysis tool within SimCYP. Input parameters chosen for sensitivity analysis include PRO OAT1 and OAT3 inhibitor rate constant (K i) and TFV OAT1 and OAT3 intrinsic clearance (Clint).
Results
Base model verification and PRO dose optimization
Initial TFV model development
Predicted TFV and TFV‐DP concentration‐time profiles for a single dose of 300 mg TDF in the plasma and PBMC, respectively, are shown in Figure 2. The visual predictive checks for both TFV and TFV‐DP were accurate. Notably, large variability was seen in the observed TFV‐DP concentrations. The simulated TFV plasma model also produced pharmacokinetic outcomes comparable with published data (Table S1 ).
Figure 2.
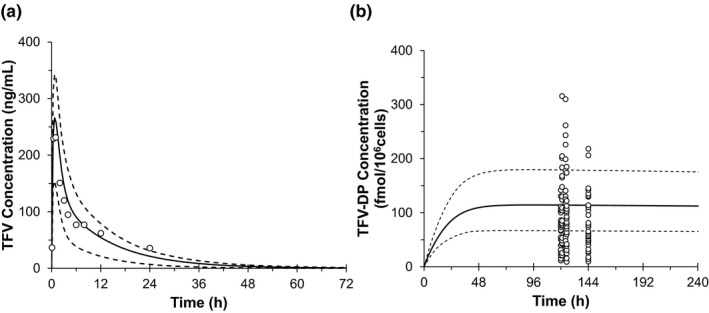
Concentration‐time profile of single oral dose 300 mg tenofovir disoproxil fumarate predicted by physiologically‐based pharmacokinetic model in the plasma—tenofovir (TFV) (a) and peripheral blood mononuclear cells—tenofovir di‐phosphate (TFV‐DP) (b) overlaid by clinical observed data (Liu et al. 21). The black open circles represent the observed data. Black solid lines represent the predicted and black dash lines show the predicted 5th and 95th confidence intervals. h, hour.
Initial PRO‐TFV model development. To optimize dose selection of PRO for the clinical study based on its in vitro OAT1 and OAT3 inhibitory effects, the DDI was simulated with both 1 and 2 g of PRO. The effects of 1 g or 2 g PRO boosted on TFV pharmacokinetic parameters are shown in Table 2. Coadministration of PRO with TDF predicted 45% and 60% increases in overall plasma exposure when given with 1 and 2 g dose of PRO, respectively. The 2 g dose of PRO was selected for the clinical study design because higher TFV exposure was predicted and 2 g is considered the maximum tolerated dose for PRO22 and thus more suitable for single‐dose boosting effects.
Table 2.
PBPK model predictions for PRO 1 and 2 g dose projections on TFV pharmacokinetic parameters in plasma
| TFV PK parameter | 1 g PRO | 2 g PRO |
|---|---|---|
| AUC0‐INF with inhibitor, ng • hour/mL | 5,064 (4,808–5,287) | 5,617 (5,290–5,964) |
| AUC0‐INF GMR | 1.46 (1.36–1.57) | 1.59 (1.55–1.64) |
| Cmax with inhibitor, ng/mL | 546 (517–574) | 566 (535–600) |
| Cmax GMR | 1.11 (1.10–1.12) | 1.13 (1.12–1.14) |
Data represented as GM (95% confidence interval).
AUC0‐INF, area under the curve from zero to infinity; Cmax, maximum concentration; GM, geometric mean; GMR, geometric mean ratio; PBPK, physiologically‐based pharmacokinetic; PK, pharmacokinetics; PRO, probenecid; TFV, tenofovir.
PRO‐TFV DDI model evaluation
PRO‐TFV plasma model
After the clinical study was completed, the predicted single‐dose 2 g PRO‐boosted TFV and multiple‐dose IPERGAY TFV plasma concentration‐time profiles were retrospectively compared with observed data (Figure 3). Visual predictive checks were accurate for the single‐dose regimen of TFV but tended to underpredict concentrations after 24 hours. Multiple‐dose TFV trough concentrations were similarly underpredicted at 24, 48, and 72 hours. Considering TDF is dosed every 24 hours and concentrations immediately prior to subsequent dosing was inaccurate, this suggests that the model may underpredict observed drug accumulation. The single‐dose effects of PRO on TFV pharmacokinetic parameters in predicted and observed data are shown in Table 3. In the presence of 2 g PRO, single‐dose TFV overall exposure area under the curve from zero to infinity (AUC0‐INF) increased by 60% and Cmax was nearly unchanged as predicted.
Figure 3.
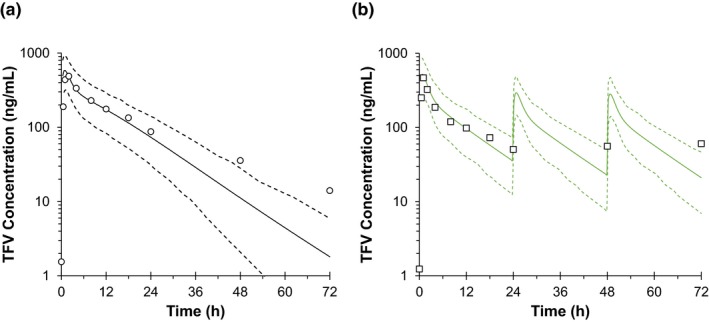
Plasma concentration‐time profiles of single‐dose probenecid‐boosted tenofovir (TFV) (a) and on‐demand IPERGAY TFV (b) predicted by physiologically‐based pharmacokinetic model and overlaid by clinical observed data (Liu et al.21). The black open circles in a and squares in b represent the observed data. Black solid lines represent the predicted and black dash lines show the 5th and 95th confidence intervals. h, hour.
Table 3.
Retrospective evaluation of PBPK model predicted TFV pharmacokinetic parameters to observed data
| PK parameters | PRED | OBS | Fold change |
|---|---|---|---|
| SD TFV + PRO | |||
| CLr, L/hour | 9.12 (8.78–9.48) | 9.48 (7.29–12.3) | 0.96 |
| Cmax, ng/mL | 566 (5,345–600) | 538 (494–585) | 1.05 |
| AUC0‐INF, ng • hour/mL | 5,617 (5,290–5,964) | 6,349 (5,598–7,200) | 0.88 |
| SD TFV–PRO | |||
| CLr, L/hour | 14.6 (14.0–15.3) | 15.4 (12.2–19.4) | 0.95 |
| Cmax, ng/mL | 502 (473–533) | 529 (451–620) | 0.95 |
| AUC0‐INF, ng • hour/mL | 3,521 (3,302–3,756) | 3,973 (3,576–4,415) | 0.89 |
| Ratio | |||
| Cmax GMR | 1.13 (1.12–1.14) | 1 (0.91–1.1) | 1.13 |
| AUC0‐INF GMR | 1.59 (1.55–1.64) | 1.6 (1.5–1.8) | 0.99 |
Data represented as GM (95% confidence interval).
AUC0‐INF, area under the curve from zero to infinity; CLr, renal clearance; Cmax, maximum concentration; GMR, geometric mean ratio; OBS, observed; PBPK, physiologically‐based pharmacokinetic; PRO, probenecid; PRED, predicted; SD, standard deviation; TFV, tenofovir.
PRO‐TFV PBMC model
Evaluations of the predicted TFV‐DP concentrations in both regimens is shown in Figure S2 . When compared with the observed data, simulated TFV‐DP concentrations were overpredicted, although the slow trend of TFV‐DP accumulation after multiple doses was captured in the model.
TFV in renal cells
The predicted amount of TFV in renal cells over time is shown in Figure 4 and Table S2 . The overall amount of TFV excreted over 72 hours remained unchanged in the presence of PRO. However, the maximum amount of TFV in renal cells was predicted to decrease by nearly threefold when given with PRO.
Figure 4.
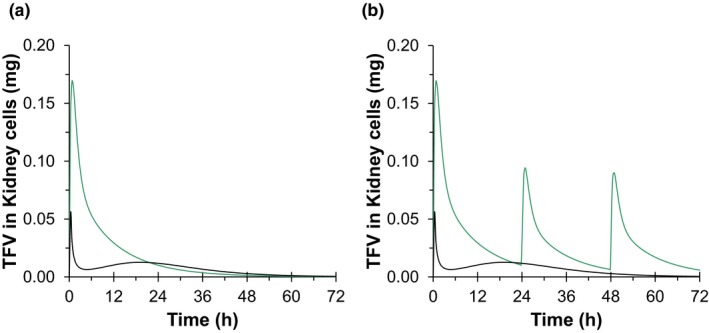
Renal cell tenofovir (TFV) concentration‐time profiles after (a) single‐dose 600 mg tenofovir disoproxil fumarate (TDF) given with (black) or without (green) 2 g probenecid (PRO) and (b) on‐demand HIV preexposure prophylaxis regimens as single‐dose 2 g PRO‐boosted 600 mg TDF (black) compared with the 3‐dose IPERGAY (green) dosing regimen predicted by the physiologically‐based pharmacokinetic model. h, hour.
Sensitivity analysis
Sensitivity analysis was performed for TFV OAT1 and OAT3 Clint (Figure S3 ) and PRO OAT1 and OAT3 K i (Figure S4 ). Effects on the TFV pharmacokinetic parameters (AUC0‐INF GMR, Cmax GMR, and Clr) are shown. Independent to input value (> 0), TFV renal secretion mediated via OAT1 and OAT3 Clint were contributors for the change in TFV AUC GMR with PRO coadministration. TFV OAT1 and OAT3 Clint had a minimal effect on Cmax GMR, as expected. Renal clearance for TFV (Clr) directly correlated with TFV OAT1 and OAT3 Clint. K i of PRO for the inhibition of OAT1 and OAT3 had impacts on AUC0‐INF GMR and Cmax GMR, whereas this inhibition constant had no impact on overall TFV renal function, Clr.
Discussion
This work is the first to publish a PBPK model to describe the transporter‐mediated interaction between TFV and PRO. This was also the first attempt to simulate the disposition of the antiretroviral in the renal cells and at the site of action (PBMCs) using a PBPK model.
Recently, a PBPK model for TFV and PRO was published as an OAT1 and OAT3 substrate and inhibitor, respectively, and studied for DDIs with a new molecule in drug development.23 The in vitro parameters for TFV were similar to ours, but PRO in vitro inhibitory constants varied, and the model underpredicted PRO effects on the new molecule in vivo. 23 In our work, the inhibition model for PRO was optimized from its effect on other molecules with similar pharmacokinetics.20 PRO has been studied previously in conjunction with other antiinfective agents, particularly those with a high propensity for renal secretion. In addition to the drug pharmacodynamics profile being directly linked to their pharmacokinetic parameters (i.e., AUC to MIC, Cmax to MIC), such pharmacokinetic properties make them excellent candidates for PRO boosting. For example, penicillin is a known OAT substrate and prescribed in combination with PRO as a boosting strategy for prolonged antibacterial coverage and serious infections such as meningitis.24 There is great benefit to providing an accurate base PBPK model to predict the mechanistic interaction between antiinfectives and PRO. Our work with TFV and PRO may serve as a prototype for this concept.
Our accurate PBPK model predicted DDI effects between PRO and TFV in plasma (Cmax and AUC0‐INF) within 15% of observed. A single dose of 2 g PRO increased TFV overall plasma exposure by 60%. Our clinical study and the PBPK model in this article evaluated the drug exposure of TFV in fasted healthy volunteers. Other studies found that TDF given with high‐fat or light meals increases TFV systemic exposure by ~ 35%. Thus, the inhibitory effects of PRO along with the food effect may result in much higher exposure of TFV than is predicted using fasting conditions. The change observed in TFV exposure from PRO alone correlates well with the drug's ADME properties, making it a suitable agent for the mechanistic underpinnings in a PBPK model. TFV has low bioavailability (~ 25%), high fraction unbound (f u > 0.9) and is entirely cleared by the kidneys with no known enzyme metabolism involvement.25, 26, 27 TFV is eliminated through both renal filtration and active secretion via influx‐mediated OAT1 and OAT3 and efflux‐mediated MRP4 transporter involvement.13, 15 Because TFV is highly unbound in the blood, a greater portion of its clearance is perhaps through glomerular filtration. Only 20–30% of TFV in circulation is available for transporter‐mediated secretion.27 Thus, the inhibitory effects of PRO on the renal transporters OAT1 and OAT3 altered a considerable portion of TFV total apparent clearance as seen by the 60% change in AUC0‐INF. Although PRO boosted TFV in plasma well, there were smaller changes at the intracellular level (~ 30% increase at 24 hours)21 that were not accurately reflected in the PBPK model. In healthy volunteers, a single PRO‐boosted TDF dose resulted in TFV‐DP concentrations that stayed above the EC90 (16 fmol/106 cells) for more than 72 hours.28 This may be adequate coverage for single‐dose HIV PrEP but requires further study.
Although TDF is generally well tolerated, long‐term adverse events have been associated with its use, which include nephrotoxicity29 and osteopenia.30 Tenofovir‐induced renal proximal tubulopathy, or Fanconi syndrome, has been suggested as a potential mechanism leading to both adverse events.29, 30, 31 By blocking entry of TFV into the renal proximal tubule, PRO use may limit the risk of renal toxicity by limiting TFV exposure to the proximal tubular cells without diminishing efficacy. In a case report, the PRO‐TDF combination was studied in three patients and found to be clinically beneficial.32 The HIV‐infected patients were adequately treated for HIV infection and provided renal protection from Fanconi syndrome when coprescribed PRO and TDF.32 This model may serve as a clinical predictive tool for TFV‐PRO use in these clinical scenarios where tailoring care is crucial for the relief of toxic adverse events. TDF‐induced bone loss may also occur via alternative mechanisms, including increased TFV plasma exposure. However, these mechanisms are not expected to be exacerbated by PRO boosting as overall plasma exposure is similar to that of previously evaluated regimens. In addition, any changes in renal function or bone mineral density associated with TDF use appear to be quickly reversible after TDF cessation, suggesting that TDF‐based PrEP does not lead to permanent dysfunction.33, 34 As expected, our model shows PRO inhibition of OAT‐mediated TFV renal uptake reduced the amount of TDF exposure in the kidneys. Peak TFV kidney exposure following a PRO‐boosted regimen is expected to be reduced on average by more than threefold when compared with the IPERGAY regimen, with a substantial reduction in overall exposure.
There is an increasing use of PBPK models for enzyme‐mediated or transporter‐mediated DDIs. PBPK models are especially touted for the ability to extrapolate beyond the data set available and in some cases be used in lieu of clinical data for product labeling, designing clinical studies, and first‐in‐human projections.6 Although CYP‐mediated DDIs are common, transporter‐mediated DDIs are less well studied because of the uncertainty in the human physiology of transporters and the general assumption that transporter‐mediated DDIs have less clinically significant interactions. However, PRO can be a strong inhibitor for certain OAT1 and OAT3 substrates and evidently beneficial for some patients. An accurate PBPK model has been created for the single‐dose interaction between TFV and PRO in the systemic circulation. This model may serve to inform other OAT1 and OAT3 substrates under future investigation. In this context, the US Food and Drug Administration recommends new molecular entities with considerable renal clearance and shown to be in vitro OAT1 and OAT3 to be studied with PRO.35 In addition, this model also provides a mechanism‐based evaluation for proximal tubule cell drug uptake. The developed PBPK model is a translational approach from in vitro TFV and PRO parameters to predict the underlying mechanistic and physiologic components of the interaction. In this work, the model was used as a predictive tool for the boosting effects of PRO on TFV for the exploration of a single‐dose HIV prevention regimen.
Often, model predictions correlated poorly to observed in the PBMC compartment, limiting its use for accurate quantitative predictions only to the plasma compartment. This may be in part because of the nonlinear kinetics of the TFV‐DP in PBMCs after multiple doses. The rate of TFV‐DP entering and leaving PBMCs is inconsistent and varies from a single dose to steady‐state TDF administration.4 Because the model was developed using steady‐state TFV‐DP administration, this may account for some of the inaccuracy in TFV‐DP predictions because the TDF regimens evaluated in our work did not achieve steady‐state levels. In addition, because the rates of TFV‐DP kinetics in PBMCs were unavailable in healthy volunteers, the PBMC compartment in our model was generated using a data set derived from HIV‐infected individuals. The plasma pharmacokinetics in healthy volunteers and HIV‐1 infected individuals are thought to be similar. However, intracellular kinetics are generally less understood on a mechanistic level, which may require further study. In addition, the multiple‐dose TFV plasma trough concentrations seen in the IPERGAY regimen were underpredicted. This is most likely attributed to the incorrect capture of TFV accumulation after multiple doses. TFV demonstrates biphasic elimination in vivo with a long distribution phase, probably because of the high fraction unbound of TFV, making the drug available for tissue uptake. We think the model is misinterpreting the delayed elimination phase for the distribution phase and thus miscalculating the true elimination phase, although the impact of the DDI was still accurately predicted. This is an inherent limitation to our model and is difficult to resolve with the current modeling software and must be taken into account if applying this model to daily dosing.
In conclusion, this is the first attempt to model the effect of PRO on plasma TFV and intracellular exposure of its active metabolite, TFV‐DP, in the context of inhibition of renal transporters (OAT1 and OAT3). An accurate single‐dose PRO‐TDF PBPK model was developed. PRO boosted TFV plasma exposure by 60% and increased TFV‐DP in PBMC by approximately threefold. Although further refinement would be required, this initial work suggests that a PBPK model can be used to evaluate the safety and efficacy of DDIs involving inhibition of OAT1 and OAT3.
Funding
This work is supported by the Campbell Foundation for AIDS Research. S.N.L. and B.T.G. supported by the National Institute of General Medical Sciences (Grant T32 GM008425). B.T.G. was also supported by the Indiana Clinical and Translational Sciences Institute Young Investigator Award (Grant UL1 TR001108). Z.D. was also supported by the National Institute of General Medical Sciences (Grants R01s GM078501 and GM121707).
Conflict of Interest
S.N.L., Z.D., and B.T.G. declare no competing interests for this work.
Author Contributions
S.N.L., Z.D., and B.T.G. wrote the manuscript. S.N.L., Z.D., and B.T.G. designed the research. S.N.L., Z.D., and B.T.G. performed the research. S.N.L. and B.T.G. analyzed the data. B.T.G. contributed new reagents/analytical tools.
Supporting information
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Table S1.
Table S2.
Supplemental Information. Study design.
Supplemental Information. Simcyp population based simulator.
References
- 1. Molina, J.M. et al On‐demand preexposure prophylaxis in men at high risk for HIV‐1 infection. N. Engl. J. Med. 373, 2237–2246 (2015). [DOI] [PubMed] [Google Scholar]
- 2. Anderson, P.L. , Garcia‐Lerma, J.G. & Heneine, W. Nondaily preexposure prophylaxis for HIV prevention. Curr. Opin. HIV AIDS 11, 94–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sidebottom, D. , Ekström, A.M. & Strömdahl, S. A systematic review of adherence to oral pre‐exposure prophylaxis for HIV – how can we improve uptake and adherence? BMC Infect. Dis. 18, 581 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seifert, S.M. et al Intracellular tenofovir and emtricitabine anabolites in genital, rectal, and blood compartments from first dose to steady state. AIDS Res. Hum. Retroviruses 32, 981–991 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Min, J.S. & Bae, S.K. Prediction of drug‐drug interaction potential using physiologically based pharmacokinetic modeling. Arch. Pharm. Res. 40, 1356–1379 (2017). [DOI] [PubMed] [Google Scholar]
- 6. U.S. Department of Health and Human Services‐ Food and Drug Administration . Physiologically based pharmacokinetic analyses–format and content guidance for industry <https://www.fda.gov/media/101469/download> (2018). Accessed June 5, 2019.
- 7. De Sousa Mendes, M. et al Physiologically‐based pharmacokinetic modeling of renally excreted antiretroviral drugs in pregnant women. Br. J. Clin. Pharmacol. 80, 1031–1041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gilead Sciences, Inc . Truvada clinical pharmacology and biopharmaceutics review(s) <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/021752s000_Truvada_BioPharmr.pdf> (2001). Accessed June 5, 2019.
- 9. Gilead Sciences, Inc. Vemlidy prescribing information <https://www.gilead.com/-/media/files/pdfs/medicines/liver-disease/vemlidy/vemlidy_pi.pdf> (2019). Accessed June 5, 2019.
- 10. Rodgers, T. & Rowland, M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm. Res. 24, 918–933 (2007). [DOI] [PubMed] [Google Scholar]
- 11. Baheti, G. , Kiser, J.J. , Havens, P.L. & Fletcher, C.V. Plasma and intracellular population pharmacokinetic analysis of tenofovir in HIV‐1‐infected patients. Antimicrob. Agents Chemother. 55, 5294–5299 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Duwal, S. , Schutte, C. & Von Kleist, M. Pharmacokinetics and pharmacodynamics of the reverse transcriptase inhibitor tenofovir and prophylactic efficacy against HIV‐1 infection. PLoS ONE 7, e40382 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Uwai, Y. , Ida, H. , Tsuji, Y. , Katsura, T. & Inui, K. Renal transport of adefovir, cidofovir, and tenofovir by SLC22A family members (hOAT1, hOAT3, and hOCT2). Pharm. Res. 24, 811–815 (2007). [DOI] [PubMed] [Google Scholar]
- 14. Gutierrez, F. , Fulladosa, X. , Barril, G. & Domingo, P. Renal tubular transporter‐mediated interactions of HIV drugs: implications for patient management. AIDS Rev. 16, 199–212 (2014). [PubMed] [Google Scholar]
- 15. Kohler, J.J. et al Tenofovir renal proximal tubular toxicity is regulated by OAT1 and MRP4 transporters. Lab. Invest. 91, 852–858 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rowland, Y.K. , Aarabi, M. , Jamei, M. & Rostami‐Hodjegan, A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev. Clin. Pharmacol. 4, 261–274 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Wenning, L.A. et al Lack of a significant drug interaction between raltegravir and tenofovir. Antimicrob. Agents Chemother. 52, 3253–3258 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maeda, K. et al Inhibitory effects of p‐aminohippurate and probenecid on the renal clearance of adefovir and benzylpenicillin as probe drugs for organic anion transporter (OAT) 1 and OAT3 in humans. Eur. J. Pharm. Sci. 59, 94–103 (2014). [DOI] [PubMed] [Google Scholar]
- 19. Tahara, H. et al Inhibition of oat3‐mediated renal uptake as a mechanism for drug‐drug interaction between fexofenadine and probenecid. Drug Metab. Dispos. 5, 743–747 (2006). [DOI] [PubMed] [Google Scholar]
- 20. Hsu, V. et al Towards quantitation of the effects of renal impairment and probenecid inhibition on kidney uptake and efflux transporters, using physiologically based pharmacokinetic modelling and simulations. Clin. Pharmacokinet. 53, 283–293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu, S.N. et al Inhibitory effects of probenecid on pharmacokinetics of tenofovir disoproxil fumarate and emtricitabine for on‐demand HIV pre‐exposure prophylaxis. Clin. Pharmacol. Ther. (2019). 10.1002/cpt.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Selen, A. , Amidon, G.L. & Welling, P.G. Pharmacokinetics of probenecid following oral doses to human volunteers. J. Pharm. Sci. 71, 1238–1248 (1982). [DOI] [PubMed] [Google Scholar]
- 23. Ball, K. et al Prediction of renal transporter‐mediated drug‐drug interactions for a drug which is an OAT substrate and inhibitor using PBPK modeling. Eur. J. Pharm. Sci. 106, 122–132 (2017). [DOI] [PubMed] [Google Scholar]
- 24. Dacey, R.G. & Sande, M.A. Effects of probenecid on cerebrospinal fluid concentrations of penicillin and cephalosporin derivatives. Antimicrob. Agents Chemother. 6, 437–441 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Custodio, J.M. et al Effect of food on rilpivirine/emtricitabine/tenofovir disoproxil fumarate, anantiretroviral single‐tablet regimen for the treatment of HIV infection. J. Clin. Pharmacol. 54, 378–385 (2014). [DOI] [PubMed] [Google Scholar]
- 26. Chilar, T. , Ho, E.S. , Lin, D.C. & Mulato, A.S. Human renal organic anion transporter 1 (hOAT1) and its role in the nephrotoxicity of antiviral nucleotide analogs. Nucleosides Nucleotides Nucleic Acids 20, 641–648 (2001). [DOI] [PubMed] [Google Scholar]
- 27. Fernandez‐Fernandez, B. et al Tenofovir nephrotoxicity: 2011 update. AIDS Res. Treat. 2011, 354908 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anderson, P.L. et al Emtricitabine‐tenofovir concentrations and pre‐exposure prophylaxis efficacy in men who have sex with men. Sci. Transl. Med. 4, 151ra125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brim, N.M. , Cu‐Uvin, S. , Hu, S.L. & O'Bell, J.W. Bone disease and pathologic fractures in a patient with tenofovir‐induced Fanconi syndrome. AIDS Read. 17, 322–328 (2007). [PubMed] [Google Scholar]
- 30. Calmy, A. et al Low bone mineral density, renal dysfunction, and fracture risk in HIV infection: a cross‐sectional study. J. Infect. Dis. 200, 1746–1754 (2009). [DOI] [PubMed] [Google Scholar]
- 31. Izzedine, H. et al Long‐term renal safety of tenofovir disoproxil fumarate in antiretroviral‐naive HIV‐1‐infected patients. Data from a double‐blind randomized active‐controlled multicentre study. Nephrol. Dial. Transplant. 20, 743–746 (2005). [DOI] [PubMed] [Google Scholar]
- 32. Izzedine, H. et al Tenofovir/probenecid combination in HIV/HBV‐coinfected patients: how to escape Fanconi syndrome recurrence? AIDS 24, 1078–1079 (2010). [DOI] [PubMed] [Google Scholar]
- 33. Mirembe, B.G. et al Bone mineral density changes among young, healthy African women receiving oral tenofovir for HIV preexposure prophylaxis. J. Acquir. Immune Defic. Syndr. 71, 287–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Solomon, M.M. et al Changes in renal function associated with oral emtricitabine/tenofovir disoproxil fumarate use for HIV pre‐exposure prophylaxis. AIDS 28, 851–859 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. U.S. Department of Health and Human Services‐ Food and Drug Administration . In vitro metabolism‐ and transporter‐mediated drug‐drug interaction studies guidance for industry <https://www.fda.gov/media/108130/download> (2017). Accessed June 5, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Table S1.
Table S2.
Supplemental Information. Study design.
Supplemental Information. Simcyp population based simulator.