

Abstract
Osteoporosis is a highly prevalent disorder characterized by low bone mineral density and an increased risk of fracture, termed osteoporotic fracture. Notably, bone mineral density, osteoporosis and osteoporotic fracture are highly heritable; however, determining the genetic architecture, and especially the underlying genomic and molecular mechanisms, of osteoporosis in vivo in humans is still challenging. In addition to susceptibility loci identified in genome-wide association studies, advances in various omics technologies, including genomics, transcriptomics, epigenomics, proteomics and metabolomics, have all been applied to dissect the pathogenesis of osteoporosis. However, each technology individually cannot capture the entire view of the disease pathology and thus fails to comprehensively identify the underlying pathological molecular mechanisms, especially the regulatory and signalling mechanisms. A change to the status quo calls for integrative multi-omics and inter-omics analyses with approaches in ‘systems genetics and genomics’. In this Review, we highlight findings from genome-wide association studies and studies using various omics technologies individually to identify mechanisms of osteoporosis. Furthermore, we summarize current studies of data integration to understand, diagnose and inform the treatment of osteoporosis. The integration of multiple technologies will provide a road map to illuminate the complex pathogenesis of osteoporosis, especially from molecular functional aspects, in vivo in humans.
Osteoporosis, the most common bone disorder worldwide (FIG. 1), is characterized by low bone mineral density (BMD) and an increased risk of osteoporotic fracture1. According to the WHO, osteoporosis is defined as a BMD that lies 2.5 standard deviations or more below the average value for young healthy women (T-score ≤2.5)2. Consequently, the clinical diagnosis and assessment of osteoporosis is mainly based on measurements of BMD3. Of note, BMD has a heritability of 0.6–0.8, meaning that 60–80% of the variation in BMD is inherited from parents and the remainder is derived from the environment4. In addition, osteoporotic fracture, which is the end point clinical outcome of osteoporosis, has a heritability of 0.5–0.7 (REF.5). Despite this strong heritability, determining the genetic architecture (BOX 1), and especially the underlying genomic and molecular mechanisms of osteoporosis in vivo in humans, is challenging.
Fig. 1 |. Prevalence of osteoporosis in populations of age 50 years and older in selected countries.

The prevalence of osteoporosis in the non-institutionalized USA population was calculated using data collected by the National Health and Nutrition Examination Survey 2005–2010 (REF.153). The statistics for six European countries (France, Germany, Italy, Spain, Sweden and the UK) were retrieved from a report by the International Osteoporosis Foundation154. The statistics for China and Korea were obtained from a meta-analysis study published in 2016 (REF.155) and the Korea National Health and Nutrition Examination Survey 2008–2010 (REF.156), respectively. Data for Canada, Japan and Australia were obtained from a 2014 study157.
BOX 1 |. Key terms in genetic and omics studies.
Allelic heterogeneity
Multiple single nucleotide polymorphisms within the same gene and/or pathway jointly affect the same trait.
Distant gene
If a genetic variant affects the expression or otherwise interacts with genes other than the nearest gene, the target genes are referred as distant genes of the variant of interest.
Effect size
The portion of phenotypic variance that is explained by the tested variant.
Epigenomics
The study of genome-wide reversible modifications of DNA or DNA-associated proteins such as DNA methylation, histone acetylation and chromatin organization.
Expression quantitative trait loci (eQTL) analysis
A technique for assessing the associations between transcript expression and genotype to identify genetic variants that explain the variation in gene expression levels.
Fingerprint
Specific expression profiles of proteins, which can be used as characteristics to distinguish different individuals.
Genetic architecture
The characteristics of genetic variation that are responsible for heritable phenotypic variability150.
Genome-wide association studies (GWAS)
Studies using a hypothesis-free method to investigate the associations between genetic variants and traits, including diseases.
Hybrid mouse diversity panel
A collection of approximately 100 well-characterized inbred strains of mice that can be used to analyse the genetic and environmental factors underlying complex traits.
Knowledgebase
A library used to store complex structured and unstructured information by a computer system.
Long-range
The distance between regulatory regions and their target genes is considered far, usually >100 kb.
Mendelian randomization
Mendelian randomization is a method of using genetic variants to determine whether an observational association between a risk factor and an outcome is consistent with a causal effect.
Metabolomics
A field of omics science to systematically measure small molecules, commonly knowns as metabolites, within cells, biofluids, tissues or organisms.
Ome
The objects of one field of study in biology, for example, the genome, proteome or metabolome.
Proteomics
The identification and quantification of the entire protein complement of a cell, tissue or organism under a specific, defined set of conditions.
Transcriptomics
The study of the complete set of RNA transcripts (including messenger, transfer, ribosomal and non-coding regulatory RNAs) produced by a cell or tissue under specific conditions.
Weighted gene co-expression network analysis (WGCNA)
A commonly used systems biology method for studying the correlation patterns among genes. WGCNA can be used for finding clusters (referred to as modules) of genes sharing similar expression patterns across a set of samples, and for relating modules to disease status and sample traits using module behaviour and topology characteristics.
Genome-wide association studies (GWAS) have identified hundreds of susceptibility loci for osteoporosis6. However, the causal variants and/or genes and their molecular functional mechanisms are largely unknown. Other omics technologies, that is, transcriptomics, epigenomics, proteomics, metabolomics and metagenomics (BOX 1), individually might provide a useful glimpse into the windows of osteoporosis pathophysiology. However, data generated by single-omics technologies are not comprehensive enough to capture the complete pictures of the molecular events leading to osteoporosis. A change in the status quo calls for integrative multi-omics and/or inter-omics analyses with approaches in systems genetics and genomics.
Compared with individual-omics studies, multi-omics studies can provide pathways of information from the original root cause of a disease (for example, genetic variations) to functional consequences or relevant interactions7,8. Therefore, multi-omics data can provide a clearer and more comprehensive view of the pathogenesis of osteoporosis. This knowledge is important as it is not currently possible to accurately identify all patients who will experience osteoporotic fracture from measurements of BMD.
In this Review, we briefly introduce the mechanisms of bone homeostasis before providing a broad survey of GWAS and the multiple-omics studies on osteoporosis (FIG. 2). We discuss the advantages and challenges of each technology and then focus on studies in which multiple levels of omics are integrated to elucidate the potential molecular mechanisms for osteoporosis. We also discuss the current methods and challenges in combining and interpreting multi-omics data in osteoporosis studies.
Fig. 2 |. Integrating multi-omics data to elucidate the molecular mechanisms of osteoporosis.

Multiple omics technologies, including genomics (mainly refers to genome-wide association studies (GWAS)), transcriptomics, epigenomics, proteomics and metabolomics, have been applied to dissect the pathogenesis of osteoporosis. Each technology individually can only provide limited insights into the biological mechanisms of osteoporosis. By integrating multiple omics data and following-up functional experiments in cell lines and/or animal models, researchers could capture a comprehensive view of the pathogenesis of this disorder.
Bone homeostasis
Bone homeostasis is mainly controlled by the action of osteoblasts, osteocytes and osteoclasts; bone is a highly metabolically active tissue, which undergoes a continuous cycle of bone formation mediated by osteoblasts and bone resorption facilitated by osteoclasts. Importantly, disruption of bone homeostasis has a fundamental role in the pathogenesis of osteoporosis9.
The cells that comprise bone tissue have diverse origins. For example, osteoblasts are derived from mesenchymal stem cells, which can also give rise to adipocytes, chondrocytes and myocytes10 (FIG. 3). Osteoblasts produce bone by synthesizing extracellular matrix consisting of various proteins, the most abundant being type I collagen. The extracellular matrix is known as the osteoid when first deposited and is subsequently mineralized through the accumulation of calcium phosphate as hydroxyapatite (Ca10(PO4)6(OH)2). Multiple transcription factors (such as RUNX2 and OSX) and the major developmental signals (such as WNT signalling) are reported to regulate osteoblast differentiation and function11. By contrast, the bone resorbing osteoclasts are large, multinucleated cells formed by the fusion of precursors from the monocyte-derived macrophage lineage12 (FIG. 3). Osteoclasts can dissolve minerals and digest bone matrix by secreting hydrochloric acid and proteolytic enzymes13. Similar to osteoblasts, multiple factors are reported to regulate osteoclast differentiation and/or function, including macrophage colony-stimulating factor (M-CSF), receptor activator of nuclear factor-κB ligand (RANKL), cytokines (for example, IL-1) and αVβ3 integrin14.
Fig. 3 |. differentiation process of osteoblasts and osteoclasts.

Bone is a highly metabolically active tissue, which undergoes a continuous cycle of bone formation mediated by osteoblasts and bone resorption facilitated by osteoclasts. The cells that comprise bone tissue have diverse origins. Osteoblasts are derived from mesenchymal stem cells, which can also give rise to adipocytes, chondrocytes and myocytes. Osteoclasts are large, multinucleated cells formed by fusion of precursors derived from the monocyte–macrophage lineage. As the major cellular component of bone tissue, osteocytes originate from osteoblasts. We list several representative genes linked to bone metabolism by omics studies; functional experiments support their involvement in bone homeostasis. GWAS, genome-wide association studies; PTHR, parathyroid hormone receptor.
The major cellular component of bone tissue is osteocytes, which are cells that originate from osteoblasts; these cells are deeply embedded in bone tissue and comprise >90% of all bone cells15. Osteocytes can orchestrate bone homeostasis by regulating the function of bone-forming osteoblasts and bone-resorbing osteoclasts. For example, osteocytes regulate bone formation by secreting modulators of the WNT signalling pathway such as activators (nitric oxide and ATP) and inhibitors (sclerostin and DKK1)16. Moreover, osteocytes also express RANKL and M-CSF to increase osteoclast activity as well as osteoprotegerin and nitric oxide to inhibit osteoclast formation and activity16. A detailed description of the crosstalk between the three major bone cellular subsets falls outside the scope of this Review, but can be found in REF.17.
The genome influences the maintenance of bone homeostasis by encoding many factors that modulate the differentiation and activities of bone cells. In addition to several components in the aforementioned signalling pathways, hundreds of common genetic variants associated with BMD or osteoporotic fracture have been discovered. Beginning in the next section, we will present an update on what is known about genetic risk factors for osteoporosis.
Susceptibility loci derived from GWAS
Mainly through testing millions of single nucleotide polymorphisms (SNPs), GWAS have been used to identify over 500 susceptibility loci for osteoporosis-related traits18–20 (Supplementary Table 1). The sample sizes of early single-sample GWAS were fairly small (<10,000)21–24, resulting in limited statistical power; indeed, only risk loci with an effect size (BOX 1) ranging from 7.8 × 10−4 to 1.4 × 10−2 could be identified21–24 (BOX 2). However, with the increases in sample size and improvements in statistical power, GWAS meta-analyses can identify more risk loci with effect sizes of 10−4 to 10−3 (BOX 2). For example, the GEnetic Factors for OSteoporosis (GEFOS) consortium, which consists of unrelated participants and sample sizes ranging from 19,195 to 66,628, has identified dozens of new susceptibility loci (such as EN1)25 and has replicated some risk loci identified by earlier GWAS26–30 (such as ESR1 and LRP5). In mice, En1 expression was only observed in osteogenic lineages, and conditional loss of En1 resulted in low bone mass25. As the oestrogen receptor, ESR1 affects bone formation in both osteoblasts progenitors31 and mature osteoblasts32. ESR1 could also induce the transcription of Fas ligand in osteoblasts, resulting in a paracrine signal to induce osteoclast apoptosis31. In the canonical WNT signalling pathway in bone formation, the activation of β-catenin through LRP5 and Frizzled results in the upregulation of transcription factors that are crucial for osteoblast differentiation33 (FIG. 3). Together, the loci identified from GEFOS cumulatively explained ~ 6% of the phenotypic variance in BMD.
BOX 2 |. Statistical power calculations.
Statistical power is important in genome-wide association studies (GWAS) as it is the parameter that determines how effectively a given study will identify susceptibility loci. The statistical power of statistical significance is the probability that an association test statistic will reject a false null hypothesis. As an association test statistic asymptotically (that is, approaching a curve arbitrarily closely) follows a 1-degree of freedom, non-central χ2 distribution, statistical power is solely determined by the non-centrality parameter λ of this distribution (λ ≥ 0).
Statistical power is confounded by a variety of factors, including cryptic relatedness (individuals are more closely related to one another than assumed by the investigator) and population stratification151. In an ideal scenario in which an unrelated sample (that is, of individuals that are independent from each other) is drawn from a homogeneous population and under an additive mode of inheritance, the non-centrality parameter λ has a simple form152:
where N is the sample size and e is the effect size, which is defined as the portion of phenotypic variance that is explained by the target variant, that is, variant-level heritability.
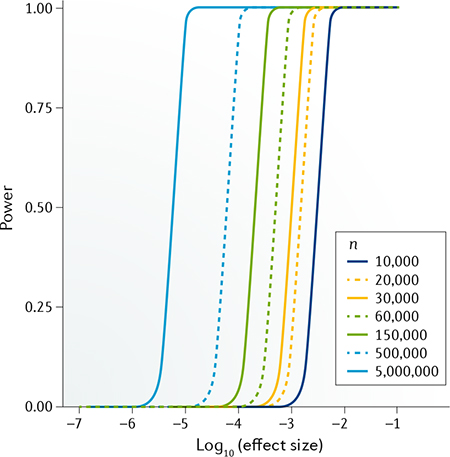
Of note, statistical power is non-relevant to variant allele frequency. Therefore, to detect associated variants of of the original effect size, the sample size should increase to n times the original sample size to reach the same level of statistical power. Refer to the figure to see the power calculated under several typical sample size settings for a GWAS scan (type I error rate threshold α = 5 × 10−8). For any given sample size, the range of effect size with intermediate power falls into a relatively narrow interval. For example, under the largest sample size shown in the figure (5,000,000), this GWAS setting has nearly 100% power to detect variants with effect size > 1 × 10−5 but nearly 0% power to detect variants with effect size < 1 × 10−6.
The UK Biobank project34 is remarkable due to its huge number of samples obtained from participants from the UK general population. Importantly, two GWAS in ~140,000 and 420,000 UK Biobank participants have identified 203 and 518 loci, respectively, associated with variation in ultrasound-based measurements of estimated heel BMD (eBMD)6,35 (FIG. 3). For example, DAAM2 was identified in a UK Biobank study; reduction of DAAM2 protein levels resulted in reduced bone mineralization6. In addition, the largest UK Biobank cohort has nearly 100% power to identify loci with an effect size of 1 × 10−5 (BOX 2). The loci identified from studying the UK Biobank cohort can cumulatively explain 20% of the phenotypic variance in eBMD, therefore substantially increasing our understanding of the genetic architecture of osteoporosis.
Several groups have also used GWAS to carry out age-specific and sex-specific analyses30,35. Variants in ESR1 and in close proximity to RANKL showed a clear age-dependent effect on BMD30. A single variant, rs17307280, at FAM9B on the X chromosome was significantly associated with eBMD in men only35. The function of FAM9B in bone homeostasis is still unknown. Of note, a direct link exists between oestrogen deficiency after menopause and the development of osteoporosis36 in women. Consequently, researchers have also attempted to investigate specific risk loci in premenopausal37 and postmenopausal38 women. SNPs in WNT16 and ESR1 were identified to be associated with lumbar spine BMD in premenopausal women37. WNT16 is a member of the WNT signalling pathway and Wnt16 knockout mice have reduced bone strength39. In postmenopausal women38, variants in TNFRSF11B, SPTBN1, ESR1 and LRP4 were reported to be susceptibility loci for osteoporotic fracture. The protein encoded by TNFRSF11B is osteoprotegerin, which functions as a decoy receptor of RANKL to inhibit bone resorption40. In vivo evidence suggested that LRP4 in osteoblasts could suppress bone formation and promote osteoclastogenesis through acting as a receptor of sclerostin41.
The missing heritability problem.
The BMD-associated loci identified so far in GWAS still cannot account for all the heritability in osteoporosis. Consideration of gene–environment interactions might explain part of this ‘missing heritability’42. Researchers could explore gene–environment interactions through investigating the effects of genetic variants on osteoporosis in specific population subgroups affected by specific environmental factors (for example, smoking). In addition, many alternative approaches have been developed to serve as complementary methods for the standard SNP-based GWAS described earlier in this Review. For example, copy number variation-based GWAS focus on identifying variation in gene copy number. The first copy number variation-based GWAS identified that a deletion variant of UGT2B17 was associated with hip osteoporotic fracture43. UGT2B17 encodes an enzyme catabolizing steroid hormones that have effects on bone formation43.
Another useful approach is that used in gene-based GWAS, which regard a gene as a basic unit for association analyses, thereby improving the statistical power (BOX 2) by overcoming the potential allelic heterogeneity (BOX 1) and reducing the number of independent statistical tests from millions (for SNPs) to tens of thousands (for genes)44. A 2018 study found three novel BMD-associated genes (UBTF, AAAS and C11orf58) through a gene-based GWAS strategy44. The roles of the proteins encoded by these three genes in bone homeostasis are still unknown. A powerful technique for identifying biological pathways that might be involved in a heritable trait is pathway-based GWAS, which considers SNPs in multiple genes in a related pathway jointly. The first pathway-based GWAS for osteoporosis identified a pathway that regulates autophagy as being associated with ultradistal radius BMD45. In addition to overcoming the effect of allelic heterogeneity and improving statistical power, gene-based GWAS and/or pathway-based GWAS also enable direct comparison between different populations with different linkage disequilibrium patterns and/or functional alleles46.
Bivariate GWAS aim to discover SNPs associated with two different traits and could identify pleiotropic genes as well as the key mechanistic links between two diseases. Any identified pleiotropic genes could provide insight into a genetic mechanism shared by both traits as well as potentially facilitating repurposing of conventional drugs used for one disease for new use in the other. A relevant example in osteoporosis research was a study identifying SNPs in SOX6 (REF.47) and the TOM1L2-SREBF1 locus48, which have pleiotropic effects on both BMD and lean mass. Finally, summary databased approaches, such as joint association studies49, pleiotropic conditional false discovery rate method50 and co-expression network prediction51, which are methods that use GWAS summary statistics alone, have also identified a large amount of novel BMD loci.
Clinical translation of GWAS findings.
To address the missing heritability problem, researchers could explore more loci under an omnigenic model52, which proposes that gene regulatory networks are so interconnected that most heritability can be explained by effects on genes outside core pathways. New findings will provide novel insights into the genetic mechanism of osteoporosis as well as osteoporosis risk prediction. For example, two studies53,54 have reported that genetic profiling of BMD-associated SNPs could improve the accuracy of osteoporotic fracture prediction. In addition, a genetic algorithm that uses SNPs as predictors also showed a strong correlation with eBMD (correlation coefficient R = 0.415)55. By contrast, for clinical applications such as drug target design, loci with large genetic effects on BMD and osteoporotic fracture risk might be more valuable. We should spare no effort to dissect the clinical applications of known risk loci, which is an ultimate goal of all GWAS.
The success of GWAS demonstrates the usefulness of disease genomics to translational medicine. Generally, compared with those without genetic support, drug targets that are reported as susceptibility loci in GWAS are twice as likely to succeed in clinical trials56. For osteoporosis, GWAS have provided genetic support for five of the eight (63%) approved anti-osteoporosis therapeutics. For example, RANKL is the drug target of denosumab and the genetic association between RANKL and BMD has been reported by GWAS18. In addition, GWAS results could provide resources for drug repositioning. Some of the susceptibility loci for BMD and osteoporotic fracture are shared with many other diseases and, consequently, some osteoporosis-associated genes have already been used as drug targets for other diseases (Supplementary Table 2). For example, the eBMD-associated gene ACHE is an approved drug (donepezil) target for Alzheimer disease. Accumulating evidence57,58 has indicated that osteoporosis and hip fracture are common complications observed in patients with Alzheimer disease, although the mechanisms underlying this association remain poorly understood. In addition, as an inhibitor of ACHE, donepezil might serve as a potential drug for osteoporosis as it could prevent RANK-induced bone loss via inhibition of osteoclast differentiation59.
The challenges of GWAS.
Several challenges exist for the use of GWAS in osteoporosis, which should be addressed in future studies. For example, diverse phenotypes (BMD at different skeletal sites using different measurements) are now included in osteoporosis GWAS, resulting in inconsistent results. The inconsistencies might be due to the heterogeneity among the different types of measurements of BMD, as the genetic correlation coefficients between the commonly used dual-energy X-ray absorptiometry and other measurements (for example, ultrasonography and quantitative CT) range from 0.505 to 0.917 (REF.60). However, even when studies use the same type of measurement, there is still heterogeneity between BMD measured at different skeletal sites. In addition, as the ultimate goal of osteoporosis research is to reduce the incidence and prevalence of osteoporotic fracture, GWAS should use osteoporotic fracture as a direct phenotype. Several novel genes (such as FAM210A, GRB10 and ETS2) have been identified as susceptibility loci by GWAS directly comparing individuals who have had an osteoporotic fracture and healthy controls61. The specific functions of these genes in bone homeostasis are still unknown. However, most of these studies combined heterogeneous forms of osteoporotic fracture at different skeletal sites, which have different aetiologies; the results obtained might therefore entail both false positive or false negative findings, which are difficult to discern. Two studies examined osteoporotic fracture at specific sites: vertebral fracture62 and hip fracture63. Two SNPs (rs2468531 and rs12742784) located in the non-coding region were found to be associated with vertebral fracture62. One SNP (rs13182402) within the ALDH7A1 gene was associated with hip fracture. The ALDH7A1 protein degrades and detoxifies acetaldehyde, which inhibits osteoblast proliferation and results in decreased bone formation63. We suggest that vertebral fracture might not be the best phenotype to study, as no consensus definition of vertebral fracture currently exists. By contrast, hip fracture has a consensus definition and might represent a feasible alternative for study.
The replication of findings in distinct GWAS is a routine process for validating the statistical results from the discovery sample64,65. However, inconsistent findings that occur between different studies might be caused by limited statistical power (BOX 2) or heterogeneous effects66 between distinct populations and ethnicities64. These effects could explain discrepancies between studies, and even the different findings between GEFOS-1 and GEFOS-2 (REFS26,28), which were conducted by largely the same research group. Future meta-analyses of GWAS should enable researchers to fully consider the heterogeneity.
Another potential issue of GWAS is the difficulty in determining the causal variants of a specific phenotype owing to complex patterns of linkage disequilibrium. Some statistical inference methods, such as PAINTOR67, GCTA-COJO68 and FINEMAP69, have been applied for the fine-mapping of plausibly causal variants for BMD with high confidence6,35,70. However, most fine-mapping algorithms need reference genotype data to estimate the linkage disequilibrium relationship between genetic variants, which introduces inescapable bias when the reference data and original GWAS data are not perfectly matched71.
Finally, although GWAS have identified hundreds of independent loci for osteoporosis-related traits (Supplementary Table 1), the underlying causal functional variants and biological regulatory mechanisms are still largely unknown. The use of GWAS alone might not be able to address these issues; however, considering functional genomics could provide new insights.
Insights from ‘omics’ studies
Functional genomics and other omics studies, which focus on, for example, global gene transcription, translation and protein–protein interactions, have identified a list of biomarkers for osteoporosis, providing insights into the biological pathways involved. Here, we review these studies, including transcriptomics, epigenomics, proteomics and metabolomics (BOX 1).
Transcriptomics.
Transcriptomics studies are used to identify differentially expressed genes between patients with osteoporosis and control individuals. Further functional experiments are used to validate the roles of these genes in osteoblastic and osteoclastic differentiation and/or activity. So far, most of the research has studied the precursor cells of osteoblasts (that is, human bone marrow mesenchymal stem cells (BMSCs)) or osteoclasts (for example, circulating monocytes) (Supplementary Table 3).
The first comparative gene expression study of circulating monocytes was carried out in individuals with high or low lumbar spine BMD measured with dual-energy X-ray absorptiometry and identified three upregulated genes (CCR3, HDC and GCR) in participants with low BMD72. The products of these genes are CC-chemokine receptor 3 (CCR3), histidine decarboxylase and glucocorticoid receptor, respectively. They are all known to be involved in osteoclastogenesis73–75. In addition to mRNA, the levels of non-coding RNA, such as microRNA76, long non-coding RNA (lncRNA)77 and circular RNA78, have also been studied. For example, a 2019 study77 reported that lncRNA-ORLNC1 was overexpressed in bone tissues of patients with osteoporosis compared with controls, with a T-score ≥1, and were also upregulated in bone tissue, serum and BMSCs of ovariectomy-induced osteoporotic mice compared with the sham-operated group. Further functional studies in BMSCs of mice showed that a lncRNA-ORLNC1–miR-296–PTEN signalling axis functions as a critical regulator for the switch between the osteogenesis and adipogenesis of BMSCs, that is, whether or not these cells differentiate into osteoblasts or adipocytes (FIG. 3).
Except for a few studies focused on a particular gene, for example, a synonymous variant in exon 9 of CD44 that might increase the susceptibility to osteoporosis by affecting the splicing mechanism79, transcriptomics studies of osteoporosis generally do not consider alternative splicing. Of note, alternative splicing is a ubiquitous regulatory mechanism of gene expression and RNA mis-splicing is related to a large array of human diseases80. Therefore, transcriptomics studies that take alternative splicing into consideration are needed in the field of bone research.
Epigenomics.
Epigenetic factors, mainly DNA methylation and histone modification, have considerable effects on the differentiation and activities of bone cells81 and might contribute to pathogenetic mechanisms of osteoporosis82. The early epigenetic studies of osteoporosis often focused on a few candidate genes with known importance in bone biology (Supplementary Table 4). For example, one study compared the DNA methylation levels at the SOST promoter region in bone biopsy samples from postmenopausal women with osteoporosis (n = 4) and healthy controls (n = 4)83. They found that the SOST promoter showed higher DNA methylation in patients with osteoporosis than in controls, which was replicated in an independent cohort of 63 postmenopausal women (27 with osteoporosis and 36 without osteoporosis). Interestingly, the authors also observed strong positive associations between bone SOST mRNA and serum levels of sclerostin with BMD. Because sclerostin is known as an inhibitor of bone formation84, the authors speculated that the increase in SOST promoter methylation and the reduction of SOST expression in patients with osteoporosis might represent a compensatory mechanism to counteracting osteoporosis-associated bone loss. However, conflicting results were reported by a 2019 study comparing femoral bone tissues from 16 Chinese patients with osteoporosis who had femoral neck and/or trochanter fractures and 16 controls (patients with traumatic factures but normal BMD), wherein SOST gene expression (at both mRNA and protein levels) was significantly increased and SOST promoter was slightly hypo-methylated in patients with osteoporosis85. Therefore, the association between SOST methylation and expression with osteoporosis warrants further investigation.
Importantly, a few groups have explored the epigenome-wide association study (EWAS) of DNA methylation with osteoporosis in humans (Supplementary Table 4). For example, in 2013, the first EWAS of human bone detected 241 differentially methylated CpG sites in femoral head trabecular bone specimens between 27 patients with osteoporotic hip fractures and 26 patients with hip osteoarthritis86. Bone samples from true control individuals were not available owing to the ethical difficulties in obtaining bone biopsy samples from healthy people. So far, the largest EWAS using bone specimens were performed in 84 postmenopausal women with substantially varied BMD87 and identified 63 differentially methylated CpGs associated with BMD.
Owing to the difficulty in obtaining human bone tissue samples, some EWAS (Supplementary Table 4) used whole blood as a proxy to test for the associations of DNA methylation with osteoporosis and BMD. One notable example study performed a large-scale EWAS for femoral neck and lumbar spine BMD in whole blood of 5,515 individuals88; however, the study did not reveal strong consistent association signals for DNA methylation at any of the >450,000 tested CpG sites. The lack of statistically significant associations between blood DNA methylation and osteoporosis was also reported in another independent EWAS89. Together, these findings suggest that blood DNA methylation patterns might not efficiently reflect the epigenomic status of bone cells if whole blood is used.
It is worth mentioning that aside from DNA methylation, other epigenetic features (for example, histone modification and high-order chromatin structure) have rarely been studied for osteoporosis82. Compared with many other human tissues and cell types, current data on the epigenetic architecture of human primary bone tissue and cells are rather scarce, even in large-scale consortium projects such as ENCODE90 and NIH Roadmap91.
Proteomics.
To further investigate the molecular determinants of BMD, proteomics studies have been carried out to catalogue proteins expressed in bone-related cells or tissues, to identify proteins that are involved in the pathogenesis of osteoporosis, and to investigate protein biomarkers that are diagnostic of osteoporosis or predictive of osteoporotic fracture. Importantly, a 2008 study identified 38 differentially expressed BMD-associated proteins from the proteome of circulating monocytes in Chinese premenopausal women with discordant hip BMD92. The significance of ANXA2 and GSN proteins to osteoporosis was later cross-validated in independent cellular proteomics studies in white women with varying BMD93,94. Of note, ANXA2 could regulate osteoclastogenesis93 (FIG. 3) and the function of osteoblasts95, and increased plasma levels of this protein were shown to serve as a risk biomarker for osteoporosis and to predict osteoporotic fracture in Chinese individuals aged over 65 years95. The first comprehensive proteome knowledgebase (BOX 1) for human monocytes, developed in 2017 (REF.96), involves a total of 2,237 unique protein-encoding genes and provides a reference map for future in-depth research on monocyte biology and osteoporosis.
In addition to protein biomarkers developed from cellular proteomics studies, a number of potential protein biomarkers have been identified directly from plasma or serum proteomics studies. For example, one group identified a diagnostic fingerprint (BOX 1) for bone turnover in the serum of postmenopausal women, which could serve as a reflection of the increased osteoclast activity, leading to increased bone turnover, that is associated with decreasing BMD97. Besides, multiple layers of evidence were integrated to further ascertain the significance of proteins and genes of interest for osteoporosis. For example, a prior proteomics study discovered that superoxide dismutase 2 (SOD2) in circulating monocytes was significantly upregulated at protein level in vivo in Chinese individuals with low versus high hip BMD92. By integrating evidence from DNA, RNA and protein levels, a pursuant study ascertained SOD2 as a susceptibility gene for osteoporosis in Chinese populations98. Furthermore, proteome-based network and pathway studies identified important groups of proteins for osteoporosis99,100. For example, a mass spectrometry-based quantitative proteomics study integrated with network analysis identified two novel pathways, that is, regulation of actin cytoskeleton and “leukocyte transendothelial migration” as being related to osteoporosis96.
Some serum or plasma-derived protein biomarkers with diagnostic potential for osteoporotic fracture were identified from cross-sectional studies (Supplementary Table 5). Furthermore, serum-derived protein biomarkers predictive of incident osteoporotic fracture were identified from prospective studies (Supplementary Table 6). For example, a prospective study101 suggested that increased serum levels of sclerostin are associated with increased risk of hip fracture. Excitingly, the antibody of sclerostin, that is, romosozumab (Evenity), has been approved by the FDA as a novel drug to treat osteoporosis in postmenopausal women with high risk of fracture.
Metabolomics.
The human metabolome (BOX 1) reflects downstream changes in gene sequence and gene and/or protein expression. Thus, small changes in genes, their transcription and the translation of proteins that are hard to detect in other omics studies can be amplified at the metabolomic level102, enabling the most powerful screening of biomarkers or therapeutic targets closely related to disease or phenotype. Of note, preclinical studies carried out in postmenopausal osteoporosis animal models have shown different metabolomic profiles compared with control animals, especially regarding changes in amino acid and lipid metabolism103,104.
The first metabolomics study of osteoporosis in humans compared high and low BMD groups and reported four distinguishing metabolites: lactate, acetone, acetate and glutamine105. Subsequently, eight metabolomics studies investigating osteoporosis in humans have been published and have reported >100 metabolites associated with BMD (Supplementary Table 7). In line with the findings of animal studies, human studies also highlight the importance of amino acid and lipid metabolism in bone. In particular, many amino acids were reported to be associated with BMD by multiple independent studies105–109. For example, glutamine is increased107,110 and proline is decreased in menopausal women with decreased BMD109. Moreover, carbohydrate108 and nucleoside106 metabolism are also altered in women with decreased BMD. Finally, BMD-related metabolic pathways have been reported, including bile acid biosynthesis, which might affect intestinal calcium absorption107, and the tricarboxylic acid cycle, which might be related to increased oxidative stress in patients with low BMD108.
Metabolomics research of osteoporosis is in the early stages. For example, all the current studies (Supplementary Table 7) are cross-sectional. Although two studies used Mendelian randomization (BOX 1) to provide potential evidence for a causal relationship of identified metabolites with osteoporosis110,111, well designed longitudinal studies are necessary. In addition, potential sex differences in osteoporosis-related metabolites should be investigated, as most published studies only include women. For meaningful data generation, metabolite identity validation and absolute quantification should follow untargeted metabolomics analysis, which was used in all the current studies. These steps are necessary for conducting functional research of identified metabolites to derive meaningful biological knowledge112.
Summary.
Currently, functional genomics and other omics studies of osteoporosis have limitations, for example, the use of inappropriate tissue and/or cell specimens, small sample sizes and subsequent low statistical power, poorly characterized cohorts, and/or lack of healthy control individuals. Normal bone samples from healthy individuals are difficult to obtain as bone biopsy is invasive and carries a small risk of bleeding, fracture and/or infection. Consequently, bone samples obtained by biopsy from patients with osteoarthritis have been used as surrogate controls in several studies of osteoporosis86. However, the use of osteoarthritis bone samples might not be an ideal study design, as it is questionable to what extent cells from osteoarthritis bone resemble normal bone cells. Several studies have suggested that the trabecular structure and gene expression of osteoarthritis bone, at least in the vicinity of articular tissue, might differ from those of normal healthy bone113,114.
In contrast to genetic variants, functional genomic and other omics changes are cell and tissue specific and therefore studies must be performed in bone or bone lineage cells. Moreover, bone itself is a heterogeneous tissue, composed of a number of different cell types (FIG. 3) and failure to account for the cellular heterogeneity might lead to false-positive or false-negative results115,116. A large consortium aiming to generate large-scale data for bone or bone lineage cells, such as the Cancer Genome Atlas Program for cancer, would greatly improve our ability to understand, diagnose, treat and prevent osteoporosis.
An integrative approach
Although each omics approach has provided useful information to begin to understand the pathophysiology of osteoporosis, no single omics approach (BOX 1) can capture the entire biological complexity of osteoporosis. Thus, approaches to integrate multi-omics data have emerged to create a holistic picture of osteoporosis. Next, we describe ways in which integrative multi-omics can help GWAS loci interpretation and the identification and prioritization of osteoporosis-related genes and pathways.
Integrating multi-omics data to interpret GWAS.
The function of genetic variants located in coding regions of DNA, that is, in exons, is straightforward to study. However, the majority of GWAS susceptibility loci are located in non-coding or intergenic regions117, which poses challenges to the functional interpretation of risk variants and the translation of GWAS findings into clinical application. Therefore, the first crucial step is to prioritize and/or pinpoint the true causal and/or functional variants118.
A complementary approach to fine-mapping is to leverage multiple layers of epigenomic or functional data to assign potential functions for genetic variants identified by GWAS. For example, the assay for transposase-accessible chromatin using sequencing (known as ATAC-seq) is widely used to evaluate the accessibility of chromatin at the location of possible functional GWAS variants. Databases like RegulomeDB119 can also be used to annotate SNPs with known and predicted regulatory elements such as regions of DNase I hypersensitivity and binding sites of transcription factors. The systematic analyses of the epigenetic features in all known osteoporosis risk loci identified by GWAS could identify common regulatory elements for osteoporosis-associated genes and might predict new osteoporosis-linked genes with similar regulatory features120. Using machine learning algorithms, researchers can extract intricate regulatory information underlying GWAS variants and use this knowledge to predict new osteoporosis susceptibility variants121. For example, one study predicted 37,584 new candidate osteoporosis-associated SNPs by applying a random forest model121. However, epigenetic regulatory analysis on GWAS variants have largely relied on reference epigenomic data (for example, NIH Roadmap Epigenomics Project) in which epigenomic profiles of each unique type of human tissues or cells were mostly generated from only one individual. Therefore, the allelic effects of GWAS variants on the inter-individual variation of cell-specific and/or tissue-specific epigenomic profiles (for example, individuals with different genotypes at a specific loci might have different levels of chromatin accessibility at the corresponding region in a specific tissue or cell of interest) are largely unknown. Future investigation with genomic and epigenomic data generated from the same set of a large number of participants should help uncover the allelic regulatory effects of osteoporosis-associated genetic variants on the epigenome.
Expression quantitative trait loci (eQTL) analysis (BOX 1) in osteoblasts122 and osteoclasts123 has helped to characterize the function of causal genetic variants located outside of exons and their downstream target genes in osteoporosis. For example, microarray profiles of undifferentiated osteoblasts from 95 unrelated donors of Swedish origin were used to identify eQTL for BMD GWAS122. Data in cultured primary osteoclasts from 158 individuals were used to identify colocalizing eQTL for genes associated with BMD123. Moreover, eQTL analysis on other bone-related cells and tissues, such as circulating monocytes120, lymphocytes124 or whole blood124,125, have also been widely applied to decipher the regulatory roles of GWAS variants in osteoporosis. For example, lymphocytes and peripheral blood eQTL were used to identify LINC00339 as a potentially causal gene for BMD GWAS124. The current sample sizes of eQTL studies on bone tissues are fairly small (<200); future studies in large samples are needed to increase the statistical power. In addition, the observed eQTL mapping on osteoporosis-associated GWAS variants might be accidental owing to high linkage disequilibrium with the true causal variant. To address this problem, several colocalization analysis approaches126,127 have been developed to determine whether susceptibility loci identified by GWAS and eQTL share the same causal variant.
Importantly, studies in 3D chromatin interactions, which are measured by high-throughput chromatin interaction capture (known as Hi-C) analysis, have revealed a new regulatory mechanism by which functional GWAS variants could regulate distant gene expression via the formation of long-range (BOX 1) loops6. For example, the first Hi-C analysis in human osteoblasts and osteocytes revealed that the genes that had chromatin interactions with causal eBMD-associated variants were strongly enriched for osteocyte signature genes6, suggesting that osteoporosis-associated genetic variants affect crucial bone-related genes by long-range regulation. A 2019 study further demonstrated that 57% of the identified chromatin interaction pairs linking osteoporosis-associated SNPs to gene promoters fell in distant genes only128. In one notable study, researchers found that five functional SNPs were located in an intergenic super-enhancer that could regulate the expression of RANKL (FIG. 3) via long-range chromosomal interactions (>100 kb)129. In addition, the intergenic non-coding osteoporosis-associated GWAS variant rs6426749 at 1p36.12 could regulate LINC00339 expression via long-range loop formation (~ 360 kb)124. Downregulation of LINC00339 significantly increases the expression of CDC42 in osteoblasts, which is a pivotal regulator involved in bone metabolism124.
When integrating multi-omics data to interpret osteoporosis GWAS susceptibility loci, the most commonly used method is to build evidence for a functional variant signal through the pairwise analyses of data sets6,124. Multi-dimensional methods that can analyse three or more data sets simultaneously (for example, Bayesian models130) will further advance our understanding of osteoporosis aetiology.
Gene and network-based studies.
As genes do not function in isolation, the integration of different omics datasets at the level of genes and gene networks represents a critical approach to further increase our system-level understanding of the genetic basis of osteoporosis. For example, one group applied the weighted gene co-expression network analysis (WGCNA)131 tool to transcriptomic expression data from bone tissues obtained from the Hybrid Mouse Diversity Panel132 (BOX 1) and identified gene co-expression networks (termed as modules) that are highly associated with BMD133. From a specific BMD-associated module, they selected two ‘hub’ genes (that is, genes that are highly correlated with many other genes in a given module), Maged1 and Pard6g, for subsequent in vitro and in vivo molecular function analyses. Specifically, knocking down the expression of Maged1 and Pard6g in osteoblasts lead to altered osteoblast proliferation and differentiation, and mice deficient in Maged1 had decreased BMD133. These results demonstrated the power of using a systems biology approach to discover novel genes and regulatory mechanisms involved in osteoporosis.
The network information obtained from the above analyses can be used to identify or prioritize novel disease-related genes that might not reach genome-wide significance in conventional GWAS analysis. For example, one group constructed a WGCNA network for 1,574 genes that had GWAS nominal evidence of association (P < 0.05) with BMD using gene expression data from transiliac bone biopsy samples obtained from 84 postmenopausal white women (39 with BMD T-score ≤ −1.0 and 45 with BMD T-score > −1.0) and discovered one module containing 44 highly interconnected genes significantly associated with BMD134. Further detailed submodule analyses suggested several novel candidate genes (for example, HOMER1 and SPTBN1) that are important for bone metabolism134. HOMER1 encodes a member of the Homer family of dendritic proteins and has an important role in glutamate signalling, which is involved in regulation of bone remodelling135. Interestingly, a 2017 study using a similar strategy also predicted the gene SPTBN1 as a causal GWAS gene51. SPTBN1 encodes a molecular scaffolding protein and is broadly expressed in many tissues and cell types, including high expression in osteoblasts51. Moreover, Sptbn1 heterozygous knockout (Sptbn1+/−) mice displayed significantly altered whole body BMD compared with the controls51, confirming the critical role of this gene in the regulation of BMD. In addition to WGCNA, other more sophisticated algorithms for network construction and analysis of multi-omics data are quickly emerging and therefore provide exciting opportunities for future osteoporosis research136,137.
Validation through functional studies
With numerous genes identified from GWAS, different omics studies and integrative analyses, the next step is to validate their functional roles in the pathogenesis of osteoporosis. The development of the CRISPR–Cas9 genome editing tool138 has revolutionized molecular biology and genetics. For osteoporosis-associated gene functional studies, CRISPR–Cas9 has been used for coding-gene knockout139, gene mutation repair140 and validation of the effects of non-coding enhancers124,129. For example, CRISPR–Cas9-mediated knockouts of DAAM2 in osteoblast cell lines resulted in a marked reduction in inducible mineralization6.
Databases that record the consequences of disrupting each of the protein-coding genes in mice provide rich resources for validating the functional effects of candidate genes derived from GWAS and different omics studies. For example, The Origins of Bone and Cartilage Disease project, which is part of the International Knockout Mouse Consortium, aims to test over 1,600 mutant C57BL/6 strains with systemic single gene deletions for abnormalities of bone and cartilage. Although not specific to bone, similar databases, such as Mouse Genome Informatics and International Mouse Phenotyping Consortium, also provide integrated genetic, genomic and biological data to facilitate the study of osteoporosis. In addition to acting as validation resources, the identification of extreme skeletal phenotypes in mutant mouse lines in these databases can also serve as a new approach for osteoporosis-associated gene discovery141. For example, a 2014 study142 analysed high-throughput screening data for 3,762 distinct global gene knockout mouse lines and identified multiple genes affecting bone mass.
Conclusions
As mentioned above, functional genomics studies of osteoporosis need to be conducted using skeletal samples and should also account for cellular heterogeneity, both of which pose obvious difficulties for human studies. The recent rapid development of single-cell omics technologies provides us with powerful new tools to individually or even simultaneously (that is, single-cell multi-omics approaches) assess various omics features at the resolution of individual cells143, which can revolutionize our understanding of cellular heterogeneity. For example, using single-cell RNA sequencing, a 2018 study was able to successfully identify the long-sought multipotent human skeletal stem cell that exclusively differentiates into bone, cartilage and stroma but not adipose144. Application of the single-cell omics approaches to the field of osteoporosis research will enable us to explore known and unknown cellular heterogeneity in bone cells and tissues and dissect the cell-intrinsic and cell-specific mechanisms underlying osteoporosis.
To bridge the gap between the statistical associations and biological regulatory mechanisms, various experimental approaches can be used to test the function of osteoporosis susceptibility loci. Rather than testing the function of genetic variants one-by-one, high-throughput methods developed in 2017 (REF.145) (for example, massively parallel reporter assay and self-transcribing active regulatory region sequencing) can be used to examine thousands of variants in a single experiment. Applying such high-throughput methods into the post-GWAS studies of osteoporosis will lay functional evidence and mechanisms over the association results.
Another area for future osteoporosis research is the study of the human gut microbiome146 — a number of studies in animal models147 and humans148 have provided compelling evidence for the importance of the gut microbiome in bone metabolism and health. With the strong interactions known to exist between the gut microbiome and the host genome149, future host and microbiome multi-omics integration studies might lead to a major breakthrough in the prediction and therapeutic treatment of osteoporosis.
Supplementary Material
Key points.
Osteoporosis, which is the most common bone disorder worldwide, and its related traits (low bone mineral density and osteoporotic fracture) are highly heritable.
Multiple omics technologies, including genomics, transcriptomics, epigenomics, proteomics and metabolomics, have been applied to identify the molecular factors contributing to the pathogenesis of osteoporosis.
Building upon the success in single-omics discovery research, studies have integrated data from different omics levels to better elucidate the molecular and functional mechanisms for osteoporosis.
Integration of omics approaches can provide a holistic road map to comprehensively illuminate the complex pathogenesis of osteoporosis and fulfil the potential of personalized disease risk prediction, intervention and treatment as well as drug development or re-purposing.
Acknowledgements
T.L.Y. and S.S.D. acknowledge the support of the National Natural Science Foundation of China (31771399, 31970569 and 81573241), the Innovative Talent Promotion Plan of Shaanxi Province for Young Sci-Tech New Star (2018KJXX-010) and the special guidance funds for the construction of world-class universities (disciplines) and characteristic development in central universities. H.S. and H.W.D. acknowledge the support of grants from the National Institutes of Health (R01AR059781, P20GM109036, R01MH107354, R01MH104680, R01GM109068, R01AR069055, U19AG055373, R01DK115679), the Edward G. Schlieder Endowment and the Drs. W. C. Tsai and P. T. Kung Professorship in Biostatistics from Tulane University.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Endocrinology thanks D. Karasik, J. Tobias and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41574-019-0282-7.
References
- 1.Kanis JA Diagnosis of osteoporosis. Osteoporos. Int 7, S108–S116 (1997). [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis : report of a WHO study group [meeting held in Rome from 22 to 25 June 1992] (WHO, 1994). [PubMed] [Google Scholar]
- 3.Johnell O et al. Predictive value of BMD for hip and other fractures. J. Bone Miner. Res 20, 1185–1194 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Peacock M, Turner CH, Econs MJ & Foroud T Genetics of osteoporosis. Endocr. Rev 23, 303–326 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Deng HW et al. Relevance of the genes for bone mass variation to susceptibility to osteoporotic fractures and its implications to gene search for complex human diseases. Genet. Epidemiol 22, 12–25 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Morris JA et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat. Genet 51, 258–266 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; As the largest GWAS of BMD until 2019, this paper performed analyses in over 426,000 individuals from the UK Biobank and identified 518 loci associated with BMD, 301 of which were novel.
- 7.Sun YV & Hu YJ Integrative analysis of multi-omics data for discovery and functional studies of complex human diseases. Adv. Genet 93, 147–190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hasin Y, Seldin M & Lusis A Multi-omics approaches to disease. Genome Biol. 18, 83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raisz LG Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J. Clin. Invest 115, 3318–3325 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pittenger MF et al. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Long F Building strong bones: molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol 13, 27–38 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Udagawa N et al. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl Acad. Sci. USA 87, 7260–7264 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyle WJ, Simonet WS & Lacey DL Osteoclast differentiation and activation. Nature 423, 337–342 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Teitelbaum SL Bone resorption by osteoclasts. Science 289, 1504–1508 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Knothe Tate ML, Adamson JR, Tami AE & Bauer TW The osteocyte. Int. J. Biochem. Cell Biol 36, 1–8 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Dallas SL, Prideaux M & Bonewald LF The osteocyte: an endocrine cell and more. Endocr. Rev 34, 658–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sims NA & Walsh NC Intercellular cross-talk among bone cells: new factors and pathways. Curr. Osteoporos. Rep 10, 109–117 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Sabik OL & Farber CR Using GWAS to identify novel therapeutic targets for osteoporosis. Transl. Res 181, 15–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards JB, Zheng HF & Spector TD Genetics of osteoporosis from genome-wide association studies: advances and challenges. Nat. Rev. Genet 13, 576–588 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Liu YJ, Zhang L, Papasian CJ & Deng HW Genome-wide association studies for osteoporosis: a 2013 update. J. Bone Metab 21, 99–116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Styrkarsdottir U et al. Multiple genetic loci for bone mineral density and fractures. N. Engl. J. Med 358, 2355–2365 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Richards JB et al. Bone mineral density, osteoporosis, and osteoporotic fractures: a genome-wide association study. Lancet 371, 1505–1512 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Styrkarsdottir U et al. New sequence variants associated with bone mineral density. Nat. Genet 41, 15–17 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Xiong DH et al. Genome-wide association and follow-up replication studies identified ADAMTS18 and TGFBR3 as bone mass candidate genes in different ethnic groups. Am. J. Hum. Genet 84, 388–398 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng HF et al. Whole-genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature 526, 112–117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; By using Whole-genome sequencing, this study identified a low-frequency non-coding variant near the novel locus (EN1) associated with lumbar spine BMD and the risk of fracture.
- 26.Rivadeneira F et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat. Genet 41, 1199–1206 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richards JB et al. Collaborative meta-analysis: associations of 150 candidate genes with osteoporosis and osteoporotic fracture. Ann. Intern. Med 151, 528–537 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Estrada K et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet 44, 491–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moayyeri A et al. Genetic determinants of heel bone properties: genome-wide association meta-analysis and replication in the GEFOS/GENOMOS consortium. Hum. Mol. Genet 23, 3054–3068 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Medina-Gomez C et al. Life-course genome-wide association study meta-analysis of total body bmd and assessment of age-specific effects. Am. J. Hum. Genet 102, 88–102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parikka V et al. Estrogen responsiveness of bone formation in vitro and altered bone phenotype in aged estrogen receptor-α-deficient male and female mice. Eur. J. Endocrinol 152, 301–314 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Maatta JA et al. Inactivation of estrogen receptor α in bone-forming cells induces bone loss in female mice. FASEB J. 27, 478–488 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Crockett JC, Rogers MJ, Coxon FP, Hocking LJ & Helfrich MH Bone remodelling at a glance. J. Cell Sci 124, 991–998 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Collins R What makes UK Biobank special? Lancet 379, 1173–1174 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Kemp JP et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat. Genet 49, 1468–1475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riggs BL, Khosla S & Melton LJ 3rd. A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J. Bone Miner. Res 13, 763–773 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Koller DL et al. Meta-analysis of genome-wide studies identifies WNT16 and ESR1 SNPs associated with bone mineral density in premenopausal women. J. Bone Miner. Res 28, 547–558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C et al. Susceptibility genes for osteoporotic fracture in postmenopausal Chinese women. J. Bone Miner. Res 27, 2582–2591 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Zheng HF et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLOS Genet. 8, e1002745 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simonet WS et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89, 309–319 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Xiong L et al. Lrp4 in osteoblasts suppresses bone formation and promotes osteoclastogenesis and bone resorption. Proc. Natl Acad. Sci. USA 112, 3487–3492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaprio J Twins and the mystery of missing heritability: the contribution of gene-environment interactions. J. Intern. Med 272, 440–448 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang TL et al. Genome-wide copy-number-variation study identified a susceptibility gene, UGT2B17, for osteoporosis. Am. J. Hum. Genet 83, 663–674 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first study to investigate the contribution of copy number variation to osteoporosis identified a deletion variant of UGT2B17 associated with osteoporotic fracture.
- 44.Zhu W et al. Gene-based GWAS analysis for consecutive studies of GEFOS. Osteoporos. Int 29, 2645–2658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang LS et al. Pathway-based genome-wide association analysis identified the importance of regulation-of-autophagy pathway for ultradistal radius BMD. J. Bone Miner. Res 25, 1572–1580 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang H, Chanda P, Alonso A, Bader JS & Arking DE Gene-based tests of association. PLOS Genet. 7, e1002177 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu YZ et al. Powerful bivariate genome-wide association analyses suggest the Sox6 gene influencing both obesity and osteoporosis phenotypes in males. PLOS ONE 4, e6827 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Medina-Gomez C et al. Bivariate genome-wide association meta-analysis of pediatric musculoskeletal traits reveals pleiotropic effects at the SREBF1/TOM1L2 locus. Nat. Commun 8, 121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pei YF et al. Joint association analysis identified 18 new loci for bone mineral density. J. Bone Miner. Res 34, 1086–1094 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Greenbaum J et al. Increased detection of genetic loci associated with risk predictors of osteoporotic fracture using a pleiotropic cFDR method. Bone 99, 62–68 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calabrese GM et al. Integrating GWAS and co-expression network data identifies bone mineral density genes SPTBN1 and MARK3 and an osteoblast functional module. Cell Syst. 4, 46–59.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boyle EA, Li YI & Pritchard JK An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee SH et al. Prediction of future osteoporotic fracture occurrence by genetic profiling: a 6-year follow-up observational study. J. Clin. Endocrinol. Metab 101, 1215–1224 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Ho-Le TP, Center JR, Eisman JA, Nguyen HT & Nguyen TV Prediction of bone mineral density and fragility fracture by genetic profiling. J. Bone Miner. Res 32, 285–293 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Kim SK Identification of 613 new loci associated with heel bone mineral density and a polygenic risk score for bone mineral density, osteoporosis and fracture. PLOS ONE 13, e0200785 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nelson MR et al. The support of human genetic evidence for approved drug indications. Nat. Genet 47, 856–860 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Xia WF et al. Swedish mutant APP suppresses osteoblast differentiation and causes osteoporotic deficit, which are ameliorated by N-acetyl-L-cysteine. J. Bone Miner. Res 28, 2122–2135 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cornelius C et al. Osteoporosis and Alzheimer pathology: role of cellular stress response and hormetic redox signaling in aging and bone remodeling. Front. Pharmacol 5, 120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sato T et al. Donepezil prevents RANK-induced bone loss via inhibition of osteoclast differentiation by downregulating acetylcholinesterase. Heliyon 1, e00013 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karasik D et al. Heritability and genetic correlations for bone microarchitecture: the framingham study families. J. Bone Miner. Res 32, 106–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trajanoska K et al. Assessment of the genetic and clinical determinants of fracture risk: genome wide association and mendelian randomisation study. BMJ 362, k3225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nielson CM et al. Novel genetic variants associated with increased vertebral volumetric BMD, reduced vertebral fracture risk, and increased expression of SLC1A3 and EPHB2. J. Bone Miner. Res 31, 2085–2097 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo Y et al. Genome-wide association study identifies ALDH7A1 as a novel susceptibility gene for osteoporosis. PLOS Genet. 6, e1000806 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greene CS, Penrod NM, Williams SM & Moore JH Failure to replicate a genetic association may provide important clues about genetic architecture. PLOS ONE 4, e5639 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kraft P, Zeggini E & Ioannidis JP Replication in genome-wide association studies. Stat. Sci 24, 561–573 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu YJ, Zhang L, Pei YF, Papasian CJ & Deng HW On genome-wide association studies and their meta-analyses: lessons learned from osteoporosis studies. J. Clin. Endocrinol. Metab 98, E1278–E1282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kichaev G et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLOS Genet. 10, e1004722 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benner C et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Greenbaum J & Deng HW A statistical approach to fine mapping for the identification of potential causal variants related to bone mineral density. J. Bone Miner. Res 32, 1651–1658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Benner C et al. Prospects of fine-mapping trait-associated genomic regions by using summary statistics from genome-wide association studies. Am. J. Hum. Genet 101, 539–551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu YZ et al. A novel pathophysiological mechanism for osteoporosis suggested by an in vivo gene expression study of circulating monocytes. J. Biol. Chem 280, 29011–29016 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Mohan S, Hu Y & Edderkaoui B Chemokine receptor 3 is a negative regulator of trabecular bone mass in female mice. J. Cell. Biochem 120, 13974–13984 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Fitzpatrick LA et al. Targeted deletion of histidine decarboxylase gene in mice increases bone formation and protects against ovariectomy-induced bone loss. Proc. Natl Acad. Sci. USA 100, 6027–6032 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takuma A et al. Dexamethasone enhances osteoclast formation synergistically with transforming growth factor-β by stimulating the priming of osteoclast progenitors for differentiation into osteoclasts. J. Biol. Chem 278, 44667–44674 (2003). [DOI] [PubMed] [Google Scholar]
- 76.Eskildsen T et al. MicroRNA-138 regulates osteogenic differentiation of human stromal (mesenchymal) stem cells in vivo. Proc. Natl Acad. Sci. USA 108, 6139–6144 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang L et al. The long non-coding RNA-ORLNC1 regulates bone mass by directing mesenchymal stem cell fate. Mol. Ther 27, 394–410 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao K et al. Hsa_Circ_0001275: a potential novel diagnostic biomarker for postmenopausal osteoporosis. Cell Physiol. Biochem 46, 2508–2516 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Vidal C, Cachia A & Xuereb-Anastasi A Effects of a synonymous variant in exon 9 of the CD44 gene on pre-mRNA splicing in a family with osteoporosis. Bone 45, 736–742 (2009). [DOI] [PubMed] [Google Scholar]
- 80.Scotti MM & Swanson MS RNA mis-splicing in disease. Nat. Rev. Genet 17, 19–32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marini F, Cianferotti L & Brandi ML Epigenetic mechanisms in bone biology and osteoporosis: can they drive therapeutic choices? Int. J. Mol. Sci 17, E1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van Meurs JB, Boer CG, Lopez-Delgado L & Riancho JA Role of epigenomics in bone and cartilage disease. J. Bone Miner. Res 34, 215–230 (2019). [DOI] [PubMed] [Google Scholar]
- 83.Reppe S et al. Methylation of bone SOST, its mRNA, and serum sclerostin levels correlate strongly with fracture risk in postmenopausal women. J. Bone Miner. Res 30, 249–256 (2015). [DOI] [PubMed] [Google Scholar]
- 84.Li X et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem 280, 19883–19887 (2005). [DOI] [PubMed] [Google Scholar]
- 85.Cao Y & Wang B Expression of sclerostin in osteoporotic fracture patients is associated with DNA methylation in the CpG island of the SOST gene. Int. J. Genomics 2019, 7076513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Delgado-Calle J et al. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheum. 65, 197–205 (2013). [DOI] [PubMed] [Google Scholar]; The first epigenome-wide association study of human bone detected 241 differentially methylated CpG sites in femoral head trabecular bone specimens between 27 patients with osteoporotic hip fractures and 26 patients with hip osteoarthritis.
- 87.Reppe S et al. Distinct DNA methylation profiles in bone and blood of osteoporotic and healthy postmenopausal women. Epigenetics 12, 674–687 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morris JA et al. Epigenome-wide association of DNA methylation in whole blood with bone mineral density. J. Bone Miner. Res 32, 1644–1650 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fernandez-Rebollo E et al. Primary osteoporosis is not reflected by disease-specific DNA methylation or accelerated epigenetic age in blood. J. Bone Miner. Res 33, 356–361 (2018). [DOI] [PubMed] [Google Scholar]
- 90.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deng FY et al. Proteomic analysis of circulating monocytes in Chinese premenopausal females with extremely discordant bone mineral density. Proteomics 8, 4259–4272 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Deng FY et al. Peripheral blood monocyte-expressed ANXA2 gene is involved in pathogenesis of osteoporosis in humans. Mol. Cell Proteom 10, M111.011700 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Deng FY et al. Is GSN significant for hip BMD in female Caucasians? Bone 63, 69–75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou X et al. Anxa2 attenuates osteoblast growth and is associated with hip BMD and osteoporotic fracture in Chinese elderly. PLOS ONE 13, e0194781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zeng Y et al. Mass spectrometry based proteomics profiling of human monocytes. Protein Cell 8, 123–133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first comprehensive proteome knowledgebase for human monocytes was developed in 2017; it involves a total of 2,237 unique protein-encoding genes and provides a reference map for future in-depth research on monocyte biology and osteoporosis.
- 97.Bhattacharyya S, Siegel ER, Achenbach SJ, Khosla S & Suva LJ Serum biomarker profile associated with high bone turnover and BMD in postmenopausal women. J. Bone Miner. Res 23, 1106–1117 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Deng FY et al. An integrative study ascertained SOD2 as a susceptibility gene for osteoporosis in Chinese. J. Bone Miner. Res 26, 2695–2701 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang L et al. Network-based proteomic analysis for postmenopausal osteoporosis in Caucasian females. Proteomics 16, 12–28 (2016). [DOI] [PubMed] [Google Scholar]
- 100.Zeng Y et al. Quantitative proteomics and integrative network analysis identified novel genes and pathways related to osteoporosis. J. Proteomics 142, 45–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Arasu A et al. Serum sclerostin and risk of hip fracture in older Caucasian women. J. Clin. Endocrinol. Metab 97, 2027–2032 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Johnson CH, Ivanisevic J & Siuzdak G Metabolomics: beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol 17, 451–459 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wu QY et al. Long non-coding RNAs: a new regulatory code for osteoporosis. Front. Endocrinol 9, 587 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ma B et al. Metabolomic profiles delineate signature metabolic shifts during estrogen deficiency-induced bone loss in rat by GC-TOF/MS. PLOS ONE 8, e54965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.You YS et al. Association between the metabolome and low bone mineral density in Taiwanese women determined by 1H NMR spectroscopy. J. Bone Miner. Res 29, 212–222 (2014). [DOI] [PubMed] [Google Scholar]; The first metabolomics study of osteoporosis in humans compared high and low BMD groups and reported four distinguishing metabolites: lactate, acetone, acetate and glutamine.
- 106.Miyamoto T et al. Metabolomics-based profiles predictive of low bone mass in menopausal women. Bone Rep. 9, 11–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao Q et al. Metabolomic profiles associated with bone mineral density in US Caucasian women. Nutr. Metab 15, 57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yu L et al. Association between metabolic profiles in urine and bone mineral density of pre- and postmenopausal Chinese women. Menopause 26, 94–102 (2019). [DOI] [PubMed] [Google Scholar]
- 109.Cabrera D et al. Association of plasma lipids and polar metabolites with low bone mineral density in Singaporean-Chinese menopausal women: a pilot study. Int. J. Environ. Res. Public Health 15, E1045 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moayyeri A et al. Metabolomic pathways to osteoporosis in middle-aged women: a genome-metabolome-wide Mendelian randomization study. J. Bone Miner. Res 33, 643–650 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu L et al. Assessing the associations of blood metabolites with osteoporosis: a Mendelian randomization study. J. Clin. Endocrinol. Metab 103, 1850–1855 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD & McLean JA Untargeted metabolomics strategies — challenges and emerging directions. J. Am. Soc. Mass Spectrom 27, 1897–1905 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ding C, Cicuttini F & Jones G Tibial subchondral bone size and knee cartilage defects: relevance to knee osteoarthritis. Osteoarthritis Cartilage 15, 479–486 (2007). [DOI] [PubMed] [Google Scholar]
- 114.Chou CH et al. Genome-wide expression profiles of subchondral bone in osteoarthritis. Arthritis Res. Ther 15, R190 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jaffe AE & Irizarry RA Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 15, R31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Teschendorff AE & Zheng SC Cell-type deconvolution in epigenome-wide association studies: a review and recommendations. Epigenomics 9, 757–768 (2017). [DOI] [PubMed] [Google Scholar]
- 117.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schaid DJ, Chen W & Larson NB From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet 19, 491–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Boyle AP et al. Annotation of functional variation in personal genomes using regulomeDB. Genome Res. 22, 1790–1797 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Guo Y et al. Integrating epigenomic elements and gwass identifies bdnf gene affecting bone mineral density and osteoporotic fracture risk. Sci. Rep 6, 30558 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yao S et al. Regulatory element-based prediction identifies new susceptibility regulatory variants for osteoporosis. Hum. Genet 136, 963–974 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Grundberg E et al. Population genomics in a disease targeted primary cell model. Genome Res. 19, 1942–1952 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mullin BH et al. Expression quantitative trait locus study of bone mineral density gwas variants in human osteoclasts. J. Bone Miner. Res 33, 1044–1051 (2018). [DOI] [PubMed] [Google Scholar]
- 124.Chen XF et al. An osteoporosis risk SNP at 1p36.12 acts as an allele-specific enhancer to modulate LINC00339 expression via long-range loop formation. Am. J. Hum. Genet 102, 776–793 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provides a mechanistic explanation for the influence of the 1p36.12 locus on human osteoporosis, using multiple omics technologies to bridge GWAS results to physiology.
- 125.Meng XH et al. Integration of summary data from GWAS and eQTL studies identified novel causal BMD genes with functional predictions. Bone 113, 41–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Giambartolomei C et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLOS Genet. 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hormozdiari F et al. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet 99, 1245–1260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chesi A et al. Genome-scale Capture C promoter interactions implicate effector genes at GWAS loci for bone mineral density. Nat. Commun 10, 1260 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhu DL et al. Multiple functional variants at 13q14 Risk locus for osteoporosis regulate RANKL expression through long-range super-enhancer. J. Bone Miner. Res 33, 1335–1346 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Wang Q et al. A Bayesian framework that integrates multi-omics data and gene networks predicts risk genes from schizophrenia GWAS data. Nat. Neurosci 22, 691–699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lusis AJ et al. The Hybrid Mouse Diversity Panel: a resource for systems genetics analyses of metabolic and cardiovascular traits. J. Lipid Res 57, 925–942 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Calabrese G et al. Systems genetic analysis of osteoblast-lineage cells. PLOS Genet. 8, e1003150 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen YC et al. Integrative analysis of genomics and transcriptome data to identify potential functional genes of BMDs in females. J. Bone Miner. Res 31, 1041–1049 (2016). [DOI] [PubMed] [Google Scholar]
- 135.Chenu C, Serre CM, Raynal C, Burt-Pichat B & Delmas PD Glutamate receptors are expressed by bone cells and are involved in bone resorption. Bone 22, 295–299 (1998). [DOI] [PubMed] [Google Scholar]
- 136.Zhang JG et al. Integrative analysis of transcriptomic and epigenomic data to reveal regulation patterns for BMD variation. PLOS ONE 10, e0138524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Al-Barghouthi BM & Farber CR Dissecting the genetics of osteoporosis using systems approaches. Trends Genet. 35, 55–67 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Duan X et al. Deficiency of ATP6V1H causes bone loss by inhibiting bone resorption and bone formation through the TGF-β1 pathway. Theranostics 6, 2183–2195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Saito A et al. Targeted reversion of induced pluripotent stem cells from patients with human cleidocranial dysplasia improves bone regeneration in a rat calvarial bone defect model. Stem Cell Res. Ther 9, 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Freudenthal B et al. Rapid phenotyping of knockout mice to identify genetic determinants of bone strength. J. Endocrinol 231, R31–R46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Brommage R et al. High-throughput screening of mouse gene knockouts identifies established and novel skeletal phenotypes. Bone Res. 2, 14034 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chappell L, Russell AJC & Voet T Single-cell (multi)omics technologies. Annu. Rev. Genomics Hum. Genet 19, 15–41 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Chan CKF et al. Identification of the human skeletal stem cell. Cell 175, 43–56.e21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Santiago-Algarra D, Dao LTM, Pradel L, Espana A & Spicuglia S Recent advances in high-throughput approaches to dissect enhancer function. F1000Res 6, 939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen YC, Greenbaum J, Shen H & Deng HW Association between gut microbiota and bone health: potential mechanisms and prospective. J. Clin. Endocrinol. Metab 102, 3635–3646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jones RM, Mulle JG & Pacifici R Osteomicrobiology: the influence of gut microbiota on bone in health and disease. Bone 115, 59–67 (2018). [DOI] [PubMed] [Google Scholar]
- 148.Wang J et al. Diversity analysis of gut microbiota in osteoporosis and osteopenia patients. PeerJ 5, e3450 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Goodrich JK, Davenport ER, Clark AG & Ley RE The relationship between the human genome and microbiome comes into view. Annu. Rev. Genet 51, 413–433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Timpson NJ, Greenwood CMT, Soranzo N, Lawson DJ & Richards JB Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat. Rev. Genet 19, 110–124 (2018). [DOI] [PubMed] [Google Scholar]
- 151.Price AL, Zaitlen NA, Reich D & Patterson N New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet 11, 459–463 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang L et al. A new method for estimating effect size distribution and heritability from genome-wide association summary results. Hum. Genet 135, 171–184 (2016). [DOI] [PubMed] [Google Scholar]
- 153.Wright NC et al. The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J. Bone Miner. Res 29, 2520–2526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.International Osteoporosis Foundation. Broken bones, broken lives: a roadmap to solve the fragility fracture crisis in Europe (IOF, 2018). [Google Scholar]
- 155.Chen P, Li Z & Hu Y Prevalence of osteoporosis in China: a meta-analysis and systematic review. BMC Public Health 16, 1039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lee KS, Bae SH, Lee SH, Lee J & Lee DR New reference data on bone mineral density and the prevalence of osteoporosis in Korean adults aged 50 years or older: the Korea National Health and Nutrition Examination Survey 2008–2010. J. Korean Med. Sci 29, 1514–1522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wade SW, Strader C, Fitzpatrick LA, Anthony MS & O’Malley CD Estimating prevalence of osteoporosis: examples from industrialized countries. Arch. Osteoporos 9, 182 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.