

Abstract
Stress impairs extinction learning, and these deficits depend, in part, on stress-induced norepinephrine (NE) release in the basolateral amygdala (BLA). For example, systemic or intra-BLA administration of propranolol reduces the immediate extinction deficit (IED), an impairment in extinction learning that occurs when extinction trials are administered soon after fear conditioning. Here, we explored whether locus coeruleus (LC)-NE regulates stress-induced changes in spike firing in the BLA and consequent extinction learning impairments. Rats were implanted with recording arrays in the BLA and, after recovery from surgery, underwent a standard auditory fear conditioning procedure. Fear conditioning produced an immediate and dramatic increase in the spontaneous firing of BLA neurons that persisted (and in some units, increased further) up to an hour after conditioning. This stress-induced increase in BLA firing was prevented by systemic administration of propranolol. Conditioning with a weaker footshock caused smaller increases in BLA firing rate, but this could be augmented by chemogenetic activation of the LC. Conditioned freezing in response to a tone paired with a weak footshock was immune to the IED, but chemogenetic activation of the LC before the weak conditioning protocol increased conditioned freezing behavior and induced an IED; this effect was blocked with intra-BLA infusions of propranolol. These data suggest that stress-induced activation of the LC increases BLA spike firing and causes impairments in extinction learning. Stress-induced increases in BLA activity mediated by LC-NE may be a viable therapeutic target for individuals with stress- and trauma-related disorders.
SIGNIFICANCE STATEMENT Patients with post-traumatic stress disorder (PTSD) show heightened amygdala activity; elevated levels of stress hormones, including norepinephrine; and are resistant to the extinction of fear memories. Here, we show that stress increases basolateral amygdala (BLA) spike firing. This could be attenuated by systemic propranolol and mimicked by chemogenetic activation of the locus coeruleus (LC), the source of forebrain norepinephrine (NE). Finally, we show that LC-NE activation is sufficient to produce extinction deficits, and this is blocked by intra-BLA propranolol. Stress-induced increases in BLA activity mediated by LC-NE may be a viable therapeutic target for individuals with PTSD and related disorders.
Keywords: fear conditioning, extinction, locus coeruleus, norepinephrine, stress, amygdala
Introduction
Stress contributes to a number of psychiatric disorders, and it is well known that stress influences aversive learning processes that contribute to the development and maintenance of post-traumatic stress disorder (PTSD; O'Donnell et al., 2004; Morilak et al., 2005; Arnsten, 2009, 2015; Milad et al., 2009; Pervanidou and Chrousos, 2010; Parsons and Ressler, 2013; Raio et al., 2014; Arnsten et al., 2015; Maren and Holmes, 2016). For example, numerous studies demonstrate that either acute or chronic stress impairs the extinction of fear after Pavlovian conditioning (Izquierdo et al., 2006; Miracle et al., 2006; Chang and Maren, 2009; Wilber et al., 2011; MacPherson et al., 2013; Merz et al., 2014; Raio et al., 2014; Raio and Phelps, 2015; Maren and Holmes, 2016). Extinction learning is thought to mediate, in part, cognitive-behavioral therapies for PTSD, including exposure therapy, and patients with PTSD exhibit deficits in extinction learning (Wessa and Flor, 2007; Pitman et al., 2012; Garfinkel et al., 2014; Giustino et al., 2016; Maren and Holmes, 2016). In the laboratory, we, and others, have shown that extinction learning is impaired in humans and rodents when it occurs within minutes to hours of fear conditioning (Maren and Chang, 2006; Chang et al., 2010; Kim et al., 2010; Fitzgerald et al., 2015; Hollis et al., 2016; Maren and Holmes, 2016; Merz et al., 2016). Considerable evidence suggests that this “immediate extinction deficit” (IED) is mediated by footshock stress during fear conditioning itself (Maren and Chang, 2006; Chang et al., 2010; Fitzgerald et al., 2015; Maren and Holmes, 2016; Giustino et al., 2017). Importantly, the IED models extinction learning impairments in both rodents and humans in the aftermath of acute trauma, as well as extinction impairments associated with symptomatic stress in patients with PTSD (Maren and Chang, 2006; Rauch et al., 2006; Wessa and Flor, 2007; Milad et al., 2009; Raio et al., 2014; Fitzgerald et al., 2015; Giustino et al., 2016, 2017; Merz et al., 2016).
Previous work indicates that stress-induced extinction deficits are mediated by forebrain norepinephrine release in the medial prefrontal cortex (mPFC) and amygdala (Arnsten, 2009, 2015; Kim et al., 2010; Giustino et al., 2016, 2017; Giustino and Maren, 2018). Indeed, individuals with PTSD and related disorders present with elevated amygdala activity as well as heightened levels of neuromodulators, including norepinephrine (Southwick et al., 1999a,b; Milad et al., 2009; Giustino et al., 2016; Krystal et al., 2018). The locus coeruleus–norepinephrine (LC-NE) system heavily innervates the amygdala and is highly responsive to stress (Fallon et al., 1978; Foote et al., 1980; Loughlin et al., 1986; Jodo et al., 1998; Quirarte et al., 1998; Passerin et al., 2000; Buffalari and Grace, 2007; Chen and Sara, 2007; McCall et al., 2015, 2017; Naegeli et al., 2018; Giustino and Maren, 2018). Past work has demonstrated that LC projections to the amygdala are associated with increased fear and anxiety-like behavior (McCall et al., 2017; Uematsu et al., 2017), and noradrenergic blockade in the amygdala is sufficient to rescue stress-induced deficits in fear extinction (Giustino et al., 2017).
These data suggest the LC-NE system critically regulates amygdala activity, which may ultimately drive stress-induced extinction deficits via interactions with the mPFC (Giustino et al., 2019). To address this possibility, we combined single-unit basolateral amygdala (BLA) recordings with systemic pharmacology in freely moving rats to examine whether β-adrenoceptors mediate stress-induced changes in amygdala firing rates. We next combined single-unit amygdala recordings with LC-specific chemogenetic manipulations to determine whether LC-NE drives changes in amygdala single-unit activity. Last, we directly examined the contribution of the LC-NE system to the immediate extinction deficit.
Materials and Methods
Subjects
Eighty-one experimentally naive, adult, male, Long–Evans Blue Spruce rats (weighing 200–224 g; 50–57 d old) were obtained from a commercial supplier (Envigo). Upon arrival and throughout the experiments, rats were individually housed in cages within a humidity- and temperature-controlled vivarium and kept on a 14/10 h light/dark cycle (lights on at 7:00 A.M.) with ad libitum access to food and water. All experiments were conducted in the daytime during the light phase. Rats were handled for ∼30 s/d for 5 d to habituate them to the experimenter before any behavioral testing or surgical procedures were carried out. All procedures were conducted at Texas A&M University and were performed in strict accordance with the guidelines and regulations set forth by the National Institutes of Health and Texas A&M University with full approval from its Animal Care and Use Committee.
Experimental design and statistical analyses
The work reported here was conducted in a series of four independent experiments.
Experiment 1.
In the first experiment, we examined whether fear conditioning alters BLA spike firing rates and whether those changes are mitigated by systemic propranolol, a β-adrenoceptor antagonist. Rats were implanted with a 16-channel microelectrode array targeting the BLA. After recovery from surgery, the animals were transported to the recording room, and after a 3 min stimulus-free baseline period, rats were injected with vehicle (n = 5, 143 neurons) or propranolol (10 mg/kg, i.p., n = 5, 137 neurons). Twenty minutes later, rats received five conditioned stimulus–unconditioned stimulus (CS–US) pairings and remained in the chamber for 60 min following the last footshock.
A modified rodent behavioral chamber (30 × 24 × 21 cm, Med Associates) enclosed in a sound-attenuating cabinet was used for recording experiments (Experiments 1 and 2). This chamber was modified to allow for freely moving electrophysiological recordings as well. The chamber was composed of two aluminum side walls, a Plexiglas rear wall, a hinged Plexiglas door, and an open top. The grid floor consisted of 19 stainless steel rods (4 mm diameter) spaced 1.5 cm apart (center to center). A loudspeaker attached to the outside of a grating in one aluminum wall was used to play auditory tones. Locomotor activity of the rat was transduced by a load cell under the floor of the chamber, and the output of the load cell was recorded by an OmniPlex recording system (Plexon). Thus, all behavioral and neural activity was recorded automatically with this system.
Each rat was individually fear conditioned in Context A. In this procedure, the rat was transported to the room in a black plastic box, connected to a headstage with a flexible cable (OmniPlex, Plexon), and placed in the behavioral chamber. The chamber had been cleaned with 1% ammonium hydroxide to provide a distinct olfactory cue, and a black pan containing a thin layer of the same solution had been placed under the grid floor. The room was illuminated with red ambient lights (Context A). After a 3 min stimulus-free baseline period, the system was briefly paused while the animal remained plugged in and was injected with either vehicle or propranolol (10 mg/kg, i.p.). The rat was placed back in the chamber for 20 min to allow adequate time for the drug to take effect. The rat then received five auditory tone–footshock pairings. Recordings did not occur during shock presentation due to electrical noise. The tones (CS) were 2 s, 80 dB, 2 kHz; the shocks (US) were 0.5 s and 1 mA, where shock onset occurred at tone offset. There was a 1 min intertrial interval (ITI) between shocks. The session continued for 60 min after the final shock, and then the rat was returned to its home cage.
Extracellular single-unit activity was recorded with a multichannel neurophysiological recording system (Plexon). Wideband signals recorded on each channel were referenced to one of the recording wires (resulting in a maximum of 15 channels of activity per rat), amplified (8000×), digitized (40 kHz sampling rate), and saved on a PC for offline sorting and analysis. The recording reference wire we chose for each session was selected to optimize the quality of the recordings. After high-pass filtering the signal at 600 Hz, waveforms were manually sorted using two-dimensional principal component analysis (Offline Sorter, Plexon). Only well-isolated units were used in our analysis. If two units with similar waveforms and identical time stamps appeared on adjacent electrodes, we only used one unit. We then imported sorted waveforms and their time stamps to NeuroExplorer (Nex Technologies) for further analysis.
Experiment 2.
The second experiment examined whether chemogenetic activation of the LC fosters increases in BLA spike firing after footshock. This experiment followed a nearly identical protocol except that animals received infusions of an adenoviral vector expressing Gq-coupled designer receptor exclusively activated by designer drugs (DREADDs) into the LC (Vazey and Aston-Jones, 2014), in addition to a 16-channel microelectrode array targeting the BLA. We and others have previously validated this virus in vivo and have reported clozapine N-oxide (CNO)-mediated increases in LC firing lasting for up to 1 h (Vazey and Aston-Jones, 2014; Giustino et al., 2019). Animals were injected with vehicle (n = 5, 117 neurons) or CNO (3 mg/kg, i.p., n = 5, 116 neurons) after a 3 min baseline period and conditioned with five weak CS–US pairings 20 min later, and remained in the chamber for 60 min following the last shock to monitor neural and freezing behavior. The tones (CS) were 2 s, 80 dB, 2 kHz; the shocks (US) were 0.5 s, 0.5 mA, where shock onset occurred at tone offset.
The data were analyzed with conventional parametric statistics (StatView, SAS Institute). One-way and two-way ANOVA and repeated-measures ANOVA were used to assess general main effects and interactions (α = 0.05). Results are shown as mean ± SEM. χ2 analyses were used to examine differences in neuronal populations (i.e., neuron count split by excited and suppressed). Analyses for Experiments 1 and 2 used repeated-measures ANOVA with a within-subject variable of time and a between-subject variable of systemic drug for both freezing behavior and spontaneous firing rates. The spontaneous firing rate data were plotted in 20 s bins for the duration of the session, and the z-score was normalized to the entire preshock period.
Experiment 3.
The third experiment examined whether chemogenetic activation of the LC is sufficient to induce an immediate extinction deficit using a weak conditioning procedure that itself does not produce an extinction deficit. Rats received systemic vehicle (VEH) or clozapine N-oxide (CNO) injections ∼10 min before conditioning (Context A). After conditioning, animals underwent immediate (or no extinction (NO EXT)—context exposure only) procedures (Context B). Rats were tested for extinction (EXT) retrieval (Context B) 48 h later in a drug-free state. Group sizes were as follows: VEH-EXT, n = 11; CNO-EXT, n = 12; VEH-NO EXT, n = 8; CNO-NO EXT, n = 8.
Animals underwent similar behavioral procedures as described above for the recording experiments. On Day 1, rats were transported to the conditioning context (Context A) in squads of eight. Rats received either vehicle or CNO (3 mg/kg, i.p.) ∼5–10 min before fear conditioning. After a 3 min stimulus-free baseline period, all animals were conditioned with a single CS–US pairing using a weak footshock (0.5 s, 0.5 mA shock). Rats were removed from the conditioning chambers and underwent immediate extinction or no-extinction (i.e., Context B exposure, no CS presentation) procedures in Context B. Rats were transported to Context B in white transport boxes. The chamber was cleaned with 3% acetic acid, and a metal pan containing a thin layer of the same solution was placed under the grid floor. Ambient white lights illuminated the room. For animals undergoing immediate extinction, 45 CS-alone trials were presented (30 s ITI) after a 3 min stimulus-free baseline period. Rats undergoing no-extinction procedures remained in the chambers for the same amount of time, but no CS was presented. Animals were returned to their home cages at the end of the session. Animals were tested for extinction retrieval 48 h later. This extinction retrieval test was identical to the initial extinction session.
Experiment 4.
The fourth and last experiment examined whether the extinction deficit produced by chemogenetic LC activation is mediated by β-adrenoceptors in the BLA. Experiment 4 followed procedures nearly identical to Experiment 3, except that intracranial drug infusions occurred before fear conditioning. Animals were transported to an infusion room where they received intra-BLA vehicle or propranolol (PROP) intracranially (5 μg/μl, 0.5 μl/hemisphere) and systemic VEH or CNO infusions (in a 2 × 2 factorial design). Dummies were removed, and stainless steel injectors (33 gauge) connected to Hamilton syringes mounted to an infusion pump were inserted into the guide cannula for intracranial infusions. All infusions were administered ∼20 min before conditioning. Infusions were administered at a rate of 0.25 μl/min for 2 min, and the injectors were left in place for 1 min to allow for diffusion. After the infusions, clean dummies were secured to the guide cannula. Animals were then treated with either VEH or CNO (intraperitoneally) and underwent fear conditioning (Context A) 10 min later. Conditioning consisted of a 3 min preshock baseline period followed by three CS–US trials (0.5 s, 0.5 mA shocks). Immediate extinction (20 min after conditioning) and extinction retrieval (Context B) procedures were identical to the third experiment. Group sizes were as follows: VEH-VEH, n = 5; PROP-VEH, n = 6; VEH-CNO, n = 4; and PROP-CNO, n = 7.
The data were analyzed with conventional parametric statistics (StatView, SAS Institute). One-way and two-way ANOVA and repeated-measures ANOVA were used to assess general main effects and interactions (α = 0.05). Results are shown as mean ± SEM. Analyses for Experiments 3 and 4 examined differences in freezing behavior with a within-subject variable of time and between-subject variable of drug treatment (both systemic and intracranial).
Surgeries
Rats were anesthetized with isoflurane (5% induction, 2% maintenance) and placed in a stereotaxic apparatus (Kopf Instruments). The scalp was incised and retracted. For rats receiving viral infusions into the LC (Experiments 2–4), the head was tilted downward at a 15° angle such that the bregma skull surface was 2 mm below intersectional lambda in the horizontal plane. Burr holes were drilled above the LC in each hemisphere, and injectors were lowered into the LC to infuse AAV9-PRSx8-hM3Dq-HA (1.214e14 GC/ml, University of Pennsylvania Vector Core), an LC-specific Gq-coupled DREADD. The PRSx8 promoter is a synthetic dopamine-β-hydroxlyase promoter that restricts expression to noradrenergic neurons (Vazey and Aston-Jones, 2014). Viral infusions were made with a hypodermic injector (Small Parts/Amazon) that was connected to a Legato 101 infusion pump (KD Scientific) and a 10 μl syringe (Hamilton Company) using polyethylene tubing (Braintree Scientific). Virus was infused at a rate of 0.25 μl/min, and the injector was removed 5 min after the infusion ended to allow for adequate diffusion. The coordinates for each infusion (relative to intersection lambda) were as follows: anteroposterior (AP), −3.8 mm; mediolateral (ML), ±1.4 mm; and dorsoventral (DV), −7.0, −6.5, and −6.0 mm. We infused 0.5 μl at these three depths in each hemisphere due to the specificity of the virus and variability from animal to animal in terms of the DV location of the LC. At least 2 weeks were allowed following viral infusions before beginning experiments.
For rats that received a microelectrode array targeting the BLA (regardless of whether this was preceded by LC viral infusions, Experiments 1 and 2), the skull was leveled in the horizontal plane. Three to five burr holes were drilled for anchor screws. Burr holes overlying the BLA in the right hemisphere were also made for the microelectrode array. The animal was then implanted with a 16-channel microelectrode array (Innovative Neurophysiology). The 4 × 4 array was constructed of 16 individual 50 μm diameter tungsten wires of equal length (10.5 mm). The wires in each row and the rows themselves were spaced 200 μm apart (center to center). The array was positioned with its long axis parallel to the anteroposterior plane. Coordinates for the center-most wires relative to the bregma skull surface were as follows: AP, −2.9; ML, +4.8; DV, −8.55. One electrode was selected to be used as a ground to optimize recordings, resulting in a maximum of 15 channels per animal. The array was then secured to the skull with dental acrylic. Animals were given at least 1 week (2 weeks if viral manipulations were involved) to recover before undergoing behavioral and recording procedures.
For animals that received bilateral BLA cannula (Experiment 4), surgical procedures were similar to those described above. In brief, the skull was leveled in the horizontal plane. Three to five burr holes were drilled for anchor screws. Burr holes overlying the BLA were also made for the cannula implantation (10 mm, 26 gauge). The animal was then implanted with a bilateral cannula (Plastics One) using the following coordinates: AP, −2.9; ML, ±4.8; DV, −8.55. The cannulae were secured to the skull with dental acrylic. Stainless steel dummies (30 gauge, extending 1 mm beyond the guide cannula) were inserted into the guide cannula to prevent clogging. Animals were given at least 2 weeks to recover before behavioral procedures.
Drugs
d,l-Propranolol hydrochloride was obtained from a commercial supplier (Sigma-Aldrich). The drug was dissolved in distilled water and injected systemically (10 mg/kg, i.p.) or intracranially (5 μg/μl, 0.5 μl/hemisphere). Clozapine N-oxide was obtained from the NIH and dissolved in 2.5% DMSO in distilled water and injected systemically (3 mg/kg, i.p.).
Histology
After completion of the experiments, the rats were overdosed with sodium pentobarbital (100 mg/kg; Fatal-Plus, Vortex Pharmaceuticals). For rats implanted with a BLA array, electrolytic lesions were created by passing electrical current (80 μA, 10 s; A365 stimulus isolator, World Precision Instruments) through four of the recording wires (the four corners of the 4 × 4 array). Rats were then perfused transcardially with 0.9% saline followed by 10% formalin. Brains were extracted from the skull and postfixed in a 10% formalin solution for 24 h, followed by a 30% sucrose solution, where they remained for a minimum of 48 h. Coronal brain sections of the BLA (40 μm thickness) were cut on a cryostat (−20°C; Leica Microsystems), mounted on subbed microscope slides, and stained with thionin (0.25%) to visualize electrode placements or cannula tracks.
To visualize LC viral expression, brains were sectioned (40 μm thickness) in the coronal plane with a cryostat and stored in a 0.01% sodium azide solution until further processing. Sections were blocked in PBS with 0.1% Triton X-100 (TX) and 3% normal donkey serum (NDS; 2 ml/well) for 1 h. All steps occurred in this PBS-TX-NDS solution at room temperature. Sections were then incubated in primary antibodies [mouse anti-tyrosine hydroxylase (TH; 1:2000) and rabbit anti-HA (1:1000)] for 24 h. Sections were then rinsed three times (10 min each). Sections were then incubated in secondary antibodies [donkey anti-mouse Alexa Fluor 488 (1:500; for TH) and donkey anti-rabbit Alexa Fluor 594 (1:500; for HA)] for 3 h. Afterward, sections were rinsed three times (10 min each). Next, the sections were mounted on microscope slides using PBS and coverslipped using Fluoromount (Diagnostic Biosystems). Images were obtained using a Zeiss AXIO Imager M2. The following suppliers were used for the above materials: NDS, EMD Millipore; Triton X-100, Sigma-Aldrich; mouse anti-TH, ImmunoStar; rabbit anti-HA, Cell Signaling Technology; Invitrogen donkey anti-mouse IgG (H+L) Alexa Fluor 488, Thermo Fisher Scientific; Invitrogen donkey anti-rabbit IgG (H+L) Alexa Fluor 594, Thermo Fisher Scientific.
Results
Experiment 1: propranolol reduces footshock-induced freezing and mitigates BLA firing
Recent work suggests that intra-BLA propranolol reduces the IED and enables extinction learning under stress (Giustino et al., 2017). We sought to examine whether footshock stress alters single-unit firing in the BLA and contributes to extinction deficits (Fig. 1A, experimental design). Animals were implanted with a 16-channel microelectrode array targeting the amygdala (Fig. 1B, example histology and schematic representation of electrode placements). Animals were transported to the recording room for fear conditioning (Context A). After a 3 min stimulus-free baseline period, animals were injected with either VEH (n = 5) or PROP (n = 5). The animals remained in the chamber for 20 min to allow sufficient time for drug to take effect. Animals then received five CS–US pairings (see Materials and Methods for details) and remained in the chamber for 60 min following the last footshock. As expected, vehicle-treated animals exhibited sustained increases in freezing behavior, and this was limited by propranolol treatment (Fig. 2A, main effect of drug; F(1,8) = 19.30, p = 0.0023). During this session, we recorded from a total of 280 single units in the BLA (VEH: n = 143, baseline firing rate = 2.89 ± 0.16; PROP: n = 137, baseline firing rate = 3.00 ± 0.15). Both groups had relatively minor changes in firing rate after the systemic injections: VEH, postdrug firing rate = 3.05 ± 0.15; PROP, postdrug firing rate = 2.85 ± 0.13. Thus, the neural data were z-score-normalized to the entire preshock period. Fear conditioning produced a dramatic increase in spontaneous firing rate in the population of neurons recorded in the BLA, an effect that was attenuated by propranolol (Fig. 2B, main effect of drug; F(1,278) = 19.26, p < 0.0001).
Figure 1.
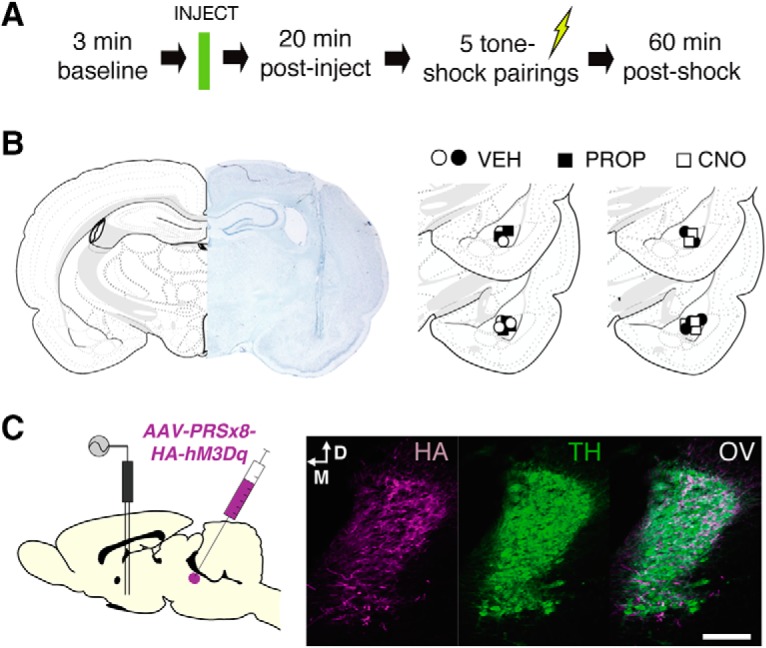
Experimental design and representative histology. A, Experimental design for recording experiments (Experiments 1 and 2). B, Nissl-stained tissue showing representative electrode placement in the amygdala alongside schematic histology depicting location of the center of the recording array for all rats split by recording experiment (n = 5 rats/group, per experiment). C, Experimental approach for the recording experiment in animals expressing hM3Dq in the LC (Experiment 2); injection of AAV-PRSx8-hM3Dq-HA into the LC yielded robust expression of hM3Dq among LC neurons. HA, HA tag immunolabeled for viral expression; OV, overlay. Scale bar, 100 μm.
Figure 2.

Propranolol reduces footshock-induced freezing and mitigates shock-induced increases in BLA spike firing. A, Percentage of freezing (mean ± SEM) across the duration of the session split by drug group (VEH, n = 5; PROP, n = 5). Systemic PROP treatment produced a reliable decrease in postshock freezing throughout the session (t = 0 is immediately after the last conditioning trial). B, Average spontaneous BLA firing rate over the course of the session split by drug treatment (20 s bins). Footshock produced rapid and sustained changes in amygdala firing rates that were mitigated by PROP treatment. C, Pie charts showing the percentage of neurons (split by drug) that increased or decreased in firing rate after the last conditioning trial. No difference between drug treatment was observed in the proportion of single units showing footshock-induced changes in firing rate. Exc, Excited; Supp, suppressed. D, Heat maps depicting normalized firing rate for every neuron recorded in vehicle- and propranolol-treated rats. E, F, Average firing rate over the course of the session in the two groups comparing “excited” and “suppressed” neuronal populations (20 s bins). Propranolol treatment limited both increases and decreases in BLA spike firing.
Although footshock stress increased the average firing rate of BLA neurons, there was considerable heterogeneity in the response of individual BLA neurons. We therefore divided the single units into two populations based on the direction of their postshock firing rate change in the first 20 s bin after footshock; based on this criterion, neurons were classified as either “excited” (z > 0) or “suppressed” (z < 0) immediately after the last footshock. Figure 2C demonstrates that no difference was observed when comparing drug treatment in terms of the proportion of neurons showing shock-induced increases or decreases in firing rate (χ2(1) = 0.94, p = 0.33). However, we did observe differences in the magnitude of both excited (Fig. 2E, main effect of drug, F(1,133) = 13.92, p = 0.0003) and suppressed (Fig. 2F, main effect of drug, F(1,143) = 4.92, p = 0.028) population activity based on drug treatment. These differences are further demonstrated by the heat maps (Fig. 2D) depicting each neuron across the entire session. That is, propranolol treatment limited both shock-induced increases and decreases in BLA firing rates. These data suggest that footshock stress induces rapid and sustained changes in the magnitude of BLA spontaneous firing rates, and this is regulated by the action of norepinephrine at β-adrenoceptors.
Experiment 2: LC-NE activation paired with weak footshocks induces sustained increases in freezing and BLA firing
Because propranolol mitigated stress-induced alterations in BLA firing, and past work has shown a role for LC projections to the amygdala in fear conditioning (Uematsu et al., 2017), we hypothesized that the LC-NE system was driving changes in BLA activity. To examine this possibility, we examined whether pairing LC-NE activation with a weaker, and presumably less stressful, footshock (see Materials and Methods for details) would recapitulate our previous findings (Fig. 1, experimental approach and histology).
As shown in Figure 3A, systemic administration of CNO produced sustained increases in freezing behavior relative to vehicle-treated animals after conditioning with weak footshocks (main effect of drug, F(1,8) = 6.27, p = 0.037). During this session, we recorded from a total of 233 single units in the BLA (VEH: n = 117, baseline firing rate = 2.34 ± 0.21; CNO: n = 116, baseline firing rate = 2.57 ± 0.26). Before conditioning, injections of either VEH or CNO did not reliably alter firing rates (VEH: postdrug firing rate = 2.48 ± 0.20; CNO: postdrug firing rate = 2.82 ± 0.33). After conditioning with weak footshocks, VEH-treated rats showed modest increases in BLA spike firing that were much reduced relative to those observed with stronger footshocks (e.g., Fig. 2). However, BLA spike firing rates after weak footshocks were dramatically increased in rats treated with CNO (Fig. 3B; main effect of drug, F(1,231) = 12.67, p = 0.0005). This suggests that increasing LC-NE release can drive BLA spike firing under conditions that produce minimal changes in BLA firing alone.
Figure 3.

LC-NE activation paired with weak footshocks induces sustained freezing and increases BLA spike firing. A, Percentage of freezing (mean ± SEM) across the duration of the session split by drug group (VEH, n = 5; PROP, n = 5). Animals expressing hM3Dq in the LC that were treated with CNO (to activate LC-NE release) showed sustained freezing relative to vehicle controls (t = 0 is immediately after the last conditioning trial). B, Average firing rate over the course of the session split by drug treatment (20 s bins). Neurons recorded from vehicle-treated rats showed moderate increases in BLA firing, whereas CNO rats showed a marked increase in firing rate for the duration of the session. C, Pie charts showing the percentage of neurons (split by drug) that increased or decreased firing rate after the last conditioning trial. CNO treatment resulted in a larger proportion of recorded units showing increased firing rate in the postshock period. Exc, Excited; Supp, suppressed. D, Heat maps depicting normalized firing rate for every neuron recorded split by drug treatment. E, F, Average firing rate over time split by drug comparing excited and suppressed neuronal populations. CNO treatment produced a marked increase in the magnitude of excitation, but not inhibition, within the BLA.
To determine whether LC-NE activation altered the proportion of neurons showing increased amygdala firing, we classified neurons as described above. Single units were considered to be either excited or suppressed if they showed an increase or decrease following the last footshock, respectively. A χ2 analysis revealed that CNO-mediated LC-NE activation resulted in a larger proportion of BLA neurons showing excitation after fear conditioning relative to vehicle-treated animals (Fig. 3C; χ2 (1) = 10.83, p = 0.001). Moreover, LC-NE activation resulted in a larger magnitude of stress-induced BLA firing in the excited neuronal population (Fig. 3E; main effect of drug, F(1,158) = 10.35, p = 0.0016) but not in the suppressed population (Fig. 3F). The heat maps showing each recorded neuron across the entire session further demonstrate this observation (Fig. 3D). These data suggest that the LC-NE drives stress-induced increases in amygdala firing rates as well as freezing behavior.
Experiment 3: LC-NE activation after a weak footshock induces an immediate extinction deficit
Although the LC-NE system regulates both amygdala firing rates and freezing behavior, the LC itself has not been directly implicated in extinction deficits. We hypothesized that we could induce an IED by pairing LC activation with a weak conditioning protocol that normally does not induce an IED. For this experiment, animals expressing excitatory LC DREADDs received a single conditioning trial with a weak footshock followed by an immediate extinction procedure 15–20 min after conditioning. VEH or CNO was administered 10 min before conditioning.
As shown in Figure 4A, all groups exhibited similar increases in freezing during conditioning, which was confirmed by a repeated-measures ANOVA (main effect of time, F(1,35) = 173.81, p < 0.0001). During the extinction session, VEH-treated animals exhibited a reduction in freezing across the session; however, this was greatly reduced in CNO-treated rats, regardless of their extinction group (i.e., EXT or NO EXT; Figure 4B; main effect of drug, F(1,35) = 86.38, p < 0.0001). Forty-eight hours later, VEH-treated rats that underwent extinction exhibited low levels of freezing behavior during the retention test (Fig. 4C), indicating that the immediate extinction procedure failed to yield an IED with the weak conditioning protocol. However, rats that underwent extinction after CNO treatment exhibited a deficit in extinction retrieval and showed significantly greater freezing in response to the CS during the test, similar to animals that did not undergo extinction (NO EXT). These observations were confirmed with a repeated-measures ANOVA (Fig. 4C; time × extinction × drug interaction, F(5,175) = 2.45, p = 0.036). These data are summarized in Figure 4D, which depicts the average freezing in the first nine-trial block of the extinction retrieval session (F(1,35) = 4.47, p = 0.042). Of course, it is possible that preconditioning CNO administration affected fear memory consolidation rather than extinction learning per se. However, VEH- and CNO-treated control groups that did not undergo the extinction procedure showed similar levels of freezing during the retention test, as seen in Figure 4, C and D.
Figure 4.

Chemogenetic LC activation induces an immediate extinction deficit after a weak fear conditioning procedure. Rats expressing hM3Dq in the LC were injected with VEH or CNO 10 min before fear conditioning with a single weak footshock. The rats subsequently received either immediate extinction (VEH-EXT, n = 11; CNO-EXT, n = 12) or context exposure alone (VEH-NO EXT, n = 8; CNO-NO EXT, n = 8). A, CNO administration did not affect postshock freezing during the fear conditioning session. B, CNO administration dramatically elevated freezing behavior during the extinction and context exposure sessions. C, During a retrieval test 48 h after extinction, rats that were treated with CNO before extinction exhibited greater CS-elicited freezing than VEH-treated rats, suggesting that LC-NE activation impaired extinction learning. This effect was not simply the result of LC activation increasing the consolidation of the fear memory, because CNO- and VEH-treated rats that did not undergo extinction exhibited similar levels of CS-evoked freezing during the retrieval test. D, Summary of the retrieval data showing the average freezing for the first nine-trial block split by group. VEH-treated rats that underwent extinction showed minimal freezing relative to the NO EXT groups and CNO-treated rats that received extinction training, demonstrating that LC activation drove an extinction deficit. Open circles represent data from individual subjects. Percentage of freezing (mean ± SEM) is shown for all sessions.
Experiment 4: intra-BLA propranolol blocks the LC-induced immediate extinction deficit
We have shown that (1) footshock increases BLA spike firing, and that this can be reduced with systemic propranolol (Experiment 1); (2) chemogenetic activation of the LC increases BLA spike firing rates after weak shocks (Experiment 2); and (3) chemogenetic activation of the LC induces an IED with a weak conditioning procedure that normally does not yield an IED (Experiment 3). Here, we examined whether β-adrenoceptors in the BLA are required for LC-induced extinction impairments. Animals expressing excitatory DREADDs in the LC underwent a weak conditioning procedure after intracranial infusions of VEH or PROP into the BLA followed by systemic injections of either VEH or CNO in a 2 × 2 factorial design (Fig. 5A shows a representative micrograph and schematized cannula placements).
Figure 5.

Intra-BLA propranolol blocks LC-NE-induced extinction deficits. Rats expressing hM3Dq in the LC received intra-BLA infusions of VEH or PROP approximately 20 min prior to fear conditioning. The rats were then systemically injected with VEH or CNO 10 min prior to conditioning (VEH-VEH, n = 5; PROP-VEH, n = 6; VEH-CNO, n = 4; and PROP-CNO, n = 7). A, Representative and schematic histology of BLA cannula placements. B, All groups conditioned similarly as evidenced by an increase in freezing behavior following footshocks. C, Animals underwent immediate extinction in a new context 15–20 min after conditioning. Rats treated with CNO showed elevated freezing relative to vehicle controls and this was not reduced by intra-BLA propranolol. D, Animals underwent an identical extinction retrieval session 48 hrs after extinction in a drug-free state. Rats that received systemic injections of CNO and intra-BLA VEH infusions showed elevated freezing throughout this session, replicating our prior finding (i.e., LC-NE activation induces an immediate extinction deficit when paired with a weak shock). Intra-BLA PROP reduced freezing during this session similar to rats treated with systemic VEH and either intra-BLA VEH or PROP, suggesting that intra-BLA PROP mitigates LC-induced extinction deficits. E, The retrieval data are summarized in panel E, which shows the average freezing for the first 9-trial block split by group. Rats that were administered systemic VEH and either intra-BLA VEH or PROP showed minimal freezing, suggesting that this protocol is not sufficient to induce an IED. Rats treated with systemic CNO alongside intra-BLA VEH displayed elevated freezing levels, indicative of an IED and this was blocked by intra-BLA PROP. Open circles represent data from individual subjects. Percentage of freezing (mean ± SEM) is shown for all sessions.
As shown in Figure 5B, all groups conditioned similarly. This was confirmed by a repeated-measures ANOVA, which revealed a main effect of time (F(1,18) = 64.11, p < 0.0001). During the immediate extinction session, animals treated with CNO showed elevated freezing behavior independent of intracranial infusions (Fig. 5C; main effect of systemic drug, F(1,18) = 15.68, p = 0.0009). Forty-eight hours later, animals that underwent the extinction procedure after CNO treatment combined with an intra-BLA vehicle infusion exhibited an IED; they showed elevated freezing during the retention test relative to controls receiving systemic and intra-BLA vehicle (Fig. 5D). However, intra-BLA infusion of propranolol eliminated the LC-induced IED. These observations were confirmed in an ANOVA, which revealed a significant systemic × intra-BLA drug interaction (F(1,18) = 7.98, p = 0.0112). These data are summarized in Figure 5E, which depicts the average freezing in the first nine-trial block of the extinction retrieval session (F(1,18) = 9.26, p = 0.007). These data demonstrate that intra-BLA propranolol limits LC-driven extinction deficits, suggesting a critical role for β-adrenoceptors in the amygdala.
Discussion
Here, we show that footshock stress induces rapid and sustained increases in the spontaneous firing rate of BLA single units. These increases in BLA firing were blocked by systemic propranolol administration and potentiated by chemogenetic activation of LC-NE, suggesting a key role for β-adrenoceptors. These stress-related changes in BLA activity persisted for up to an hour after footshock, a time window that corresponds to stress-induced deficits in extinction learning. Last, we demonstrate that LC-NE activation impairs extinction learning, and this was blocked by intra-BLA infusions of propranolol, presumably by exciting BLA circuits that promote fear expression at the expense of extinction learning (Fig. 6 for a proposed circuit mechanism).
Figure 6.

Proposed circuit mechanism underlying stress-induced extinction deficits. Under low levels of stress, the LC-NE system is minimally engaged. This enables successful extinction learning via IL-mediated feedforward inhibition of the BLA. In contrast, this circuit reverses under high levels of stress, and this reversal is driven by LC-NE. Activation of the BLA results in shunted IL firing via feedforward inhibition, thereby interfering with extinction learning.
We, along with others, have suggested that stress acts to impair extinction learning by altering mPFC function, and this may be mediated by NE (Chang et al., 2010; Kim et al., 2010; Fitzgerald et al., 2015; Maren and Holmes, 2016). Activity in the infralimbic (IL) subdivision of the mPFC is thought to regulate successful extinction learning (Milad and Quirk, 2002; Maren and Quirk, 2004; Giustino and Maren, 2015). We have previously shown that noradrenergic blockade enables extinction learning under stress, and we initially hypothesized that this was mediated by footshock-induced changes in mPFC single-unit activity (Fitzgerald et al., 2015). Consistent with this, we observed that footshock stress resulted in rapid and sustained decreases in IL firing rates, and this could be blocked by systemic propranolol. We surmised that this decreased IL activity was an underlying factor in the immediate extinction deficit, but intra-IL infusions of propranolol had no effect on stress-induced extinction deficits (Giustino et al., 2017). However, both the mPFC and BLA are highly sensitive to stress, and it has been suggested that stress impairs prefrontal function while enhancing BLA activity, a state that may limit extinction learning (Arnsten, 2009, 2015; Arnsten et al., 2015; Giustino and Maren, 2018). Indeed, intra-BLA propranolol, on the other hand, rescued the immediate extinction deficit (Giustino et al., 2017). We now show that footshock stress dramatically increases spontaneous firing rates among neurons in the BLA, and this is dependent upon the action of NE at β-adrenoceptors insofar as propranolol limits these changes.
Past work has demonstrated that BLA projections to the mPFC are involved in both the conditioning and extinction of fear (Senn et al., 2014; Burgos-Robles et al., 2017; Klavir et al., 2017). It is possible that the observed NE-dependent changes in BLA firing also mediated stress-induced decreases in IL firing via these direct projections. Senn et al. (2014) showed that differing levels of activity in BLA projections to prelimbic (PL) and inframlimbic (IL) influence extinction retention. This is in line with our current and past work in which we show footshock-induced suppression of IL firing that outlasts changes in PL firing rates (Fitzgerald et al., 2015). Our current data now demonstrate that footshock stress produces rapid and prolonged activation of the BLA, a change in firing that is opposite to that observed in IL after footshock. These data suggest a circuit mechanism by which BLA projections to the IL may mediate extinction deficits (Fig. 6). It is possible that LC-NE drives increases and decreases in the BLA and IL via direct projections, respectively. In line with this idea, a recent study has shown that LC projections to the mPFC mediate aversion and increase anxiety-like behavior, although it is not known whether this was due to a suppression of mPFC activity (Hirschberg et al., 2017). Another possibility is that, in addition to direct projections, LC-NE may act to enhance BLA firing in amygdala neurons that project to the IL and synapse on inhibitory interneurons to promote feedforward inhibition, thus resulting in impaired extinction. Indeed, BLA neurons projecting to the IL have been shown to dampen IL firing via a feedforward inhibitory mechanism (McGarry and Carter, 2016), and IL projections to the BLA mediate both the acquisition and recall of extinction learning (Cho et al., 2013). Under conditions of high stress, it seems likely that these circuits are tilted in favor of BLA-mediated suppression of IL, thus promoting a high fear state while simultaneously leading to poor extinction.
Extinction deficits may be mediated, in part, by the LC-NE system prioritizing consolidation of the recent fear memory at the expense of a new extinction memory, particularly when extinction learning occurs soon after fear conditioning. Indeed, BLA NE has long been implicated in fear memory consolidation (Cahill et al., 1994; McGaugh, 2000; McGaugh and Roozendaal, 2002; Roozendaal et al., 2006; McIntyre et al., 2012; Giustino and Maren, 2018). Along these lines, recent work has shown that optogenetic stimulation of LC projections to the BLA mediate fear consolidation, whereas inhibiting this pathway reduced the strength of fear memories (Uematsu et al., 2017). However, it is unknown how these manipulations affected single-unit and population-level dynamics in both the BLA and mPFC. McCall et al. (2017) demonstrated that optogenetic activation of LC projections to the BLA resulted in a majority of responsive BLA neurons showing increased firing, whereas a smaller proportion of BLA neurons were suppressed. These data align nicely with our current findings, which show an overall excitatory population response in the BLA that is amplified by chemogenetic LC activation.
Interestingly, others have shown that footshock as well as infusion of adrenoceptor agonists into the BLA suppress firing rates in anesthetized animals (Buffalari and Grace, 2007; Chen and Sara, 2007). However, these authors also noted that a small subpopulation of neurons in the BLA showed increased spiking in response to stimulation of amygdala adrenoceptors. Of course, it is has been shown that anesthesia influences basal NE activity in a way that might influence pharmacological manipulations of adrenergic receptors within the BLA (Vazey and Aston-Jones, 2014). In addition, pharmacological manipulations, whether locally infused or systemically administered, may not entirely replicate chemogenetic activation of the LC or its terminals in downstream brain regions, which may help explain some of these discrepant findings. Our current work shows a robust footshock-induced increase in population activity within the BLA, although several neurons were also suppressed. When these footshocks were then paired with LC-NE stimulation, this further augmented BLA excitability in terms of the magnitude as well as the proportion of neurons showing increased firing, suggesting that elevated levels of NE facilitate BLA spiking. In line with this, we found that the LC-induced extinction deficit was blocked by intra-BLA infusions of propranolol. This suggests that the IED is mediated by shock-induced increases in BLA NE, which increase the firing of BLA neurons that promote fear expression at the expense of extinction learning. Further work examining the effects of stress on extinction impairments at more remote time points will be an important area of research given that our manipulations of the NE system occurred before conditioning. This may shed light on stressor- and trauma-related disorders for which individuals often seek treatment at varying delays from the initial traumatic experience.
In humans, individuals experiencing PTSD and related disorders present with heightened amygdala activity (Debiec and LeDoux, 2006; Rauch et al., 2006; Milad et al., 2009; Giustino and Maren, 2018), elevated NE (Yehuda et al., 1992; Southwick et al., 1999a; Arnsten, 2009; Giustino et al., 2016; Giustino and Maren, 2018; Krystal et al., 2018), and extinction impairments (Milad et al., 2009; Garfinkel et al., 2014; Giustino et al., 2016). The current data strongly suggest that extinction deficits may result from elevated LC-NE activity that, in turn, increases BLA firing rates to promote fear at the expense of forming a new extinction memory. Recent advantages in neuroimaging technology now allow researchers to measure activity in the human LC, an advance that will further our understanding of the role of LC-NE in PTSD (Liu et al., 2017; Krystal et al., 2018; Priovoulos et al., 2018). Although pharmacological manipulations of NE transmission have shown some promise for the treatment of PTSD, the literature is largely split on their efficacy and utility (Giustino and Maren, 2018). Our data suggest that both stress and proximity to trauma are key factors that influence how the LC and BLA interact to influence extinction learning; this interaction is critical for designing interventions that are appropriately timed to yield the most effective clinical outcomes. Future work in humans will likely shed light on the role of the LC-NE system on an individual-to-individual basis, which may better allow us to appreciate when and why NE-altering drugs may be useful for reducing PTSD symptomatology.
Overall, we demonstrate that footshock stress induces prolonged increases in BLA spontaneous firing rates, and this is highly sensitive to manipulations of the NE system. That is, reducing the action of NE via propranolol eliminated these changes in firing rate, whereas selective LC-NE activation via chemogenetics enhanced stress-induced increases in BLA activity. We also show that stress and LC-NE activation induce extinction deficits, most likely due to the observed increases in amygdala firing insofar as intra-BLA propranolol blocked this deficit. These data have important clinical implications for individuals experiencing stress- and trauma-related disorders, such as PTSD, and suggest the LC-NE system may be a key regulator of heightened amygdala activity, which is observed in those with PTSD.
Footnotes
This work was supported by grants from the National Institutes of Health (R01-MH-065961 and R01-MH-117852 to S.M. and F31MH112208 to T.F.G.). We thank Dr. Gary Aston-Jones for providing the LC-specific excitatory DREADD (PRSx8-hM3Dq-HA) and Dr. Elena Vazey for guidance in using these viruses in the locus coeruleus.
The authors declare no conflicts of interest.
References
- Arnsten AF. (2009) Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci 10:410–422. 10.1038/nrn2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF. (2015) Stress weakens prefrontal networks: molecular insults to higher cognition. Nat Neurosci 18:1376–1385. 10.1038/nn.4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF, Raskind MA, Taylor FB, Connor DF (2015) The effects of stress exposure on prefrontal cortex: translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress 1:89–99. 10.1016/j.ynstr.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffalari DM, Grace AA (2007) Noradrenergic modulation of basolateral amygdala neuronal activity: opposing influences of alpha-2 and beta receptor activation. J Neurosci 27:12358–12366. 10.1523/JNEUROSCI.2007-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Kimchi EY, Izadmehr EM, Porzenheim MJ, Ramos-Guasp WA, Nieh EH, Felix-Ortiz AC, Namburi P, Leppla CA, Presbrey KN, Anandalingam KK, Pagan-Rivera PA, Anahtar M, Beyeler A, Tye KM (2017) Amygdala inputs to prefrontal cortex guide behavior amid conflicting cues of reward and punishment. Nat Neurosci 20:824–835. 10.1038/nn.4553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill L, Prins B, Weber M, McGaugh JL (1994) Beta-adrenergic activation and memory for emotional events. Nature 371:702–704. 10.1038/371702a0 [DOI] [PubMed] [Google Scholar]
- Chang CH, Maren S (2009) Early extinction after fear conditioning yields a context-independent and short-term suppression of conditional freezing in rats. Learn Mem 16:62–68. 10.1101/lm.1085009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Berke JD, Maren S (2010) Single-unit activity in the medial prefrontal cortex during immediate and delayed extinction of fear in rats. PLoS One 5:e11971. 10.1371/journal.pone.0011971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FJ, Sara SJ (2007) Locus coeruleus activation by foot shock or electrical stimulation inhibits amygdala neurons. Neuroscience 144:472–481. 10.1016/j.neuroscience.2006.09.037 [DOI] [PubMed] [Google Scholar]
- Cho JH, Deisseroth K, Bolshakov VY (2013) Synaptic encoding of fear extinction in mPFC-amygdala circuits. Neuron 80:1491–1507. 10.1016/j.neuron.2013.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE (2006) Noradrenergic signaling in the amygdala contributes to the reconsolidation of fear memory: treatment implications for PTSD. Ann N Y Acad Sci 1071:521–524. 10.1196/annals.1364.056 [DOI] [PubMed] [Google Scholar]
- Fallon JH, Koziell DA, Moore RY (1978) Catecholamine innervation of the basal forebrain. II. Amygdala, suprarhinal cortex and entorhinal cortex. J Comp Neurol 180:509–532. 10.1002/cne.901800308 [DOI] [PubMed] [Google Scholar]
- Fitzgerald PJ, Giustino TF, Seemann JR, Maren S (2015) Noradrenergic blockade stabilizes prefrontal activity and enables fear extinction under stress. Proc Natl Acad Sci U S A 112:E3729–E3737. 10.1073/pnas.1500682112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote SL, Aston-Jones G, Bloom FE (1980) Impulse activity of locus coeruleus neurons in awake rats and monkeys is a function of sensory stimulation and arousal. Proc Natl Acad Sci U S A 77:3033–3037. 10.1073/pnas.77.5.3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfinkel SN, Abelson JL, King AP, Sripada RK, Wang X, Gaines LM, Liberzon I (2014) Impaired contextual modulation of memories in PTSD: an fMRI and psychophysiological study of extinction retention and fear renewal. J Neurosci 34:13435–13443. 10.1523/JNEUROSCI.4287-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustino TF, Maren S (2015) The role of the medial prefrontal cortex in the conditioning and extinction of fear. Front Behav Neurosci 9:298. 10.3389/fnbeh.2015.00298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustino TF, Maren S (2018) Noradrenergic modulation of fear conditioning and extinction. Front Behav Neurosci 12:43. 10.3389/fnbeh.2018.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustino TF, Fitzgerald PJ, Maren S (2016) Revisiting propranolol and PTSD: memory erasure or extinction enhancement? Neurobiol Learn Mem 130:26–33. 10.1016/j.nlm.2016.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustino TF, Seemann JR, Acca GM, Goode TD, Fitzgerald PJ, Maren S (2017) β-adrenoceptor blockade in the basolateral amygdala, but not the medial prefrontal cortex, rescues the immediate extinction deficit. Neuropsychopharmacology 42:2537–2544. 10.1038/npp.2017.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustino TF, Fitzgerald PJ, Ressler RL, Maren S (2019) Locus coeruleus toggles reciprocal prefrontal firing to reinstate fear. Proc Natl Acad Sci U S A 116:8570–8575. 10.1073/pnas.1814278116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg S, Li Y, Randall A, Kremer EJ, Pickering AE (2017) Functional dichotomy in spinal- vs prefrontal-projecting locus coeruleus modules splits descending noradrenergic analgesia from ascending aversion and anxiety in rats. Elife 6:e29808. 10.7554/eLife.29808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollis F, Sevelinges Y, Grosse J, Zanoletti O, Sandi C (2016) Involvement of CRFR1 in the basolateral amygdala in the immediate fear extinction deficit. eNeuro 3:ENEURO.0084-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo A, Wellman CL, Holmes A (2006) Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci 26:5733–5738. 10.1523/JNEUROSCI.0474-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jodo E, Chiang C, Aston-Jones G (1998) Potent excitatory influence of prefrontal cortex activity on noradrenergic locus coeruleus neurons. Neuroscience 83:63–79. 10.1016/S0306-4522(97)00372-2 [DOI] [PubMed] [Google Scholar]
- Kim SC, Jo YS, Kim IH, Kim H, Choi JS (2010) Lack of medial prefrontal cortex activation underlies the immediate extinction deficit. J Neurosci 30:832–837. 10.1523/JNEUROSCI.4145-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klavir O, Prigge M, Sarel A, Paz R, Yizhar O (2017) Manipulating fear associations via optogenetic modulation of amygdala inputs to prefrontal cortex. Nat Neurosci 20:836–844. 10.1038/nn.4523 [DOI] [PubMed] [Google Scholar]
- Krystal JH, Abdallah CG, Pietrzak RH, Averill LA, Harpaz-Rotem I, Levy I, Kelmendi B, Southwick SM (2018) Locus coeruleus hyperactivity in posttraumatic stress disorder: answers and questions. Biol Psychiatry 83:197–199. 10.1016/j.biopsych.2017.09.027 [DOI] [PubMed] [Google Scholar]
- Liu KY, Marijatta F, Hämmerer D, Acosta-Cabronero J, Düzel E, Howard RJ (2017) Magnetic resonance imaging of the human locus coeruleus: a systematic review. Neurosci Biobehav Rev 83:325–355. 10.1016/j.neubiorev.2017.10.023 [DOI] [PubMed] [Google Scholar]
- Loughlin SE, Foote SL, Bloom FE (1986) Efferent projections of nucleus locus coeruleus: topographic organization of cells of origin demonstrated by three-dimensional reconstruction. Neuroscience 18:291–306. 10.1016/0306-4522(86)90155-7 [DOI] [PubMed] [Google Scholar]
- MacPherson K, Whittle N, Camp M, Gunduz-Cinar O, Singewald N, Holmes A (2013) Temporal factors in the extinction of fear in inbred mouse strains differing in extinction efficacy. Biol Mood Anxiety Disord 3:13. 10.1186/2045-5380-3-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Chang CH (2006) Recent fear is resistant to extinction. Proc Natl Acad Sci U S A 103:18020–18025. 10.1073/pnas.0608398103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Holmes A (2016) Stress and fear extinction. Neuropsychopharmacology 41:58–79. 10.1038/npp.2015.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Quirk GJ (2004) Neuronal signalling of fear memory. Nat Rev Neurosci 5:844–852. 10.1038/nrn1535 [DOI] [PubMed] [Google Scholar]
- McCall JG, Al-Hasani R, Siuda ER, Hong DY, Norris AJ, Ford CP, Bruchas MR (2015) CRH engagement of the locus coeruleus noradrenergic system mediates stress-induced anxiety. Neuron 87:605–620. 10.1016/j.neuron.2015.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall JG, Siuda ER, Bhatti DL, Lawson LA, McElligott ZA, Stuber GD, Bruchas MR (2017) Locus coeruleus to basolateral amygdala noradrenergic projections promote anxiety-like behavior. Elife 6:e18247. 10.7554/eLife.18247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry LM, Carter AG (2016) Inhibitory gating of basolateral amygdala inputs to the prefrontal cortex. J Neurosci 36:9391–9406. 10.1523/JNEUROSCI.0874-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh JL. (2000) Memory–a century of consolidation. Science 287:248–251. 10.1126/science.287.5451.248 [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Roozendaal B (2002) Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol 12:205–210. 10.1016/S0959-4388(02)00306-9 [DOI] [PubMed] [Google Scholar]
- McIntyre CK, McGaugh JL, Williams CL (2012) Interacting brain systems modulate memory consolidation. Neurosci Biobehav Rev 36:1750–1762. 10.1016/j.neubiorev.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz CJ, Hamacher-Dang TC, Wolf OT (2014) Exposure to stress attenuates fear retrieval in healthy men. Psychoneuroendocrinology 41:89–96. 10.1016/j.psyneuen.2013.12.009 [DOI] [PubMed] [Google Scholar]
- Merz CJ, Hamacher-Dang TC, Wolf OT (2016) Immediate extinction promotes the return of fear. Neurobiol Learn Mem 131:109–116. 10.1016/j.nlm.2016.03.013 [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ (2002) Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420:70–74. 10.1038/nature01138 [DOI] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, Rauch SL (2009) Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry 66:1075–1082. 10.1016/j.biopsych.2009.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miracle AD, Brace MF, Huyck KD, Singler SA, Wellman CL (2006) Chronic stress impairs recall of extinction of conditioned fear. Neurobiol Learn Mem 85:213–218. 10.1016/j.nlm.2005.10.005 [DOI] [PubMed] [Google Scholar]
- Morilak DA, Barrera G, Echevarria DJ, Garcia AS, Hernandez A, Ma S, Petre CO (2005) Role of brain norepinephrine in the behavioral response to stress. Prog Neuropsychopharmacol Biol Psychiatry 29:1214–1224. 10.1016/j.pnpbp.2005.08.007 [DOI] [PubMed] [Google Scholar]
- Naegeli C, Zeffiro T, Piccirelli M, Jaillard A, Weilenmann A, Hassanpour K, Schick M, Rufer M, Orr SP, Mueller-Pfeiffer C (2018) Locus coeruleus activity mediates hyperresponsiveness in posttraumatic stress disorder. Biol Psychiatry 83:254–262. 10.1016/j.biopsych.2017.08.021 [DOI] [PubMed] [Google Scholar]
- O'Donnell T, Hegadoren KM, Coupland NC (2004) Noradrenergic mechanisms in the pathophysiology of post-traumatic stress disorder. Neuropsychobiology 50:273–283. 10.1159/000080952 [DOI] [PubMed] [Google Scholar]
- Parsons RG, Ressler KJ (2013) Implications of memory modulation for post-traumatic stress and fear disorders. Nat Neurosci 16:146–153. 10.1038/nn.3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passerin AM, Cano G, Rabin BS, Delano BA, Napier JL, Sved AF (2000) Role of locus coeruleus in foot shock-evoked fos expression in rat brain. Neuroscience 101:1071–1082. 10.1016/S0306-4522(00)00372-9 [DOI] [PubMed] [Google Scholar]
- Pervanidou P, Chrousos GP (2010) Neuroendocrinology of post-traumatic stress disorder. Prog Brain Res 182:149–160. 10.1016/S0079-6123(10)82005-9 [DOI] [PubMed] [Google Scholar]
- Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, Milad MR, Liberzon I (2012) Biological studies of post-traumatic stress disorder. Nat Rev Neurosci 13:769–787. 10.1038/nrn3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priovoulos N, Jacobs HIL, Ivanov D, Uludağ K, Verhey FRJ, Poser BA (2018) High-resolution in vivo imaging of human locus coeruleus by magnetization transfer MRI at 3T and 7T. Neuroimage 168:427–436. 10.1016/j.neuroimage.2017.07.045 [DOI] [PubMed] [Google Scholar]
- Quirarte GL, Galvez R, Roozendaal B, McGaugh JL (1998) Norepinephrine release in the amygdala in response to footshock and opioid peptidergic drugs. Brain Res 808:134–140. 10.1016/S0006-8993(98)00795-1 [DOI] [PubMed] [Google Scholar]
- Raio CM, Phelps EA (2015) The influence of acute stress on the regulation of conditioned fear. Neurobiol Stress 1:134–146. 10.1016/j.ynstr.2014.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raio CM, Brignoni-Perez E, Goldman R, Phelps EA (2014) Acute stress impairs the retrieval of extinction memory in humans. Neurobiol Learn Mem 112:212–221. 10.1016/j.nlm.2014.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA (2006) Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research–past, present, and future. Biol Psychiatry 60:376–382. 10.1016/j.biopsych.2006.06.004 [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Hui GK, Hui IR, Berlau DJ, McGaugh JL, Weinberger NM (2006) Basolateral amygdala noradrenergic activity mediates corticosterone-induced enhancement of auditory fear conditioning. Neurobiol Learn Mem 86:249–255. 10.1016/j.nlm.2006.03.003 [DOI] [PubMed] [Google Scholar]
- Senn V, Wolff SB, Herry C, Grenier F, Ehrlich I, Gründemann J, Fadok JP, Müller C, Letzkus JJ, Lüthi A (2014) Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron 81:428–437. 10.1016/j.neuron.2013.11.006 [DOI] [PubMed] [Google Scholar]
- Southwick SM, Bremner JD, Rasmusson A, Morgan CA 3rd, Arnsten A, Charney DS (1999a) Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biol Psychiatry 46:1192–1204. 10.1016/S0006-3223(99)00219-X [DOI] [PubMed] [Google Scholar]
- Southwick SM, Paige S, Morgan CA 3rd, Bremner JD, Krystal JH, Charney DS (1999b) Neurotransmitter alterations in PTSD: catecholamines and serotonin. Semin Clin Neuropsychiatry 4:242–248. 10.153/SCNP00400242 [DOI] [PubMed] [Google Scholar]
- Uematsu A, Tan BZ, Ycu EA, Cuevas JS, Koivumaa J, Junyent F, Kremer EJ, Witten IB, Deisseroth K, Johansen JP (2017) Modular organization of the brainstem noradrenaline system coordinates opposing learning states. Nat Neurosci 20:1602–1611. 10.1038/nn.4642 [DOI] [PubMed] [Google Scholar]
- Vazey EM, Aston-Jones G (2014) Designer receptor manipulations reveal a role of the locus coeruleus noradrenergic system in isoflurane general anesthesia. Proc Natl Acad Sci U S A 111:3859–3864. 10.1073/pnas.1310025111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessa M, Flor H (2007) Failure of extinction of fear responses in posttraumatic stress disorder: evidence from second-order conditioning. Am J Psychiatry 164:1684–1692. 10.1176/appi.ajp.2007.07030525 [DOI] [PubMed] [Google Scholar]
- Wilber AA, Walker AG, Southwood CJ, Farrell MR, Lin GL, Rebec GV, Wellman CL (2011) Chronic stress alters neural activity in medial prefrontal cortex during retrieval of extinction. Neuroscience 174:115–131. 10.1016/j.neuroscience.2010.10.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R, Southwick S, Giller EL, Ma X, Mason JW (1992) Urinary catecholamine excretion and severity of PTSD symptoms in vietnam combat veterans. J Nerv Ment Dis 180:321–325. 10.1097/00005053-199205000-00006 [DOI] [PubMed] [Google Scholar]