

Abstract
Continuous flow systems for chemical synthesis are becoming a major focus in organic chemistry and there is a growing interest in the integration of biocatalysts due to their high regio‐ and stereoselectivity. Methods established for 3D bioprinting enable the fast and simple production of agarose‐based modules for biocatalytic reactors if thermally stable enzymes are available. We report here on the characterization of four different cofactor‐free phenacrylate decarboxylase enzymes suitable for the production of 4‐vinylphenol and test their applicability for the encapsulation and direct 3D printing of disk‐shaped agarose‐based modules that can be used for compartmentalized flow microreactors. Using the most active and stable phenacrylate decarboxylase from Enterobacter spec. in a setup with four parallel reactors and a subsequent palladium(II) acetate‐catalysed Heck reaction, 4‐hydroxystilbene was synthesized from p‐coumaric acid with a total yield of 14.7 % on a milligram scale. We believe that, due to the convenient direct immobilization of any thermostable enzyme and straightforward tuning of the reaction sequence by stacking of modules with different catalytic activities, this simple process will facilitate the establishment and use of cascade reactions and will therefore be of great advantage for many research approaches.
Keywords: 3D printing, biocatalysis, enzymes, flow chemistry, hydrogels
3D printing in flow chemistry: 4‐Hydroxystilbene is obtained from p‐coumaric acid employing a phenacrylate decarboxylase enzyme immobilized in 3D‐printed agarose‐based flow reactor modules and a subsequent palladium(II) acetate‐catalyzed Heck reaction with a total yield of 14.7 % on a milligram scale.
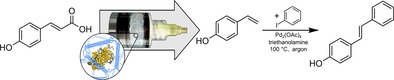
Sustainable production processes based on renewable biomass require biocatalysts with high conversion rates as well as regio‐ and stereoselectivity.1 Enzymes, as nature's main catalysts, are enabling the realization of such processes. Furthermore, they can be precisely tailored to fit with the process requirements such as substrate spectrum, regio‐ and stereoselectivity and physicochemical stability by using protein engineering.2 To improve biotechnological production processes, individual reaction steps can be compartmentalized and arranged into reaction cascades.3 This allows high process control even for chemoenzymatic routes by combining reactions that are otherwise incompatible due to reaction conditions, product inhibition or undesired side reactions.3a, 4 For the realization of such compartmentalized biocatalytic processes in continuous flow, the fast and simple immobilization of enzymes inside the microstructured flow reactors is an essential step to ensure efficient spatial separation of different reactions.5 To enable the simple rapid prototyping of biocatalytic reaction modules for compartmentalized flow microreactors, we have recently established a fabrication process using direct 3D printing of inexpensive biodegradable agarose‐based hydrogel bioinks containing thermostable enzymes (Figure 1).6
Figure 1.
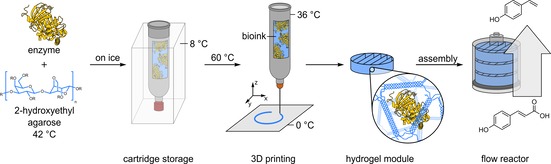
Manufacturing process of agarose‐based, compartmentalized biocatalytic flow reactors. The enzyme is mixed with a liquid agarose solution (at 42 °C) and immediately cooled down on ice and then stored at 8 °C. Using a 3D printer, the bioink is melted at 60 °C and printed onto a cooled substrate on which the bioink solidifies to form biocatalytic modules. These are integrated into flow reactors that are transfused with a substrate solution to facilitate continuous synthesis. The reactor productivity can be adjusted by variable numbers of reaction modules.6
3D printing (also known as additive manufacturing) is a fabrication process employing layer‐by‐layer deposition of materials to produce complex 3D structures with a high flexibility in design and minimal waste of material,7 which has found applications ranging from the aerospace and construction industry to smart materials, medicine and biotechnology.8 3D printing offers a simple and efficient route to rapid prototyping, the iterative process of fabricating and evaluating prototypes, dramatically reducing turnaround times in process development. Furthermore, 3D printing facilitates the production of extremely complex geometries7 enabling the fabrication of sophisticated reactor designs in chemical and biochemical engineering.9
To enhance the compatibility with biological materials a variety of methods and materials for extrusion‐based 3D bioprinting can now generate hydrogel structures customized for specific applications.10 Enzymes can be entrapped inside these hydrogels without the need for enzyme‐specific adaptations, offering protection against organic solvents6 but also modulating the reaction rate through mass transfer limitations, since the 3D‐printed structures are relatively thick.11 Such issues are less prevalent when using for example packed‐bed reactors containing 0.1–1 mm Ca‐alginate beads, packed‐bed nanofiber reactors or all‐enzyme‐hydrogel reactors, containing no carrier material at all,5a, 5e, 12 however the capability of the 3D bioprinting for the on‐demand production of biocatalytic modules, only requiring unmodified, thermostable enzymes, has great potential for fast and simple immobilization approaches.
To extend the earlier proof‐of‐concept study to other synthetically relevant enzymes, we here report on the 3D printing of biocatalytic modules to be integrated in a chemoenzymatic reaction cascade producing 4‐hydroxystilbene (3) (Scheme 1). 4‐Hydroxystilbene is an important precursor of the pharmacologically relevant class of 2,3‐dihydrobenzofuran‐based compounds.13
Scheme 1.
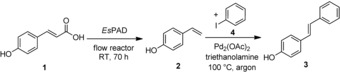
Synthesis of 4‐hydroxystilbene (3) from p‐coumaric acid (1) employing a phenacrylate decarboxylase (EsPAD) in a flow reactor setup and a subsequent Heck reaction using palladium(II) acetate.
To realize the chemoenzymatic reaction cascade (Scheme 1), we considered prokaryotic phenacrylate decarboxylases (PAD, EC 4.1.1.102) that can catalyse the formation of 4‐vinylphenols from para‐hydroxyphenacrylic acids without the supply of additional redox equivalents.14 Furthermore, unlike other cofactor‐dependent PADs, these enzymes do not require a prenylated flavin cofactor, which typically requires production by co‐expression of the flavin prenyltransferase UbiX.14, 15 In a second step of the cascade, the enzymatically produced vinylphenols can then be further functionalized, for example, by aid of other enzymes,16 oxidative dimerization17 or by organometallic coupling reactions18 as demonstrated here.
Since the printing of biocatalytic active reactor models requires an initial heating step at 60 °C for 15 min, thermostable enzymes are required, which can be obtained by utilizing naturally occurring thermostable enzymes6, 19 or enzymes thermostabilized by protein engineering.6, 19a, 20 To broaden the spectrum of available biocatalysts and to identify suitable PAD enzymes, we directly compared the enzymatic activity and thermostability of four different enzymes (Table S1) under identical conditions. To this end, we expressed and purified (Figure 2 A) the previously described EsPAD21 from Enterobacter spec. and LpPAD22 from Lactobacillus plantarum as well as LbPAD from Lactobacillus brevis, a close homologue of an already described enzyme23 (Figure S1), and the not yet described BmPAD from Bacillus megaterium.
Figure 2.
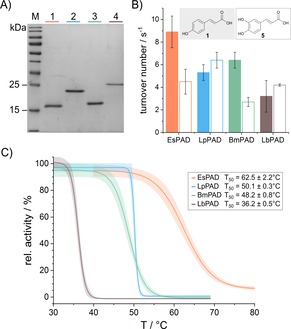
A) EsPAD (20 kDa, lane 1), LpPAD (22 kDa, lane 2), BmPAD (20 kDa, lane 3) and LbPAD (23 kDa, lane 4) were produced as pure enzymes, as indicated by the Coomassie stained 15 % SDS tris‐glycine polyacrylamide gel. M: PageRuler Prestained Protein Ladder (Thermo Scientific). B) Enzymatic activity of all four PAD enzymes at 28 °C with p‐coumaric acid (1) (filled bars) and caffeic acid (5) (empty bars) as substrate. C) Average thermal inactivation curves with 95 % confidence interval of all four purified PAD enzymes after 10 min incubation and their corresponding T50 values (for individual data sets see Figures S2–S5). Analyses B and C were carried out in technical duplicates and biological triplicates.
To enable direct comparison of the four PAD variants, their substrate specificity and activity were initially studied. Thus, trans‐cinnamic acid, the simplest phenacrylic acid without any aromatic substitution, and the hydroxy‐substituted phenacrylic acids, p‐coumaric acid (1) and caffeic acid (5), were tested as substrates. Their enzymatic activity at 28 °C was determined by the decrease in absorbance at the substrate‐specific wavelengths of 274, 294 and 311 nm, respectively. As expected, all four PAD enzymes only showed activity with the para‐hydroxy‐substituted phenacrylic acids, p‐coumaric acid and caffeic acid, confirming that the para‐hydroxy functional group is essential for the formation of the para‐quinone methide intermediate, which leads to decarboxylation of the substrate.14 The activity of all four enzymes was found to be on the same order of magnitude under the conditions examined here (Figure 2 B and Table S2), with EsPAD converting p‐coumaric acid (1) three times faster than LbPAD, and LpPAD converting caffeic acid (5) twice as fast as BmPAD.
Since the activities of all enzymes are in the same range, we focused on the enzyme thermostability as the essential parameter for the intended use in the 3D printing process. Therefore, the temperature‐dependent loss of activity of the enzymes was characterized by incubating 1 μm enzyme solutions for 10 min at variable temperatures in the range between 30–80 °C. The remaining enzyme activity was then determined at 25 °C using p‐coumaric acid as substrate. Plots of the resulting activities against incubation temperature allowed to determine T50 values, defined as the temperature at which 50 % of the initial enzyme activity still remained (Figure 2 C and Figures S2–S5). From these analyses, EsPAD was found to be the most thermostable enzyme, which also showed the highest activity with p‐coumaric acid as substrate.
To confirm that the determined T50 value is a well suited measure for prediction of the applicability of the enzymes for the 3D printing process, agarose bioinks were prepared from the four enzymes (2 μm) and used for printing of reaction modules (0.97 nmol enzyme per module) with geometries as described previously.6 By employment of the flow reactor setup (Figure 3 A), the reaction modules were perfused with 0.125 mm p‐coumaric acid in reaction buffer at a constant flow rate of 12.5 μL min−1. Samples of the reactor outflow were taken at regular time intervals and analysed by HPLC (Figure S6). As expected from the T50 measurements, the reactor modules containing LpPAD, BmPAD or LbPAD produced no detectable amounts of 4‐vinylphenol from p‐coumaric acid, whereas the EsPAD‐containing modules (with 38±15 % enzyme activity remaining after the printing process) converted up to 78 % of the available p‐coumaric acid under these conditions (Figure 3 B). The observed time‐dependent decrease of the conversion rate has been observed before and is related to the leaching of the encapsulated enzyme.6
Figure 3.
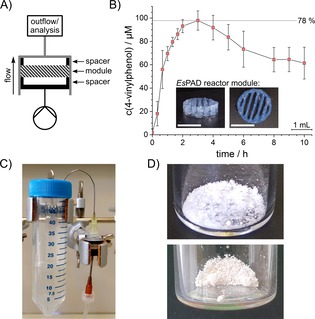
A) Schematic setup of the flow reactor for flow experiments with one reactor module. B) Conversion of p‐coumaric acid (1) in a biocatalytic flow setup employing EsPAD containing reaction modules at a constant flow rate of 12.5 μL min−1 to 4‐vinylphenol (2) quantified by HPLC in the outflow. Inset: 3D‐printed reactor modules prior to assembly into the reactor. White bar indicates 10 mm. In a reaction employing 0.125 mm substrate, conversion rates of 50–78 % were observed for more than 10 h after an initial equilibration phase. The standard deviation indicates two independent experiments. C) Flow reactor setup of one out of four parallel reactors to produce 4‐vinylphenol. The flow reactor next to the 50 mL collection tube is perfused with substrate solution by a syringe pump. For more detailed information see Figure S8. D) The purified 4‐vinylphenol (2) (top) and, after subsequent Heck‐coupling, the purified 4‐hydroxystilbene (3) (bottom) are obtained as white crystals.
To improve the system towards full conversion, we increased the enzyme concentration inside the bioink to 100 μm EsPAD. Under these reaction conditions, 30 mL of 1 mm p‐coumaric acid were converted over 40 h with efficiencies of up to 98 % (Figure S7). After this process optimization, we could simply parallelize four identical flow reactors, each equipped with a 100 μm EsPAD reactor module (48.3 nmol enzyme per module, Figures 3 C and S8). This increase in scale by numbering‐up led to a conversion of 35 mg p‐coumaric acid (1) to 4‐vinylphenol (2) in a total volume of 211 mL and with an average total turnover of 590 4‐vinylphenol molecules per enzyme.
Following to extraction of the collected outflows and a quick chromatographic purification of the raw product, solid 4‐vinylphenol (2) was obtained with an isolated yield of 54 % (Figure 3 D). According to the reaction shown in Scheme 1, the purified 4‐vinylphenol (2) was then coupled with iodobenzene (4) by using a palladium‐catalysed Heck reaction. This led to the formation of 4‐hydroxystilbene (3), obtained as white crystals in a total yield of about 15 % (Figure 3 D).
In summary, we here demonstrate for the first time that unmodified EsPAD can be used in the production of bioinks, which are suitable for direct 3D printing of reactor modules using additive manufacturing processes without post‐processing steps. We also directly compared four different phenacrylate decarboxylases and identified EsPAD as the enzyme with the highest thermostability and the greatest turnover of the PAD enzymes tested. After optimization of reaction parameters, 3D‐printed modules were readily prepared and used for continuous flow production of 4‐vinylphenol (2) from p‐coumaric acid (1). Scale‐up of the reaction was conveniently achieved by numbering‐up of the reactors without further process optimization. Such processes can be integrated into chemoenzymatic reaction cascades, as shown here using the example of the subsequent palladium‐catalysed formation of 4‐hydroxystilbene (3). As shown here, new enzyme classes can be easily screened for enzymes compatible with the 3D bioprinting presented. The high modularity as well as scalability of the reactor system and the ease of its manufacturing can then be utilized to introduce these new enzyme classes into flow reaction systems.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Helmholtz programme BioInterfaces in Technology and Medicine. We thank Manfred Maier for the support, advice and assistance with the 3D printing process and the fluidic setup.
M. Peng, E. Mittmann, L. Wenger, J. Hubbuch, M. K. M. Engqvist, C. M. Niemeyer, K. S. Rabe, Chem. Eur. J. 2019, 25, 15998.
Contributor Information
Martin Peng, https://www.ibg.kit.edu/ibg1/.
Dr. Kersten S. Rabe, Email: kersten.rabe@kit.edu.
References
- 1. Straathof A. J., Chem. Rev. 2014, 114, 1871–1908. [DOI] [PubMed] [Google Scholar]
- 2. Bornscheuer U. T., Huisman G. W., Kazlauskas R. J., Lutz S., Moore J. C., Robins K., Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Rabe K. S., Müller J., Skoupi M., Niemeyer C. M., Angew. Chem. Int. Ed. 2017, 56, 13574–13589; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13760–13777; [Google Scholar]
- 3b. France S. P., Hepworth L. J., Turner N. J., Flitsch S. L., ACS Catal. 2017, 7, 710–724; [Google Scholar]
- 3c. Quin M. B., Wallin K. K., Zhang G., Schmidt-Dannert C., Org. Biomol. Chem. 2017, 15, 4260–4271; [DOI] [PubMed] [Google Scholar]
- 3d. Wheeldon I., Minteer S. D., Banta S., Barton S. C., Atanassov P., Sigman M., Nat. Chem. 2016, 8, 299–309; [DOI] [PubMed] [Google Scholar]
- 3e. Küchler A., Yoshimoto M., Luginbuhl S., Mavelli F., Walde P., Nat. Nanotechnol. 2016, 11, 409–420; [DOI] [PubMed] [Google Scholar]
- 3f. Chen Z., Zeng A. P., Curr. Opin. Biotechnol. 2016, 42, 198–205. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Sheldon R. A., van Pelt S., Chem. Soc. Rev. 2013, 42, 6223–6235; [DOI] [PubMed] [Google Scholar]
- 4b. Meller K., Szumski M., Buszewski B., Sens. Actuators B 2017, 244, 84–106. [Google Scholar]
- 5.
- 5a. Peschke T., Bitterwolf P., Gallus S., Hu Y., Oelschlaeger C., Willenbacher N., Rabe K. S., Niemeyer C. M., Angew. Chem. Int. Ed. 2018, 57, 17028–17032; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 17274–17278; [Google Scholar]
- 5b. Peschke T., Bitterwolf P., Hansen S., Gasmi J., Rabe K. S., Niemeyer C. M., Catalysts 2019, 9, 164; [Google Scholar]
- 5c. Peschke T., Skoupi M., Burgahn T., Gallus S., Ahmed I., Rabe K. S., Niemeyer C. M., ACS Catal. 2017, 7, 7866–7872; [Google Scholar]
- 5d. Contente M. L., Paradisi F., Nat. Catal. 2018, 1, 452–459; [Google Scholar]
- 5e. Bitterwolf P., Gallus S., Peschke T., Mittmann E., Oelschlaeger C., Willenbacher N., Rabe K. S., Niemeyer C. M., Chem. Sci. 2019, 10, 9752–9757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maier M., Radtke C. P., Hubbuch J., Niemeyer C. M., Rabe K. S., Angew. Chem. Int. Ed. 2018, 57, 5539–5543; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5638–5642. [Google Scholar]
- 7. Ngo T. D., Kashani A., Imbalzano G., Nguyen K. T. Q., Hui D., Composites Part B 2018, 143, 172–196. [Google Scholar]
- 8.
- 8a. Joshi S. C., Sheikh A. A., Virtual and Physical Prototyping 2015, 10, 175–185; [Google Scholar]
- 8b. Wu P., Wang J., Wang X., Autom. Construct. 2016, 68, 21–31; [Google Scholar]
- 8c. Khoo Z. X., Teoh J. E. M., Liu Y., Chua C. K., Yang S., An J., Leong K. F., Yeong W. Y., Virtual and Physical Prototyping 2015, 10, 103–122; [Google Scholar]
- 8d. Prasad L. K., Smyth H., Drug Dev. Ind. Pharm. 2016, 42, 1019–1031; [DOI] [PubMed] [Google Scholar]
- 8e. Krujatz F., Lode A., Seidel J., Bley T., Gelinsky M., Steingroewer J., New Biotechnol. 2017, 39, 222–231. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Parra-Cabrera C., Achille C., Kuhn S., Ameloot R., Chem. Soc. Rev. 2018, 47, 209–230; [DOI] [PubMed] [Google Scholar]
- 9b. Kazenwadel F., Biegert E., Wohlgemuth J., Wagner H., Franzreb M., Eng. Life Sci. 2016, 16, 560–567. [Google Scholar]
- 10. Ozbolat I. T., Hospodiuk M., Biomaterials 2016, 76, 321–343. [DOI] [PubMed] [Google Scholar]
- 11. Schmieg B., Döbber J., Kirschhöfer F., Pohl M., Franzreb M., Front. Bioengin. Biotechnol. 2018, 6, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Lee B. B., Ravindra P., Chan E. S., Chem. Eng. Technol. 2013, 36, 1627–1642; [Google Scholar]
- 12b. Nair S., Kim J., Crawford B., Kim S. H., Biomacromolecules 2007, 8, 1266–1270; [DOI] [PubMed] [Google Scholar]
- 12c. Nunes M. A. P., Martins S., Rosa M. E., Gois P. M. P., Fernandes P. C. B., Ribeiro M. H. L., Bioresour. Technol. 2016, 213, 208–215. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Bhusainahalli V. M., Spatafora C., Chalal M., Vervandier-Fasseur D., Meunier P., Latruffe N., Tringali C., Eur. J. Org. Chem. 2012, 5217–5224; [Google Scholar]
- 13b. Bassanini I., D′Annessa I., Costa M., Monti D., Colombo G., Riva S., Org. Biomol. Chem. 2018, 16, 3741–3753. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Rodríguez H., Angulo I., d. Las Rivas B., Campillo N., Páez J. A., Muñoz R., Mancheño J. M., Proteins Struct. Funct. Bioinf. 2010, 78, 1662–1676; [DOI] [PubMed] [Google Scholar]
- 14b. Gu W., Yang J., Lou Z., Liang L., Sun Y., Huang J., Li X., Cao Y., Meng Z., Zhang K.-Q., PLoS One 2011, 6, e16262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aleku G. A., Prause C., Bradshaw-Allen R. T., Plasch K., Glueck S. M., Bailey S. S., Payne K. A. P., Parker D. A., Faber K., Leys D., ChemCatChem 2018, 10, 3736–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Furuya T., Kuroiwa M., Kino K., J. Biotechnol. 2017, 243, 25–28; [DOI] [PubMed] [Google Scholar]
- 16b. Ni J., Wu Y. T., Tao F., Peng Y., Xu P., J. Am. Chem. Soc. 2018, 140, 16001–16005. [DOI] [PubMed] [Google Scholar]
- 17. Chen P.-Y., Wu Y.-H., Hsu M.-H., Wang T.-P., Wang E.-C., Tetrahedron 2013, 69, 653–657. [Google Scholar]
- 18. Gómez Baraibar A., Reichert D., Mugge C., Seger S., Groger H., Kourist R., Angew. Chem. Int. Ed. 2016, 55, 14823–14827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15043–15047. [Google Scholar]
- 19.
- 19a. Sterner R., Liebl W., Crit. Rev. Biochem. Mol. Biol. 2001, 36, 39–106; [DOI] [PubMed] [Google Scholar]
- 19b. Solanki K., Abdallah W., Banta S., Biotechnol. J. 2016, 11, 1483–1497; [DOI] [PubMed] [Google Scholar]
- 19c. Li G., Rabe K. S., Nielsen J., Engqvist M. K. M., ACS Synth. Biol. 2019, 8, 1411–1420. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Buss O., Muller D., Jager S., Rudat J., Rabe K. S., ChemBioChem 2018, 19, 379–387; [DOI] [PubMed] [Google Scholar]
- 20b. Yang H., Liu L., Li J., Chen J., Du G., ChemBioEng. Rev. 2015, 2, 87–94; [Google Scholar]
- 20c. Modarres H. P., Mofrad M. R., Sanati-Nezhad A., RSC Adv. 2016, 6, 115252–115270; [Google Scholar]
- 20d. Steipe B., Methods Enzymol. 2004, 388, 176–186; [DOI] [PubMed] [Google Scholar]
- 20e. Bommarius A. S., Paye M. F., Chem. Soc. Rev. 2013, 42, 6534–6565; [DOI] [PubMed] [Google Scholar]
- 20f. Lehmann M., Wyss M., Curr. Opin. Biotechnol. 2001, 12, 371–375; [DOI] [PubMed] [Google Scholar]
- 20g. Steiner K., Schwab H., Comput. Struct. Biotechnol. J. 2012, 2, e201209010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20h. Wijma H. J., Floor R. J., Janssen D. B., Curr. Opin. Struct. Biol. 2013, 23, 588–594. [DOI] [PubMed] [Google Scholar]
- 21. Gu W., Li X., Huang J., Duan Y., Meng Z., Zhang K.-Q., Yang J., Appl. Microbiol. Biotechnol. 2011, 89, 1797–1805. [DOI] [PubMed] [Google Scholar]
- 22. Rodríguez H., Landete J. M., Curiel J. A., d. Las Rivas B., Mancheño J. M., Muñoz R., J. Agric. Food Chem. 2008, 56, 3068–3072. [DOI] [PubMed] [Google Scholar]
- 23. Landete J. M., Rodríguez H., Curiel J. A., d. Las Rivas B., Mancheño J. M., Muñoz R., J. Ind. Microbiol. Biotechnol. 2010, 37, 617–624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary