

Abstract
Cholinesterase inhibitor plays an important role in the treatment of patients with Alzheimer’s disease (AD). Herein, we report the medicinal chemistry efforts leading to a new series of 1,3-dimethylbenzimidazolinone derivatives. Among the synthesised compounds, 15b and 15j showed submicromolar IC50 values (15b, eeAChE IC50 = 0.39 ± 0.11 µM; 15j, eqBChE IC50 = 0.16 ± 0.04 µM) towards acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). Kinetic and molecular modelling studies revealed that 15b and 15j act in a competitive manner. 15b and 15j showed neuroprotective effect against H2O2-induced oxidative damage on PC12 cells. This effect was further supported by their antioxidant activity determined in a DPPH assay in vitro. Morris water maze test confirmed the memory amelioration effect of the two compounds in a scopolamine-induced mouse model. Moreover, the hepatotoxicity of 15b and 15j was lower than tacrine. In summary, these data suggest 15b and 15j are promising multifunctional agents against AD.
Keywords: Alzheimer’s disease; cholinesterase inhibitor; 1,3-dimethylbenzimidazolinone derivatives; molecular docking; structural modification; structure-activity relationship
1. Introduction
Alzheimer’s disease (AD) is a chronic and progressive neurodegenerative disorder that is characterised by memory loss, disorientation, deficits in cognitive functions, and behavioural abnormalities1–3. Today, it is estimated that over 50 million people worldwide live with dementia, and this number is estimated to increase to more than 152 million by 2050. The current cost of the disease is about a trillion US dollars a year, and that is forecast to double by 20304. Although the exact cause of AD is not yet understood, numerous literatures have suggested the involvement of several factors for the development of the disease, including low levels of acetylcholine (ACh) in the hippocampus and cortex area of the brain, the deposition of amyloid β (Aβ) peptide, the neurofibrillary tangles (p-Tau), and the oxidative stress, which is recently proved to play vital roles in the pathogenesis of AD5–7. Various drugs have been used over the past several years for the treatment of AD. Currently, there are only four drugs against AD in clinical, namely donepezil, rivastigmine, galantamine, and memantine8. Therefore, it is necessary to develop novel compounds with potential therapeutic value9,10.
Cholinergic system, which plays an important role in the regulation of learning, cognition, and memory processes11, has been extensively studied for the design of anti-AD drugs12,13. AChE contains two distinct binding sites: the catalytic active site (CAS), located at the bottom of the gorge, is the binding site for both substrates and inhibitors; the peripheral anionic site (PAS), situated at the entrance of the gorge, is the binding site of enzyme inhibitors; a mid-gorge recognition site between CAS and PAS is also identified14,15. BChE is expressed in neuroglia and is also found in the intestine, liver, kidney, heart, lung, and serum. It plays a major role in the metabolism of ester-containing compounds16. It can also hydrolyse ACh. In progressed AD patient, the level of AChE dramatically decreases due to the loss of neuron. However, it has been discovered that the level of BChE does not decline, or may even increase in progressed AD17. Therefore, BChE is a potential therapeutic target for restoring ACh levels in the brain, improving cognitive impairment, reducing adverse effects, especially in progressed AD patients. In summary, both acetylcholinesterase inhibitors (AChEIs) and butyrylcholinesterase inhibitors (BChEIs) are urgently needed for the treatment of AD18–21.
In our previous study, we discovered several new hits as ChEs inhibitors through pharmacophore-based virtual screening. G801-0274 is one of the active hits19. With the aim to understand the structure-activity relationship (SAR), as well as to identify the pharmacophoric scaffold of this compound, structural modifications (Figure 1) and biological activity evaluations were performed and reported in this paper. Our data revealed a new type of ChEI with both in vitro and in vivo activity. It merits further medicinal chemistry efforts to identify therapeutic agents against AD.
Figure 1.

Design of a series of 1,3-dimethylbenzimidazolinone derivatives as cholinesterase inhibitors.
2. Results and discussion
2.1. Chemistry
The routes for the synthesis of the desired compounds 9a–9p, 11a–11f, 15a–15j are illustrated in Scheme 1. Firstly, 4-piperidinecarboxamide (1) was treated with substituted benzyl bromides (2a–2p) using potassium carbonate in methanol to get 1–(1-substituted phenyl)piperidine-4-carbox-amides (3a–3p). 3a–3p were treated with LiAlH4 in dry THF at 75 °C for 4 h to afford (1-benzylpiperidin-4-yl)methanamine derivatives (4a–4p). Secondly, compound 5 was treated with di(1H-imidazol-1-yl)methanone in chloroform at 60 °C for 12 h to obtain compound 6, which was condensed with methyl iodide to yield compound 7. Compound 7 was treated with lithium hydroxide to afford carboxylic acids 8, which was condensed with (1-benzylpiperidin-4-yl)methanamine derivatives (4a–4p) to acquire target compounds 9a–9p.
Scheme 1.

Reagents and conditions: (a) K2CO3, CH3OH, 85 °C, 5 h; (b) LiAlH4, THF, reflux, 5 h; (c) di(1H-imidazol-1-yl)methanone, CHCl3, 60 °C, 12 h; (d) NaH, MeI, DMF, r.t.; (e) THF, H2O, LiOH, 50 °C 2 h; (f) PyBOP, DIPEA, DMF, r.t, 4 h; (g) Chlorosulfonic acid, rt, 6 h; and (h) Et3N, CH2Cl2, r.t, 4 h.
Compounds 10a–10f were condensed with 4a, 4g, and 4m to yield target compounds 11a–11f. Compound 12 was reacted with methyl iodide to yield compound 13, which was then transformed to 14 using chlorosulfonic acid. The target compounds 15a–15j were successfully obtained through condensation of 14 and different piperidine including 4a–4d, 4f, 4i, and 4l–4o.
2.2. Pharmacology
2.2.1. AChE and BChE inhibitory activity of the target molecules
The synthesised compounds 9a–9p, 11a–11f, and 15a–15j were assessed to determine their anti-cholinesterase activity. AChE and BChE inhibition activities were evaluated by the method described by Ellman20, wherein donepezil and tacrine were used as the reference compounds. The data were expressed as IC50 values (Table 1). In vitro assays proved that most of the compounds effectively inhibited ChEs in the micromolar range. Firstly, we explored the influence of sulphonamide group on the ChEs inhibitory activity by replacing it using amide group. We synthesised compounds 9a–9p. Compared to 9a, methyl substitution (9b–9d) led to a decrease of AChE inhibitory activity. The position of the methyl group was also considered, showing that the activity was meta- > ortho- > para-. For the multi-substituted compound, the 3,4-diCH3 (9e) showed reduced inhibitory activity. Next, we explored the impact of the methoxy group on ChEs inhibitory activities. The activity on AChE was para- > meta-. Compared to 9a, 9f and 9g also showed reduced inhibitory activity.
Table 1.
Structures, eeAChE and eqBChE inhibitory activities of target compounds.
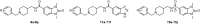 | ||||||
|---|---|---|---|---|---|---|
| Compound | R | X | Y | AChEa (IC50c, μM or IRd, %) | BChEb (IC50, μM or IR, %) | SIe |
| 9a | H | – | – | 3.14 ± 1.12 | 43.89 % | – |
| 9b | 2-CH3 | – | – | 8.08 ± 2.66 | 41.36 % | – |
| 9c | 3-CH3 | – | – | 3.83 ± 2.04 | 44.03 % | – |
| 9d | 4-CH3 | – | – | 25.71 ± 12.31 | 37.80 % | – |
| 9e | 3,4-CH3 | – | – | 9.97 ± 2.52 | 30.80 % | – |
| 9f | 3-OCH3 | – | – | 33.47 % | 25.02 % | – |
| 9g | 4-OCH3 | – | – | 5.17 ± 2.25 | 41.46 % | – |
| 9h | 2-F | – | – | 16.19 ± 4.34 | 30.01 % | – |
| 9i | 3-F | – | – | 2.93 ± 1.96 | 95.07 ± 59.26 | 0.24 |
| 9j | 4-F | – | – | 4.70 ± 2.01 | 3.71 ± 1.88 | 1.27 |
| 9k | 2-Cl | – | – | 3.92 ± 1.20 | 46.48 % | – |
| 9l | 3-Cl | – | – | 6.00 ± 3.31 | 36.93 % | – |
| 9m | 4-Cl | – | – | 0.21 ± 0.03 | 49.93 % | – |
| 9n | 2-Br | – | – | 8.87 ± 4.56 | 43.48 % | – |
| 9o | 3-Br | – | – | 3.69 ± 1.83 | 42.83 % | – |
| 9p | 4-Br | – | – | 11.06 ± 6.42 | 38.48 % | – |
| 11a | 4-OCH3 | CH | CH | 38.08 ± 12.83 | 10.75 ± 5.55 | 3.54 |
| 11b | 4-OCH3 | N | CH | 48.57 % | 44.57 % | – |
| 11c | 2-Cl | N | CH | 5.44 ± 2.22 | 21.29 ± 5.00 | 0.26 |
| 11d | 4-OCH3 | CH | N | 18.81 % | 16.51 % | – |
| 11e | 2-Cl | CH | N | 7.48 ± 3.10 | 42.12 % | – |
| 11f | H | N | N | 0.46 ± 0.40 | 43.07 % | – |
| 15a | H | – | – | 0.38 ± 0.08 | 0.45 ± 0.02 | 0.84 |
| 15b | 2-CH3 | – | – | 0.39 ± 0.11 | 0.66 ± 0.16 | 0.59 |
| – | – | 1.49 ± 0.43f | 1.33 ± 0.55g | 1.12 | ||
| 15c | 3-CH3 | – | – | 0.54 ± 0.14 | 0.47 ± 0.17 | 1.14 |
| 15d | 4-CH3 | – | – | 24.77 ± 3.48 | 100.2 ± 22.09 | 0.24 |
| 15e | 3-OCH3 | – | – | 9.35 ± 2.32 | 20.34 ± 3.08 | 0.45 |
| 15f | 3-F | – | – | 1.04 ± 0.48 | 1.37 ± 0.74 | 0.75 |
| 15g | 3-Cl | – | – | 0.15 ± 0.02 | 1.01 ± 0.46 | 0.15 |
| 15h | 4-Cl | – | – | 1.95 ± 0.28 | 2.23 ± 0.81 | 0.87 |
| 15i | 2-Br | – | – | 0.42 ± 0.06 | 0.22 ± 0.04 | 1.99 |
| 15j | 3-Br | – | – | 0.39 ± 0.15 | 0.16 ± 0.04 | 2.44 |
| – | – | 1.25 ± 0.48f | 0.66 ± 0.22g | 1.89 | ||
| Tacrine | – | – | 0.02 ± 0.01 | 0.008 ± 0.004 | 2.50 | |
| Donepezil | – | – | 0.009 ± 0.001 | 1.57 ± 0.35 | 0.006 | |
AChE (EC 3.1.1.7) from electric eel.
BChE (EC 3.1.1.8) from horse serum.
Concentration required for 50% inhibition of ChEs, data were shown in mean ± SEM of triplicate independent experiments.
Inhibitory rate of the compounds under 100 μM on ChEs.
Selectivity index (SI)=AChE IC50/BChE IC50.
AChE (EC 3.1.1.7) from human.
BChE (EC 3.1.1.8) from human.
Then, we evaluated the impact of halogen atoms on the ChEs activity. When substituted by F (9h–9j), Cl (9k–9m), Br (9n–9p) increased AChE inhibitory activity compared to compound 9a (anti-AChE IC50 = 3.14 ± 1.12 µM). Especially, 9m (anti-AChE IC50 = 0.21 ± 0.03 µM) increased 15-fold inhibitory activity towards AChE. When substituted by different halogen atoms, the activity on AChE was 4-Cl (9m) > 3-F (9i) > 3-Br (9o). And this series of structure were more selective towards AChE than BChE. However, they showed little inhibitory activity on BChE at 100 µM, except 9j (anti-BChE IC50 = 3.71 ± 1.88 µM). After the sulphonamide group was replaced by amide group, the AChE inhibitory activity can be maintained at the micromolar level, but the BChE inhibitory activity was significantly decreased, indicating that sulphonamide group was very important for maintaining the BChE inhibitory activity, which may be related to the bond angle of the sulphonamide group to the molecule.
Next, we explored the effect of the benzimidazole on ChEs inhibitory activities. we synthesised compounds 11a–11f with 1,3-dimethyl-1,3-dihydro-2H-benzo[d]imidazol-2-one replaced by indole, 1H-benzo[d]imidazole, 1H-indazole, or 1H-benzo[d][1,2,3]triazole. Simultaneously, we introduced methoxy and halogen atoms groups to the benzyloxyl moiety. We found that such structural modification resulted in a remarkably reduced activity on ChEs. We speculated that methyl groups occupy the active site pocket and interact with amino acid residues to increase inhibitory activity.
Finally, we synthesised compounds 15a–15j with various kinds of groups introduced to the benzyl moiety, to explore the impact of the benzyl moiety structural modification on ChEs inhibitory activities. Compared to 9a, 15a showed an eightfold increase in inhibitory activity towards ChEs, indicating sulphonamide group can enhanced inhibitory activity. Firstly, we introduced methyl group to the benzyl moiety. Methyl substitution at ortho- or meta- position of benzyl (15b and 15c, respectively) showed comparable activity to 15a while the para-substituted compound 15d led to reduced inhibitory activity. Next, we evaluated the impact of the methoxy group substituents on ChEs inhibitory activities. Compared to 15a, 15e showed reduced inhibitory activity. Then, compounds with different halogen substituents were also synthesised. When substituted by F (15f), it showed reduced inhibitory activity. When substituted by Cl (15g–15h), the activity on AChE was meta- > para-. When substituted by Br (15i–15j), the activity on AChE was meta- > para-. And 15j with meta substitution were more selective having an enhanced inhibitory activity on BChE than AChE. When substituted by different halogen atoms, the activity on AChE was –Br > –Cl > –F. We speculated that Br and Cl can form halogen bonds with amino acid, but F cannot form halogen bond. Therefore, the compounds with Br and Cl substituents were more potent than F.
To further validated the inhibitory activities of synthesised compounds on human ChEs, the representative compounds, 15b and 15j were selected for determination (Table 1). 15b exhibited huAChE IC50 = 1.49 ± 0.43 µM, huBChE IC50 = 1.33 ± 0.55 µM; 15j exhibited huAChE IC50 = 1.25 ± 0.48 µM, huBChE IC50 = 0.66 ± 0.22 µM. The results showed that the synthesised compounds efficiently inhibited the activities of human ChEs, further confirrmed their activities as ChEs inhibitors.
2.2.2. Kinetic studies of AChE and BChE inhibition
In order to determine the kinetic type of AChE and BChE inhibition, compound 15b and compound 15j were selected for the kinetic study. In each case, the kinetic type of enzyme inhibition was obtained through the modified Ellman’s method and Lineweaver–Burk secondary plots21. As shown in Figure 2, the results showed that the plots of 1/V versus 1/[S] gave straight lines with different slopes depending on concentrations of the inhibitor, and the lines intersected on the vertical axis. The Lineweaver–Burk plot showed that Vmax is the same regardless of concentration of the inhibitor, and Km increases with increasing concentration of the inhibitor. This behaviour indicates that 15b and 15j inhibit the ChEs in a competitive manner.
Figure 2.

(A) Lineweaver–Burk plot for the inhibition of eeAChE by compound 15b at different concentrations of substrate. (B) Lineweaver–Burk plot for the inhibition of eqBChE by compound 15j at different concentrations of substrate.
2.2.3. Docking studies
To further investigate the binding modes of 15b and 15j with ChEs, molecular docking was performed using the Discovery Studio software 2016, BIOVIA, San Diego, CA. The predicted binding mode for the compound 15b was shown in Figure 3(A). 15b simultaneously occupied both the CAS and PAS of AChE. The benzyl ring of N-benzylpiperidine moiety interacted with Trp86 in CAS via aromatic π–π interaction. The benzimidazole ring interacted with Trp286 and Tyr341 in PAS via π–π stacking interactions. The sulphonyl group formed two hydrogen bonds with Phe295 and Arg296. Besides, the sulphur formed two π–sulphur interactions with Phe297 and Phe338. Meanwhile, the methyl groups moiety of 15b formed some π–alkyl interactions with Tyr72, Ser293, Tyr337, and His447. All these facts provide an explanation for micromolar inhibitory activity of compound 15b against AChE.
Figure 3.

(A) Binding mode prediction of 15b with huAChE (PDB id: 4EY7). (B) Binding mode prediction of 15j with huBChE (PDB id: 4TPK). Colour coding: green, hydrogen bond; red, π–π stacking; orange, π–sulphur; purple, π–alkyl.
Molecular docking of 15j in the active site of BChE has been shown in Figure 3(B). 15j exhibited a U-shaped conformation. The benzimidazole ring interacted with Trp82 and His438 via π–π stacking interactions. The sulphur formed a π–sulphur interaction with Trp82. The charged nitrogen of piperidine ring formed a π–cation interaction with Asp70. And some π–alkyl interactions were formed between Tyr332, Tyr440, Phe329, Trp430, and the methyl, bromine groups in 15j, which increased inhibitory activities by strengthening the binding affinity.
2.2.4. Cell toxicity studies
We focussed on the cell toxicity of the synthesised compounds. Compounds 15b and 15j were selected as representative compounds for the evaluation of their potential cytotoxic effects. 15b and 15j exhibited no cytotoxicity to PC-12 cells at different concentrations (10, 20, 30, and 50 µM), indicating its low cell toxicity. Results are shown in Figure 4(A).
Figure 4.

(A) In vitro cell toxicity of 15b and 15j on PC-12 cell line. (B) In vitro hepatotoxicity of 15b and 15j on HepG2 cell line. Data were expressed as mean ± SD (n = 3).
Next, MTT assay was performed using the human hepatoma cell line HepG2 to evaluate preliminary the hepatotoxicity of 15b and 15j in vitro, which were incubated with 15b and 15j of 5, 15, 50 µM respectively for 24 h. Compared to donepezil, the two compounds showed low toxicity on cell viability (Figure 4(B)).
2.2.5. Inhibition of self-induced Aβ1–42 aggregation
Compounds 15b and 15j were evaluated for their inhibitory capacity on self-induced Aβ1–42 aggregation based on a thioflavin T-based fluorometric assay. All the tested compounds showed no inhibitory activities under 50 µM (Supplementary Figure S1(A)).
2.2.6. DPPH radical scavenging activity (DPPH)
Compounds 15b and 15j were selected to be evaluated for their 1,1-diphenyl-2-picrylhydrazyl radical scavenging activity (DPPH) compared with trolox as reference compound12. 15b showed a better inhibitory rate than (12.2%) 15j (6.3%) at 1 mM. Therefore, they showed weak antioxidant activities (Supplementary Figure S1(B)).
2.2.7. Neuroprotective effect against H2O2-induced cell death in PC12 neurons
We evaluated the cytoprotective effects of 15b and 15j on H2O2-induced cell damage. Treatment with 800 µM H2O2 for 24 h caused over 55% death rate of PC12 cells compared with the control group (Supplementary Figure S2(A)). Then we pre-treated PC12 cells with 15b and 15j for 24 h, the mortality rate of PC12 cells caused by H2O2 was reduced. Such protective effect exhibited dose-dependent manner for both the two compounds. 15b showed a better cytoprotective effect than 15j. These results indicated that 15b and 15j had a potential in antagonising the oxidative stress.
2.2.8. Behavioural studies
Compound 15b and 15j were selected for in vivo behavioural study22. The ameliorating potential of 15b and 15j against scopolamine-induced cognition impairment in ICR mice were investigated in Morris water maze test. Tacrine (20 mmol kg−1 body weight) was used as positive control. 15b, 15j (15 mg kg−1) and tacrine were orally administered to the ICR mice 30 min before intraperitoneal (ip) administration of scopolamine (1 mg kg−1) or saline solution for 10 consecutive days to adapt the apparatus. The test included 5 days of learning and memory training and a probe trial on the sixth day. The mean escape latency values of all the groups on the sixth day were shown in Table 2 and Figure 5(A). Compared to the control group, scopolamine led to a remarkable delay of the latency to target (8.34 ± 0.53 s vs. 25.83 ± 0.75 s), indicating that the cognitive impairment mouse model was successfully built. Treatment of tacrine ameliorated the impairment and the latency to target reduced to 12.06 ± 0.37 s. Compared to tacrine, 15b reduced the latency to target (9.29 ± 0.31 s), but 15j exhibited a comparable activity (12.85 ± 0.72 s), indicating that they considerably ameliorated the cognitive impairment of the treated mice.
Table 2.
Effects of oral administration of 15b and 15j (15 mg kg−1) on scopolamine-induced memory impairment in ICR mice evaluated by the Morris water maze test. Tacrine (15 mg kg−1) was used as positive control. Data are presented as the mean ± SEM (n = 6).
| Group | Latency to target (s) | Distance to target (cm) |
|---|---|---|
| Control | 8.34 ± 0.53 | 216.23 ± 13.62 |
| Model | 25.83 ± 0.75 | 657.70 ± 27.28 |
| Tacrine | 12.06 ± 0.37 | 310.01 ± 9.54 |
| 15b | 9.29 ± 0.31 | 252.32 ± 8.42 |
| 15j | 12.85 ± 0.72 | 350.27 ± 16.59 |
Figure 5.

Effects of oral administration of tacrine (15 mg kg−1), 15b (15 mg kg−1), and 15j (15 mg kg−1) on scopolamine-induced cognitive impairment in ICR mice determined by the Morris water maze test. (A) The latency to target; (B) the distance to target; and (C) the trajectories of mice. Data are presented as the mean ± SEM (n = 6; ***p < 0.001 vs. scopolamine group).
The distance to target (Table 2; Figure 5(C)) and the trajectories of the mice in each group were also analysed. Compared to the control group, the administration of scopolamine remarkably led to the extended distance to target (657.70 ± 27.28 cm vs. 216.23 ± 13.62 cm). When treated with tacrine, 15b and 15j the distance to target were significantly shortened. These results were supported by trajectory analysis. As shown in Figure 5(C), the trajectory of the mice in scopolamine model group was very long and disordered, while tacrine, 15b and 15j groups showed shortened distances. Mice treated with 15b and 15j almost recovered to the normal cognition, with a similar orientation and distance to that of the normal mice. Taken together, these results supported that 15b and 15j remarkably ameliorated the cognition impairment caused by scopolamine.
2.2.9. Hepatotoxicity studies
We next investigated the possible drug-induced hepatotoxicity by comparing their toxic profile to tacrine in the in vivo assays. The alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured, as shown in Table 3 and Figure 6. Heparinised serum was obtained after the treatment of 15b and 15j at 8, 22, and 36 h, respectively. The serum levels of ALT and AST are directly proportional to the severity of the liver damage. Compared to the control group, after the treatment of 15b and 15j, the levels of ALT and AST were slightly induced at 22 h, but in general, no remarkable damage was observed. The level of ALT and AST slightly reduced at 36 h, compared to those of tacrine group, which suggested that 15b and 15j had preliminary safety.
Table 3.
ALT and AST activity after the administration of 15b and 15j. Tacrine (30 mg kg−1) was used as the reference compound. Values are expressed as the mean ± SEM of six independent experiments.
| ALT(U/L) |
|||
|---|---|---|---|
| Group | 8 h | 22 h | 36 h |
| Control | 25.4 ± 0.7 | 27.0 ± 1.2 | 34.3 ± 0.9 |
| Model | 34.4 ± 1.2 | 32.6 ± 1.9 | 33.7 ± 1.2 |
| Tacrine | 32.7 ± 1.7 | 31.7 ± 1.2 | 35.8 ± 6.7 |
| 15b | 29.1 ± 0.4 | 32.1 ± 1.0 | 30.2 ± 1.1 |
| 15j | 28.1 ± 1.1 | 32.2 ± 5.3 | 27.3 ± 3.9 |
| AST(U/L) |
|||
| Group |
8 h |
22 h |
36 h |
| Control | 86.4 ± 13.3 | 83.4 ± 9.8 | 98.7 ± 7.4 |
| Model | 91.9 ± 5.0 | 85.6 ± 6.8 | 92.1 ± 13.7 |
| Tacrine | 95.1 ± 9.5 | 108.9 ± 6.1 | 110.4 ± 9.4 |
| 15b | 92.1 ± 8.3 | 88.4 ± 11.2 | 101.0 ± 10.1 |
| 15j | 96.5 ± 6.8 | 88.6 ± 4.9 | 88.4 ± 6.3 |
Figure 6.

The ALT and AST levels. (A) The ALT levels of five subgroups. (B) The AST levels of five subgroups. Data are presented as the mean ± SEM (n = 6; ns p > 0.05).
To further analyse, the hepatotoxicity of 15b and 15j, morphologic studies by immunohistochemical staining were applied. Treatment of tacrine (Figure 7(B)), 15b (Figure 7(C)) or 15j (Figure 7(D)) did not result in remarkable morphologic changes in liver compared to the control group (Figure 7(A)). In summary, the results indicated that 15b and 15j had a low hepatotoxicity profile.
Figure 7.

Histopathological study of livers of male mice after treatment with the solvent only (control, A), or 36 h after administration of tacrine (B), 15b (C), or 15j (D). HE staining, original magnification ×200.
3. Conclusion
In summary, in the present studies, a series of benzylpiperidine-linked 1,3-dimethylbenzimidazolinone derivatives were designed and synthesised for the treatment of AD. In vitro assays proved that most of the compounds effectively inhibited ChEs in the micromolar range. 15b and 15j showed the most potent activity (15b, anti-AChE IC50 = 0.39 ± 0.11 µM, anti-BChE IC50 = 0.66 ± 0.16 µM; 15j, anti-AChE IC50 = 0.39 ± 0.15 µM, anti-BChE IC50 = 0.16 ± 0.04 µM). Kinetic studies indicated the competitive inhibition manner of 15b and 15j on AChE and BChE. Cell toxicity assay using HepG2 cells showed compounds 15b and 15j had a lower hepatotoxicity profile with respect to donepezil. In addition, no significant increases in ALT and AST levels were observed after the administration of 15b and 15j in mice. Furthermore, administration of 15b and 15j considerably ameliorated the cognition impairment in the scopolamine treated ICR mice in Morris water maze test. 15b exhibited cytoprotective effects against H2O2 induced cell damage. 15b and 15j also showed antioxidant activity according to the DPPH assay. Finally, 15b and 15j can be a promising lead compound for further development of newer anti-AD drugs.
4. Experimental section
4.1. Chemistry
All the chemicals were purchased from commercial companies and used without purification. All reactions were monitored by analytical thin layer chromatography (TLC) on silica gel 60 F254 precoated plates (purchased from Qingdao Haiyang Inc., Qingdao, China). Visualisation was achieved using UV light (254 nm and 365 nm), Flash column chromatography was performed with silica gel (200–300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. Melting points were determined using a Mel - TEMP II melting point apparatus. 1H NMR spectra are recorded on Bruker AV-500 or AV-300 MHz instruments in CDCl3 or DMSO-d6. Chemical shifts are reported in parts per million (d) downfield from the signal of tetramethylsilane (TMS) as the internal standard. Coupling constants are reported in Hz. All chemical shifts are reported in parts per million (ppm), relative to the internal standard. The following abbreviations are used to indicate the multiplicities of the respective signals: s – singlet; bs – broad singlet; d – doublet; dd – doublet of doublets; t – triplet; and m – multiplet. High-resolution mass spectrometry (HRMS) was performed on a Mariner Mass Spectrum (ESI) or an LC/MSD TOF HR-MS Spectrum.
4.1.1. General procedure for the synthesis of intermediates 4a–4p
To a solution of 4-piperidinecarboxamide (1) (2.0 g, 15.6 mmol) in methanol, potassium carbonate (4.3 g, 31.2 mmol) and substituted benzyl bromide (2a–2p, 1.5 equiv.) were added, and the reaction mixture was refluxed for 6–8 h. Solvent was distilled off, and addition of crushed ice to the residue led to the formation of white Coloured precipitates of 1-(substituted benzyl)-piperidine-4-carboxamides (3a–3p)5. Then, to a suspension of 3a–3p (1.0 g, 4.5 mmol) in dry THF (20 ml) was added slowly to a solution of LiAlH4 (0.8 g, 22.9 mmol, 5 equiv.) in dry THF (30 ml). The mixture was stirred under reflux and argon atmosphere for 4 h. After cooling, water was added at 0 °C, the precipitate was filtered and washed with Et2O. The filtrate was extracted with Et2O (×2), dried over MgSO4 and the solvent was evaporated under vacuum to afford compounds 4a–4p as a white solid23.
4.1.2. General procedure for the synthesis of intermediate 6
To a solution of methyl 3,4-diaminobenzoate (5) (1.0 g, 0.6 mmol, 1.0 equiv.) in chloroform (12 ml), di(1H-imidazol-1-yl)methanone (1.4 g, 9.0 mmol, 1.5 equiv.) was added. The mixture was heated to 60 °C for 12 h under a nitrogen atmosphere. After cooling the reaction to room temperature, the white precipitate was collected, washed with chloroform (8 ml × 2), and dried in vacuo to give intermediate 6 (0.8 g, 68%) as a white solid24. 1H NMR (300 MHz, DMSO-d6): δ 11.03 (s, 1H), 10.87 (s, 1H), 7.63 (dd, J1 = 8.2, J2 = 1.6 Hz, 1H), 7.47 (s, 1H), 7.02 (d, J = 8.2 Hz, 1H), 3.82 (s, 3H).
4.1.3. General procedure for the synthesis of intermediate 7
To a stirred solution of intermediate 6 (1.0 g, 5.2 mmol, 1.0 equiv.) in DMF (10 ml) at 0 °C, NaH (60% dispersion in mineral oil, 1.8 g, 13.0 mmol, 2.5 equiv.) was added, and the mixture was stirred for 15 min. Methyl iodide (1.8 g, 13.0 mmol, 2.5 equiv.) was added dropwise, and the mixture was stirred at room temperature for an additional 12 h. Water (30 ml) was added and extracted with EtOAc (30 ml × 3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel chromatography (petroleum ether/EtOAc = 8:1) to give intermediate 7 (0.7 g, 60%) as a white solid25. 1H NMR (300 MHz, DMSO-d6): δ 7.74 (dd, J1 = 8.2, J2 = 1.6 Hz, 1H), 7.66 (s, 1H), 7.23 (d, J = 8.2 Hz, 1H), 3.85 (s, 3H), 3.36 (d, J = 3.6 Hz, 6H).
4.1.4. General procedure for the synthesis of intermediate 8
To a solution of the intermediate 7 (1.0 g, 4.5 mmol, 1.0 equiv.) in THF (10 ml)/H2O (10 ml), LiOH (0.5 g, 22.7 mmol, 5.0 equiv.) was added, and the mixture was stirred at 50 °C for 2 h, followed by evaporation of THF under reduced pressure. To the residue solution, 10 ml H2O was added, and then 6 N hydrochloric acid was utilised to adjust the pH to 2.0–3.0. The resulting precipitate was collected by filtration, washed with water and dried to afford the intermediate 8 as white solid (0.85 g, 94%)26. 1H NMR (300 MHz, DMSO-d6): δ 12.69 (s, 1H), 7.73 (dd, J1 = 8.2, J2 = 1.5 Hz, 1H), 7.65 (d, J = 1.3 Hz, 1H), 7.20 (d, J = 8.2 Hz, 1H), 3.36 (d, J = 3.6 Hz, 6H).
4.1.5. General procedure for the synthesis of target compounds 9a–9p
Compound 8 (0.2 g, 0.9 mmol, 1.0 equiv.) and PyBOP (0.6 g, 1.2 mmol, 1.3 equiv.) and DIPEA (0.2 g, 1.9 mmol, 2.0 equiv.) were added to 10 ml of DMF and stirred at room temperature for 20 min. Then, intermediates 4a–4p (1.2 equiv.) was added and stirred at room temperature for 4 h. After completion of the reaction, the reaction mixture was quenched with water. The aqueous phase was extracted with CH2Cl2 (3 × 15 ml). The CH2Cl2 layer was then washed with brine and dried over anhydrous Na2SO4. After concentration, the crude product was purified by silica gel column chromatograph (CH2Cl2/methanol = 80:1) to give target compounds 9a–9p.
4.1.5.1. N-((1-benzylpiperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9a)
White solid, yield: 66%, m.p. 121–123 °C. 1H NMR (300 MHz, CDCl3): δ 7.52 (d, J = 1.4 Hz, 1H), 7.47 (dd, J1 = 8.1, J2 = 1.6 Hz, 1H), 7.33 (d, J = 4.5 Hz, 4H), 6.97 (d, J = 8.1 Hz, 1H), 6.26 (s, 1H), 3.52 (s, 2H), 3.46 (d, J = 2.4 Hz, 6H), 3.40 (t, J = 6.1 Hz, 2H), 2.94 (d, J = 11.7 Hz, 2H), 2.00 (dd, J1 = 11.7, J2 = 9.6 Hz, 2H), 1.75 (s, 4H), 1.42 (td, J = 12.1, 3.5 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.68, 154.57, 132.41, 129.85, 128.88, 127.87, 121.16), 107.34, 107.08, 52.80, 44.84, 27.58, 27.48. HRMS (ESI): calcd. for C23H28N4O2 [M + H]+ 393.2290, found 393.2283.
4.1.5.2. 1,3-dimethyl-N-((1–(2-methylbenzyl)piperidin-4-yl)methyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9b)
White solid; yield: 58%, m.p. 131–133 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.92 (s, 1H), 8.49 (d, J = 6.0 Hz, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 7.2 Hz, 1H), 7.33 (dd, J1 = 11.6, J2 = 6.6 Hz, 2H), 7.21 (d, J = 8.1 Hz, 1H), 4.30 (d, J = 5.2 Hz, 2H), 3.36 (d, J = 1.8 Hz, 6H), 3.21 (s, 2H), 3.09 (s, 2H), 2.40 (s, 3H), 1.89 (d, J = 12.8 Hz, 3H), 1.44 (d, J = 12.9 Hz, 2H), 1.23 (d, J = 1.7 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.78, 154.56, 138.98, 132.37, 131.36, 130.08, 129.86, 128.84, 127.74, 126.67, 121.18, 107.19, 57.25, 52.30, 46.32, 44.35, 34.26, 27.51, 26.37, 19.78. HRMS (ESI): calcd. for C24H30N4O2 [M + H]+ 407.2447, found 407.2441.
4.1.5.3. 1,3-dimethyl-N-((1–(3-methylbenzyl)piperidin-4-yl)methyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9c)
White solid, yield: 78%, m.p. 109–110 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.11 (s, 1H), 8.45 (t, J = 5.7 Hz, 1H), 7.65 (d, J = 7.6 Hz, 2H), 7.36 (d, J = 7.3 Hz, 1H), 7.29 (d, J = 5.4 Hz, 2H), 7.22 (d, J = 8.8 Hz, 1H), 4.24 (d, J = 5.0 Hz, 2H), 3.37 (d, J = 1.8 Hz, 6H), 3.21 (d, J = 6.1 Hz, 2H), 2.92 (d, J = 12.1 Hz, 2H), 2.35 (s, 3H), 1.88 (d, J = 14.1 Hz, 3H), 1.74 (s, 2H), 1.40 (d, J = 11.8 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.80, 154.56, 138.60, 132.48, 132.19, 130.64, 129.86, 129.21, 128.77, 127.73, 121.17, 107.20, 59.83, 52.04, 46.34, 44.31, 34.26, 27.52, 21.36. HRMS (ESI): calcd. for C24H30N4O2 [M + H]+ 407.2447, found 407.2440.
4.1.5.4. 1,3-dimethyl-N-((1–(4-methylbenzyl)piperidin-4-yl)methyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9d)
White solid, yield: 85%, m.p. 151–153 °C. 1H NMR (300 MHz, CDCl3): δ 7.52 (d, J = 1.4 Hz, 1H), 7.48 (dd, J1 = 8.1, J2 = 1.5 Hz, 1H), 7.21 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 8.1 Hz, 2H), 6.95 (d, J = 8.1 Hz, 1H), 6.38 (s, 1H), 3.54 (s, 2H), 3.45 (s, 6H), 3.38 (t, J = 6.1 Hz, 2H), 2.98 (d, J = 11.6 Hz, 2H), 2.35 (s, 3H), 2.02 (dd, J1 = 19.6, J2 = 7.4 Hz, 3H), 1.76 (d, J = 12.7 Hz, 2H), 1.45 (td, J = 12.2, 3.3 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.59, 154.55, 136.31, 135.64, 132.33, 129.81, 129.23, 129.13, 127.95, 121.17, 107.19, 62.51, 53.31, 45.31, 36.21, 30.20, 27.50, 21.15. HRMS (ESI): calcd. for C24H30N4O2 [M + H]+ 407.2447, found 407.2454.
4.1.5.5. N-((1–(3,4-dimethylbenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9e)
White solid, yield: 64%, m.p. 164–165 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.07 (s, 1H), 8.45 (d, J = 5.9 Hz, 1H), 7.65 (d, J = 7.4 Hz, 1H), 7.28 – 7.13 (m, 4H), 4.19 (d, J = 5.0 Hz, 2H), 3.36 (d, J = 1.8 Hz, 6H), 3.23 – 3.15 (m, 2H), 2.90 (d, J = 12.4 Hz, 2H), 2.25 (s, 6H), 1.88 (d, J = 13.8 Hz, 3H), 1.40 (d, J = 12.3 Hz, 2H), 1.21 (d, J = 18.4 Hz, 3H). 13 C NMR (126 MHz, DMSO-d6): δ 166.79, 154.56, 138.34, 137.21, 132.63, 132.48, 130.31, 129.87, 129.14, 127.73, 127.42, 121.16, 107.36, 107.06, 59.64, 51.92, 44.32, 34.27, 27.59, 27.48, 27.33, 19.83, 19.62. HRMS (ESI): calcd. for C25H32N4O2 [M + H]+ 421.2603, found 421.2607.
4.1.5.6. N-((1–(3-methoxybenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9f)
White solid, yield: 56%, m.p. 178–179 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.12 (s, 1H), 8.45 (t, J = 6.0 Hz, 1H), 7.65 (d, J = 7.6 Hz, 2H), 7.40 (t, J = 7.8 Hz, 1H), 7.22 (d, J = 8.7 Hz, 1H), 7.09 – 7.02 (m, 2H), 4.25 (d, J = 4.9 Hz, 2H), 3.80 (s, 3H), 3.37 (d, J = 1.9 Hz, 6H), 3.21 (t, J = 6.0 Hz, 2H), 2.93 (d, J = 12.4 Hz, 2H), 1.89 (d, J = 13.8 Hz, 2H), 1.39 (t, J = 12.4 Hz, 2H), 1.26 (d, J = 10.7 Hz, 3H). 13 C NMR (126 MHz, DMSO-d6): δ 166.80, 159.85, 154.57, 132.49, 130.48, 129.88, 127.73, 123.66, 121.17, 117.20, 115.38, 107.36, 107.06, 55.67, 52.11, 46.34, 34.28, 32.00, 29.89, 27.54. HRMS (ESI): calcd. for C24H30N4O2 [M + H]+ 423.2396, found 423.2394.
4.1.5.7. N-((1–(4-methoxybenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9g)
White solid, yield: 67%, m.p. 111–113 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.03 (s, 1H), 8.45 (d, J = 5.7 Hz, 1H), 7.64 (s, 1H), 7.41 (d, J = 8.6 Hz, 2H), 7.21 (d, J = 8.8 Hz, 1H), 7.03 (d, J = 8.6 Hz, 2H), 4.21 (d, J = 4.6 Hz, 2H), 3.79 (s, 3H), 3.36 (d, J = 1.8 Hz, 6H), 3.20 (t, J = 5.9 Hz, 2H), 2.89 (d, J = 11.6 Hz, 2H), 1.85 (t, J = 20.0 Hz, 4H), 1.38 (d, J = 10.9 Hz, 2H), 1.20 (dd, J1 = 15.9, J2 = 8.6 Hz, 1H). 13 C NMR (126 MHz, DMSO-d6): δ 166.79, 160.57, 154.56, 133.22, 132.48, 129.87, 127.73, 121.16, 114.65, 107.35, 107.06, 59.29, 55.69, 51.71, 44.31, 34.30, 27.53, 27.36. HRMS (ESI): calcd. for C24H30N4O2 [M + H]+ 423.2396, found 423.2395.
4.1.5.8. N-((1–(2-fluorobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9h)
White solid, yield: 78%, m.p. 122–123 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.36 (s, 1H), 7.83 – 7.65 (m, 2H), 7.38 (d, J = 25.7 Hz, 2H), 7.24 (t, J = 23.5 Hz, 3H), 3.52 (s, 2H), 3.36 (d, J = 3.1 Hz, 6H), 3.16 (d, J = 5.6 Hz, 2H), 2.86 (s, 2H), 1.96 (s, 2H), 1.67 (s, 3H), 1.24 (d, J = 4.4 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.64, 162.26, 160.32, 154.56, 132.37, 129.84, 127.92, 124.73, 121.16, 115.78, 115.60, 107.31, 107.08, 53.01, 45.05, 35.70, 29.77, 27.56, 27.47. HRMS (ESI): calcd. for C23H27FN4O2 [M + H]+ 411.2196, found 411.2191.
4.1.5.9. N-((1–(3-fluorobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9i)
White solid, yield: 78%, m.p. 128–130 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.36 (t, J = 5.6 Hz, 1H), 7.66 (d, J = 4.9 Hz, 2H), 7.35 (dd, J1 = 14.3, J2 = 7.8 Hz, 1H), 7.20 (d, J = 8.6 Hz, 1H), 7.10 (dd, J1 = 17.3, J2 = 8.1 Hz, 3H), 3.46 (s, 2H), 3.36 (d, J = 3.4 Hz, 6H), 3.17 (t, J = 6.0 Hz, 2H), 2.79 (d, J = 11.2 Hz, 2H), 1.91 (t, J = 10.9 Hz, 2H), 1.63 (t, J = 16.9 Hz, 3H), 1.21 (d, J = 11.6 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.58, 163.65, 161.72, 154.55, 142.47, 132.34, 130.45, 129.82, 127.96, 124.99, 121.15, 115.51, 114.04, 107.30, 107.09, 62.13, 53.43, 45.33, 36.23, 30.36, 27.55, 27.46. HRMS (ESI): calcd. for C23H27FN4O2 [M + H]+ 411.2196, found 411.2190.
4.1.5.10. N-((1–(4-fluorobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9j)
White solid, yield: 82%, m.p. 165–167 °C. 1H NMR (300 MHz, CDCl3): δ 7.52 (s, 1H), 7.47 (dd, J1 = 8.2, J2 = 1.5 Hz, 1H), 7.26 (s, 1H), 6.99 (dd, J = 16.1, 8.3 Hz,: 4H), 6.27 (s, 1H), 3.47 (s, 2H), 3.46 (d, J = 1.9 Hz, 6H), 3.39 (t, J = 6.2 Hz, 2H), 2.88 (s, 2H), 1.97 (t, J = 10.6 Hz, 2H), 1.85 – 1.71 (m, 3H), 1.46 – 1.32 (m, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.57, 162.57, 160.64, 154.56, 135.32, 132.35, 130.94, 130.88, 129.83, 127.97, 121.16, 115.33, 115.16, 107.31, 107.10, 61.96, 53.36, 45.34, 36.28, 30.35, 27.57, 27.47. HRMS (ESI): calcd. for C23H27FN4O2 [M + H]+ 411.2196, found 411.2190.
4.1.5.11. N-((1–(2-chlorobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9k)
White solid, yield: 90%, m.p. 156–158 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.17 (s, 1H), 8.46 (s, 1H), 7.68 (d, J = 2.5 Hz, 1H), 7.64 (s, 1H), 7.60 (s, 1H), 7.55–7.48 (m, 2H), 7.22 (d, J = 8.6 Hz, 1H), 4.43 (d, J = 5.0 Hz, 2H), 3.44 (d, J = 11.8 Hz, 2H), 3.37 (d, J = 2.0 Hz, 6H), 3.21 (s, 2H), 1.89 (d, J = 12.6 Hz, 3H), 1.45 (d, J = 11.5 Hz, 2H), 1.21 (d, J = 17.9 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.78, 154.57, 135.30, 134.22, 132.49, 132.18, 130.43, 129.88, 128.21, 127.72, 121.18, 107.37, 107.06, 56.86, 52.54, 46.38, 33.99, 27.61, 27.50, 26.40, 26.34. HRMS (ESI): calcd. for C23H27ClN4O2 [M + H]+ 427.1901, found 427.1892.
4.1.5.12. N-((1–(3-chlorobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9l)
White solid, yield: 76%, m.p. 134–136 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.36 (t, J = 5.7 Hz, 1H), 7.65 (dd, J1 = 4.2, J2 = 2.7 Hz, 2H), 7.36 – 7.32 (m, 2H), 7.31 (s, 1H), 7.27 (s, 1H), 7.20 (d, J = 8.6 Hz, 1H), 3.45 (s, 2H), 3.37 (s, 3H), 3.35 (s, 3H), 3.17 (t, J = 6.2 Hz, 2H), 2.78 (d, J = 11.3 Hz, 2H), 1.91 (t, J = 10.6 Hz, 2H), 1.63 (t, J = 17.1 Hz, 3H), 1.27 – 1.16 (m, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.57, 154.56, 141.99, 133.33, 132.34, 130.46, 129.82, 128.69, 127.94, 127.72, 127.20, 121.16, 107.34, 107.12, 62.02, 53.41, 45.32, 36.21, 30.35, 27.58, 27.48. HRMS (ESI): calcd. for C23H27ClN4O2 [M + H]+ 427.1901, found 427.1899.
4.1.5.13. N-((1–(4-chlorobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9m)
White solid, yield: 75%, m.p. 127–129 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.36 (t, J = 5.6 Hz, 1H), 7.65 (dd, J1 = 4.3, J2 = 2.6 Hz, 1H), 7.37 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.6 Hz, 2H), 7.20 (d, J = 8.6 Hz, 1H), 3.42 (s, 2H), 3.37 (s, 3H), 3.35 (s, 3H), 3.17 (t, J = 6.2 Hz, 2H), 2.78 (d, J = 11.2 Hz, 2H), 1.90 (t, J = 10.5 Hz, 2H), 1.65 (d, J = 12.7 Hz, 3H), 1.22 (dd, J1 = 14.9, J2 = 8.5 Hz, 3H). 13 C NMR (126 MHz, DMSO-d6): δ 166.57, 154.56, 138.30, 132.35, 131.69, 130.87, 129.83, 128.52, 127.96, 121.16, 107.21, 61.94, 53.40, 45.33, 36.25, 30.36, 27.53. HRMS (ESI): calcd. for C23H27ClN4O2 [M + H]+ 427.1901, found 427.1894.
4.1.5.14. N-((1–(2-bromobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9n)
White solid, yield: 75%, m.p. 137–139 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.37 (t, J = 5.5 Hz, 1H), 7.66 (dd, J1 = 4.2, J2 = 2.7 Hz, 2H), 7.58 (d, J = 8.0 Hz, 1H), 7.50 – 7.45 (m, 1H), 7.37 (t, J = 7.4 Hz, 1H), 7.19 (dd, J1 = 8.1, J2 = 4.8 Hz, 2H), 3.51 (s, 2H), 3.37 (s, 3H), 3.36 (s, 3H), 3.18 (t, J = 6.2 Hz, 2H), 2.83 (d, J = 11.0 Hz, 2H), 2.02 (t, J = 10.7 Hz, 2H), 1.65 (t, J = 14.9 Hz, 3H), 1.22 (d, J = 9.5 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.58, 154.57, 138.16, 132.89, 132.35, 131.11, 129.83, 129.18, 128.00, 127.94, 124.28, 121.17, 107.36, 107.13, 61.95, 53.60, 45.31, 36.20, 30.39, 27.59, 27.49. HRMS (ESI): calcd. for C23H27BrN4O2 [M + H]+ 471.1395, found 471.1384.
4.1.5.15. N-((1–(3-bromobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9o)
White solid, yield: 78%, m.p. 143–145 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.36 (t, J = 5.6 Hz, 1H), 7.65 (dd, J1 = 4.2, J2 = 2.7 Hz, 2H), 7.48 (s, 1H), 7.42 (s, 1H), 7.29 (d, J = 6.1 Hz, 2H), 7.20 (d, J = 8.6 Hz, 1H), 3.44 (s, 2H), 3.37 (s, 3H), 3.35 (s, 3H), 3.17 (t, J = 6.1 Hz, 2H), 2.78 (d, J = 11.2 Hz, 2H), 1.91 (t, J = 10.7 Hz, 2H), 1.63 (t, J = 17.2 Hz, 3H), 1.23 (dd, J1 = 14.0, J2 = 9.1 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.57, 154.57, 142.28, 132.35, 131.59, 130.77, 130.10, 129.84, 128.11, 127.97, 122.01, 121.16, 107.33, 107.11, 61.98, 53.40, 45.32, 36.21, 30.35, 27.58, 27.48. HRMS (ESI): calcd. for C23H27BrN4O2 [M + H]+ 471.1395, found 471.1386.
4.1.5.16. N-((1–(4-bromobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-carboxamide (9p)
White solid, yield: 69%, m.p. 165–167 °C. 1H NMR (300 MHz, CDCl3): δ 7.50 (d, J = 9.3 Hz, 1H), 7.46–7.40 (m, 2H), 7.32 (d, J = 4.4 Hz, 2H), 7.20 (d, J = 8.3 Hz, 1H), 6.96 (d, J = 8.1 Hz, 1H), 6.30 (s, 1H), 3.52 (s, 2H), 3.45 (d, J = 1.6 Hz, 6H), 3.39 (t, J = 6.2 Hz, 2H), 2.98–2.85 (m, 2H), 2.04– 1.87 (m, 3H), 1.75 (d, J = 13.0 Hz, 2H), 1.38 (dd, J1 = 13.6, J2 = 9.4 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.57, 154.56, 138.71, 132.34, 131.44, 131.26, 129.83, 129.15, 128.55, 127.97, 127.22, 121.16, 120.19, 107.31, 107.10, 61.99, 53.46, 53.40, 45.33, 36.28, 36.24, 30.35, 27.57, 27.47. HRMS (ESI): calcd. for C23H27BrN4O2 [M + H]+ 471.1395, found 471.1384.
4.1.6. General procedure for the synthesis of target compounds 11a–11f
Intermediates 10a–10f (0.3 g, 1.0 equiv.) and PyBOP (1.3 equiv.) and DIPEA(2.0 equiv.) were added to 6 ml of DMF and stirred at room temperature for 20 min. Then, intermediates 4a, 4g, 4m (1.0 equiv.) were added and stirred at room temperature for 4 h. After completion of the reaction, the reaction mixture was quenched with water. The aqueous phase was extracted with CH2Cl2 (3 × 15 ml). The CH2Cl2 layer was then washed with brine and dried over anhydrous Na2SO4. After concentration, the crude product was purified by silica gel column chromatograph (CH2Cl2/methanol = 80:1) to give target compounds 11a–11f.
4.1.6.1. N-((1–(4-methoxybenzyl)piperidin-4-yl)methyl)-1H-indole-5-carboxamide (11a)
White solid, yield: 67%, m.p. 89–90 °C. 1H NMR (500 MHz, DMSO-d6): δ 8.34 (t, J = 5.4 Hz, 1H), 8.12 (s, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.41 (s, 2H), 7.34 (d, J = 7.5 Hz, 3H), 7.28 (d, J = 6.7 Hz, 1H), 6.51 (s, 1H), 3.63 (s, 3H), 3.53–3.46 (m, 2H), 3.17 (s, 2H), 2.91 (d, J = 10.0 Hz, 2H), 2.14 (s, 2H), 1.70 (d, J = 13.2 Hz, 2H), 1.64 (s, 1H), 1.29 (d, J = 11.7 Hz, 3H). 13 C NMR (126 MHz, DMSO-d6): δ 167.87, 137.77, 129.84, 128.71, 127.89, 127.41, 127.02, 126.11, 120.99, 120.25, 114.19, 111.25, 102.47, 61.96, 55.54, 52.90, 44.95, 35.69, 29.44. HRMS (ESI): calcd. for C23H27N3O2 [M + H]+ 378.2181, found 378.2179.
4.1.6.2. N-((1–(4-methoxybenzyl)piperidin-4-yl)methyl)-1H-indazole-5-carboxamide (11b)
White solid, yield: 65%, m.p. 78–79 °C. 1H NMR (500 MHz, DMSO-d6): δ 13.33 (s, 1H), 8.53 (s, 1H), 8.34 (s, 1H), 8.19 (s, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.57 (d, J = 8.7 Hz, 1H), 7.34 (d, J = 7.9 Hz, 2H), 6.92 (d, J = 8.3 Hz, 2H), 3.75 (s, 5H), 3.19 (s, 2H), 3.00 (d, J = 9.1 Hz, 2H), 2.31 (s, 2H), 1.74 (d, J = 13.0 Hz, 2H), 1.69 (s, 1H), 1.37 (d, J = 11.1 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 167.10, 159.41, 135.14, 131.68, 127.55, 125.75, 122.76, 120.93, 114.19, 55.54, 52.24, 44.76, 27.05. HRMS (ESI): calcd. for C22H26N4O2 [M + H]+ 379.2134, found 379.2124.
4.1.6.3. N-((1–(2-chlorobenzyl)piperidin-4-yl)methyl)-1H-indazole-5-carboxamide (11c)
White solid, yield: 78%, m.p. 89–90 °C. 1H NMR (500 MHz, DMSO-d6):δ 13.27 (s, 1H), 8.34 (s, 1H), 8.20 (s, 1H), 7.86 (d, J = 8.6 Hz, 1H), 7.57 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 7.2 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.32 (s, 1H), 7.27 (d, J = 7.4 Hz, 1H), 7.03 (s, 1H), 3.53 (s, 2H), 3.19 (d, J = 5.7 Hz, 2H), 2.83 (d, J = 10.8 Hz, 2H), 2.00 (t, J = 11.0 Hz, 2H), 1.68 (d, J = 12.1 Hz, 2H), 1.60 (s, 1H), 1.22 (d, J = 9.8 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 167.06, 136.55, 133.63, 131.07, 129.62, 128.85, 127.68, 127.41, 125.80, 122.80, 120.89, 110.11, 59.40, 53.61, 45.32, 36.16, 30.38. HRMS (ESI): calcd. for C21H23ClN4O [M + H]+ 383.1638, found 383.1629.
4.1.6.4. N-((1–(4-methoxybenzyl)piperidin-4-yl)methyl)-1H-benzo[d]imidazole-5-carboxamide (11d)
White solid, yield: 76%, m.p. 92–93 °C. 1H NMR (500 MHz, DMSO-d6): δ 8.84 (s, 1H), 8.48 (s, 1H), 7.93 (d, J = 5.5 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 6.91 (d, J = 8.1 Hz, 2H), 3.80 (s, 3H), 3.73 (s, 2H), 3.20 (s, 2H), 3.03 (d, J = 10.6 Hz, 2H), 2.40 (s, 2H), 1.74 (s, 3H), 1.41 (d, J = 11.2 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6):δ 166.67, 159.53, 140.23, 139.32, 132.00, 131.78, 129.35, 127.39, 126.11, 125.43, 115.74, 114.34, 114.20, 60.23, 55.54, 51.93, 44.74, 34.93, 28.29. HRMS (ESI): calcd. for C22H26N4O2 [M + H]+ 379.2134, found 379.2135.
4.1.6.5. N-((1–(2-chlorobenzyl)piperidin-4-yl)methyl)-1H-benzo[d]imidazole-5-carboxamide(11e)
White solid, yield: 45%, m.p 95–97 °C. 1H NMR (500 MHz, DMSO-d6): δ 8.51 (t, J = 5.4 Hz, 1H), 8.35 (s, 1H), 8.20 (s, 1H), 7.79 (d, J = 8.3 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.46 (d, J = 7.3 Hz, 1H), 7.38 (d, J = 7.8 Hz, 1H), 7.28 (d, J = 7.4 Hz, 1H), 7.24 (d, J = 7.5 Hz, 1H), 3.50 (s, 2H), 3.20 (d, J = 5.9 Hz, 2H), 2.80 (d, J = 11.2 Hz, 2H), 1.97 (d, J = 10.7 Hz, 2H), 1.67 (d, J = 13.0 Hz, 2H), 1.61–1.54 (m, 1H), 1.22 (d, J = 9.6 Hz, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 167.33, 144.20, 136.52, 133.65, 131.04, 129.58, 129.17, 128.78, 128.51, 127.35, 121.95, 59.40, 53.59, 45.35, 36.17, 30.38. HRMS (ESI): calcd. for C21H23ClN4O [M + H]+ 383.1638, found 383.1626.
4.1.6.6. N-((1-benzylpiperidin-4-yl)methyl)-1H-benzo[d][1,2,3]triazole-5-carboxamide(11f)
White solid, yield: 89%, m.p. 101–103 °C. 1H NMR (500 MHz, DMSO-d6): δ 8.63 (t, J = 5.5 Hz, 1H), 8.44 (s, 1H), 7.87 (d, J = 2.7 Hz, 2H), 7.47 (d, J = 7.1 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 6.9 Hz, 1H), 7.28 – 7.23 (m, 1H), 7.03 (s, 1H), 3.52 (s, 2H), 3.21 (t, J = 6.1 Hz, 2H), 3.18 (s, 1H), 2.82 (d, J = 11.3 Hz, 2H), 2.00 (t, J = 10.4 Hz, 2H), 1.68 (d, J = 12.0 Hz, 2H), 1.61 (s, 1H), 1.27 – 1.14 (m, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 166.83, 141.15, 140.74, 136.52, 133.63, 131.07, 129.60, 128.83, 127.39, 124.43, 115.72, 114.53, 59.39, 53.58, 45.41, 36.12, 30.36. HRMS (ESI): calcd. for C20H23N5O [M + H]+ 350.1981, found 350.1980.
4.1.7. General procedure for the synthesis of intermediate 13
To a stirred solution of 1,3-dihydro-2H-benzo[d]imidazol-2-one (12) (2.0 g, 14.9 mmol, 1.0 equiv.) in DMF (20 ml) at 0 °C, NaH (60% dispersion in mineral oil, 1.5 g, 37.3 mmol, 2.5 equiv.) was added, and the mixture was stirred for 15 min. Methyl iodide (5.3 g, 37.3 mmol, 2.5 equiv.) was added dropwise, and the mixture was stirred at room temperature for an additional 12 h. Water (30 ml) was added and extracted with EtOAc (30 ml × 3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel chromatography (petroleum ether/EtOAc = 8:1) to give intermediate 13 (2.0 g, 83%) as a white solid24.
4.1.8. General procedure for the synthesis of intermediate 14
Chlorosulfonic acid (3.0 ml) was added slowly to intermediate 13 (1.0 g, 6.2 mmol) at room temperature, and the reaction mixture was stirred for 18 h. The resulting mixture was added to the ice with NaCl(s) carefully, filtered, and washed with ether to collect the desired product, which was used directly to the next step (1.4 g, 87%)26.
4.1.9. General procedure for the synthesis of target compounds 15a–15j
A solution of appropriate intermediate 14 (0.3 g 1.2 mmol, 1.0 equiv.) in dichloromethane (15 ml) was slowly added to a cooled (0 °C) solution of intermediates 4a–4d, 4f, 4i, 4l–4o (1.0 equiv.) in dichloromethane (10 ml) and triethylamine (2.0 equiv.). The resulting mixture was stirred at room temperature overnight. After concentration, the crude product was purified by silica gel column chromatograph (CH2Cl2/methanol = 50:1) to give target compounds 15a–15j27.
4.1.9.1. N-((1-benzylpiperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-sulphonamide (15a)
White solid, yield: 64%, m.p. 113–114 °C. 1H NMR (500 MHz, CDCl3): δ 7.65 (dd, J1 = 8.2, J2 = 1.7 Hz, 1H), 7.49 (d, J = 1.5 Hz, 1H), 7.34 – 7.29 (m, 5H), 7.07 (d, J = 8.2 Hz, 1H), 4.43 (t, J = 6.2 Hz, 1H), 3.49 (d, J = 2.3 Hz, 6H), 2.88 (d, J = 11.7 Hz, 2H), 2.83 (t, J = 6.7 Hz, 2H), 1.92 (t, J = 11.0 Hz, 2H), 1.64 (s, 6H), 1.47 (m, 1H). 13 C NMR (126 MHz, DMSO-d6): δ 154.54, 133.39, 133.02, 130.05, 129.19, 128.56, 127.26, 120.54, 107.96, 106.37, 62.79, 53.23, 48.56, 36.08, 29.94, 27.70, 27.64. HRMS (ESI): calcd. for C22H28N4O3S [M + H]+ 429.1960, found 429.1965.
4.1.9.2. 1,3-dimethyl-N-((1–(2-methylbenzyl)piperidin-4-yl)methyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-sulphonamide (15b)
White solid, yield: 73%, m.p. 68–70 °C. 1H NMR (500 MHz, CDCl3): δ 7.67 (s, 1H), 7.51 (s, 1H), 7.17 (d, J = 11.8 Hz, 3H), 7.08 (d, J = 6.8 Hz, 1H), 4.59 (s, 1H), 3.49 (s, 6H), 3.13 (d, J = 7.3 Hz, 2H), 2.92 (s, 2H), 2.84 (t, J = 6.5 Hz, 2H), 2.35 (s, 3H), 2.01 (s, 2H), 1.67 (s, 2H), 1.43 (t, J = 7.3 Hz, 2H), 1.27 (s, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 154.52, 137.65, 133.42, 133.01, 130.56, 130.05, 127.69, 125.88, 120.56, 107.90, 106.36, 60.25, 53.13, 48.38, 35.79, 29.50, 27.66, 27.61, 19.36. HRMS (ESI): calcd. for C23H30N4O3S [M + H]+ 443.2117, found 443.2111.
4.1.9.3. 1,3-dimethyl-N-((1–(3-methylbenzyl)piperidin-4-yl)methyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-sulphonamide (15c)
White solid, yield: 58%, m.p. 79–80 °C. 1H NMR (500 MHz, CDCl3): δ 7.65 (dd, J1 = 8.2, J2 = 1.7 Hz, 1H), 7.49 (d, J = 1.6 Hz, 1H), 7.21 (dd, J1 = 9.8, J2 = 5.2 Hz, 1H), 7.11 (s, 1H), 7.07 (t, J = 7.7 Hz, 3H), 4.45 (t, J = 6.4 Hz, 1H), 3.49 (d, J = 2.5 Hz, 6H), 3.44 (s, 2H), 2.87 (d, J = 11.6 Hz, 2H), 2.83 (t, J = 6.7 Hz, 2H), 2.35 (s, 3H), 1.90 (dd, J1 = 11.7, J2 = 9.8 Hz, 2H), 1.65 (s, 4H), 1.52–1.42 (m, 1H). 13 C NMR (126 MHz, DMSO-d6): δ 154.55, 137.55, 133.45, 133.03, 130.07, 129.78, 128.42, 127.87, 126.27, 120.54, 107.93, 106.36, 62.86, 53.29, 48.58, 36.11, 29.97, 27.69, 27.63, 21.46. HRMS (ESI): calcd. for C23H30N4O3S [M + H]+ 443.2117, found 443.2119.
4.1.9.4. 1,3-dimethyl-N-((1–(4-methylbenzyl)piperidin-4-yl)methyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-sulphonamide (15d)
White solid, yield: 77%, m.p. 150–152 °C. 1H NMR (500 MHz, CDCl3) δ 7.65 (dd, J1 = 8.2, J2 = 1.5 Hz, 1H), 7.49 (d, J = 1.2 Hz, 1H), 7.20 (d, J = 8.5 Hz, 1H), 7.17 (d, J = 8.2 Hz, 1H), 7.13 (s, 1H), 7.06 (d, J = 8.2 Hz, 1H), 6.85 (d, J = 8.5 Hz, 1H), 4.58 (d, J = 5.9 Hz, 1H), 3.81 (s, 2H), 3.48 (d, J = 4.4 Hz, 6H), 3.43 (d, J = 10.7 Hz, 2H), 2.85 (d, J = 11.5 Hz, 2H), 2.81 (d, J = 6.6 Hz, 2H), 1.88 (t, J = 11.4 Hz, 2H), 1.64 (d, J = 12.3 Hz, 2H), 1.50 – 1.40 (m, 1H), 1.27 (s, 3H). 13 C NMR (126 MHz, DMSO-d6): δ 158.62, 154.55, 133.42, 133.03, 130.37, 130.06, 129.11, 120.54, 113.92, 107.95, 106.37, 62.21, 55.43, 53.11, 48.58, 36.15, 29.98, 27.70, 27.64, 21.15. HRMS (ESI): calcd. for C23H30N4O3S [M + H]+ 443.2117, found 443.2111.
4.1.9.5. N-((1–(3-methoxybenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-sulphonamide (15e)
White solid, yield: 55%, m.p. 80–81 °C. 1H NMR (500 MHz, CDCl3): δ 7.66 (dd, J1 = 8.2, J2 = 1.7 Hz, 1H), 7.49 (d, J = 1.5 Hz, 1H), 7.23 (t, J = 7.9 Hz, 1H), 7.07 (d, J = 8.2 Hz, 1H), 6.90 – 6.85 (m, 2H), 6.83 – 6.78 (m, 1H), 4.47 (t, J = 6.4 Hz, 1H), 3.82 (s, 3H), 3.49 (d, J = 2.5 Hz, 6H), 3.46 (s, 2H), 2.87 (d, J = 11.6 Hz, 2H), 2.83 (t, J = 6.7 Hz, 2H), 1.91 (dd, J1 = 11.6, J2 = 9.9 Hz, 2H), 1.65 (d, J = 16.5 Hz, 4H), 1.50 – 1.43 (m, 1H). 13 C NMR (126 MHz, DMSO-d6): δ 159.66, 154.55, 140.79, 133.45, 133.03, 130.07, 129.55, 121.31, 120.54, 114.55, 112.60, 107.93, 106.36, 62.73, 55.37, 53.27, 48.59, 36.10, 29.99, 27.69, 27.63. HRMS (ESI): calcd. for C23H30N4O4S [M + H]+ 459.2066, found 459.2068.
4.1.9.6. N-((1–(3-fluorobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-sulphonamide (15f)
White solid, yield: 74%, m.p. 75–76 °C. 1H NMR (500 MHz, CDCl3): δ 7.66 (dd, J1 = 8.2, J2 = 1.6 Hz, 1H), 7.49 (d, J = 1.5 Hz, 1H), 7.25 (d, J = 7.9 Hz, 1H), 7.06 (dd, J1 = 15.3, J2 = 8.8 Hz, 3H), 6.97 – 6.90 (m, 1H), 4.41 (t, J = 6.4 Hz, 1H), 3.49 (d, J = 1.7 Hz, 6H), 3.47 (s, 2H), 2.86 (d, J = 6.8 Hz, 2H), 2.83 (d, J = 6.7 Hz, 2H), 1.93 (t, J = 10.7 Hz, 2H), 1.69 – 1.59 (m, 4H), 1.53 – 1.44 (m, 1H). 13 C NMR (126 MHz, DMSO-d6): δ 163.63, 161.70, 154.53, 142.36, 133.44, 133.02, 130.44, 130.37, 130.06, 124.99, 120.54, 115.51, 115.34, 114.05, 113.88, 107.91, 106.34, 62.02, 53.21, 48.56, 36.04, 29.97, 27.67, 27.61. HRMS (ESI): calcd. for C22H27FN4O3S [M + H]+ 447.1866, found 447.1869.
4.1.9.7. N-((1–(3-chlorobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-sulphonamide (15g)
White solid, yield: 70%, m.p. 73–75 °C. 1H NMR (500 MHz, CDCl3): δ 7.66 (dd, J1 = 8.2, J2 = 1.6 Hz, 1H), 7.49 (d, J = 1.4 Hz, 1H), 7.31 (s, 1H), 7.25 – 7.21 (m, 2H), 7.19 – 7.16 (m, 1H), 7.07 (d, J = 8.2 Hz, 1H), 4.45 (t, J = 6.5 Hz, 1H), 3.49 (d, J = 1.8 Hz, 6H), 3.44 (s, 2H), 2.85 (d, J = 3.3 Hz, 2H), 2.83 (d, J = 6.5 Hz, 2H), 1.92 (t, J = 10.7 Hz, 2H), 1.63 (s, 4H), 1.52 – 1.43 (m, 1H). 13 C NMR (126 MHz, DMSO-d6): δ 154.55, 141.91, 133.46, 133.33, 133.03, 130.42, 130.07, 128.68, 127.69, 127.19, 120.54, 107.92, 106.35, 61.93, 53.20, 48.55, 36.03, 29.97, 27.69, 27.63. HRMS (ESI): calcd. for C22H27ClN4O3S [M + H]+ 463.1570, found 463.1568.
4.1.9.8. N-((1–(4-chlorobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-sulphonamide (15h)
White solid, yield: 71%, m.p. 195–197 °C. 1H NMR (500 MHz, CDCl3): δ 7.66 (d, J = 7.5 Hz, 1H), 7.51 (s, 1H), 7.39 – 7.30 (m, 4H), 7.07 (d, J = 8.2 Hz, 1H), 4.55 (s, 1H), 3.49 (s, 6H), 2.88 (s, 2H), 2.83 (t, J = 6.7 Hz, 2H), 1.96 (s, 2H), 1.76 – 1.63 (m, 4H), 1.48 (d, J = 32.9 Hz, 2H), 1.43 (t, J = 7.3 Hz, 1H). 13 C NMR (126 MHz, DMSO-d6): δ 154.55, 133.44, 133.04, 130.98, 130.07, 128.58, 120.54, 107.94, 106.36, 61.20, 53.08, 48.48, 46.03, 35.94, 29.67, 27.70, 27.64. HRMS (ESI): calcd. for C22H27ClN4O3S [M + H]+ 463.1570, found 463.1565.
4.1.9.9. N-((1–(2-bromobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-sulphonamide (15i)
White solid, yield: 81%, m.p. 134–135 °C. 1H NMR (500 MHz, CDCl3): δ 7.66 (dd, J1 = 8.2, J2 = 1.7 Hz, 1H), 7.54 (dd, J1 = 8.0, J2 = 0.9 Hz, 1H), 7.50 (d, J = 1.5 Hz, 1H), 7.46 (d, J = 7.3 Hz, 1H), 7.30 (s, 1H), 7.15 – 7.10 (m, 1H), 7.07 (d, J = 8.2 Hz, 1H), 4.55 (t, J = 6.3 Hz, 1H), 3.59 (s, 2H), 3.49 (d, J = 1.8 Hz, 6H), 2.91 (d, J = 11.1 Hz, 2H), 2.84 (t, J = 6.7 Hz, 2H), 2.07 (t, J = 11.3 Hz, 2H), 1.68 (d, J = 12.7 Hz, 2H), 1.56 – 1.49 (m, 1H), 1.27 (s, 2H). 13 C NMR (126 MHz, DMSO-d6): δ 154.52, 138.08, 133.45, 133.02, 132.89, 131.15, 130.06, 129.17, 127.96, 124.32, 120.55, 107.91, 106.35, 61.83, 53.35, 48.52, 46.16, 35.99, 30.00, 27.68, 27.63. HRMS (ESI): calcd. for C22H27BrN4O3S [M + H]+ 507.1065, found 507.1017.
4.1.9.10. N-((1–(3-bromobenzyl)piperidin-4-yl)methyl)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-5-sulphonamide (15j)
White solid, yield: 75%, m.p. 175–177 °C. 1H NMR (500 MHz, CDCl3): δ 7.67 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.1 Hz, 2H), 7.39 (dd, J1 = 20.8, J2 = 7.7 Hz, 2H), 7.20 (t, J = 7.8 Hz, 1H), 7.07 (d, J = 8.2 Hz, 1H), 4.51 (s, 1H), 3.49 (s, 6H), 3.15 (d, J = 7.3 Hz, 2H), 2.89 (s, 2H), 2.84 (t, J = 6.7 Hz, 2H), 2.00 (s, 2H), 1.72 – 1.63 (m, 2H), 1.46 (t, J = 7.3 Hz, 2H), 1.27 (s, 1H). 13 C NMR (126 MHz, DMSO-d6): δ 154.52, 133.40, 133.01, 130.81, 130.05, 128.74, 122.04, 120.55, 107.92, 106.37, 61.20, 52.91, 48.36, 46.03, 35.68, 29.67, 27.69, 27.64. HRMS (ESI): calcd. for C22H27BrN4O3S [M + H]+ 507.1065, found 507.1060.
4.2. AChE and BChE inhibition assay
The inhibitory activities of the target compounds towards AChE (from Electrophorus electricus (electric eeAChE, Sigma-Aldrich) and horse serum BChE (eqBChE, Sigma-Aldrich) or human AChE – huAChE; human BuChE – huBuChE (huAChE, huBuChE, Sigma-Aldrich) were measured by Ellman’s method20. In 96-well plates, a mixture of phosphate buffer (0.1 M, pH 8.0, 2 ml), 5,5′-dithiobis-2-nitrobenzoic acid (DTNB, 60 µL), acetylcholinestrase or butyrylcholinesterase (20 µL, 5 IU/mL) and different concentration of the compound solution (30 µL) was pre-incubated for 5 min followed by the addition of the substrate (acetylthiocholine iodide or butyrylthiocholine iodide, 20 µL). Changes in absorbance were measured at 412 nm by using microplate reader (Thermo, Varioskan Flash 3001, USA). The measurement of each concentration for each compound was detected in triplicate. GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA) was used for data processing. The inhibition curve was fitted by plotting percentage enzyme activity (100% for the reference) versus the logarithm of the concentration of tested compound. The half-maximal inhibitory concentration (IC50) values were calculated according to the inhibition curve and the data were shown in the layout of mean ± SEM by GraphPad Prism 6.0.
4.3. Kinetic studies of AChE and BChE inhibition
Kinetic studies were performed in the same manner as the determination of ChEs inhibition, while the substrate (ATC/BTC) was used in concentrations of 90, 150, 226, 452, and 904 µM. The concentrations of test compounds were set to 0, 2, 5, 10, and 20 µM for 15b and 0, 0.5, 1, 2, and 5 µM for 15j. The enzymatic reaction was extended to 7 min for eeAChE and eqBuChE before the determination of the absorption. Vmax and Km values of the Michaelis–Menten kinetics were calculated by nonlinear regression from substrate-velocity curves using GraphPad Prism 6.0. Linear regression was used for fitting the Lineweaver–Burk plots.
4.4. Molecular docking study
The crystal structures of the hAChE (PDB ID: 4EY7)28 and hBChE (PDB ID: 4TPK)29 were derived from the RCSB Protein Data Bank. Docking studies were carried out using the Discovery Studio 2016 for compound 15b and compound 15j. Two protein structures were preprocessed (i.e. protonated, removed water, added Miss sidechains, etc.) by “prepare protein” module in DS to give the structures suitable for docking. “Prepare ligands” module in DS was applied for the structural preparation of the test compounds. The native ligand in the crystal structure was used to define the binding site. The binding site was defined as a site sphere (in 10 Å radius) around the original ligands in the co-crystal structures. The docking programme CDOCKER encoded in DS 2016 was applied to identify the potential binding of compound 15b to the hAChE and compound 15j to the hBChE. Other CDOCKER parameters were set to default values. Compound 15b created 10 poses to the hAChE and compound 15j created 10 poses to the hBuChE. the poses were visually inspected, and the most suitable docking pose was selected on the basis of the score and interactions with key residues of the active site of hAChE and hBChE.
4.5. Cell studies in vitro
PC12 cell and HepG2 cell (human hepatocellular liver carcinoma cell line purchased from Institute of Materia of Chinese Academy of Medical Science), were grown in DMEM supplemented with 10% FBS at 37 °C in a humidified atmosphere containing 5% CO2. For the experiments, cells (6 × 103 cells/well) were seeded in 96-well plate in complete medium. After 24 h, the medium was removed, and cells were exposed to the increasing concentrations of compounds 15b and 15j or donepezil (10, 20, 30, and 50 µM or 5, 15, and 50 µM) in DMEM for further 24 h. Cell survival was measured through MTT assay.
4.6. Behavioural studies
Behavioural studies were performed by using adult male ICR mice (8–10 weeks old, weight 20–25 g), which were purchased from the Yangzhou University Medical Centre (Yangzhou, China). Scopolamine hydrobromide was supplied by Aladdin Reagents (H1507073, Shanghai, China). Tacrine was synthesised in our lab with >95% purity as determined by HPLC.
The mice were separated into five groups as follows: (i) vehicle as blank control, (ii) scopolamine as model group, (iii) tacrine as positive control, (iv) compound 15b as test group, and (v) compound 15j as test group. Tacrine, 15b and 15j (20 mmol kg−1 body weight) were orally administered to mice in groups (iii), (iv), and (v), respectively, 30 min before the ip administration of scopolamine (1 mg kg−1) or saline for 10 consecutive days.
Cognitive function was evaluated by the Morris water maze analysis-management system (SMART 3.0, Panlab, Madrid, Spain), according to the method previously described30.The maze was placed in a lit room with visual cues at 25 °C. An escape platform (10 cm diameter) was located in the centre of one quadrant of the circular pool (120 cm diameter, 60 cm height) with a depth of 40 cm water. The behavioural study of each mouse included 5 days of learning and memory training and a probe trial on day 6. The animal starting positions faced to the pool wall, and were pseudorandomised for each trial. For the cognitive evaluation, each mouse was individually evaluated on both visible-platform (days 1–2) and hidden-platform (days 3–5) versions of the water maze. All mice received nonspatial pretraining during the first two training days, which prepared them for the subsequent spatial learning test. During the two days, mice were trained to find the platform that was labelled by a small flag (5 cm tall). The hidden platform version was used to determine the retention of memory to find the platform. During the hidden-platform training trials, the escape platform was placed 1 cm below the surface of the water. On each day, the animal was subjected to two trials, each of which lasted for 90 s. The time for the mouse to find the platform (a successful escape) was recorded. If a mouse failed to reach the platform within 90 s, the test was terminated and the animal was gently navigated to the platform by hand. Whether a mouse was successful or failed to reach the platform within 90 s, it was kept on the platform for 30 s. On the last day (day 6), the platform was removed from its location and the animals were given a probe trial in which they had 90 s to search for the platform. The time taken to reach the missing platform and the number of times the animals crossed the platform location were recorded.
Data for the time of escape latency, the trajectory travelled, and the number of platform location crossings were recorded by SMART 3.0, Panlab, Madrid, Spain and processed by GraphPad Prism 6.0.
Supplementary Material
Acknowledgements
The authors gratefully thank the support from the grants of National Natural Science Foundation of China. The authors also thank the support from “Double First-Class” initiative Innovation team project of China Pharmaceutical University.
Funding Statement
This work was supported by the grants [Nos. 81872728 and 81573281] of National Natural Science Foundation of China and “Double First-Class” initiative Innovation team project of China Pharmaceutical University [Nos. CPU2018GF11 and CPU2018GY34].
Disclosure statement
The authors report no conflicts of interest.
References
- 1.Suh YH, Checler F.. Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer’s disease. Pharmacol Rev 2002;54:469–525. [DOI] [PubMed] [Google Scholar]
- 2.Tumiatti V, Minarini A, Bolognesi ML, et al. . Tacrine derivatives and Alzheimer’s disease. Curr Med Chem 2010;17:1825–38. [DOI] [PubMed] [Google Scholar]
- 3.Mendiola-Precoma J, Berumen LC, Padilla K, Garcia-Alcocer G.. Therapies for prevention and treatment of Alzheimer’s disease. Biomed Res Int 2016;2016:2589276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prince M, Wimo A, Guerchet M, et al. . World Alzheimer Report 2018. The global impact of dementia. An analysis of prevalence, incidence, cost and trends. London: Alzheimer’s Disease International; 2018. [Google Scholar]
- 5.Shidore M, Machhi J, Shingala K, et al. . Benzylpiperidine-linked diarylthiazoles as potential anti-Alzheimer’s agents: synthesis and biological evaluation. J Med Chem 2016;59:5823–46. [DOI] [PubMed] [Google Scholar]
- 6.Goedert M, Spillantini MG.. A century of Alzheimer’s disease. Science 2006;314:777–81. [DOI] [PubMed] [Google Scholar]
- 7.Cavalli A, Bolognesi ML, Minarini A, et al. . Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem 2008;51:347–72. [DOI] [PubMed] [Google Scholar]
- 8.Kumar A, Singh A, Ekavali . A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep 2015;67:195–203. [DOI] [PubMed] [Google Scholar]
- 9.Berk C, Paul G, Sabbagh M.. Investigational drugs in Alzheimer’s disease: current progress. Expert Opin Inv Drug 2014;23:837–46. [DOI] [PubMed] [Google Scholar]
- 10.Gocer H, Akincioglu A, Goksu S, et al. . Carbonic anhydrase and acetylcholinesterase inhibitory effects of carbamates and sulfamoylcarbamates. J Enzym Inhib Med Ch 2015;30:316–20. [DOI] [PubMed] [Google Scholar]
- 11.Ferreira-Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM.. Alzheimer’s disease: targeting the cholinergic system. Curr Neuropharmacol 2016;14:101–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie SS, Wang XB, Li JY, et al. . Design, synthesis and evaluation of novel tacrine coumarin hybrids as multifunctional cholinesterase inhibitors against Alzheimer’s disease. Eur J Med Chem 2013;64:540–53. [DOI] [PubMed] [Google Scholar]
- 13.Hosoi M, Hori K, Konishi K, et al. . Plasma cholinesterase activity in Alzheimer’s disease. Neurodegener Dis 2015;15:188–90. [DOI] [PubMed] [Google Scholar]
- 14.Ismaili L, Refouvelet B, Benchekroun M, et al. . Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog Neurobiol 2017;151:4–34. [DOI] [PubMed] [Google Scholar]
- 15.Li GL, Hong G, Li XY, et al. . Synthesis and activity towards Alzheimer’s disease in vitro: tacrine, phenolic acid and ligustrazine hybrids. Eur J Med Chem 2018;148:238–54. [DOI] [PubMed] [Google Scholar]
- 16.Mesulam M, Guillozet A, Shaw P, Quinn B.. Widely spread butyrylcholinesterase can hydrolyze acetylcholine in the normal and Alzheimer brain. Neurobiol Dis 2002;9:88–93. [DOI] [PubMed] [Google Scholar]
- 17.Li Q, He SY, Chen Y, et al. . Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur J Med Chem 2018;158:463–77. [DOI] [PubMed] [Google Scholar]
- 18.Mesulam MM, Guillozet A, Shaw P, et al. . Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002;110:627–39. [DOI] [PubMed] [Google Scholar]
- 19.El-Sayed NA, Farag AE, Ezzat MAF, et al. . Design, synthesis, in vitro and in vivo evaluation of novel pyrrolizine-based compounds with potential activity as cholinesterase inhibitors and anti-Alzheimer’s agents. Bioorg Chem 2019;93:103312. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Y, Sun W, Peng J, et al. . Design, synthesis and biological evaluation of novel copper-chelating acetylcholinesterase inhibitors with pyridine and N-benzylpiperidine fragments. Bioorg Chem 2019;93:103322. [DOI] [PubMed] [Google Scholar]
- 21.Mo J, Yang H, Chen T, et al. . Design, synthesis, biological evaluation, and molecular modeling studies of quinoline-ferulic acid hybrids as cholinesterase inhibitors. Bioorg Chem 2019;93:103310. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, Lin HZ, Yang HY, et al. . Discovery of new acetylcholinesterase and butyrylcholinesterase inhibitors through structure-based virtual screening. Rsc Adv 2017;7:3429–38. [Google Scholar]
- 23.Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM.. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95. [DOI] [PubMed] [Google Scholar]
- 24.Tamagno E, Bardini P, Obbili A, et al. . Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis 2002;10:279–88. [DOI] [PubMed] [Google Scholar]
- 25.Zhu J, Yang HY, Chen Y, et al. . Synthesis, pharmacology and molecular docking on multifunctional tacrine-ferulic acid hybrids as cholinesterase inhibitors against Alzheimer’s disease. J Enzym Inhib Med Ch 2018;33:496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lefebvre CA, Forcellini E, Boutin S, et al. . Synthesis of novel substituted pyrimidine derivatives bearing a sulfamide group and their in vitro cancer growth inhibition activity. Bioorg Med Chem Lett 2017;27:299–302. [DOI] [PubMed] [Google Scholar]
- 27.Bronner SM, Murray J, Romero FA, et al. . A Unique Approach to Design Potent and Selective Cyclic Adenosine Monophosphate Response Element Binding Protein, Binding Protein (CBP) Inhibitors. J Med Chem 2017;60:10151–71. [DOI] [PubMed] [Google Scholar]
- 28.Xu YL, Lin HY, Ruan X, et al. . Synthesis and bioevaluation of pyrazole-benzimidazolone hybrids as novel human 4-Hydroxyphenylpyruvate dioxygenase inhibitors. Eur J Med Chem 2015;92:427–38. [DOI] [PubMed] [Google Scholar]
- 29.Lin SY, Yeh TK, Kuo CC, et al. . Phenyl Benzenesulfonylhydrazides Exhibit Selective Indoleamine 2,3-Dioxygenase Inhibition with Potent in Vivo Pharmacodynamic Activity and Antitumor Efficacy. J Med Chem 2016;59:419–30. [DOI] [PubMed] [Google Scholar]
- 30.Xu LL, Zhang X, Jiang ZY, You QD.. Molecular similarity guided optimization of novel Nrf2 activators with 1,2,4-oxadiazole core. Bioorgan Med Chem 2016;24:3540–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.