

Abstract
Aim: Menopause causes arterial senescence and atherosclerotic development through decrease of estrogen. Recently, histone deacetylase SIRT1 has been reported to have protective effects against arterial senescence and atherosclerosis. However, the relationship between estrogen and SIRT1 in the context of menopause-induced arterial senescence is not well understood. The present study aims to investigate whether SIRT1 is involved in the etiology of menopause-induced arterial senescence and atherosclerotic development.
Methods: Twelve-week old female apolipoprotein E-knockout (ApoE-KO) mice underwent ovariectomy (OVX) or sham surgery.
Results: SIRT1 expression and endothelial nitric oxide synthase (eNOS) activation in the aorta were significantly lower in OVX mice than they were in sham mice (OVX vs. sham, n = 5 per group). Senescence-associated β-galactosidase activity, protein expression of Ac-p53 and PAI-1, and aortic atherosclerosis lesions were significantly greater in OVX mice than they were in sham mice. Administration of 17β-estradiol (E2) for eight weeks to OVX mice restored aortic SIRT1 expression, activated eNOS, and retarded OVX-induced arterial senescence and atherosclerotic development (E2 vs. control, n = 5 per group). The effects of E2 on SIRT1 upregulation, anti-senescence and anti-atherosclerosis were attenuated by administration of a SIRT1 inhibitor, sirtinol. In vitro experiment using human endothelial cells demonstrated that E2 also increased SIRT1 expression and retarded oxidized low density lipoprotein-induced premature senescence, which were also abolished by sirtinol. These results suggested that estrogen modulated arterial senescence and atherosclerosis through SIRT1. Additionally a selective estrogen receptor modulator (SERM), bazedoxifene, also augmented SIRT1 and inhibited arterial senescence and atherosclerotic development (SERM vs. control, n = 3 per group).
Conclusions: Downregulation of SIRT1 causes OVX-induced arterial senescence and atherosclerosis in ApoE-KO mice. Administration of estrogen or SERM enables OVX mice to restore these alterations by SIRT1 induction.
Keywords: Estrogen, eNOS, SIRT1, Atherosclerosis, Arterial senescence
See editorial vol. 27: 8–10
Introduction
Cardiovascular disease (CVD) is one of the major causes of death worldwide among older people and develops on average in women 10 years later than in men. This delay may be explained in part by the protective effect of estrogen before menopause1, 2). The Framingham study showed that the incidence of CVD in postmenopausal women was 2–6 times higher than that in premenopausal women of the same age group (age: < 40, 40–44, 45–49, 50–54)3). Therefore, it was expected that administration of estrogen might be able to prevent atherosclerotic development and reduce incidence of CVD. Indeed, animal experiments showed that female apolipoprotein E-knockout (ApoE-KO) mice subjected to ovariectomy (OVX) surgery reduced rates of atherosclerosis by administration of estrogen or 17β-estradiol (E2), which is the primary hormone used in hormone replacement therapy (HRT) in postmenopausal women4, 5). A recent clinical study also demonstrated that oral estradiol therapy retards subclinical atherosclerosis within six years of menopause6). Several other evidences support the use of HRT for primary prevention of CVD in postmenopausal women7, 8). However, there is no clear evidence that HRT alone can prevent CVD. Therefore, in order to develop the optimum method and condition of HRT, it is meaningful to understand the precise molecular mechanisms in which decrease of estrogen by menopause facilitates atherosclerosis, and in which estrogen protects arteries against atherosclerosis.
There is increasing evidence that age is an important risk factor for development of atherosclerosis and that cellular senescence promotes atherosclerosis9). It has been reported that silent information regulator 2 (Sir2) proteins, members of the sirtuin family, could prolong the lifespan of yeast; Sir2 deficiency has the opposite effect10). The effects in particular of SIRT1, a mammalian Sir2 homolog, on cellular senescence and cardiovascular diseases including atherosclerosis have been investigated. Recently, a great deal of evidence has accumulated indicating that activation of the sirtuin family, particularly SIRT1, contributes to anti-aging in the vasculature through increasing endothelial nitric oxide synthase (eNOS) activation, reducing oxidative stress, inflammation, and DNA damage11). Animal studies have demonstrated that SIRT1 plays a protective role against atherosclerosis, at least in vascular endothelial and smooth muscle cells. SIRT1 expression is decreased in ApoE-deficient mice12), and mice mated with ApoE-KO mice and smooth muscle cell-specific SIRT1-KO mice exhibited increased rates of atherosclerosis13). The dedifferentiation of vascular smooth muscle cells that occurs with aging is involved in the disease; SIRT1 activation with resveratrol treatment may promote differentiation of cells, which thus contributes to protection against atherosclerosis14). The aforementioned animal studies have revealed that SIRT1 protects the vasculature from atherosclerosis. Additionally, a great deal of evidence from clinical studies that sirtuins modulate risk factors for atherosclerosis and atherosclerosis itself has accumulated15). While the protective effects of SIRT1 in anti-atherosclerosis and age-related CVD have been studied indepth, sex differences related to SIRT1 in this disease are not well understood15). Additionally, the relationship between estrogen and SIRT1 in the context of menopause-induced arterial senescence and atherosclerotic development is not well understood.
Given the above background, we hypothesized that SIRT1 plays a crucial role in the effect of estrogen on retarding arterial senescence and atherosclerotic development.
Aim
The aim of the present study was to investigate whether SIRT1 is regulated by estrogen and is involved in the etiology of menopause-induced arterial senescence and atherosclerosis.
Methods
Animal Models
All animal experiments were conducted in compliance with protocol reviewed by the Institutional Animal Care and Use Committee and were approved by the Faculty of Medicine at the Kagoshima University. Female apolipoprotein E-knockout (ApoE-KO) mice or endothelial nitric oxide synthase-knockout (eNOS-KO) mice were compared in this study. All mice were housed in groups of one to three per cage and were maintained under a temperature-controlled environment and 12-hour light and dark cycles with food and water ad libitum. At the age of nearly 12 weeks, mice were anesthetized with a combination of 0.3 mg/kg medetomidine, 4.0 mg/kg midazolam, and 5.0 mg/kg butorphanol by intraperitoneal (i.p.) injection16) and were subjected to bilateral ovariectomy (OVX) or sham surgery. Body weight and food intake were measured every week after surgical procedures.
Experimental Procedures
To determine the effects of E2, OVX mice were subcutaneously implanted within 24 hours of OVX surgery with 60-day release E2 pellets (0.5 mg per pellet releasing 8.3 µg/day; Innovative Research of America, Sarasota, FL, USA) or control pellets for eight weeks17). To inhibit SIRT1, OVX mice implanted with an E2 pellet were treated with either sirtinol (5 mg/kg) or with same amount of dimethyl sulfoxide (DMSO) as control vehicle by i.p. injection five days per week for eight weeks. To study the effects of a selective estrogen receptor modulator (SERM), we used bazedoxifene (BZA), which was a gift from Pfizer Inc. (New York, NY, USA). OVX mice were subcutaneously treated with either BZA (0.3 mg/kg/day) or DMSO three days per week for eight weeks. To determine the role of eNOS in regulating SIRT1 expression, OVX mice treated with SERM were given water containing 1 mg/ml NG-nitro-L-arginine methyl ester (L-NAME) (Sigma, St. Luis, MO, USA), an inhibitor of eNOS, for eight weeks, as reported previously18). Additionally, we administrated control vehicle or BZA to eNOS-KO mice with OVX. Sirtinol and BZA were dissolved in DMSO.
Tissue Preparation and Lipid Analysis
All mice were sacrificed with overdose of sodium pentobarbital at eight weeks post surgery. After blood drawing, ascending aortas were immediately fixed in 4% paraformaldehyde phosphate buffer solution for immunohistochemical analysis. The aortic trees were harvested from the ascending aorta to the abdominal aorta for SA-β gal staining. For western blot analysis and real-time PCR, isolated aorta samples from ascending aortas to the bifurcation of the common iliac arteries were rinsed in phosphate buffered saline and stored at −80°C. The hearts with the aortic root were embedded in optimal cutting temperature (OCT) compound, and were frozen at −80°C for Oil Red O staining. Serum was obtained through centrifugation of blood for 10 min at 3,000 rpm at 4°C and stored −80°C until each assay was performed. The concentration of total serum cholesterol and triglycerides was measured enzymatically using a commercially available kit (FUJIFILM Wako Pure Chemical Corporation, Osaka, JPN). The blood samples and tissue samples for immunohistochemical analysis, western blot analysis, and Oil Red O staining were harvested from the same mice. In order to obtain sufficient samples, we performed SA-β gal staining or real-time PCR experiments separately from the other experiments, such as lipid analysis, immunohistochemical staining, western blot analysis, and Oil Red O staining.
Atherosclerotic Lesions
We assessed atherosclerotic lesions using Oil Red O staining according to the method described previously19). Briefly, the heart with the aortic root was embedded in OCT compound. Frozen tissue was cut into 5-µm sections and fixed on glass slides. The slides were stained with Oil Red O. All sections were examined under a microscope (Keyence, BZ-X710), and lipid staining of the aortic root in the histological sections was quantitated20). The percentage of the aortic lumen area occupied by lesions was averaged over 15 consecutive sections per rodents.
Immunohistochemistry
Immunohistochemical staining of the tissue sections was performed as described previously21). Tissue sections were stained with anti-SIRT1 rabbit polyclonal antibody (1:50; Merck Millipore, Billerica, MA, USA), anti-PECAM-1 mouse monoclonal antibody (1:50; Santa Cruz, Dallas, TX, USA), Alexa Fluor 488-conjugated goat anti-rabbit IgG (Abcam, Cambridge, GBR), Alexa Fluor 594-conjugated goat antimouse (Abcam), and Vectashield mounting medium with DAPI (Vector Laboratories, INC., Burlingame, CA, USA). Analyses were performed by fluorescence microscopy (Keyence, BZ-X710).
Western Blot Analysis
Western blotting was performed using cell lysates from mouse tissue with a NUPAGE Electrophoresis System (Invitrogen, Carlsbad, CA, USA), as reported previously22). Briefly, tissues and cells were lysed in RIPA buffer (Merck Millipore) with protease inhibitor and phosphatase inhibitor cocktail (Sigma). The protein samples were boiled at 95°C for 5 min with 4 × SDS sample buffer. The first antibodies used were as follows: eNOS; Phospho-eNOS (Ser1177); Acetylp53 (Cell Signaling Technology, Danvers, MA, USA), SIRT1 (Merck Millipore); p21; p16; PAI-1; β-actin (Santa Cruz Biotechnology); and α-tubulin (Sigma). Either horseradish peroxidase-conjugated goat antirabbit antibody (Bio-Rad, Hercules, CA, USA) or goat anti-mouse antibody (Santa Cruz Biotechnology) was then added. Densitometric analyses were performed using the ECL prime system (GE Healthcare UK Ltd, Little Chalfont, UK).
Real-Time PCR
In order to measure mRNA levels of SIRT3, p21, and p16 in aorta, we conducted real-time PCR in an isolated animal experiment. Six mice which underwent OVX surgery at the 12 weeks of age were separated into two groups; OVX and OVX + E2. Total RNA was extracted from tissues using the RNeasy Fibrous Tissue Mini Kit (Qiagen, Hilden, DEU), according to the manufacturer's protocol. Transcription into cDNA was performed using random hexamers and PrimeScript™ RT Master Mix (Takara Bio, Shiga, JPN), according to the manufacturer's protocol. All PCR reactions used SYBR Green™ Premix Ex Taq™ II (Takara Bio) to a final volume of 20 µL with each cDNA sample in the ABI PRISM 7300 Sequence Detection System (Applied Biosystems, Foster City, CA, USA), according to the manufacturer's protocol. Each gene expression was obtained from the average of triplicated PCR results and normalized to GAPDH. Sequence primers were as follows. SIRT1: forward, TGATTGGCACCGATCCTCG, reverse, CCACAG CGTCATATCATCCAG; SIRT3: forward, ATCCCG GACTTCAGATCCCC, reverse, CAACATGAAAAG GGCTTGGG; p16: forward, CCCAACGCCCCGA ACT, reverse, GCAGAAGAGCTGCTAGTGAA; p21: forward, GGCAGACCAGCCCTGACAGAT, reverse, TTCAGGGTTTTCTCTTGCAGAAG; GAPDH: forward, CTCACTCAAGATTGTCAGCA ATG, reverse, GAGGGAGATGCTCAGTGTTGG.
Cell Lines and Culture Methods
Human umbilical vein endothelial cells (HUVEC) were purchased from the Lonza Group Ltd. (Basel, CHE). They were cultured in a 100-mm collagen-coated dish for continuous growth in a humidified atmosphere of 95% air and 5% CO2 at 37°C and maintained in endothelial growth medium (EGM-2, EGM-2 singleQuots, Lonza) supplemented with 10% fetal bovine serum (FBS: Invitrogen), 10,000 units/mL penicillin, and 10 mg/mL streptomycin. When cells were at 80% confluence, the culture medium was replaced with phenol red-free Dulbecco's modified Eagle's Medium (DMEM) (GIBCO, Invitrogen) with 10% charcoal-stripped FBS (Biowest, Nuaille, FRA) and was maintained for 24 h before E2 treatment as phenol red itself is known to possess estrogenic properties23). For western blot assay, cells were treated with 10 nmol/L 17β-estradiol (E2) (Sigma) and oxidized low density lipoprotein (ox- LDL) (Alfa Aesar, Lancashire, GBR) with phenol redfree DMEM supplemented with 2% charcoal-stripped FBS for 30 min, 1 h, and 3h. Control cells were exposed to the same vehicles of E2 (10−4% ethanol) and ox-LDL. For senescence-associated β-galactosidase staining, cells were incubated with 10 nmol/L E2 and ox-LDL with or without sirtinol (50 µmol/L) (Sigma) for 24 h with phenol red-free DMEM supplemented with 5% charcoal-stripped FBS.
Senescence-Associated β-Galactosidase (SA-β Gal) Staining
The senescence of the mouse aortas was evaluated by SA-β gal staining according to the method described previously24). Briefly, aortic arches (containing the ascending aorta, arch thoracic descending aorta, and abdominal aorta) were washed with PBS on ice then were stained for 24 h at 37°C in buffer containing 1 mg/mL 5-bromo-4-chloro-3-indolyl β-D-galactoside (X-Gal) (Invitrogen), 1 mol/L MgCl2, 2% NP-40, 100 mmol/L potassium ferrocyanide (II), 100 mmol/L potassium ferrocyanide (III), and 40 mmol/L citric acid/sodium hydrogen phosphate buffer (pH 6) to visualize aortic senescence. After X-gal staining, samples were washed three times with PBS. As post-fixation to stabilize the color, the tissues were fixed with 4% paraformaldehyde.
SA-β gal activity in vitro was also assessed using a SA-β gal staining kit (Cell Signaling Technology), according to the manufacturer's protocol. SA-β gal positive cells were counted using a microscope (Keyence, BZ-X710).
Statistical Analysis
Data are presented as means ± standard deviation (S.D.). Statistical significance was evaluated using the unpaired Student's t-test for comparisons between the two groups. Differences were considered significant at P values < 0.05.
Results
Ovx Decreased SIRT1 Expression and Induced Arterial Senescence and The Development of Atherosclerosis
We performed OVX surgery on ApoE-KO mice to investigate whether OVX influenced arterial SIRT1 expression and modulated senescence and atherosclerosis. Female ApoE-KO mice were divided into two groups; sham surgery group and OVX group (Fig. 1A). There was no difference in subjects' food intake during this experiment and no differences between the two groups in body weight or the levels of total serum cholesterol and triglycerides eight weeks after surgery (Table 1). As shown in Fig. 1B, SIRT1 was mainly expressed in endothelial cells in mouse aortas. The ratio of arterial senescence and the atherosclerotic area, assessed by SA-β gal and Oil Red O staining, respectively, were greater in OVX mice than in sham mice (Figs. 1C and D). Western blot analysis revealed that the arterial expression of SIRT1 protein and the ratio of p-eNOS to eNOS were lower in OVX mice than in sham mice and the expression of Ac-p53, p21, p16, and PAI-1 was higher in OVX mice than in sham mice (Fig. 1E). These results suggest that OVX decreased aortic SIRT1 expression, accelerated arterial senescence, and induced atherosclerosis.
Fig. 1.
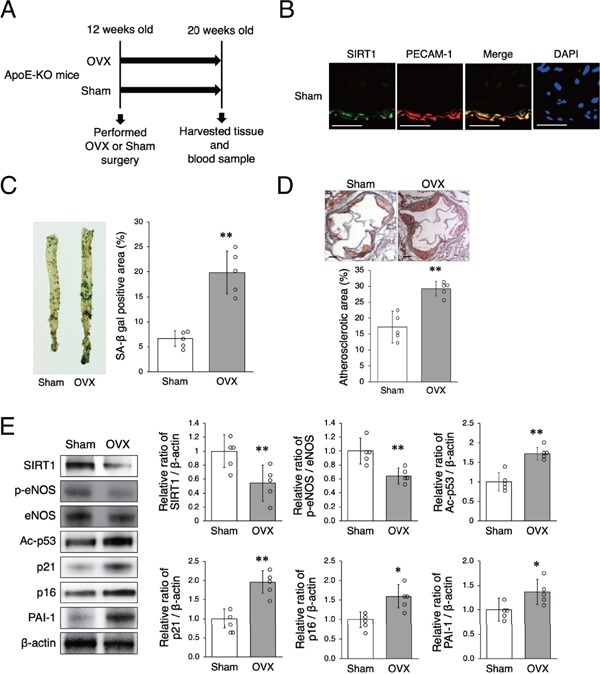
OVX decreases SIRT1 expression and facilitates arterial senescence and the development of atherosclerosis
A: Animal experiment protocol
B: Representative images of immunohistochemical staining for SIRT1 (green) and PECAM-1 (red) in the aortas of ApoE-KO mice. Nuclei were counterstained with DAPI (blue). Expression of SIRT1 was observed in the endothelial cells of sham mice. Scale bar = 50 µm
C: Representative images of SA-β gal staining of senescent cells (blue) in the aortas of sham mice and OVX mice are shown in the left panel. The aortic area of SA-β gal positive staining in OVX mice was higher than that in sham mice.
D: Representative images of Oil Red O-stained cross-sections of the aortic roots of sham mice and OVX mice are shown in the upper panel. The area of atherosclerotic lesions in OVX mice was significantly greater than that in sham mice. Scale bar = 200 µm
E: Representative band of western blot analysis and densitometric analysis in sham mice and OVX mice are shown in the left panel. The protein expression of SIRT1, p-eNOS is significantly higher in sham mice than in OVX mice and that of Ac-p53, p21, p16, and PAI-1 is lower in sham mice than in OVX mice.
All data are shown as the mean ± standard deviation (S.D.); n = 5 in each group. *P < 0.05 and **P < 0.01 vs. sham group.
Table 1. Body weight, food intake, total cholesterol, and triglyceride.
| Sham | OVX | P value | OVX | OVX+E2 | P value | OVX | OVX+SERM | P value | |
|---|---|---|---|---|---|---|---|---|---|
| (n = 5) | (n = 5) | (n = 6) | (n = 6) | (n = 3) | (n = 3) | ||||
| BW (g) | 23.2 ± 1.5 | 24.0 ± 0.9 | N.S. | 24.3 ± 1.2 | 24.2 ± 2.1 | N.S. | 24.0 ± 0.6 | 24.1 ± 0.5 | N.S. |
| Fl (g/day) | 3.8 ± 0.5 | 3.5 ± 0.2 | N.S. | 3.6 ± 0.3 | 3.4 ± 0.4 | N.S. | 3.5 ± 0.2 | 3.5 ± 0.1 | N.S. |
| TC (mg/dL) | 782 ± 78 | 770 ± 87 | N.S. | 747 ± 102 | 782 ± 41 | N.S. | 775 ± 242 | 763 ± 179 | N.S. |
| TG (mg/dL) | 332 ± 105 | 327 ± 80 | N.S. | 301 ± 96 | 317 ± 102 | N.S. | 338 ± 58 | 292 ± 70 | N.S. |
BW, body weight; FI, food intake; TC, total cholesterol; TG, triglyceride. N.S.; not significant
Estrogen Increased SIRT1 Expression and Retarded Arterial Senescence and The Development of Atherosclerosis
We investigated the effect of estrogen on arterial senescence and atherosclerosis. ApoE-KO OVX mice were implanted with either E2 or control pellets for eight weeks (Fig. 2A). Continuous administration of E2 from an E2 pellet increased SIRT1 expression in the endothelial cells of ApoE-KO OVX mice (Fig. 2B). There was no difference in subjects' food intake during this experiment and no differences in body weight or the levels of total serum cholesterol and triglycerides eight weeks after operation between OVX mice which were given E2 and OVX mice which were given control pellets (Table 1). The ratio of arterial senescence and the atherosclerotic area were lower in OVX + E2 mice than in OVX mice (Figs. 2C and D). In OVX + E2 mice as compared with OVX mice, the arterial expression of SIRT1 protein and the ratio of p-eNOS to eNOS were higher, and the expression of Ac-p53 and PAI-1 was lower (Fig. 2E). There were no differences in either protein or mRNA expression of SIRT3 between the two groups (Figs. 2E and F). The mRNA expression of p21 and p16 was lower in OVX + E2 mice than in OVX mice (Fig. 2F). Since E2 induced SIRT1 in the endothelial cells of aortas in OVX mice, we also conducted in vitro experiment using HUVEC with administration of ox-LDL in order to examine whether upregulation of SIRT1 was the direct effect of presence of E2. The premature senescence of HUVEC treated with ox-LDL was inhibited by administration of E2 (Fig. 2G). The expression of SIRT1 and the ratio of p-eNOS to eNOS in HUVEC treated with ox-LDL were higher with E2 treatment than without E2 treatment. There was no difference in expression of SIRT3 protein (Fig. 2H). These results suggest that E2 increased aortic and cellular SIRT1 expression and retarded arterial and cellular senescence and atherosclerotic development.
Fig. 2-1.
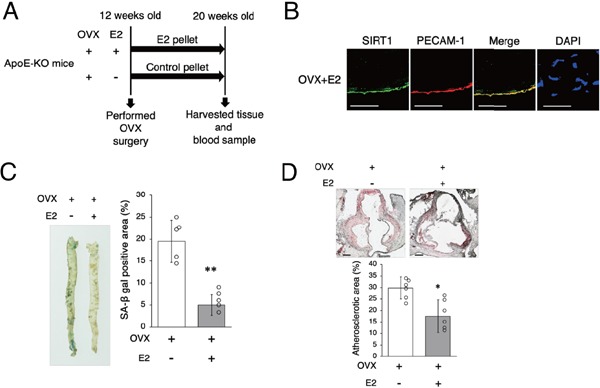
Estrogen increases SIRT1 expression and retards arterial senescence and atherosclerotic development
A: Animal experiment protocol.
B: Representative images of immunohistochemical staining for SIRT1 (green) and PECAM-1 (red) in the aortas of OVX + E2 mice. Nuclei were counterstained with DAPI (blue). E2 increased SIRT1 expression in endothelial cells in OVX + E2 mice. Scale bar = 50 µm
C: Representative images of SA-β gal staining of senescent cells (blue) in the aortas of OVX mice and OVX + E2 mice are shown in the left panel. The rate of positive SA-β gal staining in OVX + E2 mice is lower than that in OVX mice.
D: Representative images of Oil Red O-stained cross-sections of the aortic roots of OVX mice and OVX + E2 mice are shown in the upper panel. Atherosclerotic lesions in OVX mice are significantly greater than those in OVX + E2 mice. Scale bar = 200 µm.
Fig. 2-2.
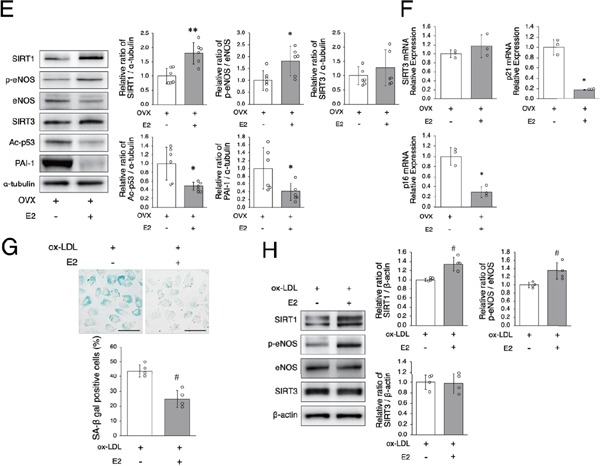
Estrogen increases SIRT1 expression and retards arterial senescence and atherosclerotic development
E: Representative band of western blot analysis and densitometric analysis in OVX mice and OVX + E2 mice is shown in the left panel. The protein expression of SIRT1 and the ratio of p-eNOS/eNOS are significantly higher and the expression of Ac-p53 and PAI-1 is lower in OVX + E2 mice as compared with OVX mice. There was no difference in SIRT3 expression between the two groups.
F: SIRT3, p21, and p16 mRNA expression in the aortas of OVX mice and OVX + E2 mice. SIRT3 mRNA expression is not significantly different between OVX mice and OVX + E2 mice. p21 and p16 mRNA expression is lower in OVX + E2 mice than in OVX mice.
G: Representative images of SA-β gal staining are shown in the upper panel. Administration of E2 decreased SA-β gal positive cells. Scale bar = 50 µm
H: Representative band of western blot analysis and densitometric analysis in HUVEC are shown in the left panel. E2 increases the protein expression of SIRT1 and the ratio of p-eNOS/eNOS. There was no difference in SIRT3 expression between the two groups.
All data are shown as the mean ± S.D.; n = 6 in each group (B, C, D, E, F), n = 4 in each group (G, H). *P < 0.05 and **P % 0.01 vs. OVX group. #P % 0.05 vs. ox-LDL group
The Beneficial Effect of Estrogen is Derived from Upregulation of SIRT1
To investigate the mechanisms in which estrogen retards arterial senescence and atherosclerosis though SIRT1 regulation in OVX mice, we performed an inhibition experiment using a SIRT1 inhibitor, sirtinol. In vitro experiment revealed that the effect of E2 on retarding cellular premature senescence was abolished by administration of sirtinol (Fig. 3A). We also performed an in vivo experiment. ApoE-KO OVX mice implanted with E2 pellets were divided into two groups; one that was given sirtinol, and the other that was given control vehicle (Fig. 3B). The atherosclerotic area was greater in OVX + E2 mice given sirtinol than in OVX + E2 mice given control vehicle (Fig. 3C). The ratio of p-eNOS to eNOS protein expression in aortas was not different and expression of Ac-p53 and PAI-1 was higher in OVX + E2 + sirtinol mice compared to OVX + E2 mice (Fig. 3D). Taken together, these results suggest that the beneficial effect of estrogen on anti-senescence and anti-atherosclerosis is derived from the upregulation of SIRT1.
Fig. 3.
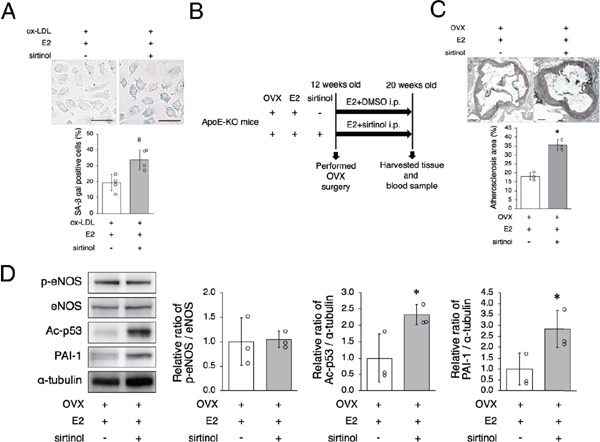
The beneficial effect of estrogen is derived from the upregulation of SIRT1
A: E2 (10 nM) + sirtinol (50 µM) increase SA-β gal positive cells relative to E2 in HUVEC with ox-LDL treatment, suggesting that sirtinol abolishes the beneficial effect of E2. Scale bar = 50 µm *P < 0.05 vs. E2 + ox-LDL group
B: Animal experiment protocol
C: Representative images of Oil Red O-stained cross-sections of the aortic roots of OVX + E2 mice and OVX + E2 + sirtinol mice are shown in the upper panel. Atherosclerotic lesions in OVX + E2 mice are significantly lower than those in OVX + E2 + sirtinol mice. Scale bar = 200 µm
D: Representative band of western blot analysis and densitometric analysis in OVX + E2 mice and OVX + E2 + sirtinol mice are shown in the left panel. The ratio of p-eNOS to eNOS is the same in both groups. Ac-p53 and PAI-1 protein expression were higher in OVX + E2 + sirtinol mice than in OVX + E2 mice.
All data are shown as the mean ± S.D.; n = 4 in each group (A). n = 3 in each group (C, D). #P < 0.05 vs. ox-LDL+E2 group, *P < 0.05 vs. OVX + E2 group
SERM Increased SIRT1 Expression and Retarded Arterial Senescence and The Development of Atherosclerosis
Recently, SERM has become available for the clinical treatment of osteoporosis and is reported to prevent CVD. Therefore, we investigated whether SERM mimics E2 to regulate arterial senescence and atherosclerotic development. BZA is a SERM that was administrated to OVX mice. DMSO was administrated to control group mice at the same volume and frequency as BZA (Fig. 4A). There was no difference in subjects' food intake during this experiment and no differences in body weight and the levels of total serum cholesterol and triglycerides eight weeks after administration of SERM or control vehicle between the two groups (Table 1). The ratio of arterial senescence and atherosclerotic area was lower in OVX + SERM mice than in OVX mice (Figs. 4B and C). Arterial protein expression of SIRT1 and the ratio of p-eNOS to eNOS were higher in OVX + SERM mice compared to OVX mice and the expression of Ac-p53, p21, and PAI-1 was lower in OVX + SERM mice compared to OVX mice (Fig. 4D). These results suggest that SERM has an effect similar to that of E2; it increases aortic SIRT1 expression and retards arterial senescence and atherosclerotic development.
Fig. 4.
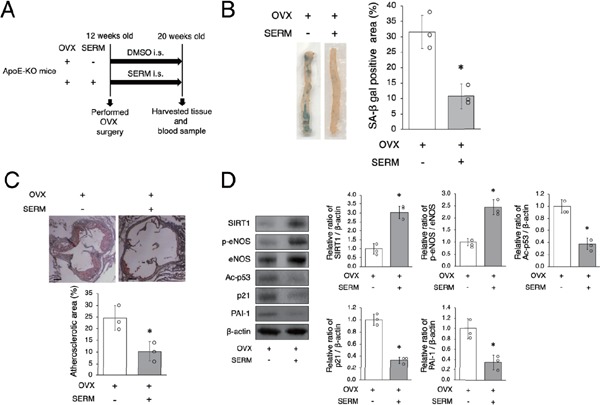
SERM increases SIRT1 expression and retards arterial senescence and atherosclerosis
A: Animal experiment protocol
B: Representative images of SA-β gal staining of senescent cells (blue) in the aortas of OVX mice and OVX + SERM mice are shown in the left panel. SA-β gal positive staining in OVX + SERM mice was lower than that in in OVX mice.
C: Representative images of Oil Red O-stained cross-sections of the aortic root of OVX mice and OVX + SERM mice are shown in the upper panel. Atherosclerotic lesions in OVX + SERM mice are significantly lower than those in OVX mice. Scale bar = 200 µm
D: Representative band of western blot analysis and densitometric analysis in OVX mice and OVX + SERM mice are shown in the left panel. SIRT1 expression and the ratio of p-eNOS to eNOS are significantly higher and the expression of Ac-p53, p21, and PAI-1 is lower in OVX + SERM mice compared to OVX mice.
All data are shown as the mean ± S.D.; n = 3 in each group. *P < 0.05 vs. OVX group
SERM-Induced SIRT1 Upregulation is Regulated by eNOS
Finally, we examined the mechanisms by which SERM regulates arterial SIRT1 expression. Since it has previously been reported that nitric oxide produced by activation of eNOS upregulates arterial SIRT1 25), we performed an inhibition experiment using an eNOS inhibitor, L-NAME (Fig. 5A). There were no differences in the ratio of arterial senescence, the atherosclerotic area, and the aortic protein expression of SIRT1, Ac-p53, p21, or PAI-1 between the two groups (Figs. 5B, C and D). We also performed eNOS inhibition experiment using eNOS-KO OVX mice and administration of either SERM or control vehicle. There were no differences in the aortic protein expression of SIRT1, Ac-p53, p21, or PAI-1 between the two groups (Fig. 5E). Taken together, these results suggest that SERM-induced SIRT1 upregulation is regulated by eNOS.
Fig. 5.
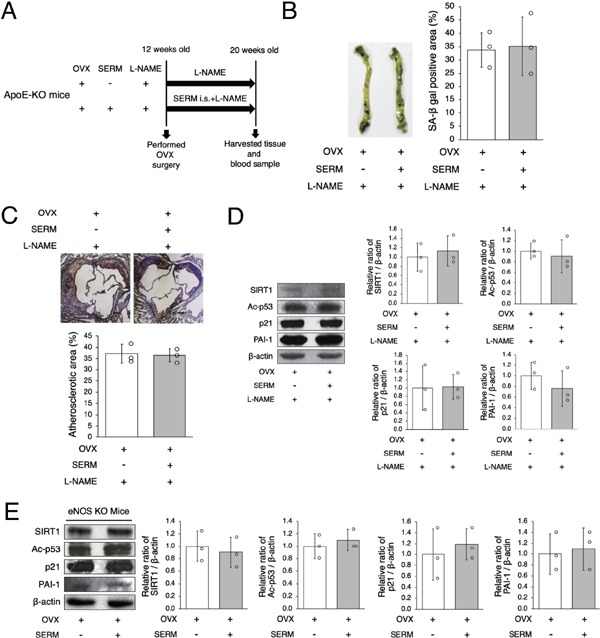
SERM-induced SIRT1 upregulation is regulated by eNOS
A: Animal experiment protocol
B: Representative images of SA-β gal staining of senescent cells (blue) in the aortas of OVX + SERM mice and OVX + SERM + L-NAME mice are shown in the left panel. There were no differences in SA-β gal positive staining in the two groups.
C: Representative images of Oil Red O-stained cross-sections of the aortic root of OVX + SERM mice and OVX + SERM + L-NAME mice are shown in the upper panel. There were no differences in atherosclerotic lesions in the two groups. Scale bar = 200 µm
D: Representative band of western blot analysis and densitometric analysis is shown in the left panel. There were no differences in SIRT1, Ac-p53, p21, and PAI-1 expression in the two groups.
E: BZA, a SERM, was administrated to eNOS-KO OVX mice. Representative band of western blot analysis and densitometric analysis is shown in the left panel. There were no differences in SIRT1, Ac-p53, p21, and PAI-1 expression between control vehicle and SERM mice.
All data are shown as the mean ± S.D.; n = 3 in each group
Discussion
The experiments described here demonstrated that arterial senescence and atherosclerotic development were facilitated by ovariectomy in ApoE-KO mice. Ovariectomy also decreased arterial SIRT1 expression in ApoE-KO mice. Administration of E2 restored SIRT1 expression and retarded arterial senescence and the development of atherosclerosis in ApoE-KO OVX mice, which was abolished by sirtinol administration. Additionally, SERM mimicked the beneficial effect of E2 that induces anti-senescence and anti-atherosclerosis through SIRT1. Taken together, these results indicate that SIRT1 plays the crucial role of estrogen in protecting arteries from senescence and atherosclerosis.
A recent study demonstrated that overexpressing SIRT1 specifically in the endothelium of ApoE-KO mice decreased atherosclerosis without changing blood lipid or glucose levels26). In the present study, E2 induced SIRT1 expression in female ApoE-KO OVX mice, but this effect was abolished by sirtinol. Calorie restriction is reported to upregulate SIRT1 27). In our experiment, body weight and food consumption were not influenced by OVX, E2 treatment, and SERM administration. These results indicate that the upregulation of SIRT1 by estrogen accompanied by antisenescence and anti-atherosclerosis is a direct effect of estrogen and is not derived from calorie restriction. Senescent cells have been found in human atherosclerotic vascular tissue and display various kinds of functional abnormalities28). SIRT1 has been shown to deacetylate p53 and forkhead transcription factors, which regulate apoptosis, stress responses, and cellular senescence28, 29). The acetylation of p53 is affected by the stress response and correlates with its activation30). Deacetylation of p53 abrogates cellular senescence and apoptosis caused by DNA damage and stress. It has been reported that p53 acetylation accelerates the expression of growth suppressive genes and induces cellular senescence31, 32). Our data suggest that SIRT1, upregulated by E2, deacetylates p53, which may contribute to retarding arterial senescence. Seven members of the sirtuin family, from SIRT1 to SIRT7, exist in mammals. Each family member has different tissue specificity, subcellular activity localization, and targets33). Evidences are also accumulating that SIRT3, which is located in the mitochondria and influences inclusive mitochondrial lysine acetylation34), is related to senescence and atherosclerosis. Mitochondrial dysfunction leads to many cardiovascular diseases, including atherosclerosis and senescence35–37). According to our data, there was no significant change in SIRT3 in either OVX or OVX + E2 mice, suggesting that SIRT1 rather than SIRT3 plays an important role related to E2 on vascular senescence and atherosclerosis.
It is well known that endogenous nitric oxide (NO) produced by eNOS plays an important role in vascular homeostasis and endothelial function, and interaction between eNOS and SIRT1 protects against endothelial senescence and atherosclerosis. SIRT1 is involved in the regulation of eNOS, leading to vascular protection. It has been reported that SIRT1 regulates eNOS activation in endothelial cells, and that decreased SIRT1 and eNOS activity causes age-related endothelial dysfunction in mice38). The expression of SIRT1 and eNOS phosphorylated at serine 1177 was lower in the aortas of 30-month-old B6D2F1 mice than in the aortas of five to seven-month-old mice. Additionally, NO may control SIRT1 expression. A previous study demonstrated that NO control of SIRT1 expression is the case in white adipose tissue and white adipocytes; calorie restriction increased SIRT1 expression in the white adipose tissue of wild type mice, and this effect was abolished in eNOS-KO mice39). Another study indicated that cilostazol, a phosphodiesterase 3 inhibitor, prevented premature senescence caused by SIRT1 upregulation through eNOS activation in vascular tissue25). In the present study, we found that either E2 or SERM administration increased SIRT1 and activated eNOS, resulting in decreased vascular senescence and atherosclerotic lesions. Furthermore, the effect of SERM on upregulating SIRT1 was abolished in eNOS-KO OVX mice. OVX + SERM mice treated with L-NAME, a NOS inhibitor, also showed no differences in arterial SIRT1 expression and senescence. These results suggest that SIRT1 expression is regulated by estrogen-induced eNOS activation.
Estrogens exert their physiological effects by binding to three known estrogen receptors (ERs): ERα; ERβ; and an orphan G-protein-coupled estrogen receptor. Several studies have indicated that ERα and ERβ differentially modulate gene expression, and the protein levels of ERα and ERβ exhibit temporal and tissue variations40, 41). It has been reported that ERα expression modulated by estrogen in endothelial cells is related to eNOS activation (phosphorylatedeNOS-Ser1177)42). Recently, several SERMs have been used widely in the prevention and treatment of postmenopausal bone fractures related to osteoporosis and breast cancer. These SERMs include tamoxifen, raloxifene, ospemifene, arzoxifene, lasofoxifene, tremifene, and bazedoxifene 43). Raloxifene has been reported to reduce the atherosclerotic lesions of ovariectomized rabbits44, 45), although its relationship with SIRT1 is unknown. Another recent study has suggested that treatment with lasofoxifene reduces the risk of stroke and coronary artery disease in randomized control trials, and that other SERMs, including lasofoxifene, may have beneficial effects on atherothrombosis46). BZA is a new third-generation SERM which has been linked to prevention of bone loss and reduced bone turnover in postmenopausal women at risk of osteoporosis. BZA binds to both ERα and ERβ, although it has a slightly higher affinity for the former47). In the present study, we used BZA to investigate whether SERMs mimic E2 and we found that administration of BZA upregulates eNOS activation and SIRT1 protein expression and retards the development of atherosclerosis and vascular senescence. These results suggest that SERMs might have potential as alternative therapeutic agents for atherosclerosis in patients who cannot be treated in other ways due to complications of HRT.
Although it is recognized that many factors independent of estrogen cause sex differences both preand post-menopause, it is clear that estrogen predominantly mediates these differences in the cardiovascular system. In the present study, we focused on the effects of estrogen on arterial senescence in postmenopausal model mice; in the future, it will be necessary to investigate whether the eNOS/SIRT1 pathway is involved in male menopause.
Limitation
Several limitations of this study must be considered. Firstly, this study had a small number of mice. Secondly, we need further experiments to examine longitudinal changes in arterial senescence and atherosclerotic development over time. Finally, we did not perform inhibition experiments of eNOS in ApoE-KO mice with E2 pellet implantation.
Conclusion
Downregulation of SIRT1 causes OVX-induced arterial senescence and atherosclerosis in ApoE-KO mice. Administration of estrogen or SERM enables OVX mice to restore these alterations by SIRT1 induction.
Acknowledgments
We thank Ms Michiko Shimokawa, Ms Ayako Kuroki, Ms Aya Yanagi for their excellent technical assistance and all the staff members of the Institute of Laboratory Animal Science, Kagoshima University (Frontier Science Research Center) who maintained the animals in good condition. We also thank the joint Research Laboratory, Kagoshima University Graduate School of Medical and Dental Science, for the use of their facilities.
Grant Support
This work was supported by in a part of Japan society for the promotion of Science KAKENHI (Grant No.JP16K09250), a Research Grant from The Japanese Geriatrics Society, and Pfizer Inc.
Conflicts of Interest
The authors report no conflicts of interest or disclosures.
References
- 1). Xing D, Nozell S, Chen YF, Hage F, Oparil S: Estrogen and mechanisms of vascular protection. Arterioscler Thromb Vasc Biol, 2009; 29: 289-295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2). Barton M, Meyer MR, Haas E: Hormone replacement therapy and atherosclerosis in postmenopausal women: Does aging limit therapeutic benefits? Arterioscler Thromb Vasc Biol, 2007; 27: 1669-1672 [DOI] [PubMed] [Google Scholar]
- 3). Kannel WB, Hjortland MC, McNamara PM, Gordon T: Menopause and risk of cardiovascular disease: the Framingham study. Ann Intern Med, 1976; 85: 447-452 [DOI] [PubMed] [Google Scholar]
- 4). Hodgin JB, Maeda N: Minireview: Estrogen and mouse models of atherosclerosis. Endocrinology, 2002; 143: 4495-4501 [DOI] [PubMed] [Google Scholar]
- 5). Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N: Estrogen receptor α is a major mediator of 17β-estradiol's atheroprotective effects on lesion size in Apoe−/−mice. J Clin Invest, 2001; 107: 333-340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6). Hodis HN, Mack WJ, Henderson VW, Shoupe D, Budoff MJ, Hwang-Levine J, Li Y, Feng M, Dustin L, Kono N, Stanczyk FZ, Selzer RH, Azen SP: Vascular effects of early versus late postmenopausal treatment with estradiol. N Engl J Med, 2016; 374: 1221-1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7). Salpeter SR, Walsh JM, Greyber E, Salpeter EE: Brief report: Coronary heart diesease events associated with hormone therapy in younger and older women. A metaanalysis. J Gen Intern Med, 2006; 21: 363-366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Schierbeck LL, Rejnmark L, Tofteng CL, Stilgren L, Elken P, Mosekilde L, Køber L, Jensen JE: Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ, 2012; 345: e6409. [DOI] [PubMed] [Google Scholar]
- 9). Wang JC, Bennett M: Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res, 2012; 111: 245-259 [DOI] [PubMed] [Google Scholar]
- 10). Kaeberlein M, McVey M, Guarente L: The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev, 1999; 13: 2570-2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11). Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A: Mechanisms of vascular aging. Circ Res, 2018; 123: 849-867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12). Rodella LF, Favero G, Rossini C, Foglio E, Bonomini F, Reiter RJ, Rezzani R: Aginig and vascular dysfunction: Beneficial melatonin effects. Age (Omaha), 2013; 35: 103-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13). Gorenne I, Kumar S, Gray K, Figg N, Yu H, Mercer J, Bennett M: Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation, 2013; 127: 386-396 [DOI] [PubMed] [Google Scholar]
- 14). Thompson AM, Martin KA, Rzucidlo EM: Resveratrol induces vascular smooth muscle cell differentiation through stimulation of SirT1 and AMPK. PLoS One, 2014; 9: 1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15). Kane AE, Sinclair D: Sirtuins and NAD+ in the Development and treatment of metabolic and cardiovascular diseases. Circ Res, 2018; 123: 868-858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Kawai S, Takagi Y, Kaneko S, Kurosawa T: Effect of three types of mixed anesthetic agents alternate to ketamine in mice. Exp Anim, 2011; 60: 481-487 [DOI] [PubMed] [Google Scholar]
- 17). Tolbert T, Thompson JA, Bouchard P, Oparil S: Estrogen-induced vasoprotection is independent of inducible nitric oxide synthase expression: Evidence from the mouse carotid artery ligation model. Circulation, 2001; 104: 2740-2745 [DOI] [PubMed] [Google Scholar]
- 18). Akasaki Y, Miyata M, Eto H, Shirasawa T, Hamada N, Ikeda Y, Biro S, Otsuji Y, Tei C: Repeated thermal therapy up-regulates endothelial nitric oxide synthase and augments angiogenesis in a mouse model of hindlimb ischemia. Circ J, 2006; 70: 463-470 [DOI] [PubMed] [Google Scholar]
- 19). Hamada N, Miyata M, Eto H, Ikeda Y, Shirasawa T, Akasaki Y, Miyauchi T, Furusho Y, Nagaki A, Aronow BJ, Tei C: Loss of clusterin limits atherosclerosis in apolipoprotein E-deficient mice via reduced expression of Egr-1 and TNF-α. J Atheroscler Thromb, 2011; 18: 209-216 [DOI] [PubMed] [Google Scholar]
- 20). Smith JD, Trogan E, Ginsberg M, Grigaux C, Tian J, Miyata M: Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci U S A, 1995; 92: 8264-8268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). Miyauchi T, Miyata M, Ikeda Y, Akasaki Y, Hamada N, Shirasawa T, Furusho Y, Tei C: Waon therapy upregulates Hsp90 and leads to angiogenesis through the Akt-endothelial nitric oxide synthase pathway in mouse hindlimb ischemia. Circ J, 2012; 76: 1712-1721 [DOI] [PubMed] [Google Scholar]
- 22). Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J: Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res, 2014; 116: 264-278 [DOI] [PubMed] [Google Scholar]
- 23). Berthois Y, Katzenellenbogen JA, Katzenellenbogen BS: Phenol red in tissue culture media is a weak estrogen: implications concerning the study of estrogen-responsive cells in culture. Proc Natl Acad Sci, 1986; 83: 2496-2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O: A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci, 1995; 92: 9363-9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Ota H, Eto M, Kano MR, Ogawa S, Iijima K, Akishita M, Ouchi Y: Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arterioscler Thromb Vasc Biol, 2008; 28: 1634-1639 [DOI] [PubMed] [Google Scholar]
- 26). Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, Liu G, Wei YS, Cai H, Liu DP, Liang CC: Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res, 2008; 80: 191-199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA: Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science, 2004; 305: 390-392 [DOI] [PubMed] [Google Scholar]
- 28). Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I: Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation, 2002; 105: 1541-1544 [DOI] [PubMed] [Google Scholar]
- 29). Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsy R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME: Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science, 2004; 303: 2011-2015 [DOI] [PubMed] [Google Scholar]
- 30). Luo J, Su F, Chen D, Shiloh A, Gu W: Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature, 2000; 408: 377-381 [DOI] [PubMed] [Google Scholar]
- 31). Bond J, Haughton M, Blaydes J, Gire V, Wynford-Thomas D, Whllie F: Evidence that transcriptional activation by p53 plays a direct role in the induction of cellular senescence. Oncogene, 1996; 13: 2097-2104 [PubMed] [Google Scholar]
- 32). Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W: Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci, 2004; 101: 2259-2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Frye RA: Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun, 2000; 273: 793-798 [DOI] [PubMed] [Google Scholar]
- 34). Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Weissman S, Verdin E, Schwer B: Mammalian Sir2 Homolog SIRT3 Regulates Global Mitochondrial Lysine Acetylation. Mol Cell Biol, 2007; 27: 8807-8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35). Madamanchi NR, Vendrov A, Runge MS: Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol, 2005; 25:29-38 [DOI] [PubMed] [Google Scholar]
- 36). Wallace DC: Mitochondrial defects in cardiomyopathy and neuromuscular disease. Am Heart J, 2000; 139: S70-85 [DOI] [PubMed] [Google Scholar]
- 37). Ikeda Y, Sciarretta S, Nagarajan N, Rubttu S, Volpe M, Frati G, Sadoshima J: New insights into the role of mitochondria dynamics and autophagy during oxidative stress and aging in the heart. Oxid Med Cell Longev, 2014; 2014: 210934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38). Donato AJ, Magerko KA, Lawson BR, Durrant JR, Lesniewski LA, Seals DR: SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J Physiol, 2011; 589: 4545-4554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39). Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO: Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science, 2005; 310: 314-317 [DOI] [PubMed] [Google Scholar]
- 40). Lindberg MK, Movérare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson J-Å, Ohlsson C: Estrogen receptor (ER)-β reduces ERα-regulated gene transcription, supporting a “ying yang” relationship between ERα and ERβ in mice. Mol Endocrinol, 2003; 17: 203-208 [DOI] [PubMed] [Google Scholar]
- 41). Couse JF, Lindzey J, Grandien K, Gustafsson JÅ, Korach KS: Tissue distribution and quantitative analysis of estrogen receptor-α (ERα) and estrogen receptor-β (ERβ) messenger ribonucleic acid in the wild-type and ERα-knockout mouse. Endocrinology, 1997; 138: 4613-4621 [DOI] [PubMed] [Google Scholar]
- 42). Gavin KM, Seals DR, Silver AE, Moreau KL: Vascular endothelial estrogen receptor α is modulated by estrogen status and related to endothelial function and endothelial nitric oxide synthase in healthy women. J Clin Endocrinol Metab, 2009; 94: 3513-3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43). Maximov PY, Lee TM JV: The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr Clin Pharmacol, 2013; 8: 135-155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Bjarnason NH, Haarbo J, Byrjalsen I, Kauffman RF, Christiansen C: Raloxifene inhibits aortic accumulation of cholesterol in ovariectomized, cholesterol-fed rabbits. Circulation, 1997; 96: 1964-1969 [DOI] [PubMed] [Google Scholar]
- 45). Bjarnason NH, Haarbo J, Byrjalsen I, Kauffman RF, Knadler MP, Christiansen C: Raloxifene reduces atherosclerosis: Studies of optimized raloxifene doses in ovariectomized, cholesterol-fed rabbits. Clin Endocrinol (Oxf), 2000; 52: 225-233 [DOI] [PubMed] [Google Scholar]
- 46). Kitagawa K: Selective Estrogen Receptor Modulators and Cardiovascular Events. Circ J, 2014; 63-64 [DOI] [PubMed] [Google Scholar]
- 47). Komm BS, Kharode YP, Bodine PVN, Harris HA, Miller CP, Lyttle CR: Bazedoxifene acetate: A selective estrogen receptor modulator with improved selectivity. Endocrinology, 2005; 146: 3999-4008 [DOI] [PubMed] [Google Scholar]