

Abstract
The Staudinger reduction and its variants have exceptional compatibility with live cells, but can be limited by slow kinetics. Here, we report novel small molecule triggers that turn on proteins via a Staudinger reduction/self-immolation cascade with substantially improved kinetics and yields. We achieved this through site-specific incorporation of a new set of azidobenzyloxycarbonyl lysine derivatives in mammalian cells. This approach allowed us to activate proteins until adding a nontoxic, bioorthogonal phosphine trigger. We applied this methodology to control a post-translational modification (SUMOylation) in live cells and using native modification machinery. This work significantly improves the rate, yield, and tunability of the Staudinger reduction-based activation, paving the way for its application in other proteins and organisms.
Keywords: amino acids, protecting groups, protein engineering, protein modifications, SUMO
Graphical Abstract
Wrestling SUMO onto proteins: We report three genetically-encoded groups to activate protein function through an optimized phosphine-based small molecule trigger. We demonstrate rapid and tunable control of protein SUMOylation and protein localization.
Introduction
Conditional control of protein function is essential to study the role that proteins play in the dynamic environment of a living cell. Pharmacological inhibitors are commonly used for this approach; while they work quickly and are usually reversible,[1] available drugs can access only a limited portion of the proteome,[2] typically can only deactivate protein function, and often have off-target effects.[1,3] Other techniques, including bump-and-hole approaches,[4–6] chemically induced dimerization[7] and dissociation,[8] and controlled degradation[9,10] have expanded conditional control over protein function. Here, we present an optimized and generally applicable approach that does not require small molecule ligand discovery, molecular engineering of the ligand, or protein of interest fusions, while enabling rapid OFF to ON switching of protein function with complete specificity.
Taking advantage of recent progress in bioorthogonal chemistry,[11] unnatural amino acids (UAAs)[12] have proven to be generalizable, site-specific switches for protein function. The UAA mutagenesis approach requires minimal engineering of the protein of interest; when the proper orthogonal translational machinery is provided, UAAs are incorporated simply by mutating the codon at the desired position to the amber stop codon.[13] UAAs allow investigators to site-specifically insert new chemical functionalities into proteins, thereby enabling small molecule control in live biological systems.[14–21] For example, palladium complexes can trigger protein function using propargylated or allylated lysine[17] and allenyl-tyrosine.[18] Additionally, tetrazine-mediated triggering of UAAs masked with trans-cyclooctene (TCO) moieties provides another clever approach to conditional control over protein function.[19] While Pd-catalyzed depropargylation and deallylation is generalizable and highly bioorthogonal, modest protein deprotection yields (≤50%) and slow kinetics (2–3 h)[17,18] limit usability for certain applications.[19] Tetrazine-mediated deprotection of TCOs is significantly faster, with rate constants exceeding 105 to 106 m−1 s−1 for optimized reagents. TCOs, however, isomerize to unreactive cis-cyclooctenes during storage or in the presence of physiologic copper[22] and thiols,[19] though half-lives on the order of hours to days are typical under physiologic conditions.[22,23] In a recent report, the Fox group demonstrated that faster TCOs could be protected from isomerization by complexation with silver nitrate, albeit with slightly reduced bioconjugation yields (possibly due to cellular instability of the particular tetrazine tested in this work).[23] Tetrazines face a trade-off between fast reaction kinetics and aqueous stability,[24] and thus must be chosen carefully.[25,26] Overall, while small-molecule triggered protein activation via UAA mutagenesis has proven useful, further development of UAAs that provide enhanced biorthogonality and/or enhanced activation kinetics while also allowing complete protein activation are needed.
Among bioorthogonal reactions, the Staudinger reduction of aryl azides by phosphines is highly compatible with live cells and organisms.[27,28] Both functional groups are abiotic. The azide is a soft electrophile that reacts preferentially with soft nucleophiles (e.g., phosphines), so the reactive pair is chemically insulated from biotic nucleophiles and electrophiles, most of which are hard.[29] Successful use of azides in photocrosslinking experiments,[30,31] glycan labeling,[25] and in pharmaceuticals[29] indicates stability and nontoxicity in physiological settings. Triaryl phosphines have also proven themselves to be safe for use in live cells and animals,[32] and cytotoxicity of phosphines is negligible below ~1 mm.[16]
Like all bioorthogonal reactions, however, the Staudinger reduction has its drawbacks. Endogenous thiols may reduce certain alkyl-[33] and aryl-azido[34] UAAs, though ambiguities remain about whether this occurs in cells or during purification and analysis.[34] Phosphines, on the other hand, are susceptible to air oxidation (and possibly metabolic oxidation[29]),[35] particularly when exposed to light (UV-Vis).[36] Despite these limitations, our group recently developed the protected lysine derivative OABK as a means of using the Staudinger reduction to turn on proteins.[16] OABK converts to lysine via self-immolation[37,38] through a 1,4-elimination-decarboxylation sequence once the phenyl azide is reduced to an aniline (Figure 1). This reaction, however, is slow (rate constants for the rate-determining[16] 1,4-elimination step are typically 10−4 to 10−3 s−1), and we observed persistence of the aniline species after the Staudinger reduction via LC-MS. Indeed, we observed a t½ value of 67 min for deprotecting OABK under biologically-relevant conditions, and t½ values for activating protein function in live cells were 98–118 min using this approach.[16] Slow kinetics represent a significant disadvantage of this initial work, which limited our ability to study proteins that act on a minute timescale.
Figure 1.

UAAs for improved Staudinger reduction-triggered protein activation in HEK293T cells. A) Structures of UAAs and the small molecule trigger 2DPBM, and a schematic of the phosphine-triggered protein activation. B) Fluorescence microscopy demonstrating UAA-dependent incorporation of PABK and MPABK (both 0.5 mm) into the mCherry-TAG-EGFP-HA reporter in HEK293T cells. C) UAA-dependent expression of the full-length protein construct was further confirmed by Western blot using an anti-HA antibody.
Thus, we developed second-generation protected lysine analogs with improved elimination kinetics, and we demonstrate that these analogues deprotect much faster and in several fold higher yield than OABK. In mammalian cells, these new UAAs reduce the time needed to conditionally trigger nuclear translocation via the Staudinger reduction by up to 81%. Furthermore, we applied these UAAs to the small molecule-control of SUMOylation, an important post-translational modification, which allowed us to visualize the functional consequences of the modification in live cells.
Results and Discussion
We hypothesized that we could improve on OABK’s deprotection kinetics by making two changes: (1) relocating the azide from the ortho to the para position, which has been reported to accelerate the self-immolation reaction of related substrates,[38,39] and (2) methylating the benzylic position to stabilize a transient (partially) positive charge here by increasing the electron density at this position.[38,40] We expected that combining these two modifications would further accelerate the amino acid deprotection and thereby protein activation. The corresponding UAAs PABK, MOABK, and MPABK (Figure 1A) were prepared by coupling Fmoc-Lysine to the appropriate NHS carbonates, followed by removal of the Fmoc group (see Supporting Information Schemes S1–S3). We determined the deprotection yield and kinetics of each UAA by LC-MS (Figure S1). Fmoc-protected analogues of each UAA (100 μm) were treated with stoichiometric amounts of 2-(diphenylphosphino)benzamide (2DPBM) in 8:2 PBS:DMSO (v/v) at room temperature.[41] 2DPBM, in our hands, activates OABK[16] and other aryl azides[42] the fastest and with the highest yield, as we determined by screening a panel of phosphines. All three second-generation UAAs deprotected in approximately 10-fold improved yield over OABK, and t½ values of 10.5, 17.7, and 30.0 min were observed for MPABK, PABK, and MOABK, respectively (Figure S1B). These results suggest that moving the azide to the para position has a stronger effect than benzylic methylation, and combining both modifications further improves the kinetics. Consumption of starting material occurred at similar rates in each case (Figure S1A), which is consistent with self-immolation and not the Staudinger reduction itself as the rate-limiting step.[16] As expected, the aniline intermediate was observed in high levels for OABK throughout the experiment. Moderate levels of this intermediate were observed for MOABK (which leveled off as the reaction progressed), while only trace levels were detected for the para-substituted isomers (data not shown). Because of the stoichiometric conditions used in this assay, a small amount of phosphine oxidation prevented full consumption of starting material. This was easily overcome in cellular experiments by using excess phosphine.
To genetically encode the UAAs, we screened a panel of pyrrolysyl-tRNA synthetase (PylRS) mutants in E. coli (see Supporting Information Table S2) using an sfGFP-Y151TAG reporter to identify mutants accepting the UAAs as substrates (Figure S2). The Y271A-Y349F double mutant (previously termed OABKRS)[16,43–45] was the most efficient synthetase for incorporating MPABK (Figure S2A). This is one of the most versatile PylRS mutants for incorporating sterically demanding lysine derivatives;[16,43–45] the Y349F mutation increases aminoacylation kinetics and yield,[43] while Y271A exposes a deep hydrophobic pocket in the PylRS catalytic site that accommodates benzyl carbamates.[43,45] This synthetase incorporated MOABK as well (Figure S3), but produced only modest protein yields with PABK (Figure S2A).[46] A PylRS mutant with L274A, C313A, and Y349F mutations was significantly more efficient at accepting PABK as a substrate (Figure S2B) and was termed PABKRS.[15] OABKRS prefers ortho- and meta-substituted Nϵ-Cbz-Lys derivatives over para-substituted substrates;[46] PABK was no exception to this rule (Figure S2A). To better understand OABKRS’s efficient incorporation of MPABK, as well as the binding modes of PABK, MOABK, and MPABK to their respective synthetases, we conducted a docking study using AutoDock Vina (see Supporting Information). Modeling revealed that MPABK’s side chain is kinked such that the benzylic methyl group fits into a space at the top of the tunnel, and the para-azido substituent is accommodated facing outward in the pocket toward D373 (Figure S4A and D), nearly identically to the azide in a published co-crystal structure of meta-azido-Nϵ-Cbz-Lys in OABKRS.[46] PABK’s azide group is positioned similarly. For MOABK, however, the azide group is not accommodated in this highly charged portion of the pocket and instead projects upward (Figure S4B and E). MOABK’s less snug fit within the enzyme likely explains the lower incorporation efficiency observed for this UAA (Figure S3A). For both OABKRS substrates, the α-carboxylate group is tucked into the tunnel away from ATP’s α-phosphate group, which is a known binding mode for Nϵ-Boc-Lys,[43] a highly efficient PylRS substrate.[47,48] PABKRS, in contrast to OABKRS, contains a C313A mutation toward the far end of the active site tunnel and an L274A mutation instead of OABKRS’s Y271A mutation at the rear of the distal hydrophobic pocket. The C313A mutation, which has been evolved for encoding other Nϵ-Cbz-Lys derivatives with electron-withdrawing para-substituents,[49] creates space in the back of tunnel so the side chain can contort backward to once again position the azide in the anterior portion of the hydrophobic pocket (Figure S4C and F). The L274A mutation appears to be an alternative to OABKRS’s Y271A mutation for expanding this pocket to accommodate the sterically-demanding benzyl substituent.[43] Overall, our docking study shows that the second-generation azidobenzyl lysine analogues in this work are likely to bind their respective PylRS mutants via well-validated binding modes, ultimately affording unnatural proteins in practical yields.
Using OABKRS, sfGFP-Y151MPABK was produced in 5.6 mg L−1 yield using standard LB media, while sfGFP-Y151MOABK expression had a lower yield of 1.4 mg L−1 culture (Figure S3A). PABKRS enabled production of sfGFP-Y151PABK at a yield of 8 mg L−1 culture (non-modified sfGFP was produced in 27 mg L−1 culture yield under identical conditions). Negligible protein expression was observed in the absence of the UAA using either synthetase. Successful incorporation of all three UAAs was confirmed by ESI-MS, which showed neither free lysine nor reduction to the aniline intermediate (Figure S3B).[33,34]
We next tested incorporation of these UAAs into proteins in mammalian cells. MOABK was surprisingly found to be highly cytotoxic (data not shown), and was excluded from further analysis as its activation kinetics were also inferior to the para-azido analogs (Figure S1B) and its incorporation efficiency was lower as well (Figure S3A). Thus, PABK and MPABK were selected for further study. We doubly-transfected HEK293T cells with two plasmids containing (1) the PylRS mutants and four copies of PylT, and (2) an mCherry-TAG-EGFP-HA reporter (Figure 1B).[16] Epifluorescence imaging of cells transfected with these plasmids showed UAA-dependent EGFP expression, whereas the truncated protein (mCherry alone) is expressed in both the presence and absence of UAAs (Figure 1B). UAA-dependent expression of the full-length fusion construct was confirmed by detecting the HA epitope tag by Western blot (Figure 1C). As in bacteria, incorporation efficiency in mammalian cells was higher for PABK than for MPABK, as observed from the higher EGFP:mCherry fluorescence ratio (Figure 1B) and higher HA:β-actin density ratio (Figure 1C). Negligible protein was produced in the absence of MPABK.
Having established that MPABK and PABK can be genetically encoded in mammalian cells, we next tested how rapidly these UAAs could control biological processes in live cells. To this end, we monitored nuclear translocation using a validated fluorescent reporter.[16,50] This reporter comprises a nuclear localization sequence (NLS) derived from SATB1 that contains an amber stop codon for the crucial lysine residue (K29)[51] and is flanked by two fluorescent proteins, N-terminal EGFP and C-terminal mCherry (Figure 2). We incubated cells transfected with this reporter with MPABK, PABK, and OABK (0.5 mm) for 40 h, then removed the excess UAA with a 1 h wash step using fresh media. After acquiring baseline images, cells were treated with 2DPBM at the limit of solubility in aqueous media (100 μm), and imaged for 205 min. Translocation was complete after 50 min, 75 min, and 195 min for MPABK, PABK, and OABK, respectively, with t½ values of 18.0, 26.7, and 94.4 min. Nuclear translocation was markedly faster for the new small molecule switches MPABK and PABK over OABK. Comparing MPABK and PABK in the 15–50 min range shows a statistically significant enhancement in case of MPABK, recapitulating the trend observed in the aforementioned LC-MS assay (Figure S1). Importantly, the fusion protein translocated completely to the nucleus, demonstrating that 2DPBM treatment leads to quantitative deprotection of protein in live cells. Based on literature reports, multiple pulses of excess phosphines may be necessary for complete Staudinger reduction to release prodrugs in cell culture,[52] but this appears to be unnecessary for our present optimized pairs (2DPBM and (M)PABK). Overall, PABK and MPABK offer a 61–81% improvement in t½ compared to OABK. The kinetics of both MPABK and PABK activation with 2DPBM rivals optical triggering of nuclear import, and these UAAs are a suitable alternative for controlling highly dynamic cellular processes.[16] Overall, MPABK and PABK have improved the kinetics of small molecule triggering via the Staudinger reduction beyond the best previously available tool. Though both are faster than existing tools, these UAAs face a trade-off between speed (where MPABK excels) and incorporation efficiency (PABK), and the proper UAA should be selected based on the research question. After this initial comparison of the UAAs, we tested a range of 2DPBM concentrations with PABK (Figure 2D). Concentrations as low as 10 μm induced a detectable translocation response. Further, both the rate and extent of nuclear translocation were tunable by varying the 2DPBM concentration in the 10–100 μm range. The t½ can be varied from 27–181 min using this system, allowing for tuning of temporal control over nearly an order of magnitude. Thus, this approach gives a high degree of control over protein function and can be used as a finely tuned dial instead of a mere OFF to ON switch.
Figure 2.

A) 2DPBM-triggered activation of the SATB1 NLS fused to EGFP and mCherry. B) Images of a representative cell expressing mCherry-SATB1-K29PABK-EGFP. At baseline, the full-length fusion construct is completely excluded from the nucleus, but it rapidly and quantitatively translocates to the nucleus upon phosphine treatment. Truncated protein (EGFP-SATB1ΔC, as the result of incomplete stop codon suppression) is small enough to diffuse through nuclear pores spontaneously and labels the entire cell. C) Nuclear translocation kinetics for mCherry-SATB1-EGFP with SATB1-K29 mutated to OABK, PABK, and MPABK. At t = 0, media was changed to DMEM containing 2DPBM (100 μm), and nuclear mCherry signal intensity (In) as a fraction of total mCherry intensity (nuclear + cytosolic, In + Ic) was quantified every five minutes. Mean ±SD (n = 3; from different fields of view) is shown along with four-parameter logistic curve models. *, significant difference between MPABK and PABK (2-way repeated measures ANOVA with Tukey’s post-hoc test, α = 0.05). D) Concentration-dependent activation of SATB1-mediated nuclear import with 2DPBM demonstrated that the kinetics and the extent of nuclear translocation are tunable. Mean ±SD (n = 3; from different fields of view) is shown.
After establishing that our second-generation UAAs enable rapid, tunable triggering of protein function in live mammalian cells, we sought to evaluate them as tools for controlling post-translational modifications. To this end, we chose to study Small Ubiquitin-Like Modifier (SUMO), which has been implicated in cancer, neurodegeneration, and the response to oxidative stress and heat shock.[53] Despite only sharing 18% sequence homology with ubiquitin, NMR structures of SUMO1 and ubiquitin are nearly identical,[54] though the surface charge distribution differs significantly.[55] SUMOylation affects protein function in a variety of ways, including localization and stability.[55] While several approaches for site-specifically SUMOylating proteins have arisen,[28,56–60] and several investigators have globally controlled SUMOylation[61,62] or turned it off at specific sites by mutating the acceptor lysine to arginine,[62–64] conditional turn-on of native SUMOylation sites in live cells with wild-type (WT) SUMO1 has not been reported to the best of our knowledge.
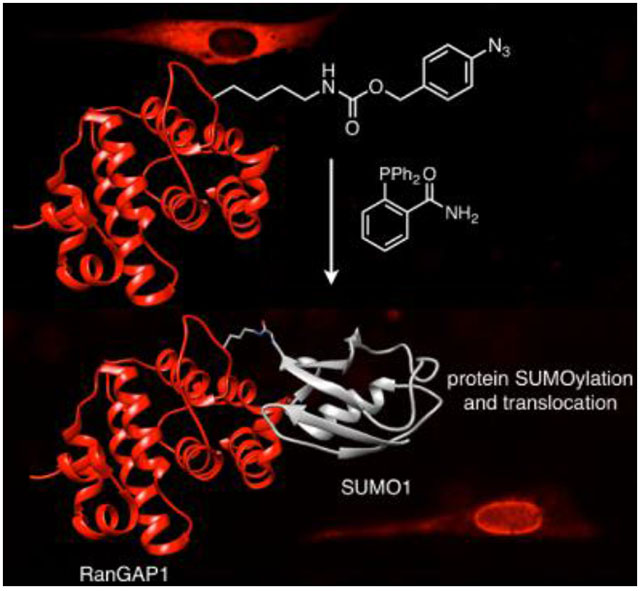
We chose Ran GTPase Activating Protein 1 (RanGAP1),[65] a key player in nucleocytosolic transport, as a proof-of-concept target. RanGAP1 exists in the cytosol in its unmodified form,[66] but is rapidly and efficiently SUMOylated,[67,68] inducing translocation to the nuclear pore complex (NPC), where it forms a ternary complex with RanBP2 and Ubc9[65,69–71] and participates in nuclear protein import.[66] Mutating the substrate lysine (K526 or K524 in the murine and human homologs, respectively[63]) to arginine blocks SUMOylation (Figure S5).[66,72] We hypothesized that the K526PABK mutation will block SUMOylation, but upon deprotection to lysine, rapid SUMOylation will occur.
We probed RanGAP1 SUMOylation using a 20 kDa C-terminal domain of murine RanGAP1 (RanGAP1ΔN or “RGΔN”; residues 400–589). This fragment is both SUMOylated and targeted to the NPC,[66] and therefore serves as a convenient means of assessing SUMOylation via Western blot. RGΔN-K526PABK was successfully expressed in a PABK-dependent manner (Figure 3B, lanes 1 and 2). Upon treatment with 2DPBM (100 μm, 12 h), unmodified FLAG-RGΔN is no longer detected (Figure 3B, lane 3). Instead, we observed SUMOylated RGΔN as a 37 kDa complex by Western blot using HA-tagged SUMO1ΔC4,[73] the mature form of SUMO1[66] (Figure 3C, lane 3). Though SUMO1 nominally has a mass of 11 kDa, the 15–17 kDa gel shift observed here is consistent with SUMOylation[59,74] and closely matches the shift seen when wild-type RGΔN is expressed in the presence of HA-SUMO1ΔC4 (Figure 3D). Taken together, these results suggest that after PABK deprotection, cells modify native FLAG-RGΔN with SUMO1. Further, it is unnecessary to overexpress SUMO1 for unmodified RGΔN-K526PABK to vanish after 2DPBM treatment (Figure 3B, lane 3), suggesting that cells modify the deprotected protein with endogenous SUMO1. Wild-type FLAG-RGΔN cannot be detected unless the K526R mutation[66] is made (Figure S5) due to rapid and quantitative SUMOylation of the substrate lysine.[67,68] Overall, our approach gives investigators a chemically-triggered “on-switch” for temporal control of a natural SUMO1 modification in mammalian cells.
Figure 3.

A) Phosphine-triggered SUMOylation of RanGAP1 and subsequent nuclear pore localization of the complex. B) RanGAP1ΔN-K526PABK is detected at ~20 kDa, but cannot be observed at this molecular weight after treatment with 2DPBM due to modification with endogenous SUMO. C) HA-SUMO1-RanGAPΔN is detected at 37 kDa by probing for the HA epitope tag after treatment with 2DPBM (100 μm, 12 h). D) A similar 37 kDa RGΔN-HA-SUMO1 complex is observed when wild-type RGΔN is co-expressed with HA-tagged SUMO1. E) NIH 3T3 cells expressing RanGAP-K524-RFP mutants show cytosolic protein for the negative control K524R mutant, whereas the wild-type RanGAP1-SUMO1-RFP (positive control) translocates to the nuclear pore complexes. The K524PABK mutant (bottom panels) matches the negative control phenotype in the absence of a phosphine trigger, but translocates to the nuclear membrane after treatment with 2DPBM, recapitulating the positive control phenotype.
To demonstrate control over SUMOylation in live cells, we imaged RanGAP1 translocation to the NPC in response to activation of K526 using a phosphine trigger. When imaging expression of a human RanGAP1-RFP fusion protein harboring a K524R mutation that permanently blocks SUMOylation,[63] the protein accumulates exclusively in the cytosol (Figure 3E, top left), as reported for the murine homolog.[66,72] By contrast, the wild-type protein is trafficked to the nucleus and accumulates on the nuclear rim (Figure 3E, top right).[66,72] A discontinuous ring of perinuclear fluorescence consistent with association to the cytosolic filaments of NPCs[71] was observed; accumulation in the cytosol — presumably once the NPCs were saturated[72] — occurred in cells with high transgene expression. Next, we incorporated PABK into this protein at K524. After a one-hour washout in fresh media to remove excess PABK, cells were treated with 2DPBM (100 μm) or DMSO for 16 h. In the absence of 2DPBM, the fusion protein (RanGAP-K524PABK-RFP) was localized exclusively in the cytosol (Figure 3e, bottom left), matching the negative control phenotype. After 2DPBM treatment, diffuse fluorescence in the nucleoplasm and bright, punctate signal on the nuclear lamina was observed, with only weak signal remaining in the cytosol, consistent with the positive control phenotype and with formation of RanBP2/Ubc9/RanGAP1-SUMO1-RFP complexes at the NPC. No fluorescence was observed for transfected cells grown in the absence of PABK (data not shown), indicating that all visible RFP fluorescence originated from unnatural protein expression. As the protected protein was still visualized at high levels 16 hours after UAA washout, unmodified RanGAP1 persists well beyond the 4 h lifespan previously confirmed with global translation inhibition by cycloheximide treatment.[75,76] The deprotected fusion protein is expressed at similar levels and with identical subcellular distribution to endogenous RanGAP1.[76]
While small molecules,[76] siRNAs,[76,77] proteins,[78] and genetic approaches[61,79] have been validated to turn off RanGAP1 SUMOylation with varying degrees of specificity, approaches for selective SUMOylation turn-on[28,56–60] remain rare and require either SUMO overexpression,[58] mutations to the SUMO modifier,[28,59] or may only be carried out in test tubes as opposed to in live cells.[49,59,60] We addressed this methodology gap through development of small molecule-triggered SUMOylation via the endogenous conjugation machinery, an approach that is applicable to other prost-translational modifications (PTMs) as well. Further, the RanGAP1 studies pave the way to applying this technique to the thousands of proteins that are conditionally SUMOylated in live cells.[80] Given the plasticity observed in SUMOylation of consensus sequences,[81] our approach will enable chemical control of SUMOylation anywhere a consensus sequence is present on a protein’s surface, thereby providing a tool to dissect the functional consequences of the PTM.
Conclusions
Variants of the Staudinger reduction are some of the most bioorthogonal reactions reported to date, and have shown the ability to function in complex biological systems where other reactions cannot.[27,29,82–84] We have demonstrated simple structural alterations to genetically encoded lysine protecting groups that rapidly accelerate deprotection via a Staudinger reduction/self-immolation sequence, thus overcoming the most significant limitation — slow kinetics[29] — while also improving deprotection yield. These UAAs are readily synthesized and incorporated into protein and have shown the fastest kinetics yet achieved in conditionally triggered nuclear translocation. Furthermore, they enabled us to externally control a native protein SUMOylation site in mammalian cells and follow the functional consequence of that activation. This approach allowed us to control protein subcellular localization via multiple mechanisms (controlling NLS function and controlling SUMOylation) and allowed us to target proteins to different subcellular locations. The methodology demonstrated here is generally applicable to other proteins, cell types,[85] and model organisms.[12,82] In contrast to light as another fast trigger of protein function,[86] our small molecule-triggered system does not require specialized equipment, is not affected by tissue opaqueness, and does not interfere with fluorescent reporters.[1] Further, it is orthogonal to visible light, potentially allowing for multiple biomolecules to be controlled in the same experiment. In choosing the appropriate tool for the research question, researchers may choose PABK when high protein yields are desired and may choose MPABK for the fastest possible activation. Although these UAAs have significantly closed the gap between the rate of small molecule triggering and optical triggering,[16] phosphines require additional time for diffusion or delivery into cells or animals. This could induce a delay in the decaging reaction compared to light stimulation, which may be pronounced in fast cellular processes, such as signal transduction As an added advantage, however, in addition to providing control over protein function via the Staudinger reduction, the azide-containing UAAs reported here may be activated with other reagents (such as TCOs[15] and Ru(II) complexes[87]). Further, they may function as vibrational reporters for IR spectroscopy,[15,88] may reveal transient or weak protein-protein interactions via photocrosslinking,[30,31,46] and could serve as handles for bioconjugation via cycloaddition reactions.[15,27] Thus, these next-generation azido-lysine derivatives serve as multifunctional, efficiently-incorporated handles for activating and probing protein structure and function in mammalian cells.
Experimental Section
Incorporation of UAAs into protein in HEK293T cells for imaging.
HEK293T cells (American Type Culture Collection, Manassas, VA) were seeded at 20,000 per well into 96-well plates (Greiner Bio-One, Frickenhausen, Germany) coated with poly-d-lysine hydrobromide (MP Biomedicals, Santa Ana, CA). Cells were grown for ~24 h (37 °C, 5% CO2) in Dulbecco’s Modified Eagle’s Medium (DMEM; GE Life Sciences, Logan, UT) supplemented with fetal bovine serum (FBS, 10% (v/v), Sigma-Aldrich, St. Louis, MO), penicillin (100 U mL−1, Corning Cellgro, Corning, NY), and streptomycin (100 μg mL−1) until reaching ~70% confluency. To transfect HEK293T cells, 1 mg mL−1 linear polyethylenimine (LPEI, Polysciences, Warrington, PA) was diluted to 0.33 mg mL−1 with prewarmed Opti-MEM media (Gibco Life Technologies, Waltham, MA). The resulting solution (2 μL) was added to solutions of plasmid DNA (200 ng total, with equal amounts of each plasmid) in Opti-MEM media (18 μL). pPB220PA-1-(U6-PylT*)4/EF1α-PABKRS was used for PABK and pPB220PA-1-(U6-PylT*)4/EF1α-OABKRS for MPABK. An mCherry-TAG-EGFP-HA reporter plasmid with an additional two copies of PylT (pMCherry-TAG-EGFP-HA-U6H1-PylT2) was used (described in the Supporting Information). The mixture was pipetted up and down five times, and was incubated for 10–15 minutes at ambient temperature. Cell culture media was replaced with 180 μL of prewarmed DMEM supplemented with FBS (10% (v/v)) and UAA (mm) or vehicle. The transfection reagent was added dropwise to each well by dispensing a small droplet of the reagent onto the pipette tip, then gently touching it to the surface of the media. The cells were grown for 48 h (37 °C, 5% CO2). Media was removed and replaced with prewarmed sterile HEPES-buffered saline (0.9% (w/v), pH 7.4). The fusion protein was visualized by epi-fluorescence microscopy (Zeiss Axio Observer Z1, 20x objective, numerical aperture 0.8 Plan-Apochromat M27) for mCherry (excitation (ex); BP550/25, emission (em), BP605/70) and for EGFP (ex, BP470/40; em, BP525/50 filter cubes).
For SATB1 nuclear translocation imaging, the cells were transfected with pCDH-EGFP-K29TAG-SATB1-mCherry (reported as eGFP-OptoNLS-SATB1-mCherry in Engelke et al., 2014)[50] and pAG-OABKRS-PylT4 (for OABK and MPABK) or pAG-PABKRS-PylT4 (for PABK) by the same method. After 36 h of incubation in DMEM supplemented with FBS (10% (v/v)) and the UAA (0.5 mm), media was changed to DMEM (high glucose, phenol red-free, HyClone Laboratories, GE Life Sciences, Logan, UT), and cells were incubated for 1 h to remove excess UAAs. The cells were placed on a WSKM Stage Top Incubator (Tokai Hit, Fujinomiya, Shizuoka Prefecture, Japan) and maintained at 37 °C in a 5% CO2 atmosphere. After acquiring baseline images, media was replaced with a freshly-prepared solution of DMEM (high glucose, phenol red-free) supplemented with 2DPBM (10–100 μm, prepared by adding a 50 mm (500 ×) stock solution of 2DPBM in degassed DMSO); this was performed on the microscope stage to avoid having to reposition the plate (since contamination was not an issue on the timescale of this experiment). Images were acquired every 5 min for 205 min. Quantification was performed as described below (see Quantification of fluorescence measurements in the Supporting Information).
Western Blot.
HEK293T cells (American Type Culture Collection, Manassas, VA) were seeded at 160,000 per well into 6-well plates (Greiner Bio-One, Frickenhausen, Germany) and grown for ~24 h (37 °C, 5% CO2) to ~80% confluency in Dulbecco’s Modified Eagle’s Medium (DMEM; HyClone Laboratories, GE Life Sciences, Logan, UT) supplemented with fetal bovine serum (FBS, 10% (v/v), Sigma-Aldrich, St. Louis, MO), penicillin (100 U mL−1, Corning Cellgro, Corning, NY), and streptomycin (100 μg mL−1). To transfect the cells, 1 mg mL−1 linear polyethylenimine (LPEI, Polysciences, Warrington, PA) was diluted to 0.33 mg mL−1 with prewarmed Opti-MEM media (Gibco Life Technologies, Waltham, MA). The resulting solution (10 μL) was added to solutions of plasmid DNA (1.5 μg each) in Opti-MEM media (200 μL). pAG vectors were used to provide PylRS mutants and PylT (4 copies) for all experiments.[16,86] MPABK and PABK were both incorporated into pMCherry-TAG-EGFP-HA. For the SUMO/RanGAP Western blots, pcDNA3-HA-SUMO1,[73] pcDNA3FLAG-RanGAPdeltaN[89] containing K526R or K526TAG mutations, and pAG-PABKRS-PylT4 were used. In all experiments, cell culture media was replaced with DMEM supplemented with FBS (10% (v/v)) and the appropriate UAA (0.5 mm) or DMSO just before transfection. After 24 h, cells were cooled on ice and washed with ice-cold PBS (2 × 1 mL). The cells were lysed in mammalian protein extraction buffer (250 μL) (GE Life Sciences, Logan, UT) supplemented with Halt Protease Inhibitor Cocktail (Thermo Scientific, Waltham, MA) on ice with orbital shaking for 20 min. The lysed cells were scraped from the plates and pipetted into microcentrifuge tubes, and were clarified by centrifugation (21,000 rcf, 20 min, 4 °C).
For the immunoblots, supernatants were mixed with Laemmli sample buffer and heated to 95 °C for 5 min. They were loaded into 1.5 mm 10% polyacrylamide gels alongside PageRuler molecular weight marker (Thermo Scientific, Waltham, MA) or dual color Precision Plus Protein Prestained Ladder (Bio-Rad, Hercules, CA). Gels were run at 60 V for 20 min followed by 120 V for 70 min. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Immobilon-P, EMD Millipore, Burlington, MA) for 2 h, 45 min at 75 V at 0 °C. The membrane was blocked with 5% (w/v) powdered nonfat milk in TBS-T for 1 h. Membranes were rinsed with TBS-T (3 × 5 min) and incubated in anti-GAPDH (Santa Cruz Biotechnology, Dallas, TX, sc-365062, mouse mAb, diluted 1:2,000 in 5% (w/v) milk in TBS-T), anti-β-actin (Santa Cruz Biotechnology, Dallas, TX, mouse mAb, diluted in 5% (w/v) milk in TBS-T)) anti-HA (Cell Signaling Technology, Danvers, MA, C29F4, rabbit mAb, diluted 1:1,500 in 5% (w/v) BSA in TBS-T) antibody solutions overnight at 4 °C. Membranes were washed with TBS-T (5 × 5 min) and incubated with secondary antibodies for 1 h at ambient temperature (1. goat anti-mouse IgG-HRP, Santa Cruz Biotechnology, sc-2005, diluted 1:20,000 in TBS-T; 2. goat anti-rabbit IgG-HRP, Santa Cruz Biotechnology, 7074S, diluted 1:5,000 in TBS-T). Membranes were washed with TBS-T (5 × 5 min) and were imaged with West Pico Chemiluminescence Substrate (Thermo Scientific, Waltham, MA) using a ChemiDoc XRS+ system with Image Lab 6.0 software (Bio-Rad, Hercules, CA).
Incorporation of UAAs into RanGAP in NIH 3T3 cells.
pDsRed1-N1-RanGAP and the K524R mutant was used in these experiments. For UAA mutagenesis, the K524TAG mutant was co-transfected with optimized synthetase plasmid pPB220PA-1-(U6-PylT*)4/EF1α-BhcKRS(IPYE) (see Supporting Information for a description of the molecular cloning procedures). NIH 3T3 cells (American Type Culture Collection, Manassas, VA) were plated at 20,000 per well on black 96-well plates (Greiner Bio-One, Frickenhausen, Germany) coated with poly-d-lysine hydrobromide (MP Biomedicals, Santa Ana, CA). When cells were ~80% confluent (approximately 18 h later), media was changed to 100 μL of DMEM (no antibiotics) supplemented with 10% FBS. For the UAA mutagenesis experiments, PABK (0.5 mm) or DMSO (0.5% (v/v)) was added. Then, DNA (200 ng total, or 100 ng of each plasmid for double transfections) was diluted in Opti-MEM media (5 μL/well), and P3000 reagent (0.2 μL/well) was added. Lipofectamine 3000 (0.2 μL/well, Invitrogen, Life Technologies, Carlsbad, CA) was diluted in Opti-MEM media (5 μL/well). The diluted DNA solution was added to the diluted reagent solution, pipetted up and down several times to mix, incubated for 10–15 min at room temperature, and added to wells (10 μL/well) by dispensing small droplets onto the pipette tip and touching them to the surface of the media one at a time, then gently swirling the plate. The cells were incubated for 48 h. For UAA experiments, media was changed to DMEM (high glucose, phenol red-free, 100 μL) 1 h before imaging to wash away excess UAAs. The fusion protein was visualized by widefield fluorescence microscopy (Zeiss Axio Observer Z1, 20x objective, numerical aperture 0.8 Plan-Apochromat M27) for mCherry (excitation (ex); BP550/25, emission (em), BP605/70). After acquiring baseline images, media was changed to DMEM (high glucose, phenol red-free, 100 μL) supplemented with 2DPBM (100 μm, prepared by adding a 50 mm (500 ×) stock solution of 2DPBM in degassed DMSO). For single-transfection experiments, 3T3 cells were plated on 8-well chamber slides (Cellvis, Mountain View, CA); the cell density and volumes of all media and reagents used for these transfections was doubled from the 96-well plate experiments.
Supplementary Material
Acknowledgements
This research was supported in part by the National Institutes of Health (R01GM112728 and T32GM008208) and the National Science Foundation (CBET-1603930). Molecular graphics were created and docking studies were performed with UCSF Chimera,[90] developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. K.M. received a Japan Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad. We thank the Chin lab (Medical Research Council Laboratory of Molecular Biology, Cambridge, UK) for plasmids encoding the PylRS and PylT genes. pcDNA3flag-RanGAPdeltaN and pcDNA3 HA-Sumo1 WT were gifts from Guy Salvesen. pDsRed1-N1 RanGAP was a gift from Mary Dasso.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Rakhit R, Navarro R, Wandless TJ, Chem. Biol 2014, 21, 1238–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Overington J, Hopkins A, Nat. Rev. Drug Discov 2006, 5, 993–996. [DOI] [PubMed] [Google Scholar]
- [3].Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, Lavan P, Weber E, Doak AK, Côté S, Shoichet BK, Urban L, Nature 2012, 486, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Islam K, Cell Chem. Biol 2018, 25, 1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wagner S, Waldman M, Arora S, Wang S, Scott V, Islam K, ChemBioChem 2019, 20, 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Luo M, ACS Chem. Biol 2012, 7, 443–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fegan A, White B, Carlson JCT, Wagner CR, Chem. Rev 2010, 110, 3315–3336. [DOI] [PubMed] [Google Scholar]
- [8].Rose JC, Foight GW, Cunningham-Bryant D, Loutey DE, Maly DJ, Dieter EM, J. Am. Chem. Soc 2019, 141, 3352–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jacobs CL, Badiee RK, Lin MZ, Nat. Methods 2018, 15, 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE, Science 2015, 348, 1376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sletten EM, Bertozzi CR, Angew. Chemie Int. Ed 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brown W, Liu J, Deiters A, ACS Chem. Biol 2018, 13, 2375–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang L, Brock A, Herberich B, Schultz PG, Science 2001, 292, 498–500. [DOI] [PubMed] [Google Scholar]
- [14].Ji H, Jing Q, Huang J, Silverman RB, Tetrahedron 2012, 68, 1359–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ge Y, Fan X, Chen PR, Chem. Sci 2016, 7, 7055–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Luo J, Liu Q, Morihiro K, Deiters A, Nat. Chem 2016, 8, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li J, Yu J, Zhao J, Wang J, Zheng S, Lin S, Chen L, Yang M, Jia S, Zhang X, Chen PR, Nat. Chem 2014, 6, 352–361. [DOI] [PubMed] [Google Scholar]
- [18].Wang J, Zheng S, Liu Y, Zhang Z, Lin Z, Li JJ, Zhang G, Wang X, Li JJ, Chen PR, J. Am. Chem. Soc 2016, 138, 15118–15121. [DOI] [PubMed] [Google Scholar]
- [19].Li J, Jia S, Chen PR, Nat. Chem. Biol 2014, 10, 1003–1007. [DOI] [PubMed] [Google Scholar]
- [20].Warner JB, Muthusamy AK, Petersson EJ, ChemBioChem 2014, 15, 2508–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang G, Li J, Xie R, Fan X, Liu Y, Zheng S, Ge Y, Chen PR, ACS Cent. Sci 2016, 2, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rossin R, Van Den Bosch SM, Ten Hoeve W, Carvelli M, Versteegen RM, Lub J, Robillard MS, Bioconjug. Chem 2013, 24, 1210–1217. [DOI] [PubMed] [Google Scholar]
- [23].Fang Y, Judkins JC, Boyd SJ, am Ende CW, Rohlfing K, Huang Z, Xie Y, Johnson DS, Fox JM, Tetrahedron 2019, 75, 4307–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Karver MR, Weissleder R, Hilderbrand SA, Bioconjug. Chem 2011, 22, 2263–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Agarwal P, Beahm BJ, Shieh P, Bertozzi CR, Angew. Chemie Int. Ed 2015, 54, 11504–11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Devaraj NK, ACS Cent. Sci 2018, 4, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR, ACS Chem. Biol 2006, 1, 644–648. [DOI] [PubMed] [Google Scholar]
- [28].Fottner M, Brunner A-D, Bittl V, Horn-Ghetko D, Jussupow A, Kaila VRI, Bremm A, Lang K, Nat. Chem. Biol 2019, 15, 276–284. [DOI] [PubMed] [Google Scholar]
- [29].Sletten EM, Bertozzi CR, Acc. Chem. Res 2011, 44, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chin JW, Santoro SW, Martin AB, King DS, Wang L, Schultz PG, J. Am. Chem. Soc 2002, 124, 9026–9027. [DOI] [PubMed] [Google Scholar]
- [31].Nguyen T-A, Cigler M, Lang K, Angew. Chemie Int. Ed 2018, 57, 2–14. [DOI] [PubMed] [Google Scholar]
- [32].Nobili S, Mini E, Landini I, Gabbiani C, Casini A, Messori L, Med. Res. Rev 2010, 30, 550–580. [DOI] [PubMed] [Google Scholar]
- [33].Kiick KL, Saxon E, Tirrell DA, Bertozzi CR, Proc. Natl. Acad. Sci. U. S. A 2002, 99, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nehring S, Budisa N, Wiltschi B, PLoS One 2012, 7, DOI 10.1371/journal.pone.0031992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Prescher JA, Bertozzi CR, Nat. Chem 2005, 1, 13–21. [DOI] [PubMed] [Google Scholar]
- [36].Geoffroy GL, Denton DA, Eigenbrot JCW, Inorg. Chem 1976, 15, 2310–2311. [Google Scholar]
- [37].Carl PL, Chakravarty PK, Katzenellenbogen JA, J. Med. Chem 1981, 24, 479–480. [DOI] [PubMed] [Google Scholar]
- [38].Alouane A, Labruère R, Le Saux T, Schmidt F, Jullien L, Angew. Chemie Int. Ed 2015, 54, 7492–7509. [DOI] [PubMed] [Google Scholar]
- [39].Erez R, Shabat D, Org. Biomol. Chem 2008, 6, 2669–2672. [DOI] [PubMed] [Google Scholar]
- [40].Mosey RA, Floreancig PE, Org. Biomol. Chem 2012, 10, 7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Saneyoshi H, Ochikubo T, Mashimo T, Hatano K, Ito Y, Abe H, Org. Lett 2014, 16, 30–33. [DOI] [PubMed] [Google Scholar]
- [42].Lukasak B, Morihiro K, Deiters A, Sci. Rep 2019, 9, 1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S, Chem. Biol 2008, 15, 1187–1197. [DOI] [PubMed] [Google Scholar]
- [44].Plass T, Milles S, Koehler C, Szymański J, Mueller R, Wießler M, Schultz C, Lemke EA, Angew. Chemie Int. Ed 2012, 51, 4166–4170. [DOI] [PubMed] [Google Scholar]
- [45].Schmidt MJ, Weber A, Pott M, Welte W, Summerer D, ChemBioChem 2014, 15, 1755–1760. [DOI] [PubMed] [Google Scholar]
- [46].Yanagisawa T, Kuratani M, Seki E, Hino N, Sakamoto K, Yokoyama S, Cell Chem. Biol 2019, 26, 1–14. [DOI] [PubMed] [Google Scholar]
- [47].Sharma V, Zeng Y, Wang WW, Qiao Y, Kurra Y, Liu WR, ChemBioChem 2018, 19, 26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bianco A, Townsley FM, Greiss S, Lang K, Chin JW, Nat. Chem. Biol 2012, 8, 748–750. [DOI] [PubMed] [Google Scholar]
- [49].Virdee S, Kapadnis PB, Elliott T, Lang K, Madrzak J, Nguyen DP, Riechmann L, Chin JW, J. Am. Chem. Soc 2011, 133, 10708–10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Engelke H, Chou C, Uprety R, Jess P, Deiters A, ACS Synth. Biol 2014, 3, 731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nakayama Y, Mian IS, Kohwi-Shigematsu T, Ogawa T, Cell Cycle 2005, 4, 1099–1106. [PubMed] [Google Scholar]
- [52].Van Brakel R, Vulders RCM, Bokdam RJ, Grüll H, Robillard MS, Bioconjug. Chem 2008, 19, 714–718. [DOI] [PubMed] [Google Scholar]
- [53].Flotho A, Melchior F, Annu. Rev. Biochem 2013, 82, 357–385. [DOI] [PubMed] [Google Scholar]
- [54].Martin S, Wilkinson KA, Nishimune A, Henley JM, Nat. Rev. Neurosci 2007, 8, 948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Geiss-Friedlander R, Melchior F, Nat. Rev. Mol. Cell Biol 2007, 8, 947–956. [DOI] [PubMed] [Google Scholar]
- [56].Lee MM, Fekner T, Tang TH, Wang L, Chan AHY, Hsu PH, Au SW, Chan MK, ChemBioChem 2013, 14, 805–808. [DOI] [PubMed] [Google Scholar]
- [57].Virdee S, Ye Y, Nguyen DP, Komander D, Chin JW, Nat. Chem. Biol 2010, 6, 750–757. [DOI] [PubMed] [Google Scholar]
- [58].Pichler A, Knipscheer P, Oberhofer E, Van Dijk WJ, Körner R, Olsen JV, Jentsch S, Melchior F, Sixma TK, Nat. Struct. Mol. Biol 2005, 12, 264–269. [DOI] [PubMed] [Google Scholar]
- [59].Pawale VS, Yadav P, Roy RP, ChemBioChem 2018, 110067, 1137–1141. [DOI] [PubMed] [Google Scholar]
- [60].Boll E, Drobecq H, Ollivier N, Blanpain A, Raibaut L, Desmet R, Vicogne J, Melnyk O, Nat. Protoc 2015, 10, 269–292. [DOI] [PubMed] [Google Scholar]
- [61].He X, Riceberg J, Pulukuri SM, Grossman S, Shinde V, Shah P, Brownell JE, Dick L, Newcomb J, Bence N, PLoS One 2015, 10, e0123882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sahin U, Lapaquette P, Andrieux A, Faure G, Dejean A, PLoS One 2014, 9, e102957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Joseph J, Tan SH, Karpova TS, McNally JG, Dasso M, J. Cell Biol 2002, 156, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Klenk C, Humrich J, Quitterer U, Lohse MJ, J. Biol. Chem 2006, 281, 8357–8364. [DOI] [PubMed] [Google Scholar]
- [65].Zhu S, Goeres J, Sixt KM, Békés M, Zhang XD, Salvesen GS, Matunis MJ, Mol. Cell 2009, 33, 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mahajan R, Gerace L, Melchior F, Delphin C, Guan T, J. Cell Biol 1998, 140, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bossis G, Chmielarska K, Gärtner U, Pichler A, Stieger E, Melchior F, Methods Enzymol 2005, 398, 20–32. [DOI] [PubMed] [Google Scholar]
- [68].Sakin V, Richter SM, Hsiao HH, Urlaub H, Melchior F, J. Biol. Chem 2015, 290, 23589–23602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Reverter D, Lima CD, Nature 2005, 435, 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Saitoh H, Pu R, Cavenagh M, Dasso M, Proc. Natl. Acad. Sci. U. S. A 2002, 94, 3736–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhang H, Saitoh H, Matunis MJ, Mol. Cell. Biol 2002, 22, 6498–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Matunis MJ, Wu J, Blobel G, J. Cell Biol 1998, 140, 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Békés M, Prudden J, Srikumar T, Raught B, Boddy MN, Salvesen GS, J. Biol. Chem 2011, 286, 10238–10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Park-Sarge O-K, Sarge KD, Methods Mol. Biol 2008, 464, 255–265. [DOI] [PubMed] [Google Scholar]
- [75].Crampton N, Kodiha M, Shrivastava S, Umar R, Stochaj U, Mol. Biol. Cell 2009, 20, 5106–5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cha K, Sen P, Raghunayakula S, Zhang XD, PLoS One 2015, 10, 8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hutten S, Flotho A, Melchior F, Kehlenbach RH, Mol. Biol. Cell 2008, 19, 2300–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ribet D, Hamon M, Gouin E, Nahori MA, Impens F, Neyret-Kahn H, Gevaert K, Vandekerckhove J, Dejean A, Cossart P, Nature 2010, 464, 1192–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hamada M, Haeger A, Jeganathan KB, van Ree JH, Malureanu L, Wälde S, Joseph J, Kehlenbach RH, van Deursen JM, J. Cell Biol 2011, 194, 597–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hendriks IA, Vertegaal ACO, Nat. Rev. Mol. Cell Biol 2016, 17, 581–595. [DOI] [PubMed] [Google Scholar]
- [81].Ulrich HD, Methods Mol. Biol. SUMO Protoc 2009, 497, 3–16. [DOI] [PubMed] [Google Scholar]
- [82].Shah L, Laughlin ST, Carrico IS, J. Am. Chem. Soc 2016, 138, 5186–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Prescher JA, Dube DH, Bertozzi CR, 2004, 430, 873–877. [DOI] [PubMed] [Google Scholar]
- [84].Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, Lo A, Bertozzi CR, Proc. Natl. Acad. Sci. U. S. A 2010, 107, 1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Aloush N, Schvartz T, König AI, Cohen S, Brozgol E, Tam B, Nachmias D, Ben-David O, Garini Y, Elia N, Arbely E, Sci. Rep 2018, 8, 14527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Luo J, Uprety R, Naro Y, Chou C, Nguyen DP, Chin JW, Deiters A, J. Am. Chem. Soc 2014, 136, 15551–15558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Chen Y, Kamlet AS, Steinman JB, Liu DR, Nat. Chem 2011, 3, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Bazewicz CG, Liskov MT, Hines KJ, Brewer SH, J. Phys. Chem. B 2013, 117, 8987–8993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mikolajczyk J, Drag M, Békés M, Cao JT, Ronai Z, Salvesen GS, J. Biol. Chem 2007, 282, 26217–26224. [DOI] [PubMed] [Google Scholar]
- [90].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, J. Comput. Chem 2004, 25, 1605–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.