

Focused ultrasound delivery of a selective TrkA agonist rescues cholinergic function in a mouse model of Alzheimer’s disease.
Abstract
The degeneration of cholinergic neurons is a prominent feature of Alzheimer’s disease (AD). In animal models of injury and aging, nerve growth factor (NGF) enhances cholinergic cell survival and function, contributing to improved memory. In the presence of AD pathology, however, NGF-related therapeutics have yet to fulfill their regenerative potential. We propose that stimulating the TrkA receptor, without p75NTR activation, is key for therapeutic efficacy. Supporting this hypothesis, the selective TrkA agonist D3 rescued neurotrophin signaling in TgCRND8 mice, whereas NGF, interacting with both TrkA and p75NTR, did not. D3, delivered intravenously and noninvasively to the basal forebrain using MRI-guided focused ultrasound (MRIgFUS)–mediated blood-brain barrier (BBB) permeability activated TrkA-related signaling cascades and enhanced cholinergic neurotransmission. Recent clinical trials support the safety and feasibility of MRIgFUS BBB modulation in AD patients. Neuroprotective agents targeting TrkA, combined with MRIgFUS BBB modulation, represent a promising strategy to counter neurodegeneration in AD.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive cognitive decline. Among the widespread neuronal and synaptic deficits in AD, degeneration of basal forebrain cholinergic neurons (BFCNs) and loss of cholinergic innervation to the cortex (CTX) and hippocampal formation (HF) substantially contribute to cognitive decline in AD (1). During the prodromal and earliest stages of AD, cholinergic neurons undergo trophic changes, including alterations in nerve growth factor (NGF) processing or transport, expression of NGF receptors, and activation of downstream NGF signaling components (2). The loss of neurotrophic support to BFCNs, beginning in early-stage AD, contributes to impaired cholinergic neurotransmission, followed by neuronal degeneration that is observed in late-stage AD (2).
In the brain, NGF promotes neuronal survival, growth, and synaptic plasticity by binding to tropomyosin receptor kinase A (TrkA) and p75 neurotrophin receptor (p75NTR) (3). NGF is produced in the medial septum (MS), diagonal band of Broca (DBB), and nucleus basalis of Meynert (NBM), which together compose the basal forebrain, HF, and CTX. In response to NGF binding, TrkA receptors expressed by BFCNs trigger activation of the tyrosine kinase domain, receptor phosphorylation, and intracellular signaling via the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/Akt cascades (3). NGF also binds p75NTR and can, in the absence of TrkA, induce neuronal apoptosis and glial inflammation through Jun N-terminal kinase (JNK) signaling (3, 4). In addition, BFCNs respond to NGF-induced activation of TrkA by enhancing their cholinergic phenotype. TrkA activation engages the cholinergic gene locus to increase choline acetyltransferase (ChAT) activity, the enzyme that synthesizes the neurotransmitter acetylcholine (ACh), which, in turn, promotes ACh release at cholinergic nerve terminals (3).
The importance of NGF in neuronal health and disease has been long recognized, yet translating a treatment strategy for AD remains a major challenge. The therapeutic efficacy of NGF is limited by its short half-life, detrimental effects related to p75NTR activation, and its inability to cross the blood-brain barrier (BBB) (5). Furthermore, assessment of the preclinical efficacy of NGF-based therapeutics has been limited to injury models, namely, the fimbria-fornix lesion-induced degeneration of BFCNs or aging models (5), in the absence of toxic amyloid-β (Aβ) peptides and tau pathology. Native NGF can bind to both TrkA and p75NTR, and in the presence of defective TrkA signaling (as observed in human AD), p75NTR activation facilitates Aβ- and tau-induced signaling and neurodegeneration (6–8). Recent clinical trials for NGF therapy in AD showed some growth of cholinergic afferents but ultimately failed to slow cognitive decline (9, 10). Further elucidation of the molecular interplay between NGF and pathological features of AD is necessary to facilitate the development of successful NGF-based therapies.
In comparison to native NGF, ligands with desirable pharmacological properties, designed to specifically activate TrkA, and not p75NTR, may be a preferable therapeutic approach to up regulate the cholinergic phenotype of BFCNs (11–14). Saragovi and colleagues (11–13) have previously demonstrated that D3, a peptidomimetic agonist of TrkA, promotes cell survival, outgrowth, cholinergic activity, and rescued learning and memory deficits in aged rats and a transgenic (Tg) mouse model of AD. However, like NGF, D3 does not cross the BBB.
In this study, we achieved noninvasive delivery of D3 to the basal forebrain in a mouse model of AD using magnetic resonance imaging–guided focused ultrasound (MRIgFUS). MRIgFUS, combined with intravenous microbubbles, allows a localized and transient increase in BBB permeability (15). Therapeutic agents injected intravenously enter MRIgFUS-targeted areas, thereby facilitating site-specific delivery of drugs, genes, and cells to the brain (16–18). The safety and feasibility of MRIgFUS BBB opening in humans with early to moderate AD was recently reported in a phase 1 clinical trial, validating its use translationally (19).
Our study was performed in the TgCRND8 murine model of AD. TgCRND8 mice harbor two human amyloid precursor protein mutations linked to familial forms of AD (i.e., Swedish, KM670/671NL; Indiana, V717F), leading to robust production of Aβ, with progressive Aβ plaque deposition and memory deficits starting at 3 months of age (20).
Deficits in several neurotransmitter systems occur in TgCRND8 mice (21, 22). Cholinergic atrophy and cell loss have been reported, coinciding with a reduction in the levels of ACh release in the prefrontal cortex of 7-month-old TgCRND8 compared to non-Tg mice (22). Consistent with TrkA deficits observed in AD postmortem tissue (23, 24), single-cell RNA sequencing also revealed lower mRNA levels of TrkA in TgCRND8 mice compared to non-Tg littermates (25).
The status of NGF signaling in relation to cholinergic function remains poorly understood in animal models of AD. Here, we report deficits in NGF levels, TrkA receptor levels, and TrkA-mediated signaling cascades in parallel with cholinergic dysfunction in the TgCRND8 model of AD, thus providing a disease-relevant phenotype to test NGF-based therapeutics. To this end, we delivered D3 to BFCNs using MRIgFUS in TgCRND8 mice. D3 was physiologically active in vivo after MRIgFUS delivery, significantly increasing TrkA phosphorylation and downstream signaling cascades in both TgCRND8 mice and non-Tg littermates. Moreover, we report that D3—and not NGF—successfully activated TrkA signaling pathways in TgCRND8 mice, representing an additional advantage of our therapeutic approach favoring TrkA activation without engaging p75NTR. MRIgFUS-mediated D3 delivery in the basal forebrain increased ChAT activity locally and enhanced ACh release from hippocampal and cortical slices. These results suggest that noninvasive, selective TrkA activation in BFCNs is sufficient to increase cholinergic function in the presence of Aβ pathology and that it can be achieved via MRIgFUS delivery of D3. TrkA receptor ligands, combined with MRIgFUS targeted delivery, merit consideration as a treatment strategy for AD.
RESULTS
NGF/TrkA signaling declines with age in TgCRND8 mice
We comprehensively evaluated age-dependent changes in NGF signaling in an AD animal model to identify a relevant, early-stage, AD phenotype for testing NGF-related therapies. We measured regional differences in the levels of NGF, TrkA, and p75NTR mRNA [quantitative real-time polymerase chain reaction (qRT-PCR) and in situ hybridization] and protein [enzyme-linked immunosorbent assay (ELISA) and Western blot] in TgCRND8 and non-Tg mice at 4, 6, and 8 months of age (Fig. 1 and fig. S1).
Fig. 1. Intraparenchymal injection of D3, a selective TrkA agonist, but not NGF, increased TrkA-dependent signaling in 6-month-old TgCRND8 mice with NGF and TrkA deficits.
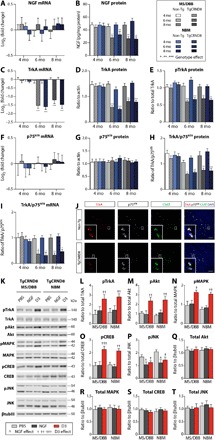
(A) No change in NGF mRNA levels was found in the MS/DBB and NBM from 4 to 8 months of age in TgCRND8 and non-Tg mice. (B) NGF protein, (C) TrkA mRNA, (D) TrkA protein, and (E) pTrkA levels were reduced in 6- and 8-month-old TgCRND8 relative to non-Tg mice. (F) p75NTR mRNA and (G) p75NTR protein levels remained stable from 4 to 8 months of age in both genotypes. The relative levels of TrkA to p75NTR (H) protein and (I) mRNA in TgCRND8 mice were decreased by 6 months of age compared to non-Tg mice. (J) Fluorescence in situ hybridization revealed down-regulation of TrkA (red) but not p75NTR (white) mRNA transcripts in ChAT+ cholinergic somata (green) of TgCRND8 mice. Scale bars, 20 μm. (K) Representative Western blots from the MS/DBB and NBM after injection of PBS, NGF, and D3 in TgCRND8 mice. D3-treated mice demonstrated increased levels of (L) pTrkA, (M) pAkt, (N) pMAPK, and (O) pCREB relative to NGF- and PBS-injected TgCRND8 mice. (P) pJNK levels were decreased following NGF and D3 treatment compared to PBS-treated mice. There were no changes in total levels of (Q) Akt, (R) MAPK, (S) CREB, and (T) JNK normalized to βtubIII. All mRNA levels were quantified by qRT-PCR. NGF protein was analyzed by ELISA. TrkA and p75NTR protein levels were measured by Western blot. All phosphorylation signal intensity values were normalized to their respective total protein levels. Statistics: Repeated-measures two-way analysis of variance (ANOVA) (A to I) and one-way ANOVA (L to T). Significance: *,^,†P < 0.05; **,††P < 0.01; ***,†††P < 0.001; * indicates comparison to age-matched non-Tg mice; ^ and † indicate comparison with PBS-treated mice. Data represent means ± SEM; n = 8 per group (A to I) and n = 4 per group (L to T).
Relative to age-matched non-Tg mice, NGF mRNA levels in the MS/DBB and NBM were unaltered in TgCRND8 mice (Fig. 1A). In contrast, NGF protein levels were reduced in 6- and 8-month-old Tg mice (Fig. 1B). Similar analysis in the HF and CTX revealed no change in NGF mRNA and protein levels between groups at all time points examined (fig. S1, A and B).
TrkA mRNA and protein levels in 6- and 8-month-old Tg mice were decreased in the basal forebrain, whereas only TrkA protein levels were reduced in the HF and CTX at these ages, consistent with anterograde transport of the receptor to projection sites of BFCNs once it is translated (Fig. 1, C and D, and fig. S1, C and D). TrkA gene expression is under positive feedback from NGF signaling and may be disrupted by reduced availability of NGF to BFCNs (2, 3). In addition, reduced levels of phosphorylated TrkA (pTrkA) were found in 6- and 8-month-old Tg mice (Fig. 1E and fig. S1E). Levels of p75NTR mRNA and protein were stable between genotypes and across all age groups and brain regions (Fig. 1, F and G, and fig. S1, F and G). These data are in line with the relative imbalance of TrkA/p75NTR at the protein level (Fig. 1H and fig. S1H) and mRNA level (Fig. 1, I and J) in 6- and 8-month-old Tg mice, as a result of a shift in the balance of NGF and TrkA expression. Our results demonstrate that deficits in the NGF/TrkA system observed in TgCRND8 mice resemble those observed in human AD and provide a relevant model to evaluate NGF-based therapeutic strategies for AD.
D3, not NGF, stimulates TrkA-dependent signaling in TgCRND8 mice
Next, we directly compared the in vivo efficacy of native NGF and compound D3, a selective TrkA agonist, to induce TrkA-dependent signaling cascades in 6-month-old TgCRND8 mice. Following direct intraparenchymal injection to the MS/DBB and NBM in TgCRND8 mice, D3, unlike native NGF, stimulated neurotrophin signaling (Fig. 1, K to T). Specifically, we found elevated levels of pTrkA, pAkt (Thr308), pMAPK (Thr202/Tyr204), and pCREB (phosphorylated cyclic adenosine monophosphate response element-binding protein; Ser133) within 90 min after D3 treatment relative to NGF-treated and phosphate-buffered saline (PBS)–treated TgCRND8 mice (Fig. 1, K to O). Downstream of p75NTR, we found reduced JNK activity (Thr183/Tyr185) in D3- and NGF-treated mice relative to PBS controls (Fig. 1, K and P). No differences were detected in total protein level normalized to β-tubulin-III (βtubIII) among all conditions (Fig. 1, Q to T). As a positive control, we performed intraparenchymal injection of D3 to the basal forebrain in age-matched, non-Tg mice to confirm signal transduction pathways activated by D3 (fig. S2, A to F) and NGF (fig. S3, A to F). Total protein levels normalized to βtubIII remained unaltered across all treatment groups (figs. S2, G to J, and S3, G to J). These findings provide experimental evidence that Aβ pathology interferes with NGF bioactivity in vivo and demonstrate that selective TrkA stimulation, in the absence of p75NTR interaction, is required to promote TrkA-dependent survival and plasticity in the presence of Aβ pathology.
Localized MRIgFUS-mediated BBB disruption in the basal forebrain
To rescue NGF/TrkA signaling deficits in the TgCRND8 model, we delivered the selective TrkA ligand, D3, to BFCNs using MRIgFUS. The MRIgFUS experimental setup is illustrated in Fig. 2A. The MS/DBB region of the basal forebrain was targeted bilaterally with one spot (Fig. 2B, green dot) and the NBM with two foci (Fig. 2B, purple dots) on T2-weighted (T2w) MR images (Fig. 2C). BBB opening was confirmed on T1w contrast-enhanced MR images, visualized by the influx of Gadovist (MRI contrast agent) from the bloodstream into sonicated areas (Fig. 2D). We found no difference in the level of contrast enhancement between TgCRND8 and non-Tg mice (Fig. 2E). The mean increase in signal intensity in the sonicated areas was 20 ± 8% and 25 ± 9% in non-Tg and TgCRND8 mice, respectively. Concomitantly with relative enhancement, no difference in the average peak pressure required to induce subharmonic emissions was observed between genotypes. The mean peak pressure reached during sonication was 1.29 ± 0.15 MPa in the TgCRND8 mice and 1.21 ± 0.11 MPa in the non-Tg controls (Fig. 2F). Relative contrast enhancement and mean acoustic pressure were comparable between treatment groups as well as subregionally in the basal forebrain (fig. S4). These data suggest that the increase in BBB permeability was consistent between genotypes, treatment groups, and distinct nuclei within the basal forebrain.
Fig. 2. Noninvasive FUS-induced BBB permeability localized to the basal forebrain under MRI guidance.
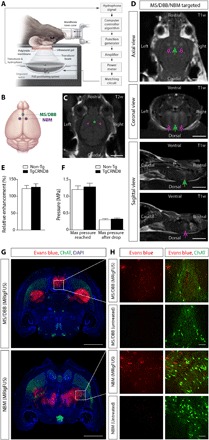
(A) Schematic of the experimental setup. Mice were positioned supine on an MRI-compatible sled with the head coupled to a polyimide membrane with ultrasound gel and placed in a tank filled with degassed, deionized water, housing the transducer and hydrophone. (B) Ultrasound foci in the MS/DBB and NBM regions of the basal forebrain were targeted on (C) axial T2w MR images. (D) BBB permeability was visualized by the influx of an MRI contrast agent (Gadovist) on contrast-enhanced T1w images in the MS/DBB (green arrows) and NBM (purple arrows). (E and F) There were no significant differences in (E) contrast enhancement or (F) pressure required to induce BBB modulation in 6-month-old TgCRND8 mice compared to non-Tg littermates. (G) Histologically, Evans blue dye (red) extravasation was detected in MRIgFUS-targeted regions of the basal forebrain (MS/DBB, top; NBM, bottom) 90 min after sonication. (H) Higher-magnification confocal micrographs in the MS/DBB (top) and NBM (bottom) revealed the entry of Evans blue in the parenchyma of animals treated with MRIgFUS compared to untreated animals, injected with Evans blue without MRIgFUS. ChAT immunoreactivity (green) was used to identify cholinergic cell bodies. Scale bars, 5 mm (C and D), 1 mm (G), and 100 μm (H). Statistics: Student’s t test (E and F). Data represent means + SEM; n = 12 per genotype.
Evans blue dye, which binds to serum albumin, and endogenous antibodies, immunoglobulin G (IgG) and IgM, proteins that normally do not cross the BBB, were used to histologically confirm the sites targeted by MRIgFUS. Immunohistochemical analyses revealed a 1-mm-wide focal spot of Evans blue dye at the level of the MS/DBB and two 1-mm-wide focal spots in the NBM (Fig. 2G). In contrast, Evans blue was not detected in brain regions nontargeted by FUS (Fig. 2G) and in control, non-FUS–treated, mice (Fig. 2H). Extravasation of blood-borne IgG and IgM into the basal forebrain was observed 90 min after MRIgFUS (fig. S5). Immunohistochemical (fig. S5A) and Western blot (fig. S5, B to E) analyses revealed greater levels of IgG and IgM in MRIgFUS-treated regions relative to unsonicated areas, in both TgCRND8 and non-Tg mice. Furthermore, there was a positive correlation between IgM levels and Gadovist contrast enhancement (fig. S5F), as previously reported in the same experimental model (26).
We found no evidence of erythrocyte extravasation or neuronal cell death in sonicated areas. Ter119-positive erythrocytes were confined to vascular structures identified by tomato lectin in MRIgFUS-targeted regions, without red blood cell infiltration into the brain parenchyma (fig. S6). We did not detect either cleaved caspase-3 or terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) immunoreactivity in neuronal nuclei (NeuN)–positive cells (fig. S7). The olfactory bulb was used as a positive control for caspase-3 and TUNEL due to the relatively high rate of granule cell turnover in this region (18). Together, these findings suggest that MRIgFUS allows BBB opening in the basal forebrain without overt tissue damage.
MRIgFUS effectively delivers D3 to the basal forebrain
Using quantitative high-performance liquid chromatography (HPLC), we measured D3 concentrations in MRIgFUS-targeted and nonsonicated brain regions of TgCRND8 mice and non-Tg controls after intravenous D3 injection. D3 was detected in D3/FUS-treated tissue 30 min after treatment (Fig. 3, A and B). The mean D3 levels in the MS/DBB were 15 ± 2 and 13 ± 3 ng/mg tissue in TgCRND8 and non-Tg mice, respectively. Similarly, D3 levels in the NBM were 16 ± 2 and 12 ± 2 ng/mg tissue in TgCRND8 and non-Tg mice, respectively. The drug concentration achieved after MRIgFUS was not significantly different between genotypes in both neuroanatomical locations. By contrast, D3 levels were below the limit of detection by HPLC in D3-treated, non-FUS controls (Fig. 3B). The mean D3 concentration in the serum and plasma was comparable between D3- and D3/FUS-treated mice for both genotypes (Fig. 3C).
Fig. 3. Enhanced D3 delivery and increased NGF protein levels in the basal forebrain following MRIgFUS-induced BBB opening.
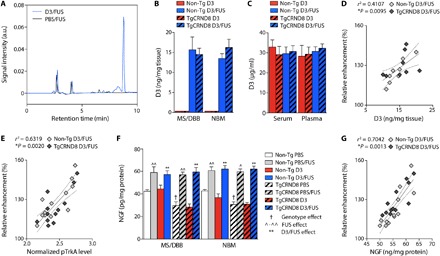
(A) Representative HPLC elution profile corresponding to D3 detection in the basal forebrain following D3/FUS treatment (blue line) relative to PBS/FUS control (black line). a.u., arbitrary units. (B) D3 levels were negligible in the brain after intravenous D3 injection without MRIgFUS (red bars). With MRIgFUS, D3 significantly entered the targeted MS/DBB and NBM in non-Tg and TgCRND8 mice (blue bars). (C) Plasma and serum D3 levels between unsonicated and sonicated animals for both genotypes were comparable. (D) Increases in D3 concentration, and (E) TrkA phosphorylation, were proportional to the contrast enhancement in MRIgFUS-treated regions for both genotypes. (F) NGF protein levels were increased in PBS/FUS- and D3/FUS-treated relative to PBS- and D3-treated animals, respectively. NGF protein levels were decreased in 6-month-old TgCRND8 mice compared to non-Tg littermates, but FUS-induced NGF protein levels were comparable between genotypes. (G) There was a positive correlation between the relative contrast enhancement and NGF levels measured. Statistics: Two-way ANOVA (B, C, and F). Significance: ^,†P < 0.05, **,^^P < 0.01; † indicates comparison with PBS-treated non-Tg mice (genotype effect); ^ indicates comparison with PBS-treated mice of the same genotype (FUS effect); * indicates comparison with D3-treated mice (i.e., intravenous D3, no MRIgFUS) of the same genotype (D3/FUS effect). (D, E, and G) Linear regression analysis. Dashed lines indicate a 95% confidence interval. Data represent means + SEM; n = 4 per group (B to D) and n = 6 per group (E to G).
The mean D3 concentration and relative contrast enhancement were positively correlated, with no apparent differences in correlation between non-Tg and TgCRND8 mice (Fig. 3D). In addition, there was a positive correlation between TrkA activation and relative contrast enhancement, consistent with enhanced D3 bioavailability in the MRIgFUS-targeted regions (Fig. 3E). Overall, these data support the feasibility of using MRIgFUS to deliver D3 locally to the basal forebrain in a noninvasive manner.
Given that the sonication scheme used to achieve MRIgFUS BBB opening in this study would potentially allow the entry of serum NGF (26 kDa), or factors stabilizing NGF and/or its release, we evaluated NGF protein levels by ELISA. Elevated levels of NGF were found in PBS/FUS relative to PBS-treated mice and D3/FUS relative to D3-treated animals in the MS/DBB and NBM for both TgCRND8 and non-Tg mice (Fig. 3F). There was a positive correlation between NGF levels and contrast-enhanced T1w voxel intensity in MRIgFUS-treated non-Tg and TgCRND8 mice, indicating that Gadovist enhancement may provide a means of estimating NGF alterations in the targeted tissue volume (Fig. 3G). The elevated local NGF levels resulting from MRIgFUS lend support to the hypothesis that our therapeutic paradigm may be used to promote endogenous brain plasticity and repair for the treatment of AD.
MRIgFUS-mediated delivery of D3 activates TrkA-dependent signaling pathways
To further elucidate TrkA signaling deficits in the TgCRND8 mouse model of AD, we compared signal transduction targets modulated downstream of TrkA phosphorylation in the basal forebrain (Fig. 4). In an independent cohort of 6-month-old mice, we confirmed the reduction in pTrkA levels in TgCRND8 relative to non-Tg mice in the MS/DBB and NBM (Fig. 4B). Parallel to reduced levels of pTrkA, pAkt, pMAPK, and pCREB were also decreased in TgCRND8 mice (Fig. 4, C to E). Downstream of p75NTR, we found increased JNK activity in TgCRND8 compared to non-Tg mice (Fig. 4F).
Fig. 4. MRIgFUS delivery of D3 increased TrkA-dependent signaling cascades in the basal forebrain 90 min after treatment.

(A) Representative Western blots from the MS/DBB and NBM after PBS, PBS/FUS, D3, or D3/FUS treatment. Blots were probed for pTrkA, pAkt, pMAPK, pCREB, pJNK, the corresponding total proteins, and βtubIII to control for loading. Levels of (B) pTrkA, (C) pAkt, (D) pMAPK, and (E) pCREB were reduced in PBS-treated TgCRND8 mice relative to non-Tg mice and stimulated in D3/FUS-treated mice compared to PBS- and D3-treated (no MRIgFUS) controls in the MS/DBB and NBM for both genotypes. (F) Elevated levels of pJNK were found in control TgCRND8 mice relative to non-Tg mice. pJNK was decreased in TgCRND8 mice after D3/FUS treatment compared to control conditions in Tg mice, but levels remained unaltered in non-Tg mice. Statistics: Two-way ANOVA. Significance: *,†P < 0.05; **,††P < 0.01, ***,^^^P < 0.001; † indicates comparison with PBS-treated non-Tg mice (genotype effect); ^ indicates comparison with PBS-treated mice of the same genotype (FUS effect); * indicates comparison with D3-treated group (i.e., intravenous D3, no MRIgFUS) of the same genotype (D3/FUS effect). Data represent means + SEM; n = 6 per group.
Thus, we assessed the functional effects of MRIgFUS delivery of D3 on TrkA-dependent signaling pathways in 6-month-old TgCRND8 and non-Tg littermates (Fig. 4). We found elevated pTrkA levels in the MS/DBB and NBM within 90 min after D3/FUS treatment relative to PBS/FUS-, PBS-, and D3-treated mice in both genotypes (Fig. 4, A and B). TrkA activation was achieved in 6-month-old TgCRND8 mice in the presence of decreased TrkA expression, and the level of pTrkA was not significantly different from D3/FUS-treated non-Tg mice (Fig. 4B).
We observed elevated pAkt in PBS/FUS- and D3/FUS-treated mice compared to PBS- and D3-treated animals, respectively (Fig. 4C). These data are in line with recent studies, which demonstrate that MRIgFUS BBB opening leads to elevated pAkt levels, 90 min after sonication (27, 28). Similarly, we found an increase in MAPK activation in D3/FUS-treated compared to PBS/FUS-, D3-, and PBS-treated mice (Fig. 4D). CREB function was acutely increased after D3/FUS treatment (Fig. 4E), likely to be due to phosphorylation at Ser133 by multiple kinases, including MAPK and Akt (3). JNK activity was decreased after D3/FUS relative to control conditions in TgCRND8 mice (Fig. 4F). For each protein in the NGF/TrkA signaling pathway, no difference was detected in total protein level normalized to βtubIII among all treatment groups (fig. S8).
MRIgFUS-mediated delivery of D3 enhances cholinergic function
We established whether deficits in TrkA signaling correspond to diminished cholinergic function in TgCRND8 mice. ChAT activity in the basal forebrain (enriched in cholinergic cell bodies) and ACh release in the HF and CTX (enriched in cholinergic axon terminals) were measured (Fig. 5). In 6-month-old TgCRND8 mice, ChAT activity was reduced by 30 to 40% relative to non-Tg littermates (Fig. 5, A and B). Furthermore, basal and potassium-induced ACh release was lower in TgCRND8 compared to non-Tg slices (Fig. 5, C and D), consistent with the loss of enzymatic activity related to ACh synthesis in TgCRND8 mice. These results indicate that 6-month-old TgCRND8 and non-Tg mice represent valid experimental models of impaired and unimpaired cholinergic function, respectively.
Fig. 5. MRIgFUS delivery of D3 enhances cholinergic function 3 days after treatment.
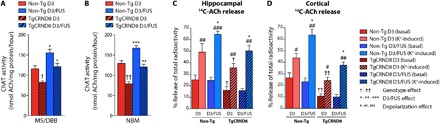
(A and B) ChAT enzymatic activity was significantly reduced in TgCRND8 relative to non-Tg littermates in the (A) MS/DBB and (B) NBM. ChAT activity was significantly increased in D3/FUS-treated compared to D3-treated (no MRIgFUS) controls in the MS/DBB and NBM for both TgCRND8 and non-Tg mice. (C) Basal and potassium depolarization–induced ACh release from hippocampal slices was significantly reduced in TgCRND8 mice compared to non-Tg controls. Evoked hippocampal ACh release was increased after D3/FUS treatment compared to D3-treated controls in both genotypes. Basal release of ACh was not significantly different in D3/FUS-treated compared to D3-treated animals for both genotypes. (D) TgCRND8 mice had decreased levels of basal and potassium-evoked cortical ACh release relative to non-Tg littermates. Evoked ACh release was also elevated in cortical slices from both TgCRND8 and non-Tg mice following D3/FUS treatment, but basal levels of ACh release remained unaltered in both genotypes. Statistics: Two-way ANOVA. Significance: *,†,#P < 0.05; **,††, ##P < 0.01, ***,###P < 0.001; † indicates comparison with D3-treated non-Tg mice (genotype effect); # indicates comparison with basal levels of ACh release of the same treatment and genotype (depolarization effect); * indicates comparison with D3-treated group (i.e., intravenous D3, no MRIgFUS) of the same genotype (D3 /FUS effect). Data represent means + SEM; n = 5 per group.
Thus, we evaluated the potential of MRIgFUS delivery of D3 to increase cholinergic function in TgCRND8 and non-Tg mice. Three days after sonication, elevated ChAT activity was found in the D3/FUS-treated MS/DBB (Fig. 5A) and NBM (Fig. 5B) relative to animals that received intravenous D3 administration only, for both genotypes. In addition, we examined the impact of D3/FUS treatment on basal and depolarization-evoked ACh release in hippocampal and cortical slices, also at 3 days after FUS. Potassium-evoked ACh release was elevated in D3/FUS compared to intravenous D3 treated animals of both genotypes in hippocampal (Fig. 5C) and cortical preparations (Fig. 5D). Together, our data demonstrate that a single D3/FUS treatment can stimulate cholinergic function in TgCRND8 mice up to 3 days after sonication.
Overall, these findings highlight the in vivo efficacy of our treatment paradigm—that is, to selectively engage TrkA receptors and regulate critical intracellular signaling cascades that are sufficient to promote cholinergic function in the presence of Aβ pathology (Fig. 6). Thus, enhancing cholinergic neurotransmission by MRIgFUS-mediated delivery of D3 represents a possible therapeutic approach to limit cognitive decline in AD.
Fig. 6. MRIgFUS delivery of D3, a selective TrkA agonist, to the basal forebrain rescued neurotrophin signaling and cholinergic function in the TgCRND8 model of AD.
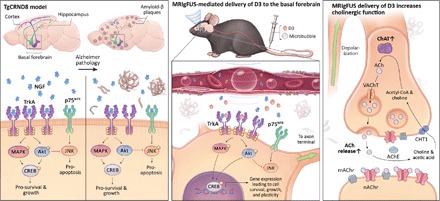
(Left) By 6 months of age, TgCRND8 mice demonstrated deficits in the NGF/TrkA signaling system, similar to what is observed in human AD. TgCRND8 mice had reduced NGF protein levels, TrkA expression, and activation of downstream signaling cascades, coupled with increased JNK activity in the basal forebrain. (Middle) MRIgFUS was used to noninvasively and locally increase BBB permeability in the basal forebrain, enhancing the bioavailability of intravenous D3 to cholinergic cell bodies in the MS/DBB and NBM. Using this therapeutic approach in TgCNRD8 mice, we stimulated TrkA and downstream signaling effectors—pAkt, pMAPK, and pCREB—while simultaneously decreasing pJNK known to be triggered by p75NTR activation. (Right) MRIgFUS delivery of D3 to the basal forebrain led to enhanced cholinergic neurotransmission in widespread axon terminal regions of the CTX and HF, as evidenced by enhanced ChAT activity, the enzyme that synthesizes ACh, and ACh release.
DISCUSSION
There is an urgent need for therapeutics and delivery methods that slow or reverse the degeneration of neurons in AD, including cholinergic neurons. Acetylcholinesterase inhibitors, which represent the most common pharmacological compounds used for the treatment of AD, do not restore dying or degenerating neurons. In contrast, NGF-related therapeutics bode well for the maintenance of cholinergic morphology, survival, and function. In particular, strategies in which NGF-mediated activation of BFCN function can be safely and effectively accomplished hold therapeutic potential for AD.
In this study, we show that MRIgFUS delivery of D3 is a novel therapeutic approach to enhance cholinergic function, addressing fundamental limitations of current NGF-based treatment modalities for AD. MRIgFUS-mediated delivery of D3 led to selective TrkA signaling in BFCNs while reducing p75NTR activation, and elevated ChAT activity and ACh release in a mouse model of amyloidosis with deficits in NGF, TrkA and cholinergic function—pathologies that are present in human AD. Our findings are consistent with the suggestion that NGF administration directly to cholinergic cell bodies, rather than in axon terminals, bypasses transport defects and allows NGF/TrkA signaling at the soma (29–31). Even in the presence of reduced TrkA levels, D3/FUS treatment increased TrkA signaling in TgCNRD8 mice, as assessed by activation of TrkA and downstream effectors including Akt, MAPK, and CREB (Fig. 4). Although p75NTR expression levels remain stable, downstream p75NTR activity decreased in response to D3/FUS treatment as measured by JNK activation. TrkA signaling has been shown to silence p75NTR-mediated pathways such as JNK via PI3K/Akt (3). Because D3 does not interact with p75NTR directly, Akt activation may represent a potential mechanism by which D3/FUS treatment decreases JNK phosphorylation. Moreover, MRIgFUS delivery of D3 rescued cholinergic function, i.e., depolarization-induced ACh release in amyloid-burdened hippocampal and cortical regions, and ChAT activity in the basal forebrain. The levels of evoked ACh release and ChAT activity for up to 3 days after D3/FUS treatment were similar to those observed in untreated, non-Tg controls, which have levels of cholinergic signaling sufficient to support normal cognitive function (Fig. 5). In this way, localized D3 delivery to the basal forebrain by MRIgFUS led to widespread restoration of cholinergic transmission in the associated projection regions of the HF and CTX.
To date, NGF-based therapies have been validated in preclinical models of aging and injury. Although these studies lend further support to the rationale of NGF as a therapeutic agent for AD, the predictive validity of these models was limited by the absence of Aβ and tau pathology. Multiple lines of evidence suggest that Aβ and tau are inextricably linked to NGF-related signaling cascades in vitro and in vivo (6–8). Here, we discovered alterations in the status of NGF bioactivity related to NGF mRNA and protein levels, NGF receptor levels, and downstream signaling components in a preclinical model of amyloidosis, which, taken together, appear to recapitulate the signaling alterations reported in early-stage AD patients (2). Evidence from preclinical and human studies indicate that the shift in TrkA/p75NTR levels may represent a switch favoring the binding of NGF to p75NTR, possibly activating apoptosis over survival pathways in BFCNs and contributing to cholinergic dysfunction before cell loss in AD (2, 3). Our signaling data also suggest a TrkA/p75NTR signaling imbalance, which can contribute to cholinergic dysfunction in AD. Previous studies suggest that increased p75NTR activity coupled with increased Aβ load exacerbates Aβ-induced neurodegeneration (6–8). Moreover, our data in the TgCRND8 model indicate that the amount of ACh synthesized (Fig. 5, A and B) and released (Fig. 5, C and D) is impaired with disease progression and may, in part, underlie the cognitive deficits observed in this model of AD (20). Likewise, Bellucci et al. (22) reported decreased ChAT immunoreactivity in the NBM and release of cortical ACh in 7-month-old TgCRND8 mice relative to non-Tg controls. To this end, the TgCRND8 model is suitable for testing NGF-related therapeutic agents as a means to improve cholinergic function in the presence of Aβ pathology.
Within this framework, selective activation of TrkA, avoiding p75NTR, was necessary to induce TrkA-related signaling pathways and sufficient to up-regulate cholinergic function in the context of amyloid pathology. Our results demonstrate that D3, in contrast to native NGF, stimulates TrkA signaling in the TgCRND8 mice (Fig. 1, K to T). Our findings draw attention to a previously unknown role of amyloid on NGF bioactivity in vivo, including the relative imbalance in TrkA/p75NTR signaling, amyloid pathology, and potentially other mechanisms affecting NGF stability, and could partly explain the limited efficacy of NGF-directed treatments for AD to date (9, 10, 32).
A major concern for NGF-based therapy is that undesirable pleiotropic effects can reduce the therapeutic benefit of NGF. For example, p75NTR activation can negate the benefit of engaging TrkA (14). Delivery of D3, a highly selective and potent partial TrkA agonist, is a promising approach to promote TrkA signaling (11), and studies in animal models of aging and AD have shown improvements in memory performance (12, 13). Clinically, D3 has been shown to be a well-tolerable and effective treatment for dry eye, without inducing potential treatment-related side effects such as aberrant growth of sensory fibers (33).
In addition, the delivery of NGF-related therapeutic agents to the brain, to date, required invasive surgery due to their limited ability to cross the BBB (5). We demonstrate that MRIgFUS is an effective method for targeted drug delivery to the basal forebrain in a noninvasive manner with real-time feedback monitoring. In human AD, clinical trials for NGF and vector-based NGF delivery all require complex intracranial surgical procedures that produce BBB breakage and tissue damage and carry substantial risk for severe hemorrhages and infection. Moreover, these surgeries pose a higher risk of adverse events among elderly individuals. These detrimental effects could rapidly outweigh the potential benefit of NGF therapy (9, 10, 32). The MRIgFUS parameters used here did not induce erythrocyte extravasation (fig. S6) or neuronal apoptosis (fig. S7). These data are in line with previous studies, which demonstrated that these MRIgFUS settings are below the threshold of damage (34, 35). Focused ultrasound combined with MRI as an imaging method for targeting the brain allows ultrasound to be applied throughout the entire basal forebrain, without significant diffusion of the drug to distal brain regions, especially in the ventricles and meninges containing cerebrospinal fluid. Here, an intravenous dose of 500 μg of D3 was sufficient to activate TrkA in the basal forebrain targeted by MRIgFUS, without drug diffusion to other brain regions (D3 levels below the detection threshold by HPLC).
Further work to optimize experimental design parameters should be conducted with the goal of achieving consistent BBB modulation of permeability and simultaneously minimizing potential damage. Factors such as sonication scheme, microbubble composition, and brain subregion can influence the extent of BBB permeability and associated biological effects following FUS treatment. Microbubble dose, size, shell properties, and half-life circulation can affect the degree of BBB permeability (36). In this study, we used the recommended clinical imaging dose of Definity microbubbles (20 μl/kg) to approximate an equivalent microbubble dose between animals. Recently, McMahon and Hynynen (34) reported that Definity at 20 μl/kg compared to 100 μl/kg (10 times higher than the clinical imaging dose) can be used to increase BBB permeability without activation of inflammatory markers (i.e., nuclear factor κB signaling pathway genes), tissue damage, or MRI evidence of edema and hemorrhage. The probability of FUS-induced BBB permeability using a clinical imaging dose of Definity and Optison is similar across a range of pressure amplitudes, as the ones achieved in our study and established previously by McDannold et al. (37).
We monitored and controlled FUS-mediated BBB modulation in real time using an acoustic controller. The applied acoustic pressure was incrementally increased until subharmonic emissions (characteristic of stable cavitation) were detected, thereby increasing BBB permeability below the threshold for tissue damage (35). This method has been shown to improve sonication consistency and safety. A similar approach was used in the first clinical trial for BBB opening in AD using MRIgFUS with no adverse events after sonication (19). Immunohistochemical analysis in our study revealed no neuronal loss or erythrocyte extravasation in the basal forebrain 90 min after sonication (figs. S6 and S7). These findings are in accordance with other preclinical studies that demonstrate increased BBB permeability in the absence of edema, neuronal damage, and minimal to no erythrocyte extravasation using real-time feedback monitoring (34, 35, 38, 39).
Previous evidence suggests that local differences in vasculature and brain region can lead to variable effects of FUS-induced BBB permeability (40). Here, we show that similar pressure amplitudes were required to induce subharmonic emissions in the subregions of the basal forebrain (fig. S4), and acoustic pressures were comparable to those achieved following FUS-mediated BBB opening in the hippocampus of TgCRND8 mice and non-Tg littermates using an identical transducer/hydrophone configuration and feedback controller (38). We observed no differences in BBB opening between TgCRND8 and non-Tg mice (Fig. 2F) despite vascular dysfunction in TgCRND8 mice by 6 months of age (41). Future work is necessary to evaluate whether BBB recovery varies temporally in different basal forebrain regions and due to the presence of AD pathology.
In recent years, the potential use of MRIgFUS for AD has evolved beyond a method for drug delivery. In several mouse models of AD, FUS-mediated BBB opening reduced amyloid and tau pathology, even in the absence of drug delivery (26, 38, 39, 42, 43). Here, we combined FUS, as a delivery tool with disease-modulatory effects, with a pharmacological agent that is designed to enhance plasticity and resilience of BFCNs vulnerable to degeneration in AD. In this way, our therapeutic approach targets multiple pathologies implicated in AD and thus may prove more efficacious than a single target paradigm to slow or reverse disease progression. We found elevated levels of NGF in the brain parenchyma following MRIgFUS (Fig. 3, F and G). The source of increased NGF levels at sites of FUS-BBB modulation remains to be identified. Candidates include circulating NGF present in the bloodstream, cells capable of rapid NGF release residing on the parenchymal side of the BBB and known to be first responders [e.g., mast cells; reviewed in (44)], and microglia and astrocytes known to be activated by FUS (26, 28) that secrete NGF as early as 3 hours after stimulation in vitro (45). However, it remains to be tested whether glial cells can release sufficient amounts of NGF within 90 min in vivo, to account for the levels detected here.
In the adult mouse, basal NGF levels in the serum equivalent to 50 to 100 pg/ml may be physiologically meaningful in view of NGF binding affinity for the TrkA receptor [Kd (dissociation constant) of ~10−11 M] (3, 11, 46). Our results demonstrate that elevated levels of NGF are not sufficient to induce TrkA phosphorylation in non-Tg and TgCRND8 mice at 90 min following MRIgFUS alone. This result may be explained by NGF instability in solution and sensitivity to proteolysis and supported by the fact that NGF bioactivity is optimal at nanomolar concentrations (11). Nevertheless, this finding has important implications for the therapeutic action of D3 in MRIgFUS-treated areas. Because NGF and D3 can bind TrkA concomitantly, D3 can potentiate TrkA signals activated by low picomolar levels of NGF (11, 47). Therefore, MRIgFUS-induced increase in NGF and D3 may together further potentiate TrkA-dependent signaling cascades in the context of Aβ pathology.
An important part of preclinical development toward clinical translation will be dosing optimization for D3/FUS treatment. Previous work has shown that a single intraparenchymal injection of NGF in young adult and aged rats induces a rapid TrkA tyrosine phosphorylation response that persists for up to 5 days after injection (48, 49). Notably, our results demonstrate that BFCNs remain responsive to TrkA stimulation in the presence of amyloid pathology and impaired NGF-related signaling mechanisms. They further suggest that the biological response of a single D3/FUS treatment, at the level of TrkA receptor phosphorylation, is similar in healthy, adult cholinergic neurons and those burdened by amyloid pathology. For successful clinical application of D3, it will be essential to define the dosing interval needed to elicit TrkA phosphorylation leading to sustained cholinergic function and cognitive benefit. Prolonged TrkA signaling may be required to rescue degenerating BFCNs (i.e., for maintenance and remodeling of cell bodies, axons, and dendrites of BFCNs) and affect cognition. Preclinical studies demonstrated that administration of NGF and specific agonists of TrkA for at least 2 weeks in aged or lesioned animal models reversed BFCN atrophy and ameliorated behavioral impairments; however, the deficits reappeared after cessation of treatment (5, 12, 50, 51).
The first AD BBB modulation clinical trial recently revealed that single and repeated FUS treatments, monitored in real time using passive cavitation detection, are well tolerated in AD patients (19), supporting the feasibility of FUS delivery of D3 in AD patients. Subsequent larger clinical trials will help determine the potential therapeutic benefit of FUS-induced BBB permeability alone. An important question for future work will be to assess whether repeated administration of a TrkA agonist using FUS can act as a combination therapy to restore cholinergic function and reduce Aβ deposition and tau pathology in ways that promote cognitive function.
Evidence suggests that administration of NGF or TrkA ligands can, on one hand, improve performance in cognitively impaired rodents and, on the other hand, compromise intact function in healthy animals (12, 13, 51–53). This differential effect in diseased compared to healthy brains has to be taken into consideration when treatment strategies are planned. NGF modulation of cholinergic function is tightly regulated such that either excess or lack of the neurotrophin can be detrimental. Excessive and adverse neuronal plasticity as a result of TrkA activation is unlikely to occur in the presence of toxic Aβ species that are negatively influencing TrkA signaling continuously; however, this remains to be tested.
Pharmacological treatment of AD with D3 will require personalized strategies to address the disease process of each individual. Promising advances in neuroimaging may be eventually used to identify patients who might be most responsive to a therapeutic regimen incorporating agonists to TrkA. For instance, Trk-targeted radiotracers for positron emission tomography imaging may reveal spatiotemporal changes in TrkA receptor expression level—an early molecular alteration in mild cognitive impairment (MCI) and AD correlated with cognitive impairment (54). Alternatively, radioligands designed to quantify and map in vivo degeneration of cholinergic terminals in AD patients could be used for clinical identification, as well as monitoring the efficacy of TrkA-based interventions (55).
In conclusion, our study demonstrates that, in contrast to native NGF, only a selective TrkA agonist, without engaging p75NTR, can stimulate prosurvival and regenerative signaling in the context of Aβ pathology. The compound D3, and not NGF, activated TrkA-dependent signaling cascades in the TgCRND8 model of AD. Our results reveal a relative imbalance of TrkA/p75NTR activity in TgCRND8 mice, as observed in human AD, which may partly explain the failure of NGF reported here and, more broadly, why NGF therapy in AD clinical trials failed to display the efficacy observed in preclinical animal models without Aβ and tau pathology. As an alternative therapeutic strategy, we demonstrated that noninvasive MRIgFUS-mediated delivery of D3 localized to BFCNs stimulated TrkA signaling, and it was sufficient to rescue cholinergic function, even in the presence of Aβ pathology. Further investigation of the long-term effects of D3 delivery to the basal forebrain using MRIgFUS and after repeated treatments may reveal a promising therapy to promote cognition regulated by cholinergic neurotransmission in AD. Overall, our study highlights an alternative and complementary approach to amyloid- and tau-targeted therapies to counter neurodegeneration in AD.
MATERIALS AND METHODS
Study design
Our study aimed to (i) investigate how the NGF/TrkA signaling system is altered with disease progression in a mouse model of AD, (ii) compare the efficacy of native NGF and selective TrkA agonist D3 in a disease-relevant phenotype, and (iii) establish the feasibility of noninvasive and localized D3 delivery to the basal forebrain with MRIgFUS. Accordingly, we treated and analyzed four cohorts of TgCRND8 and non-Tg littermates. A first cohort of sex-balanced TgCRND8 and non-Tg mice at 4, 6, and 8 months of age (n = 8 per group) was used to characterize the genotype- and age-associated alterations in the NGF/TrkA signaling system using qRT-PCR, in situ hybridization, ELISA, Western blotting, and confocal microscopy.
In a second cohort, sex-balanced, 6-month-old TgCRND8 and non-Tg mice were randomly assigned to receive PBS, 2 μg of NGF, or 2 μg of D3 injected to the basal forebrain via intraparenchymal injection. Mice were sacrificed 90 min after injection (n = 4 per group). Using immunoprecipitation and Western blotting, we measured TrkA-dependent signaling pathways activated in the basal forebrain following PBS, NGF, and D3 administration.
A third cohort of sex-balanced, 6-month-old TgCRND8 and non-Tg littermates was tested. Mice were randomly assigned to treatment groups as follows: (i) D3/FUS-treated (500 μg, intravenous dose of D3 with MRIgFUS), (ii) PBS/FUS-treated (intravenous PBS with MRIgFUS), (iii) D3-treated (500 μg, intravenous dose of D3, no MRIgFUS), or (iv) PBS-treated (intravenous PBS, no MRIgFUS). Using histological methods, Western blotting, ELISA, and confocal microscopy, we evaluated the extent of BBB permeability in the basal forebrain 90 min following MRIgFUS (n = 6 PBS/FUS- and PBS-treated non-Tg and Tg mice). HPLC was used to measure the concentration of D3 delivered to the basal forebrain targeted with MRIgFUS, and mice were sacrificed 30 min after treatment to maximize therapeutic detection (n = 4 per group). Using immunoprecipitation and Western blotting, we evaluated the effect of D3/FUS treatment on TrkA signaling cascades 90 min after sonication (n = 6 per group). Treatment allocation was kept blinded until analysis. All animals were included in the analysis.
In the fourth cohort, sex-balanced, 6-month-old TgCRND8 and non-Tg mice were randomly assigned to either a D3/FUS-treated (500 μg, intravenous dose of D3 with MRIgFUS) or D3-treated (500 μg, intravenous dose of D3, no MRIgFUS) experimental group. Mice were sacrificed 3 days following treatment (n = 5 per group) for the assessment of ChAT activity in the basal forebrain, in addition to basal and depolarization-evoked ACh release in cholinergic terminal regions following D3 or D3/FUS treatment.
Animals
TgCRND8 mice and their age-matched non-Tg littermates were used in this study (20). All procedures were conducted in accordance with guidelines established by the Canadian Council on Animal Care and protocols approved by the Sunnybrook Research Institute Animal Care Committee.
Intracranial injections
Anesthesia was induced and maintained with isoflurane in oxygen. Mice were placed in a stereotaxic apparatus (David Kopf Instruments). The skull was exposed, and burr holes were made with a Stoelting drill to the following coordinates: MS/DBB: anteroposterior, +0.8 mm; mediolateral, +0.3 mm; dorsoventral, −4.4 mm (measured from bregma with a 4° vertical angle alignment); NBM: anteroposterior, −0.3 mm; mediolateral, +1.5 mm; dorsoventral, −5.6 mm (measured from bregma). A 33-gauge needle attached to a 5-μl Hamilton syringe containing the solution to be injected was lowered to the injection site, and 1-μl injection was made over 10 min. Following each injection, the needle was held in place at the injection site for 5 min and then removed from the brain.
MRI-guided focused ultrasound
Mice were anesthetized with 5% isoflurane and maintained at 2% isoflurane in medical air. Head fur was removed with depilatory cream, and a 26-gauge angiocatheter was inserted into the tail vein. The MRIgFUS experimental setup is illustrated in Fig. 2A. MRIgFUS was performed using the in-house build prototype system (similar to LP100, FUS Instruments Inc.), and animals were imaged using a 7.0-T MRI (Bruker). The acoustic beam was generated using a spherically focused transducer (1.68-MHz frequency, 75 mm diameter, 60 mm radius of curvature), with standard BBB disruption parameters (10 ms bursts, 1 Hz burst repetition frequency, 120-s duration). Axial T2w MR images were used to target three focal spots, covering the basal forebrain bilaterally.
At the time of sonication, mice received an intravenous injection of D3 (500 μg) diluted in PBS, or PBS only, Definity microbubbles (0.02 ml/kg; Lantheus Medical Imaging), and Gadovist (0.2 ml/kg, Schering AG) via the tail vein. For animals that received Evans blue, the dye (2% solution in PBS; 4 ml/kg; Sigma-Aldrich) was injected immediately after the sonication. Each injectable was administered individually, separated by a 150 μl saline flush to ensure transfer into the bloodstream.
Acoustic emissions were recorded using a polyvinylidene fluoride (PVDF) hydrophone coupled to an acoustic controller (34). The applied acoustic pressure was increased incrementally after each pulse until subharmonic emissions were detected, then reduced to 25%, and maintained for the remainder of the sonication. BBB permeability was visualized by an increase in Gadovist signal intensity on T1w MR images acquired 5 min after sonication. Contrast enhancement was assessed by measuring pixel intensity in a 2 mm × 2 mm target region normalized to a reference region of the brain. A custom image analysis program was used to calculate relative contrast enhancement (MATLAB, MathWorks).
Biodistribution and pharmacokinetic analysis of D3
D3 was quantified using HPLC versus standard curves as previously described (11). Mice were intracardially perfused with saline 30 min after treatment, and brains were removed and flash-frozen in liquid nitrogen. Tissue was homogenized in methanol using a mini-pestle fitted in a pellet pestle motor (Kontes) for 1 min at 4°C and centrifuged at 12,000 rpm for 15 min at 4°C for phase separation. The organic phase was collected and evaporated using nitrogen gas and resuspended in 40 μl of methanol for further analysis.
RNA isolation, reverse transcription, and qRT-PCR
Mice were intracardially perfused with ice-cold saline, and brains were removed, flash-frozen in liquid nitrogen, and stored at −80°C until analysis. Samples were homogenized (Minilys homogenizer, Bertin Technologies) in 1 ml of TRIzol (Invitrogen), and RNA was isolated with the PureLink RNA Mini Kit (Invitrogen). RNA concentration and purity were confirmed by spectrophotometry (NanoDrop Technologies). Synthesis of complementary DNA (cDNA) was performed using the SuperScript III First-Strand Synthesis SuperMix Kit (Invitrogen), followed by qRT-PCR using the ViiA 7 Real-Time PCR System (Applied Biosystems) with SYBR Green qPCR Master Mix (Applied Biosystems). Relative gene expression of NGF, TrkA, and p75NTR was normalized to the mean Ct value of three housekeeping genes (b-actin, GAPDH, and HPRT1), using the ΔΔCt method. All samples were run in triplicate. Primers used in this study are listed in table S1.
In situ hybridization
Mice were intracardially perfused with ice-cold saline, followed by 4% paraformaldehyde (PFA). Brains were isolated, postfixed in 4% PFA for 24 hours, and equilibrated in 30% sucrose (0.1 M PO4 buffer). Brains were sectioned axially at 40 μm and stored in cryoprotectant [50 mM phosphate buffer, 30% ethylene glycol, and 25% glycerin (pH 7.4)] at −20°C. Fluorescence in situ hybridization was performed using RNAscope (ACD Newark, CA, USA) according to the manufacturer’s instructions with slight modifications. Briefly, brain sections were washed in PBS, and endogenous peroxidase activity was quenched with 3% (v/v) H2O2 in PBS for 45 min at room temperature (RT). Antigen retrieval was performed at 95°C for 20 min using ACD antigen retrieval buffer. Sections were washed with PBS, mounted on Superfrost Plus slides (Fisher Scientific), and dried overnight at 37°C. Sections were then permeabilized with 0.3% Triton X-100 (Sigma-Aldrich) in PBS for 30 min at RT and then washed in double-distilled H2O. Probes for TrkA (ACD ref.: 435791-C1), ChAT (ACD ref.: 408731-C2), and p75NTR (ACD ref.: 494261-C3) were added in a 50:1:1 ratio. Sections were covered with parafilm and incubated overnight at 40°C in a humidified chamber (HybEZ oven, ACD). Subsequent washes were performed with 0.025% Tween 20 in 0.1× SSC (SSCT). Incubation times for additional steps were as follows, each succeeded by three washes in SSCT for 2 min at RT: Amp1-3 for 60 min at 40°C, horseradish peroxidase (HRP)–C1-C3 for 25 min at 40°C, TSA Plus fluorophore (TSA dyes, PerkinElmer) for 30 min at 40°C, and HRP blocker (ACD) for 20 min at 40°C; nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI; ACD).
Protein extraction
Mice were intracardially perfused with ice-cold saline, and brains were removed, flash-frozen in liquid nitrogen, and stored at −80°C until analysis. Tissue lysates were prepared by homogenizing tissue in ice-cold lysis buffer containing 20 mM tris-HCl (pH 7.5), 137 mM NaCl, 2 mM EDTA, 4% NP-40 detergent, and protease and phosphatase inhibitors (Sigma-Aldrich), using the Minilys homogenizer (Bertin Technologies), and then centrifuged at 12,000 rpm for 15 min at 4°C. The supernatant was collected and used to measure total protein concentration with the BCA assay (Pierce Biotechnology).
Western blotting
Brain homogenates were resolved by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to activated PVDF membranes. The membranes were blocked with 5% bovine serum albumin in tris-buffered saline/Tween 20 (TBST). The following primary antibodies were incubated overnight at 4°C: TrkA (1:1000; Cell Signaling, 2505), p75NTR (1:1000; Santa Cruz Biotechnology, sc-271708), pAkt (1:1000; Cell Signaling, 4060), Akt (1:1000; Cell Signaling, 9272), pCREB (1:1500; Cell Signaling, 9198), CREB (1:1500; Cell Signaling, 9104), pMAPK (1:2000; Cell Signaling, 9101), MAPK (1:2000; Cell Signaling, 9102), pJNK (1:1000; Cell Signaling, 9251), JNK (1:1000; Cell Signaling, 9252), βtubIII (1:5000; Covance, MMS-435P), and β-actin (1:5000; Sigma-Aldrich, A5316). Membranes were washed in TBST, incubated with corresponding IRDye secondary antibodies at 1:15,000 for 1 hour at RT (LI-COR Biosciences), and imaged with an Odyssey infrared imaging system (LI-COR Biosciences). Densitometry analysis was performed using ImageStudio software.
NGF ELISA
NGF levels were quantified using a commercially available ELISA kit from RayBiotech (ELM-bNGF-1) according to the manufacturer’s protocol. Samples (1:5 dilution in tissue lysis buffer) and controls were analyzed in triplicates.
Immunoprecipitation
Tissue lysates containing 500 μg of protein were left shaking overnight at 4°C with 5 μg of TrkA antibody (Santa Cruz Biotechnology, sc-118). Protein A agarose beads (50 μl; Santa Cruz Biotechnology, sc-2001) were added and left shaking at 4°C for 4 hours. The beads were washed with lysis buffer. SDS-PAGE and Western blotting were performed as described above. TrkA phosphorylation was measured by blotting for phosphotyrosine (1:1000; Upstate, 16-103) and total TrkA (1:1000; Cell Signaling, 2505).
Immunohistochemistry
Mice were intracardially perfused with saline, followed by 4% PFA. Brains were isolated, postfixed in 4% PFA for 24 hours, and equilibrated in 30% sucrose. Brains were sectioned axially at 40 μm and stored in cryoprotectant at −20°C. Sections were rinsed with 0.1 M PBS and then incubated for 1 hour at RT in 10% donkey serum and 0.3% Triton X-100 in PBS. Sections were incubated with mouse anti-IgM (1:400; Jackson ImmunoResearch, C839P24), mouse anti-IgG (1:400; Chemicon, AP192C), goat anti-ChAT (1:500; EMD Millipore, AB144P), tomato lectin (1:400; Vector Laboratories, DL1174), or Ter119 (1:400; BioLegend, 116207) for 72 hours at 4°C. Sections were incubated with fluorescently labeled secondary antibodies appropriate for the primary antibodies used (1:200; Jackson ImmunoResearch) for 2 hours at RT and then counterstained with DAPI. For TUNEL, we used the DeadEnd Fluorometric TUNEL System (Promega) as per the manufacturer’s instructions. For double immunostaining with cleaved caspase-3 and NeuN, TUNEL-stained sections were incubated in blocking solution (10% donkey serum, 0.3% Triton X-100 in PBS) for 2 hours at RT and then with anti-cleaved caspase-3 (1:300; Cell Signaling, 9661) and anti-NeuN (1:500; EMD Millipore, ABN90) for 24 hours at 4°C. Sections were rinsed with PBS and then incubated with fluorescent secondary antibodies (1:200; Jackson ImmunoResearch) for 24 hours at 4°C.
Confocal microscopy
Immunofluorescence was detected by laser scanning confocal microscopy (Nikon A1) using 20× and 60× magnification objectives and visualized using NIS-Elements (Nikon). Z-stack images were acquired with an optical thickness of 1 or 0.2 μm using the 20× and 60× objectives, respectively. Maximum intensity projections were generated from Z-stacks.
[14C]ACh release and ChAT activity
Three days following D3/FUS or D3 (i.e., intravenous D3, no MRIgFUS) treatment, mice were deeply anesthetized, decapitated, and selected brain regions (i.e., MS/DBB, NBM, HF, and CTX) were dissected rapidly on ice. The MS/DBB and NBM were flash-frozen in liquid nitrogen and kept at −80°C until ChAT activity quantification. Tissue was homogenized in 5% (w/v) buffer containing 50 mM phosphate buffer, 10 mM EDTA, 1× protease inhibitor, and 5% Triton X-100 (pH 7.4). ChAT activity was measured by incorporation of [14C]acetyl–coenzyme A into [14C]ACh as described previously (56). ChAT activity is expressed as nanomoles of radioactive ACh formed per milligram of protein per hour. To measure [14C]ACh release, the freshly dissected CTX and HF were cut into 500-μm-thick traverse slices. The release of labeled ACh was determined following a preincubation with labeled choline as described previously (56). [14C]ACh release is expressed as a percentage of total radioactivity. The total radioactivity was measured using liquid scintillation counting (Beckman Coulter). The BCA assay was used to determine the protein content of the samples (Pierce Biotechnology).
Statistical analysis
Prism 7 software (GraphPad) was used for statistical analysis and graphical presentation. All values are presented as the means ± SEM. Differences between two groups were assessed using unpaired Student’s t tests. Three groups were compared using a one-way ANOVA. Correlation analyses were performed using linear regression analysis with 95% confidence intervals. All other analyses were performed using a two-way ANOVA (with or without repeated measures, as indicated). Multiple comparisons between groups were performed with Bonferroni’s post hoc analysis for significant ANOVAs. In all cases, P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank P. Fraser, D. Westaway, and P. St George-Hyslop for supplying breeding pairs of TgCRND8 mice. We acknowledge K. Mikloska and S. Seerala for assistance with MRIgFUS; S. Rideout-Gros, V. Chan, and M. Theodore for animal care; and H.Y. Lin for graphics. S. Josephy offered insights on signaling pathways. P. Nagy provided helpful advice on cholinergic assays. Funding: This work was supported by the Canadian Institutes of Health Research (grants FRN 93603 to I.A. and FDRN 154272119312 to K.H.). Additional funding was received from the FDC Foundation, the WB Family Foundation, Gerald and Carla Connor, the Weston Brain Institute (TR130117 to I.A.), The National Institute of Biomedical Imaging and Bioengineering of the NIH (R01 EB003268 to K.H.), the Canadian Consortium on Neurodegeneration and Ageing (to H.U.S.), and the Canada Research Chair Program (to K.H.). The LI-COR Odyssey CLx was purchased with funds given by the James H. Cummings Foundation. K.X. was awarded a Frederick Banting and Charles Best Canada Graduate Scholarship (GSD 152271). Author contributions: K.X., H.U.S., K.H., and I.A. conceived and planned the experiments. K.X., K.M.-C., H.N., and S.H. performed the experiments. K.X. drafted the manuscript. All authors discussed the results and edited the manuscript. Competing interests: K.H. is a founder and a board member of FUS Instruments, a startup company manufacturing preclinical focused ultrasound devices. He receives research support from the company. K.H. and H.U.S. are inventors on patents related to this work (nos. EP1265605B1 and US6514221B2). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The drug-agents used, and related analogs or monoclonal antibodies, can be provided by H.U.S. pending scientific review and a completed material transfer agreement with the Lady Davis Institute. Requests for any agents should be submitted to uri.saragovi@mcgill.ca. In accordance with the material transfer agreement between the University of Toronto and Sunnybrook Health Sciences Centre, TgCRND8 mice can be provided by P. Fraser pending scientific review and a completed material transfer agreement. Requests for TgCRND8 mice should be submitted to paul.fraser@utoronto.ca.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/4/eaax6646/DC1
Fig. S1. Age-dependent NGF and TrkA deficits in the HF and CTX of TgCRND8 mice.
Fig. S2. Intraparenchymal injection of D3-induced TrkA-dependent signaling in the basal forebrain of 6-month-old non-Tg mice.
Fig. S3. Intraparenchymal injection of NGF-stimulated TrkA signaling pathways in the basal forebrain of 6-month-old non-Tg mice.
Fig. S4. MRIgFUS-mediated BBB disruption in the basal forebrain after intravenous administration of PBS or D3.
Fig. S5. Increased extravasation of endogenous immunoglobulins into the basal forebrain following MRIgFUS.
Fig. S6. MRIgFUS BBB disruption in the basal forebrain did not cause erythrocyte extravasation.
Fig. S7. MRIgFUS-BBB disruption in the basal forebrain did not induce neuronal apoptosis.
Fig. S8. Western blot analysis of total Akt, MAPK, CREB, and JNK protein levels after MRIgFUS-mediated delivery of D3.
Table S1. Primer sequences for qRT-PCR.
REFERENCES AND NOTES
- 1.Bartus R. T., Dean R. L. III, Beer B., Lippa A. S., The cholinergic hypothesis of geriatric memory dysfunction. Science 217, 408–414 (1982). [DOI] [PubMed] [Google Scholar]
- 2.Williams B. J., Eriksdotter-Jonhagen M., Granholm A.-C., Nerve growth factor in treatment and pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 80, 114–128 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Deinhardt K., Chao M. V., Trk receptors. Handb. Exp. Pharmacol. 220, 103–119 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Bai Y., Dergham P., Nedev H., Xu J., Galan A., Rivera J. C., ZhiHua S., Mehta H. M., Woo S. B., Sarunic M. V., Neet K. E., Saragovi H. U., Chronic and acute models of retinal neurodegeneration TrkA activity are neuroprotective whereas p75NTR activity is neurotoxic through a paracrine mechanism. J. Biol. Chem. 285, 39392–39400 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuszynski M. H., Nerve growth factor gene delivery: Animal models to clinical trials. Dev. Neurobiol. 67, 1204–1215 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Matrone C., Ciotti M. T., Mercanti D., Marolda R., Calissano P., NGF and BDNF signaling control amyloidogenic route and Aβ production in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 13139–13144 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knowles J. K., Rajadas J., Nguyen T.-V., Yang T., LeMieux M. C., Vander Griend L., Ishikawa C., Massa S. M., Wyss-Coray T., Longo F. M., The p75 neurotrophin receptor promotes amyloid-β(1-42)-induced neuritic dystrophy in vitro and in vivo. J. Neurosci. 29, 10627–10637 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen L.-L., Mañucat-Tan N. B., Gao S.-H., Li W.-W., Zeng F., Zhu C., Wang J., Bu X.-L., Liu Y.-H., Gao C. Y., Xu Z. Q., Bobrovskaya L., Lei P., Yu J.-T., Song W., Zhou H.-D., Yao X.-Q., Zhou X.-F., Wang Y.-J., The ProNGF/p75NTR pathway induces tau pathology and is a therapeutic target for FTLD-tau. Mol. Psychiatry 23, 1813–1824 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Rafii M. S., Tuszynski M. H., Thomas R. G., Barba D., Brewer J. B., Rissman R. A., Siffert J., Aisen P. S.; AAV2-NGF Study Team , Adeno-associated viral vector (serotype 2)-nerve growth factor for patients with Alzheimer disease: A randomized clinical trial. JAMA Neurol. 75, 834–841 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tuszynski M. H., Yang J. H., Barba D., U H.-S., Bakay R. A. E., Pay M. M., Masliah E., Conner J. M., Kobalka P., Roy S., Nagahara A. H., Nerve growth factor gene therapy: Activation of neuronal responses in Alzheimer disease. JAMA Neurol. 72, 1139–1147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maliartchouk S., Feng Y., Ivanisevic L., Debeir T., Cuello A. C., Burgess K., Saragovi H. U., A designed peptidomimetic agonistic ligand of TrkA nerve growth factor receptors. Mol. Pharmacol. 57, 385–391 (2000). [PubMed] [Google Scholar]
- 12.Bruno M. A., Clarke P. B. S., Seltzer A., Quirion R., Burgess K., Cuello A. C., Saragovi H. U., Long-lasting rescue of age-associated deficits in cognition and the CNS cholinergic phenotype by a partial agonist peptidomimetic ligand of TrkA. J. Neurosci. 24, 8009–8018 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aboulkassim T., Tong X.-K., Tse Y. C., Wong T.-P., Woo S. B., Neet K. E., Brahimi F., Hamel E., Saragovi H. U., Ligand-dependent TrkA activity in brain differentially affects spatial learning and long-term memory. Mol. Pharmacol. 80, 498–508 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Josephy-Hernandez S., Jmaeff S., Pirvulescu I., Aboulkassim T., Saragovi H. U., Neurotrophin receptor agonists and antagonists as therapeutic agents: An evolving paradigm. Neurobiol. Dis. 97, 139–155 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Hynynen K., McDannold N., Vykhodtseva N., Jolesz F. A., Noninvasive MR imaging–guided focal opening of the blood-brain barrier in rabbits. Radiology 220, 640–646 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Jordão J. F., Ayala-Grosso C. A., Markham K., Huang Y., Chopra R., McLaurin J., Hynynen K., Aubert I., Antibodies targeted to the brain with image-guided focused ultrasound reduces amyloid-β plaque load in the TgCRND8 mouse model of Alzheimer’s disease. PLOS ONE 5, e10549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burgess A., Ayala-Grosso C. A., Ganguly M., Jordão J. F., Aubert I., Hynynen K., Targeted delivery of neural stem cells to the brain using MRI-guided focused ultrasound to disrupt the blood-brain barrier. PLOS ONE 6, e27877 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xhima K., Nabbouh F., Hynynen K., Aubert I., Tandon A., Noninvasive delivery of an α-synuclein gene silencing vector with magnetic resonance–guided focused ultrasound. Mov. Disord. 33, 1567–1579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lipsman N., Meng Y., Bethune A. J., Huang Y., Lam B., Masellis M., Herrmann N., Heyn C., Aubert I., Boutet A., Smith G. S., Hynynen K., Black S. E., Blood–brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat. Commun. 9, 2336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chishti M. A., Yang D.-S., Janus C., Phinney A. L., Horne P., Pearson J., Strome R., Zuker N., Loukides J., French J., Turner S., Lozza G., Grilli M., Kunicki S., Morissette C., Paquette J., Gervais F., Bergeron C., Fraser P. E., Carlson G. A., George-Hyslop P. S., Westaway D., Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Krantic S., Isorce N., Mechawar N., Davoli M. A., Vignault E., Albuquerque M., Chabot J.-G., Moyse E., Chauvin J.-P., Aubert I., McLaurin J., Quirion R., Hippocampal GABAergic neurons are susceptible to amyloid-β toxicity in vitro and are decreased in number in the Alzheimer’s disease TgCRND8 mouse model. J. Alzheimers Dis. 29, 293–308 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Bellucci A., Luccarini I., Scali C., Prosperi C., Giovannini M. G., Pepeu G., Casamenti F., Cholinergic dysfunction, neuronal damage and axonal loss in TgCRND8 mice. Neurobiol. Dis. 23, 260–272 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Mufson E. J., Counts S. E., Ginsberg S. D., Gene expression profiles of cholinergic nucleus basalis neurons in Alzheimer’s disease. Neurochem. Res. 27, 1035–1048 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Ginsberg S. D., Che S., Wuu J., Counts S. E., Mufson E. J., Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 97, 475–487 (2006). [DOI] [PubMed] [Google Scholar]
- 25.McKeever P. M., Kim T., Hesketh A. R., MacNair L., Miletic D., Favrin G., Oliver S. G., Zhang Z., St George-Hyslop P., Robertson J., Cholinergic neuron gene expression differences captured by translational profiling in a mouse model of Alzheimer's disease. Neurobiol. Aging 57, 104–119 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Jordão J. F., Thévenot E., Markham-Coultes K., Scarcelli T., Weng Y.-Q., Xhima K., O’Reilly M., Huang Y., McLaurin J., Hynynen K., Aubert I., Amyloid-β plaque reduction, endogenous antibody delivery and glial activation by brain-targeted, transcranial focused ultrasound. Exp. Neurol. 248, 16–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jalali S., Huang Y., Dumont D. J., Hynynen K., Focused ultrasound-mediated BBB disruption is associated with an increase in activation of AKT: Experimental study in rats. BMC Neurol. 10, 114 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kovacs Z. I., Kim S., Jikaria N., Qureshi F., Milo B., Lewis B. K., Bresler M., Burks S. R., Frank J. A., Disrupting the blood–brain barrier by focused ultrasound induces sterile inflammation. Proc. Natl. Acad. Sci. U.S.A. 114, E75–E84 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kordower J. H., Winn S. R., Liu Y. T., Mufson E. J., Sladek J. R. Jr., Hammang J. P., Baetge E. E., Emerich D. F., The aged monkey basal forebrain: Rescue and sprouting of axotomized basal forebrain neurons after grafts of encapsulated cells secreting human nerve growth factor. Proc. Natl. Acad. Sci. U.S.A. 91, 10898–10902 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper J. D., Salehi A., Delcroix J.-D., Howe C. L., Belichenko P. V., Chua-Couzens J., Kilbridge J. F., Carlson E. J., Epstein C. J., Mobley W. C., Failed retrograde transport of NGF in a mouse model of Down’s syndrome: Reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc. Natl. Acad. Sci. U.S.A. 98, 10439–10444 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conner J. M., Darracq M. A., Roberts J., Tuszynski M. H., Nontropic actions of neurotrophins: Subcortical nerve growth factor gene delivery reverses age-related degeneration of primate cortical cholinergic innervation. Proc. Natl. Acad. Sci. U.S.A. 98, 1941–1946 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Honig L. S., Gene therapy in Alzheimer disease—It may be feasible, but will it be beneficial? JAMA Neurol. 75, 791–793 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Meerovitch K., Torkildsen G., Lonsdale J., Goldfarb H., Lama T., Cumberlidge G., Ousler G. W. 3rd, Safety and efficacy of MIM-D3 ophthalmic solutions in a randomized, placebo-controlled phase 2 clinical trial in patients with dry eye. Clin. Ophthalmol. 7, 1275–1285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McMahon D., Hynynen K., Acute inflammatory response following increased blood-brain barrier permeability induced by focused ultrasound is dependent on microbubble dose. Theranostics 7, 3989–4000 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Reilly M. A., Hynynen K., Blood-brain barrier: Real-time feedback-controlled focused ultrasound disruption by using an acoustic emissions–based controller. Radiology 263, 96–106 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McMahon D., Poon C., Hynynen K., Evaluating the safety profile of focused ultrasound and microbubble-mediated treatments to increase blood-brain barrier permeability. Expert Opin. Drug Deliv. 16, 129–142 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDannold N., Vykhodtseva N., Hynynen K., Use of ultrasound pulses combined with Definity for targeted blood-brain barrier disruption: A feasibility study. Ultrasound Med. Biol. 33, 584–590 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burgess A., Dubey S., Yeung S., Hough O., Eterman N., Aubert I., Hynynen K., Alzheimer disease in a mouse model: MR imaging–guided focused ultrasound targeted to the hippocampus opens the blood-brain barrier and improves pathologic abnormalities and behavior. Radiology 273, 736–745 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poon C. T., Shah K., Lin C., Tse R., Kim K. K., Mooney S., Aubert I., Stefanovic B., Hynynen K., Time course of focused ultrasound effects on β-amyloid plaque pathology in the TgCRND8 mouse model of Alzheimer’s disease. Sci. Rep. 8, 14061 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi J. J., Feshitan J. A., Baseri B., Wang S., Tung Y.-S., Borden M. A., Konofagou E. E., Microbubble-size dependence of focused ultrasound-induced blood–brain barrier opening in mice in vivo. IEEE Trans. Biomed. Eng. 57, 145–154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lai A. Y., Dorr A., Thomason L. A., Koletar M. M., Sled J. G., Stefanovic B., McLaurin J., Venular degeneration leads to vascular dysfunction in a transgenic model of Alzheimer’s disease. Brain 138, 1046–1058 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Leinenga G., Götz J., Scanning ultrasound removes amyloid-β and restores memory in an Alzheimer’s disease mouse model. Sci. Transl. Med. 7, 278ra33 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Pandit R., Leinenga G., Götz J., Repeated ultrasound treatment of tau transgenic mice clears neuronal tau by autophagy and improves behavioral functions. Theranostics 9, 3754–3767 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silver R., Curley J. P., Mast cells on the mind: New insights and opportunities. Trends Neurosci. 36, 513–521 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Sofroniew M. V., Howe C. L., Mobley W. C., Nerve growth factor signaling, neuroprotection, and neural repair. Annu. Rev. Neurosci. 24, 1217–1281 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Ruberti F., Capsoni S., Comparini A., Di Daniel E., Franzot J., Gonfloni S., Rossi G., Berardi N., Cattaneo A., Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen, and skeletal muscle dystrophy. J. Neurosci. 20, 2589–2601 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maliartchouk S., Debeir T., Beglova N., Cuello A. C., Gehring K., Saragovi H. U., Genuine monovalent ligands of TrkA nerve growth factor receptors reveal a novel pharmacological mechanism of action. J. Biol. Chem. 275, 9946–9956 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Knusel B., Kaplan D. R., Hefti F., Intraparenchymal NGF injections in adult and aged rats induce long-lasting Trk tyrosine phosphorylation. Exp. Neurol. 139, 121–130 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Venero J. L., Hefti F., Knusel B., Trophic effect of exogenous nerve growth factor on rat striatal cholinergic neurons: Comparison between intraparenchymal and intraventricular administration. Mol. Pharmacol. 49, 303–310 (1996). [PubMed] [Google Scholar]
- 50.Lucidi-Phillipi C. A., Clary D. O., Reichardt L. F., Gage F. H., TrkA activation is sufficient to rescue axotomized cholinergic neurons. Neuron 16, 653–663 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frick K. M., Price D. L., Koliatsos V. E., Markowska A. L., The effects of nerve growth factor on spatial recent memory in aged rats persist after discontinuation of treatment. J. Neurosci. 17, 2543–2550 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Josephy-Hernandez S., Pirvulescu I., Maira M., Aboulkassim T., Wong T. P., McKinney R. A., Saragovi H. U., Pharmacological interrogation of TrkA-mediated mechanisms in hippocampal-dependent memory consolidation. PLOS ONE 14, e0218036 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markowska A. L., Koliatsos V. E., Breckler S. J., Price D. L., Olton D. S., Human nerve growth factor improves spatial memory in aged but not in young rats. J. Neurosci. 14, 4815–4824 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schirrmacher R., Bailey J. J., Mossine A. V., Scott P. J. H., Kaiser L., Bartenstein P., Lindner S., Kaplan D. R., Kostikov A., Fricker G., Mahringer A., Rosa-Neto P., Schirrmacher E., Wängler C., Wängler B., Thiel A., Soucy J.-P., Bernard-Gauthier V., Radioligands for tropomyosin receptor kinase (Trk) positron emission tomography imaging. Pharmaceuticals 12, 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aghourian M., Legault-Denis C., Soucy J.-P., Rosa-Neto P., Gauthier S., Kostikov A., Gravel P., Bédard M.-A., Quantification of brain cholinergic denervation in Alzheimer’s disease using PET imaging with [18F]-FEOBV. Mol. Psychiatry 22, 1531–1538 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Nagy P. M., Aubert I., Overexpression of the vesicular acetylcholine transporter increased acetylcholine release in the hippocampus. Neuroscience 218, 1–11 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/4/eaax6646/DC1
Fig. S1. Age-dependent NGF and TrkA deficits in the HF and CTX of TgCRND8 mice.
Fig. S2. Intraparenchymal injection of D3-induced TrkA-dependent signaling in the basal forebrain of 6-month-old non-Tg mice.
Fig. S3. Intraparenchymal injection of NGF-stimulated TrkA signaling pathways in the basal forebrain of 6-month-old non-Tg mice.
Fig. S4. MRIgFUS-mediated BBB disruption in the basal forebrain after intravenous administration of PBS or D3.
Fig. S5. Increased extravasation of endogenous immunoglobulins into the basal forebrain following MRIgFUS.
Fig. S6. MRIgFUS BBB disruption in the basal forebrain did not cause erythrocyte extravasation.
Fig. S7. MRIgFUS-BBB disruption in the basal forebrain did not induce neuronal apoptosis.
Fig. S8. Western blot analysis of total Akt, MAPK, CREB, and JNK protein levels after MRIgFUS-mediated delivery of D3.
Table S1. Primer sequences for qRT-PCR.