

Abstract
Despite widespread use as an immunosuppressant, the therapeutic efficacy of the undecapeptide cyclosporine A (CyA) is compromised when given by the oral route because of the innate hydrophobicity of the drug molecule, potentially leading to poor aqueous solubility and bioavailability. The aim of this study was to develop and characterize nanofibers based on the water-miscible polymer polyvinylpyrrolidone (PVP), incorporating CyA preloaded into polymeric surfactants so as to promote micelle formation on hydration; therefore, this approach represents the novel combination of three dissolution enhancement methodologies, namely solid dispersion technology, micellar systems, and nanofibers with enhanced surface area. The preparation of the nanofibers was performed in two steps. First, mixed micelles composed of the water-soluble vitamin E derivative d-α-tocopheryl poly(ethylene glycol) 1000 succinate and the amphiphilic triblock polymer Pluronic F127 (Poloxamer 407) were prepared. The micelles were characterized in terms of size, surface charge, drug loading, and encapsulation efficiency using transmission electron microscopy, dynamic light scattering, Fourier-transform infrared spectroscopy, high-performance liquid chromatography, and scanning electron and atomic force microscopy analysis. Nanofibers composed of PVP and the drug-loaded surfactant system were then prepared via electrospinning, with accompanying thermal, spectroscopic, and surface topological analysis. Dissolution studies indicated an extremely rapid dissolution profile for the fibers compared to the drug alone, while wettability studies also indicated a marked decrease in contact angle compared to the drug alone. Overall, the new approach appears to offer a viable means for considerably improving the dissolution of the hydrophobic peptide CyA, with associated implications for improved oral bioavailability.
1. Introduction
Cyclosporine A (CyA; Figure 1) is a cyclic undecapeptide, originally derived from the fungus Tolypocladium inflatum, and is used extensively as a therapeutic immunosuppressant because of its selective and reversible inhibition of T-lymphocytes, accompanied by low cytotoxicity.1 More specifically, CyA is indicated for the treatment of conditions such as allograft rejection in transplantation, autoimmune diseases including psoriasis and rheumatoid arthritis, and dry eye disease. However, the drug is classified as Biopharmaceutics Classification System (BCS) class IV, with both low water solubility and membrane permeability, hence adsorption following oral administration is a major challenge because of both difficulties associated with transfer to the aqueous phase (a prerequisite for absorption) as well as limitations to subsequent transfer across the gastrointestinal wall. These absorption characteristics are associated with the physicochemical characteristics of the molecule, including high molecular weight (1203 kDa), low water solubility (27.67 μg/mL at 25 °C) coupled with high lipophilicity (log P = 2.92 at pH 7.4), and the very rigid cyclic structure of the molecule which provides a further architectural barrier to membrane permeation.2
Figure 1.

Chemical structures of CyA, vitamin E TPGS, Pluronic F127, and PVP.
As a consequence of these absorption difficulties, the first commercial formulations of CyA (Sandimmune, approved in 1983) were emulsion-based, either for intravenous (with Cremophor EL and ethanol) or oral administration (in the form of soft gelatin capsules or an oral solution). These formulations were in themselves problematic; for example, coadministration of Cremophor EL is associated with a wide range of adverse effects, including anaphylactic shock, hypersensitivity reactions, and variation in blood pressure, hyperlipidemia, and peripheral neuropathy.1−4 Consequently, there has been considerable research into improving CyA water miscibility and membrane permeability via a number of routes and approaches,2−5 including the use of colloidal delivery systems including solid lipid nanoparticles, liposomes, nanosuspensions, and micelles.3−6 Among these, polymeric micellar-based drug delivery system is considered particularly promising and has been explored for ocular formulations, allowing enhanced water miscibility without the need for inclusion of an oily phase that would impede vision, while also enhancing permeability across the cornea.7 However, for the oral route, there is an additional challenge that solid dosage forms are generally preferred.
Recently, significant attention has been drawn to the possibility of using nanofibers for oral delivery, particularly as the manufacture of such systems at a scale commensurate with commercialization is now possible.8 In addition, nanofibers have been investigated as a means of enhancing delivery and uptake of CyA into cells in vitro.8−10 Taken together, these studies suggest that a suitable nanofiber-based formulation could potentially achieve the twin goals of being the basis for a solid oral dosage form while also improving dissolution and potentially uptake.
In this study, we investigate the possibility of combining micellar and nanofiber delivery technologies as a means of enhancing, in the first instance, the dissolution rate of the drug into relevant aqueous media. This is a prerequisite to any enhancement of uptake because of the necessity of any drug being in a molecular disperse form prior to absorption. The ability of micellar systems to improve drug solubility via incorporation into those regions of the structure that are dielectrically compatible is well known. However, more recently, the possibility of using nanofibrous systems as alternatives to matrix solid dispersions for the dissolution enhancement of poorly water soluble drugs has been outlined;11 not only may the fibers act as solid dispersions to present the drug to the aqueous media in a molecularly disperse form within a water-miscible base but the high surface area of the fibers further enhances the dissolution rate from the solid surface. Here, we propose to incorporate the CyA into micelles that are themselves incorporated into nanofibers, thereby providing a novel approach for dissolution enhancement combining micellar, nanofiber, and solid dispersion technologies.
We examine the use of d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS, Figure 1), an amphiphilic nonionic water-soluble derivative of vitamin E formed by conjugation of vitamin E succinate with polyethylene glycol (PEG). TPGS has generally regarded as safe (GRAS) status and is an FDA-approved pharmaceutical excipient; notably, it offers numerous advantageous features including solubilization of hydrophobic drugs, enhanced cellular uptake of the drug payload, and inhibition of P-glycoprotein to prevent drug efflux.13 However, it also has a relatively high critical micelle concentration (cmc) of circa 0.02% w/w, rendering TPGS micelles prone to dissociation on dilution, thereby potentially losing the solubilization advantage in the gastrointestinal tract. Consequently, generating mixed or composite structures with another copolymeric material with a suitable cmc value represents a means of overcoming this difficulty. In order to achieve this, we explore the use of Pluronic F127 (henceforth referred to as F127) triblock copolymers, consisting of hydrophilic polyethylene oxide (PEO) and hydrophobic polypropylene oxide (PPO) segments arranged in a triblock structure (PEO100–PPO65–PEO100; Figure 1) with relatively extended PEO blocks. This material is commonly used as a low-cmc micellar carrier (0.0031%, w/w12,13) while also having high biocompatibility and an acceptable safety profile and solubilization capacity. F127 has little effect on P-glycoprotein and does not significantly enhance membrane transport because of its relatively hydrophilic nature.
Our proposal is that by combining TPGS and F127 in mixed micelles, we will generate a carrier system that is suitable for CyA from both a loading and a delivery perspective, while subsequent incorporation into nanofibers composed of the water-miscible polymer polyvinylpyrrolidone (PVP)14 (Figure 1) will generate a highly hydrophilic system that not only offers a high surface area for dissolution but may also be further processed into a solid dosage form for oral delivery (this polymer being a common component of a range of oral dosage forms). In the first instance, the solubility of CyA in different Pluronic F127/TPGS ratios and concentrations was established, followed by characterization of the drug-loaded micelles in terms of particle size, zeta potential, morphology, encapsulation, and surface morphology. The micellar formulation was then loaded into PVP nanofibers and the product characterized in terms of physicochemical properties, in vitro release, and wettability studies. In this manner, the possibility of using micelle-forming nanofibers as the potential carrier system for CyA may be effectively evaluated.
2. Materials and Methods
2.1. Materials
CyA (C62H111N11O12, m.wt. 1.2 kDa) was procured from Santa Cruz Biotechnology, UK. TPGS (m.wt. 574.874 g/mol), pyrene (m.wt. 202.25 g/mol), and Poloxamer 407 (Pluronic F127 m.wt. 12 500 g/mol) were purchased from Sigma-Aldrich UK. PVP (high m.wt. 146 000–186 000) was purchased from Sigma-Aldrich, UK. Coumarin-6 was procured from Sigma-Aldrich UK. Phosphate buffer solution (PBS) tablets biotech grade E404 (VWR Life Science, UK) were used to prepare the pH 7.4 and 6.8 buffer solutions. The solvents including Milli-Q water and high-performance liquid chromatography (HPLC) grade chemicals were all of analytical grade.
2.2. Development of the HPLC-Based CyA Detection Method
The quantification of CyA was performed utilizing an HPLC-UV system with a Zorbax Eclipse Plus reverse phase C-18 column (dimension 4.6 × 250 mm, 5 μm) (Agilent Technologies Santa Clara, USA), thermostated at 68 °C. The mobile phase used was an 80:20 (v/v) mixture of acetonitrile and HPLC grade water using a flow rate of 1 mL/min.15 The injection volume used was 20 μL, and absorbance was monitored at 210 nm. The calibration curve was constructed over a concentration range of 0.5–250 μg/mL with an R2 value of 0.998 (Figure S1).
2.3. CyA Equilibrium Solubility Studies
In order to evaluate the drug solubility, the CyA was dissolved in ethanol and the solution added in excess to the aqueous systems under study. The mixed systems were incubated to allow equilibration and the ethanol evaporated so that any excess CyA not incorporated into the aqueous solvent system (including the micelles) would precipitate and be filtered out. To ensure ethanol evaporation over the incubation period, pierced aluminum foil was used as a covering for the reaction container. The CyA content was the measured using the HPLC method described above. More specifically, a 1 mg/mL ethanolic solution of CyA was added to the range of molar concentrations (0.0005–10 mM) of surfactant mixture (F127/TPGS; 3:2 molar ratio) in aqueous solution and incubated for 12 h at 37 °C at 100 rpm. The F127/TPGS 3:2 molar ratio was chosen based on literature precedent,12,13 as well as initial solubilization studies (see below), and was used throughout the study unless otherwise indicated. The suspension was then filtered (0.45 μm Millipore membrane filter), and the filtrate was diluted in acetonitrile and analyzed using the HPLC-UV system. Furthermore, the intrinsic solubility of CyA in PBS alone (pH 7.4), 30% (v/v) PBS in ethanol and 0.5% (v/v) Tween 80 in PBS were also evaluated under the same incubation conditions for a period of 72 h.
2.4. cmc Determination
In order to evaluate the cmc of the mixed micellar systems, pyrene was used as a hydrophobic fluorescence probe.13 Pyrene solution (500 μL) in acetone (0.6 μM) was added to the mixed micelle solutions (0.0005–0.1 mM) and mixed for 4–5 h followed by overnight incubation at ambient temperature; the acetone was evaporated during this process. Samples were measured for fluorescence emission using a PerkinElmer LS55 luminescence spectrometer, with the pyrene excitation wavelength being 334 nm. The ratio of first to third order emission band peak of pyrene (I372/I383) was plotted against the molar concentration of surfactant mixture (F127/TPGS). A range of molar ratios of the two surfactants (1:1, 1:4, 4:1, and 3:2) were evaluated; the 3:2 F127/TPGS molar ratio was used for subsequent studies.
2.5. Preparation of CyA-Loaded TPGS/F127 Mixed Micelles
For the preparation of the unloaded micelles following an established method,16 10 mM solutions of F127 and TPGS were mixed in a 3:2 molar ratio (F127/TPGS) followed by homogenization for 30 min using a D1 18 disperser (IKA, UK). The micellar dispersions were freeze-dried and the powder reconstituted so as to provide a standardized starting material for drug incorporation. Samples (2–50 mg) were reconstituted in 5 mL aqueous solution under stirring, and an ethanolic solution (1 mg/mL) of the drug was added in a dropwise manner followed by homogenization for 30 min. After homogenization, the sample was stirred overnight to allow equilibration and to ensure complete evaporation of ethanol. The system was subjected to dialysis [using molecular weight cut-off (MWCO) 3.5 kDa dialysis membrane] over a period of 48 h followed by filtration using a 0.45 μm Millipore membrane filter to remove any excess drug. The following w/w drug to (dried material) micelle ratios were prepared: 1:50 (F1), 1:30 (F2), 1:20 (F3), 1:10 (F4), 1:05 (F5), and 1:02 (F6); the F5 ratio was used for subsequent studies.
2.6. Evaluation of Drug-Loading Content and Entrapment Efficiency
In order to investigate the drug-loading content (LC) and encapsulation efficiency of the mixed (F127/TPGS) micelles, lyophilized systems were used. The micellar solutions were frozen at −75 °C followed by freeze-drying for at least 24 h. The mixed micelles were weighed (2–50 mg) and reconstituted in 5 mL water followed by addition of 1 mL of an ethanolic solution of drug (1 mg/mL solution) into the micellar dispersion. The solutions were sonicated for 30 min followed by overnight incubation at 37 °C under 100 rpm stirring conditions. The solutions were then dialyzed using a 3.5 kDa dialysis membrane against distilled water for 48 h, followed by further filtration (0.45 μm Millipore membrane filter) to allow removal of excess unloaded drug. The filtrate was diluted in acetonitrile to dissolve the micellar system and to release all the encapsulated drug; the product was then analyzed using the HPLC-UV system in order to evaluate the LC and EE of the micellar systems. The LC was calculated from the weight of CyA extracted from micelles/total weight of micelles added, while the EE was calculated from the weight of CyA extracted from micelles/weight of input CyA.
where the mass of input CyA is the mass of CyA used for preparation of micelles.
2.7. Characterization of CyA-Loaded Mixed Micelles and Micelle-Loaded Nanofibers
2.7.1. Assessment of Micelle Size, Polydispersity Index, and Surface Charge
A Zetasizer Nano instrument (Malvern Instruments, Malvern, UK) equipped with a 4 mW He–Ne laser (633 nm) was employed to measure the mean hydrodynamic particle size and size distribution of the unloaded and CyA-loaded formulations (F5) and electrokinetic zeta potential at 25 °C. All samples were evaluated in triplicate.
2.7.2. Morphological and Elemental Analysis of Polymeric Micelles and Nanofibers
For transmission electron microscopy (TEM) studies, diluted micellar systems were drop-casted on to copper grids and stained with 2% (w/v) phosphotungstic acid and further air-dried before observation using a Philips/FEI CM120 BioTwin microscope operated at an accelerating voltage of 100 kV to avoid any interference due to water. For nanofiber analysis, a copper grid was held directly over the aluminum foil inside the electrospinning chamber, and fibers were deposited for 5 min to ensure that a thin film was deposited for examination by TEM.
The surface morphology of the micelle-loaded electrospun fiber mats was investigated using a Jeol JSM-6480LV high-performance variable pressure analytical scanning electron microscope with a resolution of 3.0 nm, along with an energy-dispersive system and electron backscatter diffraction facilities, operated with an accelerating voltage of 5 kV and a working distance of 20 mm. A small piece of the nanofiber mat was fixed on a conductive carbon tape and mounted on the support and then sputtered with an approximately 6 nm layer of gold (Au) for 60 s with a sputter-coating unit (Bio-Rad, Polaron Division Gold Coating System). The diameter distributions of the nanofibers in the mats were determined with an ImageJ tool with sample sizes of at least 50 fibers per scanning electron microscopy (SEM) micrograph. Statistical analysis was performed with OriginPro 8 software. The instrument was equipped with an energy dispersive spectrometer to confirm the presence of drug within the CyA/micellar-loaded nanofibers.
2.7.3. Attenuated Total Reflectance Fourier Transforms Infrared Spectroscopy, X-ray Diffraction, and Differential Scanning Calorimetry Analysis
The Fourier-transform infrared (FTIR, Bruker VERTEX 70 FTIR spectrometer) spectra of pure CyA drug powder, pure polymers (TPGS, F127, and PVP), freeze-dried unloaded mixed micelles, and CyA loaded micelles were recorded over the 400–4000 cm–1 range.
X-ray diffraction [XRD, Rigaku Mini 600 (Rigaku, Tokyo, Japan)] was performed to study the physical form of the pure components, the freeze-dried micellar formulations, and the micelle-incorporated nanofibrous systems. The XRD was operated with Cu Kα radiation (1.5418 Å) at 40 kV and 15 mA), between 2 and 40° over 2θ at 2°/min speed and 0.02 step size at constant temperature 25 °C, with constant voltage and current intensity. Xpert data viewer software (PANalytical B.V., Netherland) was used to analyze the data. Modulated temperature differential scanning calorimetry (DSC) (TA Instruments Q1000, UK) was used to characterize the lyophilized micellar and micelle-loaded nanofiber samples. The micelles were placed in hermetically sealed aluminum pans and purged with 150 mL/min of nitrogen. The system was calibrated with indium, and a heating rate of 2°/min from 20 to 250 °C was used. Data analysis was performed using TA Universal Analysis software.
2.8. Electrospinning Parameters and Nanofiber Characterization
A Spraybase electrospinning system (Spraybase Instruments, Avectas, Ireland) was used for fabrication of nanofibers loaded with CyA-containing micelles. The nanofibers were prepared via single-nozzle electrospinning. PVP (25 wt %) was dissolved in double distilled water for spinning the unloaded and loaded nanofibers. CyA-loaded micelles (50 wt % freeze dried material suspended water) were mixed in a 60:40 v/v ratio (PVP/surfactant mixture) to obtain homogeneous solutions before electrospinning the loaded systems. These proportions were selected after extensive preliminary studies to both maximize drug loading and to produce good quality fibers. Note that the final composition of the dried fibers was as follows: a 100 mL spraying solution would theoretically produce 15 g of PVP and 20 g of surfactant/drug mix, in which the surfactants would be present in a 3:2 F127/TPGS molar ratio, and the drug was present in a weight ratio of 1:5 (CyA to surfactant). This in turns equates to 9.52% w/w theoretical drug loading in the dry fibers, which is a realistically high loading for a practical dosage form. The electrospinning parameters were as follows: distance between electrodes tip to collector 20–22 cm, applied voltage 18–24 kV, relative humidity 30–46%; the temperature was kept at 25 °C throughout the fabrication process. The morphology of the nanofibers was assessed by SEM, followed by ImageJ image analysis. Atomic force microscopy (AFM) (Bruker’s MultiMode 8 and JPK NanoWizard) was performed in peak-force tapping mode on thin layers of fibers deposited onto a mica surface.
2.9. Fluorescence Microscopy Examination of Micelle Incorporation into Nanofibers
Coumarin-6 was used as a hydrophobic fluorescence probe to examine the incorporation of micelles into the nanofibrous systems. The coumarin-6-loaded micelles were prepared in a similar manner to the drug-loaded micelles, with the excess coumarin-6 removed by dialysis (MWCO = 3.5 kDa) of the system for 48 h in distilled water under standard stirring conditions. The coumarin-6-loaded micelles were then blended with the aqueous solution of PVP and stirred until a homogeneous solution was obtained. The solution was then electrospun following optimized parameters: 24 kV voltage, 40% (measured) humidity, and 20 cm distance from tip to collector.
2.10. In Vitro Release Study of CyA-Loaded Micelles from Micelle-Encapsulated Nanofibers
The release of CyA from the loaded nanofibers was examined in a range of pH media (1.2 pH 0.1 N HCl, 6.8 and 7.4 pH PBS buffer). The loaded nanofiber mats were cut into circa 10 mg samples, with each experiment run in triplicate. The release study was performed at 37 °C at 100 rpm (shaking speed) in an orbital incubator shaker (SciQuip Incu Shaker MINI, UK). The nanofibers were then placed in the three dissolution media (sink conditions), and the amount of drug released was estimated at various time points up to 20 min. The drug content was measured by dilution in acetonitrile followed by HPLC-UV analysis as described above. The release of the drug from the nanofiber samples was compared to the release profile of the pure drug.
2.11. Contact Angle Measurement
The static contact angles on the drug and nanofiber mat surfaces were calculated using the sessile drop method with a drop shape analysis system-DSA10 (DSA10-Kruss, Hamburg, Germany). Deionized water (40 μL) was dropped on top of a nanofiber mat sample, and the contact angle was measured after 60 s equilibration time.
3. Results and Discussion
3.1. Incorporation of CyA into Polymeric Mixed Micelles
3.1.1. cmc Determination of Micelles and CyA Solubility Studies
The cmc values of F127/TPGS mixed micelles containing 1:1, 1:4, 4:1, and 3:2 compositional ratios (molar ratios of F127 to TPGS) were determined using the fluorescent probe pyrene that enters the hydrophobic core on formation of the micelles, thereby resulting in a decrease in the (I372/I383) fluorescence intensity ratio. The typical complete fluorescence spectra of pyrene (0.6 μM) alone and in mixed micelle suspension showed the distinct emission spectral regions of pyrene including I1 (372), I2 (380), I3 (383), I4 (391), and I5 (395) as shown in Figure S2a; a plot of the fluorescence emission of pyrene at the I372/I383 ratio in the presence of various concentrations (0.0005–0.1 mM) of copolymers F127/TPGS (3:2 molar ratio) is shown in Figure S2b. In the absence of micelles (i.e., below the cmc), the ratio of first/third emission peaks (I1/I3) was high because of the pyrene being located in a polar environment. In contrast, the presence of micelles caused a decrease in the I1/I3 emission peak ratio because of solubilization of the pyrene into the hydrophobic core of micelles, in turn leading to a reduction in the I1/I3 intensity ratio.17 The cmc values are given in Table S1. The results showed that the mixed polymeric micelles in the 3:2 and 4:1 ratios had lower cmc than the pure polymers TPGS and F123 (4:0; 0:4 ratios). The properties of these mixed micellar systems have been previously studied by Butt et al.12 who suggested that the presence of the TPGS may increase the stability of the hydrophobic core, particularly via interactions with the PPO moieties of the Poloxamer. These authors also suggested that such interactions may provide a more favorable environment for hydrophobic drug incorporation, hence the 3:2 ratio was chosen on the basis of both the stability of the micelles, as indicated by the cmc value, as well as the potential for drug incorporation.
In order to provide a baseline for comparison of solubility values for CyA, studies were conducted on the solubility of the drug in a range of solvents as indicated in Table S2. The solubility in water was found to be in good agreement with previously published values (1). The presence of the surfactants above the cmc clearly had a profound effect on the drug solubility, which may be considered to be a positive indication of the propensity for these systems to incorporate the drug.
3.1.2. Measurement of Drug Loading into Micelles
The drug loading and entrapment efficiencies of CyA using different weight ratios of drug to mixed micelles were determined. As shown in Figure 2, as the ratio of drug to micellar content increased (with the drug content being kept constant) from F1 to F6, the entrapment efficiency decreases. This is to be expected as the EE reflects the absolute amount of the drug that is able to be incorporated, hence as the relative drug loading increases (and the surfactant content decreases), the efficiency of entrapment decreases even though the absolute amount of drug increases. At the other extreme, when the ratio of drug to surfactant is very high (F6), the amount loaded will be lower as there is insufficient surfactant present to incorporate the drug effectively (hence a low EE and LC). The F5 (1:5 drug to surfactant ratio) reflects a reasonable compromise between effective incorporation while also maintaining low-surfactant concentration, in turn maximizing the loading in the final dosage form as well as reducing the possibility of issues with irritation or toxicity. This ratio (F5) was therefore used for further studies.
Figure 2.

Encapsulation efficiency (bars) and LC (square symbols) of CyA (weight kept constant at 1 mg) as a function of various weight ratios of mixed micelles composed of surfactant mixture F127/TPGS (3:2 molar ratio).
3.2. Micellar Morphological, Structural, and Thermal Studies
3.2.1. Transmission Electron Microscopy and Dynamic Light Scattering
In order to confirm the size, shape, and homogeneity of the micelles, TEM analysis of unloaded (3:2 F127/TPGS molar ratio) and loaded (F5 ratio of drug to micellar material) samples was performed. TEM micrographic images confirmed that spherical micelles were generated (Figure 3Ia,IIa). Further analysis by ImageJ and the corresponding histogram plots showed that the average size of the empty micelles was in the region of 22.33 nm (Figure 3Ia), while a significant increase in size was seen for drug-loaded micelles (in the range 100–200 nm; Figure 3IIa); micelles of similar size have been reported previously.18,19 More specifically, the size of the CyA-loaded micelles (F5) formulation was measured as 137 ± 7 nm (Figure 3I,IIb). Typically, a nonpolar drug would become incorporated into the hydrophobic core of the micelle, leading to an increase in the core size and aggregation number,20 hence the significant increase in size for the CyA-loaded micelles is in accordance with previous observations. The TEM images further indicate differences in diffraction contrast for the unloaded and drug-loaded micelles, reflecting differences in the thickness and composition of the samples.21
Figure 3.

Size and associated data for (I) unloaded and (II) CyA-loaded (F5) F127/TPGS (3:2) micellar systems. (a) TEM micrographs; (b) corresponding size distribution histograms (where A.D. = average diameter and S.D. = standard deviation); (c) DLS-based histogram with a mean average hydrodynamic size of empty micelles 68 nm and CyA-loaded micelles 180.0 nm; (d) corresponding zeta potentials with means of −3.26 and −8.7 mV, respectively.
Dynamic light scattering (DLS) studies of empty and drug-loaded micelles yielded an average hydrodynamic size of 68 ± 13 nm [polydispersity index (PDI) 0.167 (Figure 3I,IIc,d)] and 180 ± 76 nm (PDI 0.093), which was somewhat higher than the TEM determination but of the same order of magnitude; this may be a function of the low zeta potential what may have led to some degree of aggregation in the suspension prepared for the DLS studies. The PDI values are low (less than 0.2), which indicates that both populations were relatively homogeneous,22 although at the lower size range, the unloaded samples did show a bimodal distribution, possibly reflecting an element of kinetic micellar growth.23 Furthermore, the dilution and sonication stages required to reduce aggregation for the DLS studies may also have contributed to this low value. An increase in overall negative charge was observed on addition of the drug, with the electrokinetic zeta potential value increasing from −3.26 to −8.7 mV.
3.2.2. Elemental Mapping and Energy-Dispersive X-ray Spectrometry Analysis
X-ray-based elemental mapping is a useful technique for the identification of chemical components present in nanocarriers;24 here the technique was used to study freeze-dried samples so as to allow assessment of the chemical composition of the unloaded and CyA-loaded systems. In order to confirm the successful incorporation and dispersion of the drug payload within the micelle, elemental mapping (Figure 4i,ii) and point energy-dispersive X-ray spectrometry (EDS) analysis (Figure 4iii) were performed. As the polymers (TPGS, F127) used for synthesis of the micelles were devoid of any nitrogen elements, the elemental mapping images for the unloaded systems solely indicated the presence of C and O (Figure 4ia–d). In contrast, the CyA drug molecule contains a significant molecular nitrogen component (C62H11N11O12).
Figure 4.

(i) SEM–EDS elemental mapping analysis of unloaded micelles showing (a) reference SEM image, (b) carbon (C), (c) oxygen (O), (d) overlay of all elements present, and (ii) CyA-loaded (F5) micelles including the reference SEM images (a) reference SEM image, (b) carbon (C), (c) oxygen (O), (d) nitrogen (N), and (e) overlay of all elements present, (iii) point EDS analysis of (a) the unloaded mixed micelles (no nitrogen peak detected) and (b) CyA-loaded mixed micelles (nitrogen peak detected as indicated). The inset table gives weight and atomic % information (iii a,b), respectively.
The elemental map of the CyA-loaded micelles clearly indicated the presence of N (Figure 4iia–e). Furthermore, the point EDS for the unloaded micelles further confirmed the absence of the N peak in the spectrum, with elemental recordings of C 64.51% and O 35.49 wt %, respectively, as shown in the inset table (Figure 4iiia; see inset table). In contrast, 13.81 wt % N was found for the CyA-loaded micellar samples (Figure 4iiib; see inset table).
3.2.3. FTIR Spectra of CyA and Drug-Loaded Micelles
FTIR was performed to obtain further insight into the presence and possible interaction between CyA and the constituent micellar surfactants. The representative peaks of CyA were observed from 2600 to 4000 cm–1, corresponding to the −alkyl group (nCH, ∼2850–2950 cm–1) and N–H primary and secondary amine (∼3200–3600 cm–1) stretching modes (Figure 5; Table S3). In addition, the distinctive amine C–N stretching band was observed in the region of ∼1020–1300 cm–1, and the absorption bands at 1630–1680 cm–1 related to amide groups within the molecule.25
Figure 5.

FTIR spectra of CyA and F127/TPGS (3:2)-loaded micelles; peaks inside the dotted circle show peak changes highlighted in the text (a) expanded region 1750–1000 cm–1 and (b) expanded region 3600–2500 cm–1.
On incorporation of the CyA into micelles, changes in the characteristic spectra were observed although it has to be considered that the CyA peaks will not only be obscured by the presence of the F127 and TPGS but will also be of lower intensity given that the drug is a minor component of the system. More specifically, the peaks corresponding to the TPGS carbonyl band were detected at 1739 cm–1 while the backbone C–H stretching bonds are centered at 2949 and 2867 cm–1. The 3400–3650 cm–1 peaks were because of the terminal OH group, while the peak at 1045–1295 cm–1 is attributed to C–O stretching. Of these, the absorption bands from 1105 to 1242 cm–1 are attributed to the characteristic C–O–C stretching vibrations of the repeated −OCH2CH2 units of TPGS.26 The characteristic spectra of F127 showed the absorption peaks at 2884 cm–1 (C–H, stretch, aliphatic), 1342 cm–1 (in-plane O–H bend), and 1111 cm–1 (C–O stretch).27
Figure 5 shows the changes in spectra for the two characteristic regions. As mentioned above, direct comparisons with the spectrum of CyA alone may have limitations, but even so, it is of interest to note the changes in the peak position and breadth which may indicate some degree of molecular interaction with the surfactants. In the 1750–1000 cm–1 spectral region of CyA (Figure 5a), an amide peak shift from 1630 to 1625 cm–1 was observed on loading into micelles, raising the possibility of a hydrogen bonding interaction between the surfactants and the amide group of CyA, supported by the peak broadening observed as well as the change in wavenumber.28,29 A further effect was observed in spectral region 1080–1300 cm–1 with the disappearance of the 1295 and 1265 cm–1 amine peaks of CyA and observation of a new broad peak at 1280 cm–1. Figure 5b shows a similar alteration of the amine and alkyl group peaks in the 3300–2800 cm–1 regions. Overall, therefore, there is evidence to suggest an interaction between the CyA and the surfactants within the micelles; this suggestion is compatible with the marked solubilization effects noted for this mixed micelle system.
3.2.4. XRD and DSC Analysis of Freeze-Dried Drug-Loaded Micelles
The freeze-dried CyA-loaded micelles were characterized by XRD and DSC. XRD analysis enabled identification of the crystalline or amorphous state of CyA in the freeze-dried micellar polymeric dispersion. Diffraction patterns of pure TPGS, F127, and CyA were used as references for the evaluation of various micellar nanoformulations. Reference spectra indicated that CyA existed in a crystalline state before incorporation into the micellar formulations (Figure 6ia); the diffractogram of raw CyA powder showed characteristic high-intensity diffraction peaks at 2θ values of 6.86° (011), 7.8° (102), 9.26° (110), 10.76° (112), 12.58° (022), 14.48° (201), 15.04° (202), 15.2° (210), 16.78° (124), 18.6° (131), 19.38° (132), 20.84° (027), 22.14° (230), 23.28° (042), and 24.28° (119).30,31 Pure F127 also showed two characteristic peaks with the highest intensity at 2θ angles 19.13 and 23.32°, indicating that F127 is a crystalline adjuvant.32 The characteristic peaks 18.12 and 22.32° of TPGS were observed in the diffraction pattern of TPGS.33 The XRD patterns of the polymeric-mixed micelles (F127 and TPGS) and micelles loaded with CyA displayed similar peak locations in XRD patterns, with no obvious peak broadening observed (Figure 6ib). However, the diffraction patterns present in the crystalline drug were absent in the micelles, indicating that the drug was molecularly dispersed in the freeze-dried material. Further incorporation of drug loaded micelles into the amorphous PVP polymer (described in more detail below) showed the micelle-loaded PVP nanofibers to also show no evidence for crystallinity (Figure 6ic).
Figure 6.

(i) Characteristics powder XRD diffraction patterns of (a) crystalline CyA (b) F127, TPGS, unloaded, and CyA-loaded micelles with the reference to the CyA spectrum (c) PVP, unloaded, and CyA incorporated micelle-loaded nanofibrous system with reference to the CyA spectrum; (ii) DSC data for (a) Pluronic F127, TPGS powder, mixed micelles, and drug-loaded micelles (b) CyA and PVP, unloaded micelles in nanofibers (nf), drug-loaded micelles, drug-loaded micelles in nanofibers, (c) higher resolution DSC data showing transitions for CyA in comparison to formulated systems.
These findings were further confirmed by DSC analysis in that the pure CyA showed typical endothermic peaks at 111 and 122 °C,7,30 corresponding to the melting points of the drug, thus reflecting its crystalline nature. Pure PVP showed a broad endothermic peak with an onset of 50 °C, attributed to the release of absorbed water.34 F127 and TPGS 1000 exhibit sharp endothermic melting peaks at Tm of 57.28 and 39.76 °C, respectively, corresponding to the melting of PEG chains (Figure 6iia). The Tm peaks of CyA were absent in the drug-loaded micelles (and nanofiber based solid dosage form) (Figure 6iib,c), whereas all the peaks corresponding to the unloaded polymeric micelles were present, indicating again that CyA was molecularly dispersed within the micelles.
3.3. Fabrication and Characterization of Surfactant/Drug-Loaded Nanofibers
3.3.1. Morphology and Size Analysis of Nanofibers by SEM, TEM, and AFM
Unloaded PVP nanofibers and fibers incorporating the freeze-dried CyA/surfactant material were electrospun as outlined in section 2.8. The morphologies were examined using SEM (Figure 7a–d) combined with ImageJ analysis. SEM clearly showed the formation of smooth, beadless nanofibers for both the unloaded and loaded systems. Size reduction was observed on loading, with the PVP fibers having a mean diameter of 860 ± 195 nm (Figure 7a,c), while the loaded systems had a mean average diameter of 292 ± 97 nm (Figure 7b,d). A possible reason for this significant size reduction is the high proportion of surfactant present which would inevitably decrease the surface tension, one of the most critical factors contributing to nanofabrication.35 More specifically, the surface tension needs to be overcome by the electrical forces which act upon the polymer solution, leading to fine jet ejection from the nozzle, while the lower surface tension will also facilitate greater fiber stretching, enabling the formation of thinner fibers.
Figure 7.

SEM images of the (a) unloaded PVP nanofibers and (b) histogram plot showing average fiber diameter, (c) CyA/surfactant-loaded composite (F5) nanofibers, and (d) corresponding size histogram (where, S.D. = standard deviation; A.D. = average diameter).
In order to investigate the topography further, AFM in peak force tapping mode was utilized. Topographic images of the unloaded (Figure 8a) and loaded nanofibers (Figure 8b) are shown; interestingly, for the latter, there is some evidence of small distinct structures that we tentatively ascribe to small deposits of drug, given the issues that may be evident with incorporation into a nonhydrated, relatively hydrophilic matrix, although more work would be required to ascertain this for certain. The height distributions for the two sets of nanofibers, measured using an AFM depth histogram, are given in Figure S3.
Figure 8.

AFM topographic image showing (a) PVP nanofibers and (b) CyA/surfactant-loaded nanofibers; the height scale is included for the two images and a height distribution analysis given in Figure S3.
Fluorescence-based methods were used to further validate the successful incorporation of micelles into the nanofibers. Coumarin-6, a hydrophobic fluorescence dye, was selected as a surrogate for the CyA; it was observed that the nanofibers incorporating micelles loaded with coumarin-6 showed bright green fluorescence under the green fluorescence protein filter compared to the control nanofibers which showed no fluorescence signal, confirming the successful incorporation of micelles into the nanofibers (Figure S4a,c;b,d).
3.4. Release Profile and Wettability Analysis of CyA/Micelle-Loaded Nanofibers
3.4.1. In Vitro Drug Release Study
Drug solubility is an equilibrium measure but is a key determinant of the dissolution rate, itself an indication of absorption for many drugs including CyA, hence a central objective of the current work is to determine the dissolution profile of the nanofiber formulations compared to the drug alone. For dissolution testing under sink conditions (for the nanofiber systems), samples equivalent to 200 μg of CyA were used. Drug release profiles for the CyA/micelle incorporated nanofibers (F5) were compared with those from the corresponding quantity of pure crystalline CyA (Figure 9). After 14 min, drug release from the nanofiber was found to be complete, whereas there was negligible dissolution from the pure drug. The remarkable improvement in the dissolution rate for CyA via incorporation into nanofibers can be ascribed to a number of mechanisms; these include solubilization, high surface area, improved wetting, and molecular dispersion of the drug. Irrespective of the mechanism, it is clear that there is a significant dissolution advantage for the nanofiber system. A range of mathematical models have been applied to the data, and these are summarized in Table S4; while good fits were noted the complexity and multiplicity of the mechanisms involved in the dissolution from the mixed surfactant/polymer system do render such analyses of limited value, and hence they are included for completeness rather than because they necessary lend insights into the processes involved. Nevertheless, at this early stage of the development of this technology, these results are highly encouraging in relation to the possibility of improved in vivo bioavailability, given that for BCS class II and quite possibly class IV drugs, an enhancement in dissolution may be expected to result in a more rapid uptake into the bloodstream when dissolution in the gastrointestinal tract is the rate-limiting step to absorption.
Figure 9.
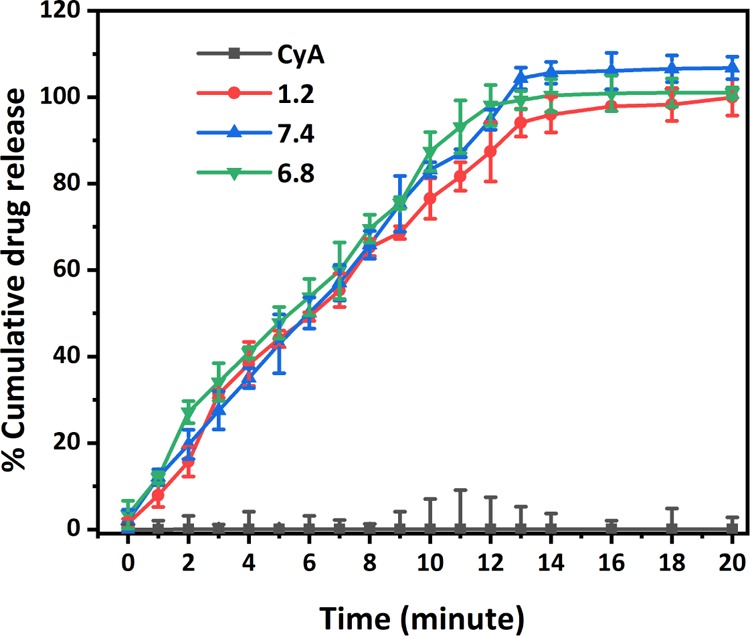
Dissolution profiles of CyA from CyA/micelle-loaded nanofibers at different pH values (1.2, 0.1 N HCl, 6.8, PBS, and 7.4, PBS) in comparison to the drug alone. The release for the nanofibrous systems after 14 min represents complete dissolution of the 200 μg of CyA.
3.4.2. Contact Angle Analysis
As mentioned above, the purpose of this study was to examine whether a combination of micellar, nanofiber, and solid dispersion technologies could provide a novel means of significantly enhancing dissolution and by implication bioavailability. The dissolution data would certainly support the approach; however there may be a number of mechanisms involved including surface area, drug dispersion, solubilization effects, and wetting effects. While it is likely that all of these are contributing, we have explored the first three of these factors in the preceding study and have noted that all three may be positively influenced by our formulation approach. However, it is well known that wettability may also be a major determinant in dissolution of pharmaceuticals,36 hence in order to explore whether the wettability may also play a role, we perform a study to establish whether the contact angle may be reduced via formulation into the nanofibers.
It was observed that CyA presents a contact angle of 113°, reflecting its highly hydrophobic nature, in contrast to the negligible contact angle shown by PVP (Figure 10). Incorporation of CyA into the micelles leads to an increase in contact angle compared to the unloaded material, reflecting the presence of highly hydrophobic drug, although the value is much smaller than for the CyA alone. The data therefore indicate that the incorporation of the CyA into the micellar and nanofiber systems leads to a marked decrease in contact angle, which may contribute to the observed enhancement in dissolution.
Figure 10.

Contact angle analyses via the sessile drop-casting method (a) CyA powder with contact angle 113°, (b) PVP powder with contact angle 5°, (c) unloaded micellar freeze-dried powder with contact angle 27°, (d) CyA loaded micelles with contact angle 39° (freeze dried powder), (e) nanofiber without micelles and (f) CyA-loaded micelle incorporated nanofibers with contact angle 23°, respectively. The upper structure seen on the images is the needle used for drop-casting.
Overall, therefore, the very significant increases in the dissolution rate observed may be reasonably ascribed to a combination of surface area, solubilization, molecular drug dispersion, and wetting effects, demonstrating that the technology described here presents multiple mechanisms of dissolution enhancement.
4. Conclusions
The study has indicated that the highly hydrophobic peptide, CyA, may be successfully formulated for enhanced dissolution using a combination strategy of first incorporating into polymeric micelles followed by further incorporation into nanofibers, using PVP as water miscible polymeric matrix materials. More specifically, mixed micelles composed of TPGS and Pluronic F127 were found to provide a suitable environment for CyA incorporation both in terms of cmc and drug incorporation, thereby presenting an initial, liquid-based system for delivery. However, in order to subsequently prepare the drug system as a solid, with advantages of surface area and solid dispersion formulation, the micellar systems were spun into nanofibers using PVP as a base. It was noted that fibers with suitable diameter and surface integrity were formed with greatly improved dissolution compared to the drug alone. Further advantages of the system include the use of materials with GRAS status and also a relatively high loading of CyA, which in turn has positive implications for the dosage requirements. The study has, therefore, indicated that by using this dual mechanism it is possible to produce solid systems that clearly enhance the dissolution, and potentially the bioavailability, of CyA. Furthermore, given the increasing number of high molecular weight and low solubility drugs that are currently in the pipeline, this technology provides a potential platform for the development of fast-dissolution solid systems for drugs, which provide particular challenges using conventional techniques.
Acknowledgments
The authors wish to thank the Interreg 2 Seas Programme 2014-2020 co-funded by the European Regional Development Fund under subsidy contract 2S01-059_IMODE for financial support for P.D. We also wish to thank the London Centre for Nanotechnology for providing research facilities for successful completion of the AFM and contact angle measurement.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b02616.
CyA calibration curve obtained by HPLC; pyrene fluorescence spectra in the absence and presence of micelles; table showing effect of surfactant composition on cmc; table showing equilibrium solubility of CyA as a function of surfactant concentration and media composition; table showing FTIR spectral information for CyA and CyA-loaded micelles; height distribution for the unloaded and CyA/micelle-loaded nanofibers measured using AFM; fluorescence microscopic imaging showing coumarin-6 loaded micelles incorporated into nanofibers; table showing kinetic models studied for CyA release from micelle-loaded nanofibers (PDF)
Author Present Address
† Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Kent ME4 4TB, United Kingdom.
The authors declare no competing financial interest.
Supplementary Material
References
- Guada M.; Beloqui A.; Kumar M. N. V. R.; Préat V.; Dios-Viéitez M. D. C.; Blanco-Prieto M. J. Reformulating Cyclosporine A (CsA): More than Just a Life Cycle Management Strategy. J. Controlled Release 2016, 225, 269–282. 10.1016/j.jconrel.2016.01.056. [DOI] [PubMed] [Google Scholar]
- Jain S.; Kambam S.; Thanki K.; Jain A. K. Cyclosporine A Loaded Self-Nanoemulsifying Drug Delivery System (SNEDDS): Implication of a Functional Excipient Based Co-Encapsulation Strategy on Oral Bioavailability and Nephrotoxicity. RSC Adv. 2015, 5, 49633–49642. 10.1039/c5ra04762e. [DOI] [Google Scholar]
- Yu H.; Xia D.; Zhu Q.; Zhu C.; Chen D.; Gan Y. Supersaturated Polymeric Micelles for Oral Cyclosporine A Delivery. Eur. J. Pharm. Biopharm. 2013, 85, 1325–1336. 10.1016/j.ejpb.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Xia D.; Yu H.; Tao J.; Zeng J.; Zhu Q.; Zhu C.; Gan Y. Supersaturated Polymeric Micelles for Oral Cyclosporine A Delivery: The Role of Soluplus-Sodium Dodecyl Sulfate Complex. Colloids Surf., B 2016, 141, 301–310. 10.1016/j.colsurfb.2016.01.047. [DOI] [PubMed] [Google Scholar]
- Tam J. M.; McConville J. T.; Williams R. O.; Johnston K. P. Amorphous Cyclosporin Nanodispersions for Enhanced Pulmonary Deposition and Dissolution. J. Pharm. Sci. 2008, 97, 4915–4933. 10.1002/jps.21367. [DOI] [PubMed] [Google Scholar]
- Aliabadi H. M.; Brocks D. R.; Lavasanifar A. Polymeric micelles for the solubilization and delivery of cyclosporine A: pharmacokinetics and biodistribution. Biomaterials 2005, 26, 7251–7259. 10.1016/j.biomaterials.2005.05.042. [DOI] [PubMed] [Google Scholar]
- Grimaudo M. A.; Pescina S.; Padula C.; Santi P.; Concheiro A.; Alvarez-Lorenzo C.; Nicoli S. Poloxamer 407/TPGS Mixed Micelles as Promising Carriers for Cyclosporine Ocular Delivery. Mol. Pharm. 2018, 15, 571–584. 10.1021/acs.molpharmaceut.7b00939. [DOI] [PubMed] [Google Scholar]
- Fülöp G.; Balogh A.; Farkas B.; Farkas A.; Szabó B.; Démuth B.; Borbás E.; Nagy Z. K.; Marosi G. Homogenization of Amorphous Solid Dispersions Prepared by Electrospinning in Low-Dose Tablet Formulation. Pharmaceutics 2018, 10, 114. 10.3390/pharmaceutics10030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holan V.; Chudickova M.; Trosan P.; Svobodova E.; Krulova M.; Kubinova S.; Sykova E.; Sirc J.; Michalek J.; Juklickova M.; Munzarova M.; Zajicova A. Cyclosporine A-Loaded and Stem Cell-Seeded Electrospun Nanofibers for Cell-Based Therapy and Local Immunosuppression. J. Controlled Release 2011, 156, 406–412. 10.1016/j.jconrel.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Sirc J.; Hampejsova Z.; Trnovska J.; Kozlik P.; Hrib J.; Hobzova R.; Zajicova A.; Holan V.; Bosakova Z. Cyclosporine A Loaded Electrospun Poly(D,L-Lactic Acid)/Poly(Ethylene Glycol) Nanofibers: Drug Carriers Utilizable in Local Immunosuppression. Pharm. Res. 2017, 34, 1391–1401. 10.1007/s11095-017-2155-x. [DOI] [PubMed] [Google Scholar]
- Marano S.; Barker S. A.; Raimi-Abraham B. T.; Missaghi S.; Rajabi-Siahboomi A.; Craig D. Q. M. Development of micro-fibrous solid dispersions of poorly water-soluble drugs in sucrose using temperature-controlled centrifugal spinning. Eur. J. Pharm. Biopharm. 2016, 103, 84–94. 10.1016/j.ejpb.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt A. M.; Amin M. C. I. M.; Katas H.; Sarisuta N.; Witoonsaridsilp W.; Benjakul R. In Vitro Characterization of Pluronic F127 and D--Tocopheryl Polyethylene Glycol 1000 Succinate Mixed Micelles as Nanocarriers for Targeted Anticancer-Drug Delivery. J. Nanomater. 2012, 2012, 1–11. 10.1155/2012/916573. [DOI] [Google Scholar]
- Meng X.; Liu J.; Yu X.; Li J.; Lu X.; Shen T. Pluronic F127 and D-α-Tocopheryl Polyethylene Glycol Succinate (TPGS) Mixed Micelles for Targeting Drug Delivery across The Blood Brain Barrier. Sci. Rep. 2017, 7, 2964. 10.1038/s41598-017-03123-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samprasit W.; Akkaramongkolporn P.; Ngawhirunpat T.; Rojanarata T.; Kaomongkolgit R.; Opanasopit P. Fast Releasing Oral Electrospun PVP/CD Nanofiber Mats of Taste-Masked Meloxicam. Int. J. Pharm. 2015, 487, 213–222. 10.1016/j.ijpharm.2015.04.044. [DOI] [PubMed] [Google Scholar]
- Xu X.; Gupta A.; Faustino P.; Sathe P. M.; Sayeed V. A.; Khan M. A. Development and Validation of a HPLC Method for Dissolution and Stability Assay of Liquid-Filled Cyclosporine Capsule Drug Products. AAPS PharmSciTech 2013, 14, 959–967. 10.1208/s12249-013-9983-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K.; Oda N.; Miyoshi K.; Funaki T.; Ida Y. Solubilization Behavior of a Poorly Soluble Drug under Combined Use of Surfactants and Cosolvents. Eur. J. Pharm. Sci. 2006, 28, 7–14. 10.1016/j.ejps.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Mitsionis A. I.; Vaimakis T. C. Estimation of AOT and SDS CMC in a Methanol Using Conductometry, Viscometry and Pyrene Fluorescence Spectroscopy Methods. Chem. Phys. Lett. 2012, 547, 110–113. 10.1016/j.cplett.2012.07.059. [DOI] [Google Scholar]
- Cai Y.; Sun Z.; Fang X.; Fang X.; Xiao F.; Wang Y.; Chen M. Synthesis, characterization and anti-cancer activity of Pluronic F68-curcumin conjugate micelles. Drug Deliv. 2016, 23, 2587–2595. 10.3109/10717544.2015.1037970. [DOI] [PubMed] [Google Scholar]
- Brandt J. V.; Piazza R. D.; Dos Santos C. C.; Vega-Chacón J.; Amantéa B. E.; Pinto G. C.; Magnani M.; Piva H. L.; Tedesco A. C.; Primo F. L.; Jafelicci M.; Marques R. F. C. Synthesis and colloidal characterization of folic acid-modified PEG-b-PCL Micelles for methotrexate delivery. Colloids Surf., B 2019, 177, 228–234. 10.1016/j.colsurfb.2019.02.008. [DOI] [PubMed] [Google Scholar]
- Rangel-Yagui C. O.; Pessoa A.; Tavares L. C. Micellar Solubilization of Drugs. J. Pharm. Pharm. Sci. 2005, 8, 147–165. [PubMed] [Google Scholar]
- Fultz B.; Howe J. M.; Fultz B.; Howe J. M.. Diffraction Contrast in TEM Images. Transmission Electron Microscopy and Diffractometry of Materials, 3rd ed.; Springer: New York, 2002; pp 377–422. [Google Scholar]
- Danaei M.; Dehghankhold M.; Ataei S.; Hasanzadeh Davarani F.; Javanmard R.; Dokhani A.; Khorasani S.; Mozafari M. R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. 10.3390/pharmaceutics10020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley E. G.; Murphy R. P.; Seppala J. E.; Smart T. P.; Hann S. D.; Sullivan M. O.; Epps T. H. Size evolution of highly amphiphilic macromolecular solution assemblies via a distinct bimodal pathway. Nat. Commun. 2014, 5, 3599. 10.1038/ncomms4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbury D. E.; Ritchie N. W. M. Elemental Mapping of Microstructures by Scanning Electron Microscopy-Energy Dispersive X-Ray Spectrometry (SEM-EDS): Extraordinary Advances with the Silicon Drift Detector (SDD). J. Anal. At. Spectrom. 2013, 28, 973–988. 10.1039/c3ja50026h. [DOI] [Google Scholar]
- Bertacche V.; Pini E.; Stradi R.; Stratta F. Quantitative Determination of Amorphous Cyclosporine in Crystalline Cyclosporine Samples by Fourier Transform Infrared Spectroscopy. J. Pharm. Sci. 2006, 95, 159–166. 10.1002/jps.20523. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Wu Y.; Chu Q.; Tan S.; Zhuang X.; Bao Y.; Wu T. D-α-Tocopherol Polyethylene Glycol Succinate-Based Derivative Nanoparticles as a Novel Carrier for Paclitaxel Delivery. Int. J. Nanomed. 2015, 10, 5219–5235. 10.2147/ijn.s82847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Mohan C.; Uma Shankar M. K. S.; Gulati M. Physiochemical Characterization and Release Rate Studies of Solid Dispersions of Ketoconazole with Pluronic F127 and PVP K-30. Iran. J. Pharm. Res. 2011, 10, 685–694. [PMC free article] [PubMed] [Google Scholar]
- Coates J.Interpretation of Infrared Spectra, A Practical Approach. Encyclopedia of Analytical Chemistry; John Wiley & Sons, Ltd., 2006; pp 1–23. [Google Scholar]
- Saliba J. B.; Silva-Cunha Junior A. d.; Silva G. R. d.; Yoshida M. I.; Mansur A. A. P.; Mansur H. S. Characterization and in vitro release of cyclosporine-A from poly(D,L-lactide-co-glycolide) implants obtained by solvent/extraction evaporation. Quim. Nova 2012, 35, 723–727. 10.1590/s0100-40422012000400013. [DOI] [Google Scholar]
- Hwang S.-J.; Karn P. R.; Jin S.-E.; Lee B. J.; Sun B. K.; Kim M.-S.; Sung J.-H. Preparation and Evaluation of Cyclosporine A-Containing Proliposomes: A Comparison of the Supercritical Antisolvent Process with the Conventional Film Method. Int. J. Nanomed. 2014, 9, 5079–5091. 10.2147/ijn.s70340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.; Lei Y.; Lu Y.; Qi J.; Nie S.; Hu F.; Pan W. Solid self-nanoemulsifying cyclosporin A pellets prepared by fluid-bed coating: preparation, characterization and in vitro redispersibility. Int. J. Nanomed. 2011, 6, 795–805. 10.2147/ijn.s17711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q.; Zhang Z.; Chen T.; Guo X.; Zhou S. Preparation and characterization of thermosensitive pluronic F127-b-poly (ε-caprolactone) mixed micelles. Colloids Surf., B 2011, 86, 45–57. 10.1016/j.colsurfb.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Cho H.-J.; Lee J.-Y.; Kang W.-S.; Piao J.; Yoon I.-S.; Kim D.-D. Soluplus/TPGS-based solid dispersions prepared by hot-melt extrusion equipped with twin-screw systems for enhancing oral bioavailability of valsartan. Drug Des., Dev. Ther. 2015, 9, 2745–2756. 10.2147/dddt.S84070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; Jain C. P. Preparation and characterization of solid dispersions of carvedilol with PVP K30. Res. Pharm. Sci. 2010, 5, 49–56. 10.4067/S0717-97072013000100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.; Myung N. V. Minimizing the Diameter of Electrospun Polyacrylonitrile (PAN) Nanofibers by Design of Experiments for Electrochemical Application. Electroanalysis 2018, 30, 2330–2338. 10.1002/elan.201800368. [DOI] [Google Scholar]
- Yang B.; Xu L.; Wang Q.; Li S. Modulation of the wettability of excipients by surfactant and its impacts on the disintegration and release of tablets. Drug Dev. Ind. Pharm. 2016, 42, 1945–1955. 10.1080/03639045.2016.1185436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.