

Abstract
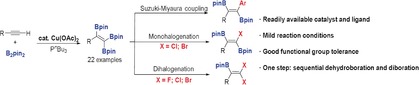
We report herein the catalytic triboration of terminal alkynes with B2pin2 (bis(pinacolato)diboron) using readily available Cu(OAc)2 and PnBu3. Various 1,1,2‐triborylalkenes, a class of compounds that have been demonstrated to be potential matrix metalloproteinase (MMP‐2) inhibitors, were obtained directly in moderate to good yields. The process features mild reaction conditions, a broad substrate scope, and good functional group tolerance. This copper‐catalyzed reaction can be conducted on a gram scale to produce the corresponding 1,1,2‐triborylalkenes in modest yields. The utility of these products was demonstrated by further transformations of the C−B bonds to prepare gem‐dihaloborylalkenes (F, Cl, Br), monohaloborylalkenes (Cl, Br), and trans‐diaryldiborylalkenes, which serve as important synthons and have previously been challenging to prepare.
Keywords: boronate esters, borylation, cross-coupling, diboration, halogenation
Three boron groups in one step: A straightforward copper‐catalyzed triboration of terminal alkynes gives access to triborylalkenes by sequential dehydroborylation and diboration. The triborylalkene products were applied in the synthesis of multisubstituted olefins with excellent regio‐ and stereoselectivity.
Organoboronic acids and their derivatives (boronate esters, trifluoroborates, and boroxines) play a critical role in organic synthesis, materials science, and pharmaceutical development.1 In particular, alkenylboron compounds have been utilized for the stereodefined construction of valuable multisubstituted alkenes, including natural products, biologically active molecules, and functional materials.2 These species can be categorized into three classes, namely monoborylalkenes, diborylalkenes, and triborylalkenes (Scheme 1).
Scheme 1.
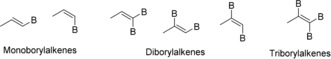
Classification of alkenylboron species.
Syntheses of monoborylalkenes and diborylalkenes have been well established. Various alkenylboronates are conventionally available through hydroboration and diboration of alkynes and dehydrogenative borylation of alkenes. Monoborylalkenes are typically synthesized by hydroboration of terminal or internal alkynes. These reactions are often promoted by metal catalysts, such as Rh,3 Ru,4 Pd,5 Ti,6 Ir,3e Cu,7 Ni,3c Fe,8 Au,9 Al,10 Co,11 or Mg,12 and, in some cases, they proceed under metal‐free conditions (Scheme 2 a).13 In addition, the metal‐catalyzed dehydrogenative borylation of alkenes has been reported as a route to monoborylalkenes or gem‐diborylalkenes (Scheme 2 a).1a, 14 The diboration of alkynes is a particularly attractive tool for the synthesis of 1,2‐diborylalkenes.2a, 15 The first metal‐catalyzed diboration of alkynes was reported by Suzuki and Miyaura in 1993 using a Pt catalyst,16 and significantly improved Pt catalyst systems were reported by our group.17 During the last few years, Pd,18 Cu,19 Co,11c, 20 Fe,21 Zn,14x and metal‐free22 systems were reported for the diboration of alkynes, which provide practical and economic alternatives to the Pt‐catalyzed processes (Scheme 2 b).16, 17, 23 However, the availability of diverse multiborylalkenes is quite limited because of the lack of efficient and versatile synthetic methods. All of these methods, albeit useful, have limitations, and therefore do not provide access to certain types of multiborylalkenes.
Scheme 2.
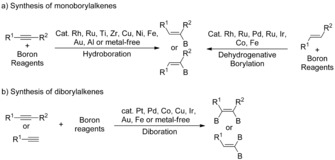
Synthesis of monoborylalkenes and diborylalkenes.
Interestingly, in 1996, in our previous study on the Pt‐catalyzed diboration of alkynes,23c we found that a novel 1,1,2‐triborylalkene was formed by desilylative borylation and subsequent diboration of bis(trimethylsilyl)acetylene with B2pin2 (Scheme 3 a). Since then, only two methods have been developed for the preparation of 1,1,2‐triborylalkenes. One is the Pt‐catalyzed diboration of alkynylboronates, which are usually synthesized using Grignard reagents or organolithium reagents (Scheme 3 b).23e, 24 Recently, Ozerov and co‐workers disclosed an Ir‐catalyzed synthesis of 1,1,2‐triborylalkenes through a two‐step reaction of terminal alkynes with HBpin under an atmosphere of CO (Scheme 3 c).25 These methods suffer from major or minor drawbacks, such as weak functional group tolerance, tedious procedures, or expensive catalysts. On the other hand, 1,1,2‐triborylalkenes (2 a and 2 r) have been shown to be potent matrix metalloproteinase (MMP‐2) inhibitors.26 Therefore, the development of efficient and versatile chemical transformations for the synthesis of diverse multiborylated alkenes from easily available starting materials is highly desirable. Herein, we report a novel and straightforward copper‐catalyzed synthesis of 1,1,2‐triborylalkenes from terminal alkynes.
Scheme 3.
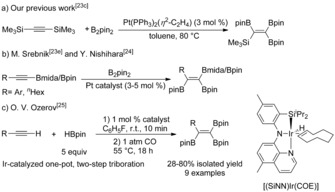
Synthesis of triborylalkenes.
Our initial studies showed that triboration of phenylacetylene (1 a) could be achieved in toluene at 80 °C in 38 % isolated yield in the presence of Cu(OAc)2, PnBu3, the diboron(4) reagent B2pin2, and iPr2EtN (Hünig's base) as a stoichiometric additive, together with 32 % monoborylalkene (Table 1), which was formed by competing hydroboration of the alkyne.
Table 1.
Optimization of the reaction conditions.[a]

|
Entry |
Catalyst |
Ligand |
Additive |
Yield 2 a [b] |
Yield 3 a |
|---|---|---|---|---|---|
|
1 |
Cu(OAc)2 |
PnBu3 |
iPr2EtN |
45 % (38 %[c]) |
32 % |
|
2 |
Cu(OTf)2 |
PnBu3 |
iPr2EtN |
0 % |
2 % |
|
3 |
CuCl2 |
PnBu3 |
iPr2EtN |
0 % |
0 % |
|
4c |
CuCl2 |
PnBu3 |
iPr2EtN |
42 % |
26 % |
|
5[c] |
CuCl |
PnBu3 |
iPr2EtN |
22 % |
34 % |
|
6 |
CuOAc |
PnBu3 |
iPr2EtN |
29 % |
20 % |
|
7 |
Cu(OAc)2 |
PPh3 |
iPr2EtN |
18 % |
40 % |
|
8 |
Cu(OAc)2 |
PCy3 |
iPr2EtN |
33 % |
23 % |
|
9 |
Cu(OAc)2 |
phen |
iPr2EtN |
trace |
8 % |
|
10 |
Cu(OAc)2 |
bpy |
iPr2EtN |
0 % |
4 % |
|
11[d] |
Cu(OAc)2 |
PnBu3 |
iPr2EtN |
14 % |
39 % |
|
12[e] |
Cu(OAc)2 |
PnBu3 |
iPr2EtN |
31 % |
18 % |
|
13 |
Cu(OAc)2 |
PnBu3 |
– |
28 % (16 %) |
28 % |
|
14 |
Cu(OAc)2 |
PnBu3 |
benzophenone |
48 % |
22 % |
|
15 |
Cu(OAc)2 |
PnBu3 |
2‐norbornene |
59 % (50 %) |
16 % |
|
16 |
Cu(OAc)2 |
PnBu3 |
acrylonitrile |
69 % (66 %) |
12 % |
|
17[f] |
Cu(OAc)2 |
PnBu3 |
acrylonitrile |
78 % (73 %) |
11 % |
|
18[f] |
Cu(OAc)2 |
– |
acrylonitrile |
0 % |
0 % |
|
19[f] |
– |
PnBu3 |
acrylonitrile |
0 % |
0 % |
[a] Reaction conditions: 1 a (0.2 mmol), B2pin2 (0.6 mmol), Cu catalyst (0.02 mmol), ligand (0.04 mmol), and additive (0.2 mmol) in toluene (2 mL) at 80 °C. [b] Yields were determined by GC/MS analysis with n‐dodecane as an internal calibration standard. Yields of isolated products are given in parentheses. [c] 20 mol % KOAc. [d] 60 °C. [e] 90 °C. [f] 4 h.
By screening Cu catalyst precursors, we identified Cu(OAc)2 as the most effective one (Table 1, entry 1). The desired product was not observed when Cu(OTf)2 or CuCl2 was used (entries 2 and 3). Addition of 20 mol % KOAc to the CuCl2 and CuCl systems was also effective, which indicated that AcO− plays an important role in this reaction and that the efficiency of a CuII precursor is somewhat higher than that of a CuI one (entries 4 and 5). Other phosphine ligands, such as PPh3 or PCy3, afforded low yields of 2 a (entries 7 and 8). Switching from phosphine ligands to nitrogen ligands (phen and bpy) gave no product (entries 9 and 10). As depicted in entries 11 and 12, the yield dropped when the reaction was conducted at either 60 °C or 90 °C. In the absence of Hünig's base, a lower yield was obtained (entry 13). To avoid the alkyne hydroboration side reaction, benzophenone, 2‐norbornene, and acrylonitrile were tested as hydrogen (B−H) acceptors instead of Hünig's base.14q, 14s, 14t The desired product was formed in good yield when acrylonitrile was used (entries 14–16). A high yield (73 %) was obtained when the reaction time was decreased from 24 h to 4 h (entry 17). As shown in entries 18 and 19, control reactions revealed that Cu(OAc)2 and the ligand were both essential for this reaction.
With optimized reaction conditions in hand, the triboration of a wide variety of terminal alkynes 1 was tested (Table 2). A range of both donor‐ and acceptor‐substituted aromatic alkynes were found to work well, affording the corresponding triborylalkenes in moderate to good yields (2 a–2 m). Arylalkynes bearing electron‐donating functional groups such as Me, OMe, and NMe2 smoothly reacted with B2pin2 to yield the corresponding triborylalkenes (isolated in 35–72 % yield). F‐, Cl‐, and CF3‐substituted arylalkynes were all viable substrates, giving moderate to high yields (47–72 %) of 2. In particular, the tolerance of halide substituents, such as F and Cl, provides possibilities for further functionalization. Unfortunately, substrates bearing strongly electron‐withdrawing groups, for example, CN and CO2Me, were not well tolerated by this system (2 h and 2 i).27 The isolated yields obtained for para‐substituted arylalkynes were higher than those for meta‐ and ortho‐substituted substrates (e.g., compare 2 b/2 c, 2 d/2 e/2 f, and 2 j/2 k). Polyaromatic and heteroaromatic substrates, for example, 2‐ethynyl‐6‐methoxynaphthalene and 3‐ethynylthiophene, reacted to give the desired products in moderate and good yields (2 n: 49 %; 2 o: 61 %).
Table 2.
Scope of the triboration of terminal alkynes.[a]
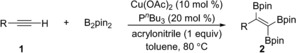
|
|
[a] Reaction conditions: 1 (0.2 mmol), B2pin2 (0.6 mmol), Cu(OAc)2 (0.02 mmol), PnBu3 (0.04 mmol), and acrylonitrile (0.2 mmol) in toluene (2 mL) at 80 °C. Yields of isolated products are given. [b] The reaction was performed on 5 mmol scale.
Furthermore, both linear‐alkyl‐ and cycloalkyl‐substituted alkynes afforded the desired products in good yields (2 p–2 u, 54–74 %). Even though it has a high degree of ring strain, a cyclopropyl moiety was retained after the reaction, providing the target product in a slightly lower yield (54 %) than its cyclopentyl and cyclohexyl analogues (64 % and 71 %, respectively). The conjugated 1,3‐enyne 1‐ethynylcyclohexene was also tested, and borylation occurred only at the triple bond, giving 2 v in 52 % yield, which indicated the high chemoselectivity of this reaction. The structure of the triborylalkene products was exemplified by a single‐crystal X‐ray diffraction study of 2 a (Table 2, bottom). To highlight the practicality of this method, this reaction was carried out on a gram scale, affording 2 a in 48 % yield.
We propose that an alkynylboronate is an intermediate in this reaction. Indeed, when using alkynylboronate 4 a as the starting material, under the standard conditions (with or without added acrylonitrile), the 1,1,2‐triborylalkene was isolated in 87 % yield and no side product was observed (see the Supporting Information, Scheme S2). Monitoring a reaction by in situ 19F NMR spectroscopy and GC/MS (Figure S1) showed that the alkyne substrate was converted into the alkynylboronate from which the final 1,1,2‐triborylalkene product was subsequently formed. Deuterium labeling studies were conducted using 1‐deutero‐2‐phenylethyne as the substrate. Under the standard conditions, 5‐d was produced from the hydroboration of acrylonitrile, which was confirmed by HRMS (Scheme S3 and Figure S4). The above result indicated that electron‐deficient alkenes were more reactive than alkynes for hydroboration and acted as a sacrificial borane (HBpin) scavenger to drive the catalysis toward alkyne triboration and away from hydroboration.
On the basis of our experimental observations (see also Schemes S2 and S3) and precedents regarding related catalytic dehydrogenative borylation processes,29 a plausible mechanism is proposed in Scheme 4. The terminal alkyne reacts with [LnCuOAc],30, 31 which is formed from Cu(OAc)2 and a phosphine ligand, followed by reduction,19b, 32 to afford alkynylcopper intermediate B.33 Intermediate B undergoes σ‐bond metathesis with B2pin2 to afford the alkynylboronate 4, as well as the copper–boryl complex C.29c, 34 Insertion of alkynylboronate 4 into a Cu−B bond in C generates alkenylcopper species D, which undergoes σ‐bond metathesis with B2pin2 to give the desired 1,1,2‐triborylalkene 2.19b Hydroboration of acrylonitrile is faster than that of alkynes, which suppresses the alkyne hydroboration side reaction and improves the efficiency of the triboration process. Byproduct 5 could be formed from alkylcopper intermediate E, which is generated by insertion of acrylonitrile into the C−B bond of C.
Scheme 4.
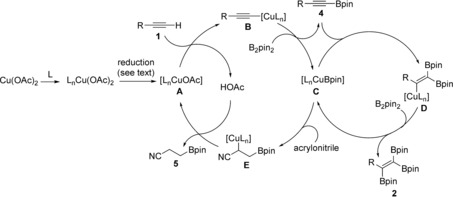
Proposed mechanism of the catalytic triboration reaction.
To explore the versatility of 1,1,2‐triborylalkenes in synthesis, we conducted a Suzuki–Miyaura cross‐coupling reaction of the triborated product 2 with aryl iodides. The 1,1,2‐triborylalkene reacted selectively to form a new C−C bond, providing trans‐diaryldiborylalkene 6 (Scheme 5 A). The E‐configuration of 6 b was confirmed by single‐crystal X‐ray diffraction (see Figure S6). Compound 2 d reacted selectively with Selectfluor, affording gem‐difluoroborylalkene 7 a in 93 % isolated yield (Scheme 5 B). Only two examples had previously been reported for the synthesis of this type of product, but small quantities of borylated fluoroalkenes were observed using polyfluoroalkenes as substrates.28 In addition, treatment of 2 with N‐chlorosuccinimide (NCS) or N‐bromosuccinimide (NBS) furnished selectively either the monohalo‐diborylated alkene (Cl and Br, 8 and 10) or the dihalo‐monoborylated alkene (Cl and Br, 9 and 11) products in good yields, depending on the amount of NCS and NBS added and the reaction time. The structure of the 10 b was confirmed by single‐crystal X‐ray diffraction (see Figure S7). To the best of our knowledge, this is the first time that products of these types (8–11) have been prepared, which clearly have potential for further use in cross‐couplings and other reactions.
Scheme 5.
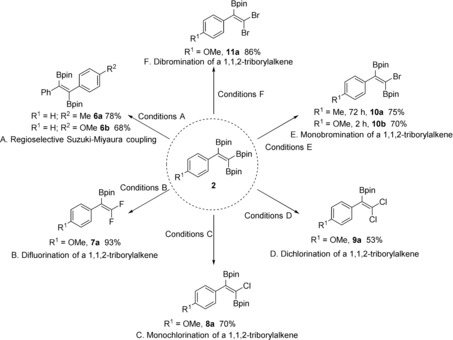
Synthetic applications of 1,1,2‐triborylalkenes with yields of isolated products. Conditions A: 4‐R2‐C6H4‐I (1 equiv), Pd(PPh3)4 (10 mol %), K3PO4 (2 equiv), H2O (7 equiv), THF, 70 °C, 24 h; conditions B: Selectfluor (3 equiv), NaHCO3 (2.2 equiv), CH3CN, room temperature, 6 h; conditions C: NCS (1.3 equiv), 60 °C, CH3CN, 12 h; conditions D: NCS (2 equiv), 60 °C, CH3CN, 48 h; conditions E: NBS (1.3 equiv), room temperature, CH3CN; conditions F: NBS (2 equiv), room temperature, CH3CN, 72 h.
In conclusion, a convenient copper‐catalyzed triboration of terminal alkynes has been developed. A variety of functional groups are tolerated, and diverse 1,1,2‐triborylalkenes were obtained in moderate to good yields. The products were applied in the synthesis of unsymmetrically substituted trans‐diaryldiborylalkenes and haloborylalkenes, which are expected to serve as useful building blocks. Additional explorations of the application of triborylalkenes and detailed mechanistic studies are currently underway.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
T.B.M. thanks the Julius‐Maximilians‐Universität Würzburg for support. X.L. and W.M. are grateful to the China Scholarship Council for providing Ph.D. scholarships. We thank AllyChem Co. Ltd. for a gift of B2pin2, and Dr. J. Zhao and Dr. X. Jia (Julius‐Maximilians‐Universität Würzburg) for helpful discussions.
X. Liu, W. Ming, A. Friedrich, F. Kerner, T. B. Marder, Angew. Chem. Int. Ed. 2020, 59, 304.
References
- 1.
- 1a. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890–931; [DOI] [PubMed] [Google Scholar]
- 1b. Boronic Acids: Preparation and Applications in Organic Synthesis Medicine and Materials, 2nd ed. (Ed.: D. G. Hall), Wiley-VCH, Weinheim, 2011; [Google Scholar]
- 1c. Synthesis and Applications of Organoboron Compounds Topics in Organometallic Chemistry, Vol. 49 (Eds.: E. Fernández, A. Whiting), Springer, Berlin, 2015; [Google Scholar]
- 1d. Neeve E. C., Geier S. J., Mkhalid I. A., Westcott S. A., Marder T. B., Chem. Rev. 2016, 116, 9091–9161. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Takaya J., Iwasawa N., ACS Catal. 2012, 2, 1993–2006; [Google Scholar]
- 2b. Zuo Z., Wen H., Liu G., Huang Z., Synlett 2018, 29, 1421–1429. [Google Scholar]
- 3.
- 3a. Männig D., Nöth H., Angew. Chem. Int. Ed. Engl. 1985, 24, 878–879; [Google Scholar]; Angew. Chem. 1985, 97, 854–855; [Google Scholar]
- 3b. Burgess K., Van der Donk W. A., Westcott S. A., Marder T. B., Baker R. T., Calabrese J. C., J. Am. Chem. Soc. 1992, 114, 9350–9359; [Google Scholar]
- 3c. Pereira S., Srebnik M., Tetrahedron Lett. 1996, 37, 3283–3286; [Google Scholar]
- 3d. Juliette J. J. J., Rutherford D., Horváth I. T., Gladysz J. A., J. Am. Chem. Soc. 1999, 121, 2696–2704; [Google Scholar]
- 3e. Ohmura T., Yamamoto Y., Miyaura N., J. Am. Chem. Soc. 2000, 122, 4990–4991; [Google Scholar]
- 3f. Endo K., Hirokami M., Shibata T., Synlett 2009, 1331–1335; [Google Scholar]
- 3g. Wang K., Bates R. W., Synthesis 2017, 49, 2749–2752. [Google Scholar]
- 4.
- 4a. Murata M., Watanabe S., Masuda Y., J. Chem. Res.-S 2002, 2002, 142–143; [Google Scholar]
- 4b. Gunanathan C., Holscher M., Pan F., Leitner W., J. Am. Chem. Soc. 2012, 134, 14349–14352; [DOI] [PubMed] [Google Scholar]
- 4c. Sundararaju B., Fürstner A., Angew. Chem. Int. Ed. 2013, 52, 14050–14054; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14300–14304. [Google Scholar]
- 5.
- 5a. Gridnev I. D., Miyaura N., Suzuki A., J. Org. Chem. 1993, 58, 5351–5354; [Google Scholar]
- 5b. Ojha D. P., Prabhu K. R., Org. Lett. 2016, 18, 432–435; [DOI] [PubMed] [Google Scholar]
- 5c. Xu S., Zhang Y., Li B., Liu S.-Y., J. Am. Chem. Soc. 2016, 138, 14566–14569; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Yang Y., Jiang J., Yu H., Shi J., Chem. Eur. J. 2018, 24, 178–186. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Hartwig J. F., Muhoro C. N., He X., Eisenstein O., Bosque R., Maseras F., J. Am. Chem. Soc. 1996, 118, 10936–10937; [Google Scholar]
- 6b. He X., Hartwig J. F., J. Am. Chem. Soc. 1996, 118, 1696–1702; [Google Scholar]
- 6c. Muhoro C. N., Hartwig J. F., Angew. Chem. Int. Ed. Engl. 1997, 36, 1510–1512; [Google Scholar]; Angew. Chem. 1997, 109, 1536–1538; [Google Scholar]
- 6d. Muhoro C. N., He X., Hartwig J. F., J. Am. Chem. Soc. 1999, 121, 5033–5046. [Google Scholar]
- 7.
- 7a. Lipshutz B. H., Boskovic Z. V., Aue D. H., Angew. Chem. Int. Ed. 2008, 47, 10183–10186; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 10337–10340; [Google Scholar]
- 7b. Jang H., Zhugralin A. R., Lee Y., Hoveyda A. H., J. Am. Chem. Soc. 2011, 133, 7859–7871; [DOI] [PubMed] [Google Scholar]
- 7c. Fujihara T., Semba K., Terao J., Tsuji Y., Catal. Sci. Technol. 2014, 4, 1699–1709; [Google Scholar]
- 7d. Yoshida H., Takemoto Y., Takaki K., Chem. Commun. 2014, 50, 8299–8302; [DOI] [PubMed] [Google Scholar]
- 7e. Feng Q., Yang K., Song Q., Chem. Commun. 2015, 51, 15394–15397; [DOI] [PubMed] [Google Scholar]
- 7f. Kim Y. E., Li D., Yun J., Dalton Trans. 2015, 44, 12091–12093; [DOI] [PubMed] [Google Scholar]
- 7g. Peck C. L., Calderone J. A., Santos W. L., Synthesis 2015, 47, 2242–2248; [Google Scholar]
- 7h. Nelson A. K., Peck C. L., Rafferty S. M., Santos W. L., J. Org. Chem. 2016, 81, 4269–4279; [DOI] [PubMed] [Google Scholar]
- 7i. Jang W. J., Lee W. L., Moon J. H., Lee J. Y., Yun J., Org. Lett. 2016, 18, 1390–1393; [DOI] [PubMed] [Google Scholar]
- 7j. Tanaka C., Nakamura K., Nishikata T., Tetrahedron 2017, 73, 3999–4003. [Google Scholar]
- 8.
- 8a. Haberberger M., Enthaler S., Chem. Asian J. 2013, 8, 50–54; [DOI] [PubMed] [Google Scholar]
- 8b. Greenhalgh M. D., Thomas S. P., Chem. Commun. 2013, 49, 11230–11232; [DOI] [PubMed] [Google Scholar]
- 8c. Tseng K.-N. T., Kampf J. W., Szymczak N. K., ACS Catal. 2015, 5, 411–415; [Google Scholar]
- 8d. Espinal-Viguri M., Woof C. R., Webster R. L., Chem. Eur. J. 2016, 22, 11605–11608; [DOI] [PubMed] [Google Scholar]
- 8e. Gorgas N., Alves L. G., Stöger B., Martins A. M., Veiros L. F., Kirchner K., J. Am. Chem. Soc. 2017, 139, 8130–8133; [DOI] [PubMed] [Google Scholar]
- 8f. Nakajima K., Kato T., Nishibayashi Y., Org. Lett. 2017, 19, 4323–4326. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Leyva A., Zhang X., Corma A., Chem. Commun. 2009, 4947–4949; [DOI] [PubMed] [Google Scholar]
- 9b. Wang Q., Motika S. E., Akhmedov N. G., Petersen J. L., Shi X., Angew. Chem. Int. Ed. 2014, 53, 5418–5422; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5522–5526. [Google Scholar]
- 10.
- 10a. Bismuto A., Thomas S. P., Cowley M. J., Angew. Chem. Int. Ed. 2016, 55, 15356–15359; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15582–15585; [Google Scholar]
- 10b. Yang Z., Zhong M., Ma X., Nijesh K., De S., Parameswaran P., Roesky H. W., J. Am. Chem. Soc. 2016, 138, 2548–2551; [DOI] [PubMed] [Google Scholar]
- 10c. Pollard V. A., Fuentes M. Á., Kennedy A. R., McLellan R., Mulvey R. E., Angew. Chem. Int. Ed. 2018, 57, 10651–10655; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10811–10815. [Google Scholar]
- 11.
- 11a. Obligacion J. V., Neely J. M., Yazdani A. N., Pappas I., Chirik P. J., J. Am. Chem. Soc. 2015, 137, 5855–5858; [DOI] [PubMed] [Google Scholar]
- 11b. Guo J., Cheng B., Shen X., Lu Z., J. Am. Chem. Soc. 2017, 139, 15316–15319; [DOI] [PubMed] [Google Scholar]
- 11c. Ferrand L., Lyu Y., Rivera-Hernández A., Fallon B. J., Amatore M., Aubert C., Petit M., Synthesis 2017, 49, 3895–3904; [Google Scholar]
- 11d. Ben-Daat H., Rock C. L., Flores M., Groy T. L., Bowman A. C., Trovitch R. J., Chem. Commun. 2017, 53, 7333–7336. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Li J., Luo M., Sheng X., Hua H., Yao W., Pullarkat S. A., Xu L., Ma M., Org. Chem. Front. 2018, 5, 3538–3547; [Google Scholar]
- 12b. Magre M., Maity B., Falconnet A., Cavallo L., Rueping M., Angew. Chem. Int. Ed. 2019, 58, 7025–7029; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7099–7103. [Google Scholar]
- 13.
- 13a. Hassner A., Soderquist J. A., J. Organomet. Chem. 1977, 131, C1–C4; [Google Scholar]
- 13b. Soderquist J. A., Colberg J. C., Delvalle L., J. Am. Chem. Soc. 1989, 111, 4873–4878; [Google Scholar]
- 13c. Hoshi M., Masuda Y., Arase A., J. Chem. Soc. Perkin Trans. 1 1990, 3237–3241; [Google Scholar]
- 13d. Shirakawa K., Arase A., Hoshi M., Synthesis 2004, 1814–1820; [Google Scholar]
- 13e. Wen K., Chen J., Gao F., Bhadury P. S., Fan E., Sun Z., Org. Biomol. Chem. 2013, 11, 6350–6356; [DOI] [PubMed] [Google Scholar]
- 13f. Ho H. E., Asao N., Yamamoto Y., Jin T., Org. Lett. 2014, 16, 4670–4673; [DOI] [PubMed] [Google Scholar]
- 13g. Warner A. J., Lawson J. R., Fasano V., Ingleson M. J., Angew. Chem. Int. Ed. 2015, 54, 11245–11249; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11397–11401; [Google Scholar]
- 13h. Fleige M., Möbus J., Vom Stein T., Glorius F., Stephan D. W., Chem. Commun. 2016, 52, 10830–10833; [DOI] [PubMed] [Google Scholar]
- 13i. McGough J. S., Butler S., Cade I. A., Ingleson M. J., Chem. Sci. 2016, 7, 3384–3389; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13j. Fritzemeier R., Gates A., Guo X., Lin Z., Santos W. L., J. Org. Chem. 2018, 83, 10436–10444; [DOI] [PubMed] [Google Scholar]
- 13k. Nagao K., Yamazaki A., Ohmiya H., Sawamura M., Org. Lett. 2018, 20, 1861–1865; [DOI] [PubMed] [Google Scholar]
- 13l. Shimoi M., Watanabe T., Maeda K., Curran D. P., Taniguchi T., Angew. Chem. Int. Ed. 2018, 57, 9485–9490; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9629–9634. [Google Scholar]
- 14.
- 14a. Westcott S. A., Marder T. B., Baker R. T., Organometallics 1993, 12, 975–979; [Google Scholar]
- 14b. Brown J. M., Lloyd-Jones G. C., J. Am. Chem. Soc. 1994, 116, 866–878; [Google Scholar]
- 14c. Motry D. H., M. R. Smith III , J. Am. Chem. Soc. 1995, 117, 6615–6616; [Google Scholar]
- 14d. Motry D. H., Brazil A. G., M. R. Smith III , J. Am. Chem. Soc. 1997, 119, 2743–2744; [Google Scholar]
- 14e. Murata M., Watanabe S., Masuda Y., Tetrahedron Lett. 1999, 40, 2585–2588; [Google Scholar]
- 14f. Kadlecek D. E., Carroll P. J., Sneddon L. G., J. Am. Chem. Soc. 2000, 122, 10868–10877; [Google Scholar]
- 14g. Coapes R. B., Souza F. E. S., Thomas R. Ll., Hall J. J., Marder T. B., Chem. Commun. 2003, 614–615; [DOI] [PubMed] [Google Scholar]
- 14h. Caballero A., Sabo-Etienne S., Organometallics 2007, 26, 1191–1195; [Google Scholar]
- 14i. Kikuchi T., Takagi J., Isou H., Ishiyama T., Miyaura N., Chem. Asian J. 2008, 3, 2082–2090; [DOI] [PubMed] [Google Scholar]
- 14j. Mkhalid I. A. I., Coapes R. B., Edes S. N., Coventry D. N., Souza F. E. S., Thomas R. Ll., Hall J. J., Bi S.-W., Lin Z., Marder T. B., Dalton Trans. 2008, 1055–1064; [DOI] [PubMed] [Google Scholar]
- 14k. Ohmura T., Takasaki Y., Furukawa H., Suginome M., Angew. Chem. Int. Ed. 2009, 48, 2372–2375; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2408–2411; [Google Scholar]
- 14l. Kondoh A., Jamison T. F., Chem. Commun. 2010, 46, 907–909; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14m. Selander N., Willy B., Szabó K. J., Angew. Chem. Int. Ed. 2010, 49, 4051–4053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4145–4147; [Google Scholar]
- 14n. Takaya J., Kirai N., Iwasawa N., J. Am. Chem. Soc. 2011, 133, 12980–12983; [DOI] [PubMed] [Google Scholar]
- 14o. Sasaki I., Doi H., Hashimoto T., Kikuchi T., Ito H., Ishiyama T., Chem. Commun. 2013, 49, 7546–7548; [DOI] [PubMed] [Google Scholar]
- 14p. Brown A. N., Zakharov L. N., Mikulas T., Dixon D. A., Liu S. Y., Org. Lett. 2014, 16, 3340–3343; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14q. Morimoto M., Miura T., Murakami M., Angew. Chem. Int. Ed. 2015, 54, 12659–12663; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12850–12854; [Google Scholar]
- 14r. Reid W. B., Spillane J. J., Krause S. B., Watson D. A., J. Am. Chem. Soc. 2016, 138, 5539–5542; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14s. Wang C., Wu C., Ge S., ACS Catal. 2016, 6, 7585–7589; [Google Scholar]
- 14t. Mazzacano T. J., Mankad N. P., ACS Catal. 2017, 7, 146–149; [Google Scholar]
- 14u. Wen H., Zhang L., Zhu S., Liu G., Huang Z., ACS Catal. 2017, 7, 6419–6425; [Google Scholar]
- 14v. Murray S. A., Luc E. C. M., Meek S. J., Org. Lett. 2018, 20, 469–472; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14w. Lu W., Shen Z., Org. Lett. 2019, 21, 142–146; [DOI] [PubMed] [Google Scholar]
- 14x. Procter R. J., Uzelac M., Cid J., Rushworth P. J., Ingleson M. J., ACS Catal. 2019, 9, 5760–5771. [Google Scholar]
- 15.
- 15a. Marder T. B., Norman N. C., Top. Catal. 1998, 5, 63–73; [Google Scholar]
- 15b. Ishiyama T., Miyaura N., Chem. Rec. 2004, 3, 271–280; [DOI] [PubMed] [Google Scholar]
- 15c. Barbeyron R., Benedetti E., Cossy J., Vasseur J. J., Arseniyadis S., Smietana M., Tetrahedron 2014, 70, 8431–8452; [Google Scholar]
- 15d. Yoshida H., ACS Catal. 2016, 6, 1799–1811; [Google Scholar]
- 15e. Zhao F., Jia X. W., Li P. Y., Zhao J. W., Zhou Y., Wang J., Liu H., Org. Chem. Front. 2017, 4, 2235–2255. [Google Scholar]
- 16. Ishiyama T., Matsuda N., Miyaura N., Suzuki A., J. Am. Chem. Soc. 1993, 115, 11018–11019. [Google Scholar]
- 17. Thomas R. Ll., Souza F. E. S., Marder T. B., J. Chem. Soc. Dalton Trans. 2001, 1650–1656. [Google Scholar]
- 18.
- 18a. Zhao T. S. N., Yang Y., Lessing T., Szabó K. J., J. Am. Chem. Soc. 2014, 136, 7563–7566; [DOI] [PubMed] [Google Scholar]
- 18b. Yang Z., Cao T., Han Y. L., Lin W. L., Liu Q., Tang Y., Zhai Y. Z., Jia M. Q., Zhang W. L., Zhu T. H., Ma S. M., Chin. J. Chem. 2017, 35, 1251–1262. [Google Scholar]
- 19.
- 19a. Lillo V., Fructos M. R., Ramirez J., Braga A. A., Maseras F., Diaz-Requejo M. M., Perez P. J., Fernandez E., Chem. Eur. J. 2007, 13, 2614–2621; [DOI] [PubMed] [Google Scholar]
- 19b. Yoshida H., Kawashima S., Takemoto Y., Okada K., Ohshita J., Takaki K., Angew. Chem. Int. Ed. 2012, 51, 235–238; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 239–242. [Google Scholar]
- 20. Krautwald S., Bezdek M. J., Chirik P. J., J. Am. Chem. Soc. 2017, 139, 3868–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Nakagawa N., Hatakeyama T., Nakamura M., Chem. Eur. J. 2015, 21, 4257–4261; [DOI] [PubMed] [Google Scholar]
- 21b. Khan A., Asiri A. M., Kosa S. A., Garcia H., Grirrane A., J. Catal. 2015, 329, 401–412. [Google Scholar]
- 22.
- 22a. Nagashima Y., Hirano K., Takita R., Uchiyama M., J. Am. Chem. Soc. 2014, 136, 8532–8535; [DOI] [PubMed] [Google Scholar]
- 22b. Morinaga A., Nagao K., Ohmiya H., Sawamura M., Angew. Chem. Int. Ed. 2015, 54, 15859–15862; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 16085–16088; [Google Scholar]
- 22c. Nagao K., Ohmiya H., Sawamura M., Org. Lett. 2015, 17, 1304–1307; [DOI] [PubMed] [Google Scholar]
- 22d. Yoshimura A., Takamachi Y., Han L. B., Ogawa A., Chem. Eur. J. 2015, 21, 13930–13933; [DOI] [PubMed] [Google Scholar]
- 22e. Kojima C., Lee K. H., Lin Z., Yamashita M., J. Am. Chem. Soc. 2016, 138, 6662–6669. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Iverson C. N., M. R. Smith III , J. Am. Chem. Soc. 1995, 117, 4403–4404; [Google Scholar]
- 23b. Ishiyama T., Matsuda N., Murata M., Ozawa F., Suzuki A., Miyaura N., Organometallics 1996, 15, 713–720; [Google Scholar]
- 23c. Lesley G., Nguyen P., Taylor N. J., Marder T. B., Scott A. J., Clegg W., Norman N. C., Organometallics 1996, 15, 5137–5154; [Google Scholar]
- 23d. Iverson C. N., M. R. Smith III , Organometallics 1996, 15, 5155–5165; [Google Scholar]
- 23e. Ali H. A., El Aziz Al Quntar A., Goldberg I., Srebnik M., Organometallics 2002, 21, 4533–4539; [Google Scholar]
- 23f. Mora-Radó H., Bialy L., Czechtizky W., Méndez M., Harrity J. P. A., Angew. Chem. Int. Ed. 2016, 55, 5834–5836; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5928–5930. [Google Scholar]
- 24. Hyodo K., Suetsugu M., Nishihara Y., Org. Lett. 2014, 16, 440–443. [DOI] [PubMed] [Google Scholar]
- 25. Lee C. I., Shih W. C., Zhou J., Reibenspies J. H., Ozerov O. V., Angew. Chem. Int. Ed. 2015, 54, 14003–14007; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14209–14213. [Google Scholar]
- 26. Ali H. A., Berkovitz R., Reich R., Srebnik M., Arch. Pharm. Pharm. Med. Chem. 2004, 337, 183–187. [DOI] [PubMed] [Google Scholar]
- 27.Only trace amounts of the desired products were observed by GC/MS.
- 28.
- 28a. Sakaguchi H., Uetake Y., Ohashi M., Niwa T., Ogoshi S., Hosoya T., J. Am. Chem. Soc. 2017, 139, 12855–12862; [DOI] [PubMed] [Google Scholar]
- 28b. Sakaguchi H., Ohashi M., Ogoshi S., Angew. Chem. Int. Ed. 2018, 57, 328–332; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 334–338. [Google Scholar]
- 29.
- 29a. Tsuchimoto T., Utsugi H., Sugiura T., Horio S., Adv. Synth. Catal. 2015, 357, 77–82; [Google Scholar]
- 29b. Pell C. J., Ozerov O. V., Inorg. Chem. Front. 2015, 2, 720–724; [Google Scholar]
- 29c. Romero E. A., Jazzar R., Bertrand G., Chem. Sci. 2017, 8, 165–168; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29d. Wei D., Carboni B., Sortais J.-B., Darcel C., Adv. Synth. Catal. 2018, 360, 3649–3654. [Google Scholar]
- 30.The exact oxidation state of Cu and indeed the nuclearity of the active catalyst are not clear, as Kleeberg has recently shown that dimeric CuI and higher-order copper–boryl clusters with Cu oxidation states between 0 and 1 are formed from LCu(OR) and diboron(4) reagents.[31]
- 31.
- 31a. Borner C., Kleeberg C., Eur. J. Inorg. Chem. 2014, 2486–2489; [Google Scholar]
- 31b. Borner C., Anders L., Brandhorst K., Kleeberg C., Organometallics 2017, 36, 4687–4690; [Google Scholar]
- 31c. Kleeberg C., Borner C., Organometallics 2018, 37, 4136–4146; [Google Scholar]
- 31d. Oschmann W., Borner C., Kleeberg C., Dalton Trans. 2018, 47, 5318–5327; [DOI] [PubMed] [Google Scholar]
- 31e. Drescher W., Kleeberg C., Inorg. Chem. 2019, 58, 8215–8229. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Hammond B., Jardine F. H., Vohra A. G., J. Inorg. Nucl. Chem. 1971, 33, 1017–1024; [Google Scholar]
- 32b. Adner D., Möckel S., Korb M., Buschbeck R., Rüffer T., Schulze S., Mertens L., Hietschold M., Mehring M., Lang H., Dalton Trans. 2013, 42, 15599–15609. [DOI] [PubMed] [Google Scholar]
- 33. Buckley B. R., Dann S. E., Heaney H., Stubbs E. C., Eur. J. Org. Chem. 2011, 770–776. [Google Scholar]
- 34.
- 34a. Zhao H., Dang L., Marder T. B., Lin Z., J. Am. Chem. Soc. 2008, 130, 5586–5594; [DOI] [PubMed] [Google Scholar]
- 34b. Romero E. A., Jazzar R., Bertrand G., J. Organomet. Chem. 2017, 829, 11–13. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary