

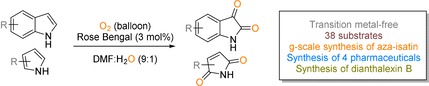
Abstract
Dearomatisation of indole derivatives to the corresponding isatin derivatives has been achieved with the aid of visible light and oxygen. It should be noted that isatin derivatives are highly important for the synthesis of pharmaceuticals and bioactive compounds. Notably, this chemistry works excellently with N‐protected and protection‐free indoles. Additionally, this methodology can also be applied to dearomatise pyrrole derivatives to generate cyclic imides in a single step. Later this methodology was applied for the synthesis of four pharmaceuticals and a pesticide called dianthalexin B. Detailed mechanistic studies revealed the actual role of oxygen and photocatalyst.
Keywords: heterocycles, homogeneous catalysis, oxygenation, pharmaceuticals, photocatalysis
Heterocycles: Dearomatisation of indole derivatives to the corresponding isatin derivatives has been achieved with the aid of visible light and oxygen. Additionally, this methodology can also be applied to dearomatise pyrrole derivatives to generate cyclic imides in a single step (see scheme). Detailed mechanistic studies revealed the actual role of oxygen and photocatalyst.
Dearomatisation of heteroaromatic compounds to the value‐added products is a powerful strategy for the synthesis of pharmaceuticals and bioactive compounds.1 In fact, most natural products contain heteroaromatic nuclei such as indole, pyrrole etc.2 Therefore, it is of significant value to dearomatise indole and pyrrole nuclei to the value‐added products, under mild reaction conditions. However, owing to their highly aromatic resonance energy, suitable tuning of the catalysts or reaction conditions is highly necessary.3 Based on this concept, dearomatisation of indole derivatives has been achieved through borylation, hydrosilylation, trifluoromethylation, arylation, allylation, etc.4 Very recently, Vincent et al. has reported an excellent electrochemical dialkoxylation method for the dearomatisation of indoles (Figure 1).5 However, all of these dearomatisation methodologies worked towards N‐protected indole derivatives and did not show any activity towards free indole or pyrrole derivatives. Additionally, an over‐stoichiometric amount of reagents or additives were required and this generated an over‐stoichiometric amount of by‐products. Therefore, to reach the practical application of this dearomatisation methodology, catalysts should work towards the free indoles (as most of the natural products contain N‐protection‐free indole or pyrrole nuclei) and the formation of by‐products should be minimized. Over the past two decades, visible‐light‐mediated organic synthesis has been of top priority for the development of sustainable chemistry under mild conditions.6 In fact, with the aid of visible light, activation of oxygen molecules and generation of reactive oxygen species (ROS) has become much easier.7 Recently, we have become interested in the visible‐light‐mediated activation of small molecules such as oxygen and carbon dioxide.8 Based on our experience in visible‐light‐mediated chemistry, we were interested in applying these ROS to dearomatise different indole and pyrrole derivatives to obtain isatin and cyclic imide derivatives in a single step. Indeed, isatins and cyclic imides are essential building blocks to the synthetic chemists as well as pharmaceutical industries.9 Many of these derivatives have been utilized as antimicrobial, antitumor, antitubercular, antimalarial, anti‐HIV, antibacterial, antiviral drugs, etc.10
Figure 1.
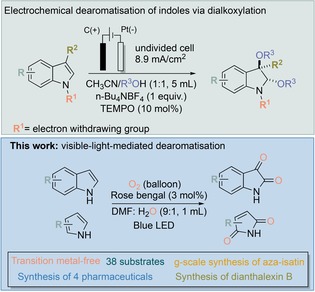
Dearomatisation of indole through electrochemistry and visible light (our work).
It should be noted that Jiang et al. has recently developed an excellent dicyanopyrazine derivative as a photocatalyst for the oxygenation of indole to isatin.11 However, the catalyst needs to be synthesized, which requires expensive starting materials and reagents and multiple steps. Therefore, development of an efficient and selective strategy for the synthesis of these scaffolds will always be welcome in organic synthesis. Additionally, if this synthetic method can be developed in a transition‐metal‐free way, it will enhance the journey of sustainability in organic synthesis.12
Based on all this information, we developed a transition metal‐free methodology for the dearomatisation of indole and pyrrole derivatives to obtain isatin and imide derivatives in a single step with the aid of visible light. At the outset of our reaction, we focused on the organic dyes, which have high reduction potential of the excited state to generate superoxide radical anion (O2/O2 .−) with the reduction potential residing at −0.56 V vs. SCE.8 In general, rose bengal, fluorenone, eosin Y, xanthone, and rhodamine 6G have high excited‐state reduction potentials for the generation of superoxide radical anion.13 Moreover, these organic dyes are cheap and commercially available. Based on this information, 1H‐indole (1 a) was taken as the model substrate to optimize the reaction conditions under oxygen atmosphere. As shown in Table S1 (see the Supporting Information), different metal‐free catalysts, such as rose bengal, fluorenone, eosin Y, xanthone, riboflavin, and rhodamine 6G, were screened with DMSO as the solvent under the irradiation of a 12 W blue LED for 18 h (Table S1, entries 1–7, Supporting Information). To our delight, rose bengal provided a 12 % yield of the corresponding isatin (1 b). Further addition of water (20 vol %) and screening of different solvents improved the yield to 30 % in DMF. Additional screening of the amount of water (10 vol %) resulted in 52 % of the corresponding product in 18 h. Finally, 93 % of the product was achieved after 24 h irradiation under blue LED.
With these optimized reaction conditions in hand, we applied our catalytic system for the dearomatisation of different indole derivatives (Scheme 1; entries 1 b–16 b). Indeed, different substitution patterns in the aromatic ring could not hinder the reaction's yield (Scheme 1; entries 3 b–11 b; 13 b). Gratifyingly, N‐protected indoles also worked splendidly and up to 84 % product was isolated (Scheme 1; entries 12 b–16 b). A longer chain and benzyl groups as the N‐protecting groups were well tolerated under the optimized reaction conditions and generated the corresponding isatin derivatives in excellent yields (Scheme 1; entries 14 b–15 b). In addition, carboxylated indole derivatives underwent decarboxylation reactions and generated the corresponding isatin derivatives with up to a 98 % yield (Scheme 2, entries 17 a–19 a). In fact, both the C2‐ and C3‐substituted indole carboxylic acid generated the corresponding isatin in high yield (Scheme 2, entries 17 a–18 a). However, in the case of N‐protected indole carboxylic acid (19 a), the decarboxylation reaction was quite slow and the corresponding N‐methyl isatin was isolated to a lower yield after 48 h.
Scheme 1.
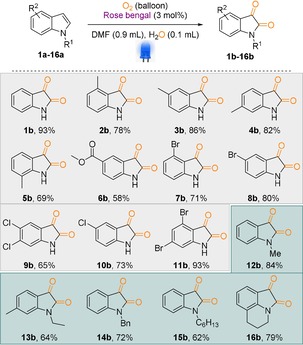
Substrates scope for the synthesis of isatin derivatives through dearomatisation of indoles. Reaction conditions: substrate (0.25 mmol), RB (3 mol %), DMF (0.9 mL), H2O (0.1 mL), reaction time 16–48 h; isolated yields.
Scheme 2.
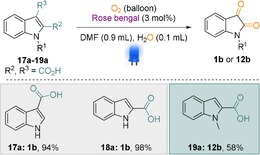
Dearomatisation of indoles via decarboxylation. Reaction conditions: substrate (0.25 mmol), RB (3 mol %), DMF (0.9 mL), H2O (0.1 mL), reaction time 48 h.
Inspired by the above substrate scope, we became interested in synthesizing 7‐azaisatin directly from 7‐azaindole (Scheme 3). 7‐Azaisatin derivatives are well‐known to exhibit anticancer or TrkA kinase inhibitory activities.14 Additionally, these compounds have been utilized in asymmetric synthesis of spirocyclic indolines and spirocyclic oxoindole compounds.15 It should be noted that 7‐azaisatin is a highly expensive molecule (486 € g−1). Therefore, direct synthesis of this compound from a cheap precursor (7‐azaindole, 12 € g−1) will be highly applicable in synthetic chemistry. To our delight, our optimized reaction conditions generated 7‐azaisatin under O2 atmosphere in 74 % isolated yield. Later, g‐scale synthesis of this compound was also achieved under the similar reaction conditions.
Scheme 3.
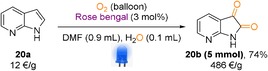
Gram‐scale synthesis of 7‐azaisatin directly from 7‐azaindole. Reaction conditions: substrate (0.25 mmol), RB (3 mol %), DMF (0.9 mL), H2O (0.1 mL), reaction time 45 h.
Following the dearomatisation of the indole derivatives, we became interested in applying our catalysts onto pyrrole derivatives to generate the corresponding cyclic imides. In general, cyclic imides and their derivatives are key building blocks for the natural product synthesis and intermediates for the synthesis of alkaloids.16 Additionally, they have been applied in pharmaceuticals, polymers, dyes and chemical industry. Despite the fact that numerous methodologies exist for the synthesis of these molecules, a novel method for the synthesis of imides directly from cheap and commercially available starting materials in a catalytic way should render a new synthetic strategy in organic synthesis.17 Based on this, we applied our optimized reaction conditions onto 3,4‐diethyl‐1H‐pyrrole and the corresponding cyclic imide was isolated in 75 % yield (Scheme 4, entry 21 b). Inspired by this result, we applied it to other pyrrole derivatives and in all cases, we isolated the corresponding products in good to excellent yield (Scheme 4, entries 22 b–26 b). It should be noted that the catalyst also worked similarly in both the symmetric and nonsymmetric backbone‐substituted pyrrole derivatives (Scheme 4, entries 21 b–23 b).
Scheme 4.
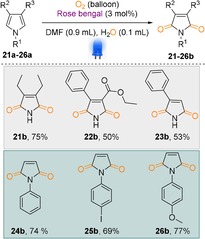
Dearomatisation of pyrroles to cyclic imides. Reaction conditions: substrate (0.25 mmol), RB (3 mol %), DMF (0.9 mL), H2O (0.1 mL), reaction time 18 h; isolated yields.
Although, different indole and pyrrole derivatives generated isatin and imide derivatives, C2‐ and C3‐substituted indole derivatives underwent oxidative cleavage products and generated ortho‐formyl/acyl anilide derivatives (Scheme 5). In fact, these ortho‐formyl/acyl anilide derivatives are highly essential structural motifs for the synthesis of natural products and bioactive compounds.18 Traditional synthesis of these compounds requires expensive transition‐metal catalysts or over‐stoichiometric amounts of oxidants.19 Based on this information, we further optimized this oxidative cleavage product and to our delight in the presence of K3PO4 (50 mol %) up to 92 % of these cleavage products were obtained. This type of cleavage worked equally in both the cases of free indole and N‐protected indole derivatives (Scheme 5, entries 27 b–33 b).
Scheme 5.
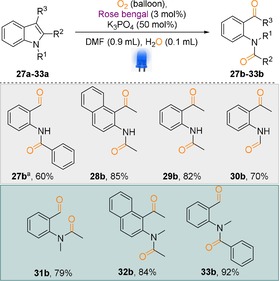
Dearomatisation of C2‐ and C3‐substituted indoles: synthesis of ortho‐formyl/acyl anilide derivatives. Reaction conditions: substrate (0.25 mmol), RB (3 mol %), K3PO4 (50 mol %), DMF (0.9 mL), H2O (0.1 mL), reaction time 45 h; isolated yields. [a] Without K3PO4.
Later, we became interested in the direct implementation of our dearomatisation protocol for the efficient transformation of indole derivatives to more complex scaffolds. To achieve this, several pharmaceuticals were synthesized (Scheme 6). Notably, the dearomatised product (1 b) from indole can be transformed into an anticancer compound (34 b) by reacting with 2‐aminobenzenethiol at 85 °C.20 Furthermore, we were able to transfer 5‐chloroindole to the corresponding steroid based antitumour compound (35 b).21 Additionally, 1H‐indole was transformed into an anticonvulsant and anti‐inflammatory compound (36 b) through dearomatisation and the addition of aniline.22 Furthermore, this anti‐inflammatory compound (36 b) was transformed into antiviral compound (37 b) in the presence of additional indole.23 Finally, we applied the oxidative cleavage protocol for the synthesis of dianthalexin B, a well‐known pesticide (Scheme 7, 38 b) and to our delight dianthalexin B was isolated with an overall yield of 48 % starting from the 2‐phenyl indole.24
Scheme 6.
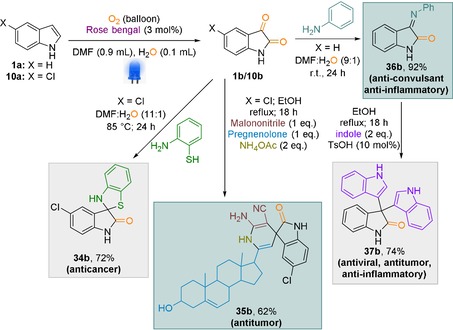
Synthesis of pharmaceuticals through dearomatisation of indole derivatives.
Scheme 7.
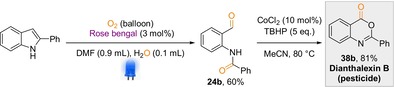
Synthesis of dianthalexin B via oxidative cleavage of 2‐phenyl indole.
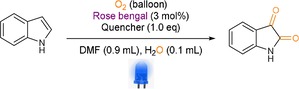
After obtaining a wide substrate scope and applications of this chemistry, we became interested in the underlying mechanism of this reaction. Initial control experiments showed that the presence of oxygen, light, and water were crucial for this reaction (Table 1).[8] Additionally, different quenchers showed that the reaction was based on the radical pathway (Table 1, entries 6 and 7). The addition of CuCl2 and benzoquinone proved the presence of a single electron transfer (SET) and superoxide radical anion species. The addition of sodium azide revealed the presence of singlet oxygen species. To investigate further, we performed Stern–Volmer quenching experiments (see the Supporting Information, Figure S3–S6).25 In fact, only oxygen and water were quenching the excited state of our photocatalyst and not the indole. Additionally, labelling experiments showed the incorporation of 18O‐labelled oxygen from 18O2 and also from H2 18O which suggested complex interaction of oxygen atmosphere and water to form the reactive oxygen species (see the Supporting Information).
Table 1.
Control and quenching experiments for the mechanistic investigation.
|
| |||
|---|---|---|---|
|
Entry |
Conditions or changes[a] |
Quencher[b] |
Yield [%][c] |
|
1 |
N2‐atmosphere |
none |
n.r. |
|
2 |
No light |
none |
n.r. |
|
3 |
No water |
none |
n.r. |
|
4 |
STD |
BHT |
35 |
|
5 |
STD |
TEMPO |
23 |
|
6 |
STD |
tert‐butanol |
87 |
|
7 |
STD |
CuCl2 |
29 |
|
8 |
STD |
benzoquinone |
32 |
|
9 |
STD |
sodium azide |
0 |
[a] Changes to standard conditions (STD); [b] 1.0 equiv. quenchers; [c] Yield determined by GC with n‐dodecane as internal standard.
Based on all these mechanistic experiments, the following reaction mechanism can be proposed (Scheme 8). Rose bengal was excited by the light source and the excited state of the photocatalyst was quenched by the oxygen to form singlet oxygen which is a well‐known reaction in photochemistry.26 The formed singlet oxygen then combined with the indole (I), to generate the corresponding peroxo species (II) which was then further oxidized to (III) through a second photocatalytic cycle and formed radical anion of the photocatalyst (RB.−). The activated peroxoindole species (III) was cleaved apart to form the corresponding cation (IV) and a hydroxy radical. Later this hydroxy radical may have abstracted the hydrogen atom from (IV) and formed (V). Rose bengal radical anion (RB.−) reacted with oxygen to form a superoxide radical anion and made the catalytic turn over. The reported value for the reduction potential of the excited state of rose bengal resides at −1.33 V vs. SCE, which is sufficient for the reduction of molecular oxygen to its superoxide radical anion (O2/O2 .−) with the reduction potential residing at −0.56 V vs. SCE.12 The generated superoxide radical anion reacted with water to form the peroxide radicals and hydroxide anions.27 The radical species (V) and hydroxy radical reacted to form (VI). Finally, (VI) was transformed into the product (VII) by releasing the water molecule. The formed peroxide radical can alternatively initiate a further reaction cycle with the starting material by a typical oxidation pathway of hydrogen peroxide (detected during the experiments; see the Supporting Information). An additional direct reaction of intermediate (IV) with water is also possible, leading to a ketol intermediate, which can be further oxidized to an isatin product. However, this pathway should only contribute to a minor degree based on the performed 18O‐labelling experiments. Based on this reaction mechanism, the complex involvement of water and oxygen, which led to the mono‐ and di‐labelled formation of product in the labelling experiments, can be explained. In case of 2,3‐disubstituted indoles, indole substrates underwent side‐on coordination with the singlet oxygen, which led to the cleavage of the C−C‐bond.28
Scheme 8.
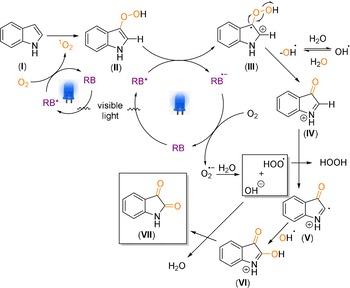
Proposed mechanism based on the experimental evidences.
In conclusion, we have provided an efficient dearomatisation methodology for the indole and pyrrole derivatives under mild reaction conditions. The products obtained in this way are highly valuable scaffold for the synthesis of fine chemicals, pharmaceuticals and bioactive compounds. Additionally, oxidative cleavage of C2‐ and C3‐substituted indole derivative generated highly important ortho‐formyl/acyl anilide derivatives. Notably, we have also shown further application of our methodology to the synthesis of pharmaceuticals and bioactive compound. At the end, detailed mechanistic experiments clearly showed the role of oxygen and water in this reaction. We are certain that synthetic chemists, as well as chemical biologists, will find this chemistry of great interest.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Fonds der Chemischen Industrie (FCI, Liebig‐Fellowship to S.D.) and Chinese Scholarship Council (CSC funding to Y.Z.) for the financial support. We are deeply thankful to Prof. Dr. Lutz Ackermann for his kind support of our work.
W. Schilling, Y. Zhang, D. Riemer, S. Das, Chem. Eur. J. 2020, 26, 390.
References
- 1.
- 1a. Eicher T., Hauptmann S., Speicher A., The Chemistry of Heterocycles: Structures, Reactions, Synthesis and Applications, Wiley-VCH, Weinheim, 2013; [Google Scholar]
- 1b. Pelletier S. W., Alkaloids: Chemical and Biological Perspective, Springer, Vol. 13, 1999. [Google Scholar]
- 2.
- 2a. Lachkar D., Denizot N., Bernadat G., Ahamada K., Beniddir M. A., Dumontet V., Gallard V., Guillot J.-F., Leblanc K., N′nang E. O., Turpin V., Kouklovsky C., Poupon E., Evanno L., Vincent G., Nat. Chem. 2017, 9, 793–798; [DOI] [PubMed] [Google Scholar]
- 2b. Tomakinian T., Guillot R., Kouklovsky C., Vinvent G., Angew. Chem. Int. Ed. 2014, 53, 11881–11885; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12075–12079; [Google Scholar]
- 2c. Ding H., DeRoy P. L., Perreault C., Larivée A., Siddiqui A., Caldwell C. G., Harran S., Harran P. G., Angew. Chem. Int. Ed. 2015, 54, 4818–4822; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4900–4904. [Google Scholar]
- 3.
- 3a. Balaban A. T., Oniciu D. C., Katritzky A. R., Chem. Rev. 2004, 104, 2777–2812; [DOI] [PubMed] [Google Scholar]
- 3b. Krygowski T. M., Cyrański M. K., Chem. Rev. 2001, 101, 1385–1420. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Chen L., Shen J.-J., Gao Q., Xu S., Chem. Sci. 2018, 9, 5855–5859; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Jayaraman A., Castro L. C. M., Desrosiers V., Fontaine F.-G., Chem. Sci. 2018, 9, 5057–5063; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Xiao Y.-C., Wang C., Yao Y., Sun J., Chen Y.-C., Angew. Chem. Int. Ed. 2011, 50, 10661–10664; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10849–10852; [Google Scholar]
- 4d. Han G., Wang Q., Chen L., Liu Y., Wang Q., Adv. Synth. Catal. 2016, 358, 561–566; [Google Scholar]
- 4e. Shen C., Liu R.-R., Fan R.-J., Li Y.-L., Xu T.-F., Gao J.-R., Jia Y.-X., J. Am. Chem. Soc. 2015, 137, 4936–4939; [DOI] [PubMed] [Google Scholar]
- 4f. Kimura M., Futamata M., Mukai R., Tamaru Y., J. Am. Chem. Soc. 2005, 127, 4592–4593. [DOI] [PubMed] [Google Scholar]
- 5. Wu J., Dou Y., Guillot R., Kouklovsky C., Vincent G., J. Am. Chem. Soc. 2019, 141, 2832–2837. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Ghosh I., Ghosh T., Bardagi J. I., König B., Science 2014, 346, 725–728; [DOI] [PubMed] [Google Scholar]
- 6b. Romero N. A., Margrey K. A., Tay N. E., Nicewicz D. A., Science 2015, 349, 1326–1330. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Nicewicz D. A., Nguyen T. M., ACS Catal. 2014, 4, 355–360; [Google Scholar]
- 7b. Nosaka Y., Nosaka A. Y., Chem. Rev. 2017, 117, 11302–11336. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Riemer D., Schilling W., Goetz A., Zhang Y., Gehrke S., Tkach I., Hollóczki O., Das S., ACS Catal. 2018, 8, 11679–11687; [Google Scholar]
- 8b. Zhang Y., Riemer D., Schilling W., Kollmann J., Das S., ACS Catal. 2018, 8, 6659–6664; [Google Scholar]
- 8c. Schilling W., Riemer D., Zhang Y., Hatami N., Das S., ACS Catal. 2018, 8, 5425–5430; [Google Scholar]
- 8d. Kollmann J., Zhang Y., Schilling W., Riemer D., Das S., Green Chem. 2019, 21, 1916–1920. [Google Scholar]
- 9.
- 9a. Varun, Sonam, Kakkar R., Med. Chem. Commun. 2019, 10, 351–368; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Moradi R., Ziarani G. M., Lashgari N., ARKIVOC 2017, 2017, 148–201; [Google Scholar]
- 9c. Hassanzadeh F., Jafari E., J. Res. Med. Sci. 2018, 23, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Sassatelli M., Debiton E., Aboab B., Prudhomme M., Moreau P., Eur. J. Med. Chem. 2006, 41, 709–716; [DOI] [PubMed] [Google Scholar]
- 10b. Vine K. L., Locke J. M., Ranson M., Pyne S. G., Bremner J. B., J. Med. Chem. 2007, 50, 5109–5117; [DOI] [PubMed] [Google Scholar]
- 10c. Sumpter W. C., Chem. Rev. 1944, 34, 393–434; [Google Scholar]
- 10d. Raj R., Carrère-Kremer S., Kremer L., Guérardel Y., Gut J., Rosenthal P. J., Forge D., Kumar V., Chem. Biol. Drug Des. 2014, 83, 622–629; [DOI] [PubMed] [Google Scholar]
- 10e. Raj R., Gut J., Rosenthal P. J., Kumar V., Bioorg. Med. Chem. Lett. 2014, 24, 756–759; [DOI] [PubMed] [Google Scholar]
- 10f. Jarrahpour A., Khalili D., De Clercq E., Salmi C., Brunel M., Molecules 2007, 12, 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang C., Li S., Bureš F., Lee R., Ye X., Jiang Z., ACS Catal. 2016, 6, 6853–6860. [Google Scholar]
- 12.
- 12a. Hirapara P., Riemer D., Hazra N., Gajera J., Das S., Green Chem. 2017, 19, 5356–5360; [Google Scholar]
- 12b. Riemer D., Hirapara P., Das S., ChemSusChem 2016, 9, 1916–1920; [DOI] [PubMed] [Google Scholar]
- 12c. Zhang Y., Schilling W., Das S., ChemSusChem 2019, 12, 2898–2910; [DOI] [PubMed] [Google Scholar]
- 12d. Sun C.-L., Shi Z.-J., Chem. Rev. 2014, 114, 9219–9280; [DOI] [PubMed] [Google Scholar]
- 12e. Riemer D., Mandaviya B., Schilling W., Götz A. C., Kühl T., Finger M., Das S., ACS Catal. 2018, 8, 3030–3034. [Google Scholar]
- 13. Romero N., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- 14. Rekulapally S., Jarapula R., Gangarapu K., Manda S., Vaidya J. R., Med. Chem. Res. 2015, 24, 3412–3422. [Google Scholar]
- 15.
- 15a. Zhou Z., Wang Z.-X., Zhou Y.-C., Xiao W., Ouyang Q., Du W., Chen Y.-C., Nat. Chem. 2017, 9, 590–594; [DOI] [PubMed] [Google Scholar]
- 15b. Cheng X., Merz K.-H., Vatter S., Christ J., Wölfl S., Eisenbrand G., Bioorg. Med. Chem. 2014, 22, 247–255. [DOI] [PubMed] [Google Scholar]
- 16. Booker-Milburn K. I., Anson C. E., Clissold C., Costin N. J., Dainty R. F., Murray M., Pantel D., Sharpe A., Eur. J. Org. Chem. 2001, 1473–1482. [Google Scholar]
- 17.
- 17a. Zhang J., Senthikumar M., Ghosh S. C., Hong S. H., Angew. Chem. Int. Ed. 2010, 49, 6391–6395; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6535–6539; [Google Scholar]
- 17b. Takebayashi S., John J. M., Bergens S. H., J. Am. Chem. Soc. 2010, 132, 12832–12834; [DOI] [PubMed] [Google Scholar]
- 17c. Driller K. M., Klein H., Jackstell R., Beller M., Angew. Chem. Int. Ed. 2009, 48, 6041–6044; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6157–6160. [Google Scholar]
- 18.
- 18a. Ogita H., Isobe Y., Takaku H., Sekine R., Goto Y., Misawa S., Hayashi H., Bioorg. Med. Chem. 2002, 10, 3473–3480; [DOI] [PubMed] [Google Scholar]
- 18b. Marco-Contelles J., Perez-Mayoral E., Samadi A., do Camo Carreiras M., Soriano E., Chem. Rev. 2009, 109, 2652–2671. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Fang P., Li M., Ge H., J. Am. Chem. Soc. 2010, 132, 11898–11899; [DOI] [PubMed] [Google Scholar]
- 19b. Chan C.-W., Zhou Z., Yu W.-Y., Adv. Synth. Catal. 2011, 353, 2999–3006; [Google Scholar]
- 19c. Yuan Y., Chen D., Wang X., Adv. Synth. Catal. 2011, 353, 3373–3379. [Google Scholar]
- 20. Suresh B., Brahmeshwary G., Swamy T., Gopi I., Ravindar V., Russ. J. Gen. Chem. 2016, 86, 1144–1150. [Google Scholar]
- 21. Zhang Y.-L., Li Y.-F., Wang J.-W., Yu B., Shi Y.-K., Liu H.-M., Steroids 2016, 109, 22–28. [DOI] [PubMed] [Google Scholar]
- 22. Vine K. L., Locke J. M., Ranson M., Pyne S. G., Bremmer J. B., Bioorg. Med. Chem. 2007, 15, 931–938. [DOI] [PubMed] [Google Scholar]
- 23. Karali N., Güzel Ö., Özsoy N., Özbey N., Salman A., J. Med. Chem. 2010, 53, 1068–1077. [DOI] [PubMed] [Google Scholar]
- 24. Yu J., Zhang-Negrerie D., Du Y., Eur. J. Org. Chem. 2016, 562–568. [Google Scholar]
- 25. Arias-Rotondo D. M., McCusker J. K., Chem. Soc. Rev. 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Bayer P., Pérez-Ruiz R., von Wangelin A. J., ChemPhotoChem 2018, 2, 559–570; [Google Scholar]
- 26b. Ghogare A. A., Greer A., Chem. Rev. 2016, 116, 9994–10034. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Vassilev P., Louwerse M. J., Baerends E. J., J. Phys. Chem. B 2005, 109, 23605–23610; [DOI] [PubMed] [Google Scholar]
- 27b. Morrison M. M., Roberts J. L., Sawyer D. T., Inorg. Chem. 1979, 18, 1971–1973. [Google Scholar]
- 28.
- 28a. Celaje J. A., Zhang D., Guerrero A. M., Selke M., Org. Lett. 2011, 13, 4846–4849; [DOI] [PubMed] [Google Scholar]
- 28b. Wasserman H. H., Lipshutz B. H., Tremper A. W., We J. S., J. Org. Chem. 1981, 46, 2991–2999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary