

SUMMARY
Somatic hypermutation (SHM) introduces point mutations into immunoglobulin (Ig) genes but also causes mutations in other parts of the genome. We have used lentiviral SHM reporter vectors to identify regions of the genome that are susceptible (“hot”) and resistant (“cold”) to SHM, revealing that SHM susceptibility and resistance are often properties of entire topologically associated domains (TADs). Comparison of hot and cold TADs reveals that while levels of transcription are equivalent, hot TADs are enriched for the cohesin loader NIPBL, super-enhancers, markers of paused/stalled RNA polymerase 2, and multiple important B cell transcription factors. We demonstrate that at least some hot TADs contain enhancers that possess SHM targeting activity and that insertion of a strong Ig SHM-targeting element into a cold TAD renders it hot. Our findings lead to a model for SHM susceptibility involving the cooperative action of cis-acting SHM targeting elements and the dynamic and architectural properties of TADs.
Graphical Abstract
In Brief
Senigl et al. show that genome susceptibility to somatic hypermutation (SHM) is confined within topologically associated domains (TADs) and is linked to markers of strong enhancers and stalled transcription and high levels of the cohesin loader NIPBL. Insertion of an ectopic SHM targeting element renders an entire TAD susceptible to SHM.
INTRODUCTION
Activated B cells diversify their antibody repertoire by both rearrangement (class switch recombination [CSR]) and somatic hypermutation (SHM) of their immunoglobulin (Ig) loci. SHM introduces point mutations into the variable region exon of Ig loci and is necessary for fine-tuning antibody specificity, including the elaboration of high-affinity antibodies in response to infection or immunization (Casellas et al., 2016; Di Noia and Neuberger, 2007; Methot and Di Noia, 2017). DNA subjected to SHM is deaminated at cytosines by activation-induced cytidine deaminase (AID). The resulting deoxyuridine lesion is resolved by error-prone base excision and mismatch repair, giving rise to mutations both at the original site of deamination and at flanking residues (Di Noia and Neuberger, 2007). Target DNA transcription is required for SHM and is thought to provide the single-strand DNA template needed for AID to act (Keim et al., 2013; Pavri and Nussenzweig, 2011). The powerful mutagenic and genome-destabilizing potential of SHM suggests the need for careful regulation of the reaction, and in fact, AID is regulated at multiple levels, including tight control of Aicda transcription, posttranslational modification, protein degradation, an extensive protein interactome, and carefully orchestrated access of the enzyme to the nucleus (Keim et al., 2013; Orthwein and Di Noia, 2012). However, none of these AID-centric mechanisms explain how AID and SHM select specific regions of the genome on which to act.
Ig loci, and in particular the region encompassing the variable region exon, are mutated by SHM at much higher frequencies than other parts of the genome (Liu and Schatz, 2009). How such Ig locus selectivity is achieved remains poorly understood. Ig loci were found to contain “mutation enhancer elements” (Kothapalli et al., 2008, 2011), and subsequent studies demonstrated that Ig enhancers and enhancer-like sequences have the ability to increase SHM of a flanking transcribed gene by two orders of magnitude or more (Blagodatski et al., 2009; Buerstedde et al., 2014). The SHM-targeting activity of these elements, which are collectively referred to as DIVAC (diversification activator), is compromised by deletion or mutation of a number of well-characterized transcription factor binding sites (TFBSs), although in most cases no single binding site was critical for activity (Blagodatski et al., 2009; Buerstedde et al., 2014). The results suggested both cooperative and redundant roles for the binding sites (and presumably the factors that bind them) in DIVAC-mediated SHM targeting. However, the mechanism by which DIVAC elements function, and hence their precise role in targeting SHM to Ig loci, remain elusive.
SHM is also detected at a subset of non-Ig genes, both in human B cell tumors (Müschen et al., 2000; Pasqualucci et al., 1998, 2001; Shen et al., 1998) and normal germinal center B cells, with some loci (e.g., Bcl6) being mutated at much higher frequencies than others (Álvarez-Prado et al., 2018; Liu et al., 2008). SHM is also associated with chromosomal translocations, such as between MYC and the Ig heavy-chain (IGH) or Ig light-chain (IGK, IGL) loci, that contribute to the development of B cell lymphoma (Janz, 2006; Nussenzweig and Nussenzweig, 2010; Robbiani et al., 2008). The existing data argue that low but variable frequency targeting of multiple non-Ig loci by AID/SHM is a routine feature of germinal center B cells.
Understanding the mechanisms responsible for the “off-target” action of AID/SHM at non-Ig loci remains a central challenge for the field. Multiple genomic and epigenomic features correlate with the action of AID, including super-enhancers, highly interconnected transcriptional regulatory elements, convergent transcription, H3K27Ac and H3K36me3 chromatin modifications, exosome substrate noncoding RNA expression, divergent transcription, and RNA polymerase II (Pol II) stalling (Álvarez-Prado et al., 2018; Meng et al., 2014; Pefanis et al., 2014; Qian et al., 2014; Wang et al., 2014). How these features may explain the pattern of SHM across the genome remains unknown. Many of these findings, however, suggest a role for enhancers, leading us to consider the possibility of mechanistic overlap between DIVAC-driven SHM targeting of Ig loci and selective SHM targeting of non-Ig loci. Specifically, we hypothesized that the targeting of SHM to Ig genes requires a specific combination of features that are also found in various combinations at other sites in the genome. Consistent with this hypothesis, Ig DIVACs, like other enhancers, are made up of a combination of widely occurring TFBSs, with their distinctive DIVAC activity likely reflecting a specific combination of such sites (Buerstedde et al., 2014).
An important architectural feature of mammalian genomes is contact domains, also referred to as topologically associated domains (TADs). TADs were identified in DNA proximity ligation assays such as Hi-C, as regions of the genome with high mutual contact probability whose boundaries often correspond to convergent CCCTC-binding factor (CTCF) binding sites (Dekker and Mirny, 2016; Krijger and de Laat, 2016; Merkenschlager and Nora, 2016; Rowley and Corces, 2016; Sexton and Cavalli, 2015; Yu and Ren, 2017). Loop extrusion mediated by the sliding of chromatin through one or a pair of cohesin rings is thought to contribute to TAD formation, establishment of TAD boundaries at CTCF binding sites (Bintu et al., 2018), and interactions between transcriptional regulatory elements (Matthews and Waxman, 2018; Merkenschlager and Nora, 2016; Vian et al., 2018). It is not known whether chromatin architecture regulates the off-target action of AID/SHM or constrains the SHM-targeting activity of Ig DIVAC elements.
We have developed lentivirus-based SHM reporter vectors and a high-throughput assay to delineate both SHM-susceptible and SHM-resistant regions in the B cell genome. This approach provides significant advantages over other assays by mapping SHM targeting potential in both active and transcriptionally silent genomic regions and circumventing biases created by the wide variation in the transcriptional and sequence features of endogenous genes. Our findings reveal that SHM-susceptible regions are contained within TADs and are strongly enriched for super-enhancers and binding of the cohesin loader NIPBL and numerous transcription factors as compared to SHM-resistant TADs. The identification of SHM-susceptible TADs allowed us to identify non-Ig enhancers that possess DIVAC activity, bind NIPBL, and are able to target SHM in various genomic locations. Insertion of a strong DIVAC element into an SHM-resistant TAD converted the TAD into one that is SHM susceptible, illustrating both the potential of DIVAC to drive SHM mistargeting and the limits imposed by chromatin loop boundaries on the spread of SHM susceptibility.
RESULTS AND DISCUSSION
Lentiviral-Based SHM-Detection Assay
To identify SHM-susceptible and SHM-resistant regions of the genome, an assay was required that could broadly and sensitively report on susceptibility to SHM independent of variations in endogenous gene transcription. To accomplish this, we developed an SHM-reporter retroviral vector (GFP7) that is conceptually similar to targeted-integration vectors previously used to identify Ig DIVAC elements in the DT40 B cell line (Buerstedde et al., 2014). GFP7 is an HIV-derived vector containing a strong cytomegalovirus promoter driving the transcription of a hypermutation target sequence (HTS7)-GFP fusion gene (Figure 1A). HTS7 contains numerous SHM hotspot motifs designed to yield stop codons upon the mutation of cytidine, allowing the vector to sensitively report SHM activity by virtue of the loss of GFP fluorescence. Blasticidin selection is used to select for vector integration and eliminate cells in which the integrated vector has become transcriptionally silenced.
Figure 1. Retroviral-Based Reporter Assay Maps SHM Susceptibility in the B Cell Genome.

(A) Map of GFP7 retroviral SHM reporter vector. Bsr, blasticidin resistance; CVM, cytomegalovirus; HTS7, hypermutation target sequence; IRES, internal ribosome entry site; SIN LTR, self-inactivating long terminal repeat; spacer, sequences that place Bsr outside of the SHM target window; T2A, self-cleaving T2A peptide; WPRE, woodchuck heptatitis virus posttranscriptional regulatory element.
(B) GFP fluorescence loss (3 weeks of culture) in WT or AID-deficient Ramos clones infected with GFP7 lacking DIVAC or containing superDIVAC or IgHi. Each point is an independent cell clone (number of clones and median of the data indicated above). Bar, data median. Red bracket, no-DIVAC GFP7 WT Ramos clones with substantial GFP loss (>1%); most data points for this sample lie close to the x axis and are not readily visible.
(C and D) Examples of DIVAC-trap HTISA data. No-DIVAC GFP7 integration site sequence read tracks for Total and GFP− populations (log scale) are shown above tracks for NIPBL, H3K4me1, super-enhancers, and GRO-seq (sense and antisense above and below the line, respectively). SHM-susceptible non-Ig (CUX1 locus, C) and SHM-resistant AGPAT3 locus (D) are shown. AGPAT3 locus data derive from a different experiment (superDIVAC chr22 TAD knockin; same as shown in Figure S3B) due to better genome coverage in that experiment.
Wild-type (WT) Ramos cells were infected with a GFP7 vector lacking an SHM-targeting element (no-DIVAC-GFP7) or containing an Ig DIVAC element, either the IGH intronic enhancer (IgHi) or superDIVAC (SD), which is composed of multiple Ig enhancers (Buerstedde et al., 2014; Williams et al., 2016). Analysis of single-cell subclones after 3 weeks of culture revealed that no-DIVAC-GFP7 yielded very few GFP− cells (median 0.04%), while the presence of IgHi or SD raised this value to 1.6% or 15.2%, respectively (Figures 1B and S1A). Virtually no GFP fluorescence loss is detected with no-DIVAC-GFP7 or SD-GFP7 in AID-deficient Ramos cells (Figure 1B). Repeating these experiments in DT40 cells revealed the same striking dependence of GFP− cell accumulation on the presence of DIVAC and AID (Figure S1C). To confirm that GFP fluorescence loss is due to mutation, we sequenced the HTS7-GFP coding sequence from sorted GFP− cells infected with no-DIVAC GFP7. This revealed that 96% (123/128) of the sequences contained ≥1 mutations (most in HTS7), with 82% containing at least 1 stop codon and another 12% containing an insertion/deletion or a missense mutation in GFP (Figure S1B). Hence, most GFP fluorescence loss with such vectors is due to the AID-dependent coding sequence mutation, which is consistent with our prior study (Buerstedde et al., 2014).
Because GFP7 vectors integrate at many positions in the Ramos genome (see below), robust mutation of the GFP7-DIVAC vectors indicates that DIVAC is capable of targeting SHM to a nearby transcription unit at many sites in the genome. These findings also raised the possibility that the no-DIVAC-GFP7 vector would function as a sensitive probe for SHM susceptibility in varied genomic environments. If this were the case, then the no-DIVAC-GFP7 reporter should be particularly susceptible to mutation when integrated in the highly SHM-susceptible Ig loci. To test this prediction, we analyzed 1,390 independent no-DIVAC-GFP7-infected Ramos single-cell clones for GFP fluorescence loss. This revealed that while the vast majority of clones exhibited no or negligible fluorescence loss, a small subset (3.8%) exhibited substantial (>1%) levels of GFP− cells after 3 weeks of culture (Figure 1B). Analysis of vector integration sites in 53 such clones revealed 17 (32%) integration sites in Ig loci and another 15 (28%) in the BCL6, PAX5, SPRED2, CXCR4, BACH2, MYC, and BCL7A loci previously documented to be SHM targets in B cells (Table S1A; Khodabakhshi et al., 2012). These data strongly argue that the no-DIVAC-GFP7 vector system is capable of identifying areas of the genome susceptible to SHM and suggest one mechanism by which SHM of the vector is activated: integration near a DIVAC element. Therefore, we refer to the use of the no-DIVAC-GFP7 vector to probe genome SHM susceptibility as the “DIVAC-trap” assay, although we recognize that the activation of SHM may occur by several mechanisms.
High-Throughput Analyses Reveal SHM-Susceptible and -Resistant Genomic Regions
We developed a method for high-throughput mapping of SHM-susceptible regions of the genome by combining GFP fluorescence loss with next-generation sequencing to identify vector integration sites (Figure S2A). Ramos cells infected with no-DIVAC-GFP7 were cultured for 3 weeks and selected in blasticidin for cells containing an actively transcribed vector. GFP− cells, which are enriched in integration sites in SHM-susceptible regions, were sorted and genomic DNA was isolated from GFP− and the “Total” (pre-sort) populations. Vector integration sites were identified by high-throughput integration site analysis (HTISA), a method we adapted from high-throughput genome translocation sequencing (HTGTS) (Chiarle et al., 2011). HTGTS and HTISA take advantage of linear-amplification PCR to reduce amplification bias, enabling estimation of the frequency of individual integration sites in the population based on the number of reads representing each integration. Amplification bias was tested by applying HTISA to a mix of equimolar amounts of genomic DNA from 12 clones whose integration site had previously been identified. This resulted in relatively uniform numbers of reads from each integration site, with differences of no more than 2-fold (Figure S2B).
The DIVAC-trap HTISA assay was performed on multiple poly-clonal populations of no-DIVAC-GFP7-infected Ramos cells (Method Details; Figure S2A). Sequence data were analyzed by dividing the genome into 25-kb bins, determining whether each bin had sufficient integration sites in the Total samples to be considered “covered,” and for each covered bin, determining whether reads were significantly enriched in each GFP− sample compared to its corresponding Total sample. Approximately 38% of reads from GFP− sample libraries derived from the Ig loci, as compared to ~1.9% from Total libraries (Figure S3A), supporting the conclusion that Ig loci represent domains that are highly susceptible to SHM. Subsequent analyses focused on the non-Ig portions of the genome.
Of the 2,264 covered bins outside the Ig loci (1.8% genome coverage), 175 (7.7%) were found to be “hot” (highly susceptible to SHM) and 1,459 (64%) were found to be “cold” (strongly resistant to SHM) (Tables S2A and S2B; see Method Details for information regarding criteria for coverage and hot and cold bins). Notably, of the 36 non-Ig integration sites identified in the analysis of highly mutating Ramos single-cell clones, 25 (69%) were within the 175 SHM-susceptible bins or a bin adjacent to one of these hot bins (Figure S2C), and none were in cold bins. To ensure that the results were not dictated by the HIV sequences in GFP7, we created vectors based on avian sarcoma and leukosis virus (ASLV), which exhibits a weak integration preference for genes and integrates more randomly than HIV (Mitchell et al., 2004; Narezkina et al., 2004). Analysis of single-cell clones infected with a no-DIVAC-GFP7 ASLV vector identified 25 non-Ig integration sites that supported substantial GFP loss. Of the 25 sites, 19 (76%) were within the 175 SHM-susceptible bins or an adjacent bin (Figure S2C), none were in cold bins, and 17 of 25 (68%) overlapped with loci identified by the HIV-based vector (Table S1B). These data indicate that the DIVAC-trap HTISA method reproducibly identifies SHM-susceptible regions.
The CUX1 locus provides an example of a region that is both well covered and highly susceptible to SHM, with many sequence reads in both the Total and GFP− populations (Figure 1C). In contrast, a broad region surrounding AGPAT3 exhibits very few GFP− reads despite containing several areas with many reads in the Total population, indicative of strong resistance to SHM (Figures 1D and S3B). IGL exhibits clustering of SHM susceptibility in several regions, the strongest corresponding to two bins surrounding the IGL enhancer (Figure S3C), a powerful DIVAC element (Buerstedde et al., 2014). Hot bins were found on all of the chromosomes except for chromosomes 15, 21, and X (Table S2A).
As noted above, only 1.8% of bins contained sufficient numbers of vector integration sites to be considered covered. This may be due to the integration preference of the HIV-derived vector and/or weak or unstable expression of the vector in some genomic region. To explore this issue, we analyzed vector integration preferences in the absence of blasticidin selection (removing the requirement for expression) by performing HTISA on WT and AID−/− Ramos cells 2 days after infection with no-DIVAC GFP7. The results revealed strong overlap with bins covered in the DIVAC-trap assay (Figure S3D). This argues that incomplete coverage of the genome is due primarily to intrinsic integration biases associated with HIV-derived vectors, which are known to prefer transcriptionally active regions (Mitchell et al., 2004). Overall, DIVAC-trap HTISA yielded large numbers of SHM-susceptible and SHM-resistant segments of the genome, which could be compared to one another to provide insight into features and factors involved in SHM susceptibility. The large portion of the genome not covered by our analysis is strongly depleted of active promoters, as assessed by levels of H3K4me3, and of active enhancers and super-enhancers, relative to the covered portion of the genome (Figure S3E); hence, they would be predicted to be generally resistant to SHM. As a result, our findings that 3.8% of vector integration sites and 7.7% of covered bins exhibit strong SHM almost certainly over-estimate the fraction of the genome that is prone to SHM.
Regions Susceptible to SHM Are Contained within Topologically Associated Domains
Analysis of the locations of hot and cold bins along the chromosomes revealed clustering of hot with hot bins and cold with cold bins at a frequency higher than expected by chance, especially for hot bins (Figure 2A). Furthermore, visual inspection revealed that many SHM-susceptible regions are delimited by a sharp drop in SHM susceptibility, giving them distinct borders (see below). This linear clustering of hot and cold bins raised the possibility that SHM susceptibility and SHM resistance are properties of TADs. To test this hypothesis, we performed Hi-C on Ramos and used the resulting data to determine the distribution of hot and cold bins in TADs in the Ramos genome. This analysis revealed a significant clustering of hot but not cold bins in TADs, with a small fraction of TADs containing ≥ 10 hot bins (Figure 2B).
Figure 2. Regions Susceptible to SHM Correspond to TADs.

(A) Clustering of bins. Proportion of bins having at least one adjacent bin of the same category (O, observed) is compared to the proportion expected for random distribution (E, expected). Proportion of covered bins with an adjacent covered bin is also shown (All). Chi-square test was used.
(B) Distribution of hot and cold bins in TADs. The distribution of hot (left) or cold (right) bins in TADs containing at least one hot or cold bin, respectively, is plotted next to the expected distribution obtained when hot or cold bins were randomly assigned to those TADs (gray bars).
(C) Number of sequence reads from Total and GFP− populations in TADs. High-confidence (HC) hot and cold TADs, dark red and blue, respectively; TADs that are hot or cold in some experiments but do not meet HC criteria, light red and light blue, respectively; covered TADs that are neither hot nor cold (neutral), black.
(D) Distribution of SHM susceptibility at hot and cold TADs. TADs and 200-kb adjacent regions were divided into 10 bins, and normalized reads per million mapped reads (RPM) is plotted for hot (top) and cold (middle) TADs. Profiles of SHM susceptibility (ratio of GFP−:Total sequence reads), bottom.
(E) Analysis of GFP7 transcription orientation bias in active genes. Top: subset of highly expressed genes analyzed. Bottom: frequencies of GFP7 vectors integrated into these genes in sense (s) or antisense (as) orientation in Total and GFP− populations. Paired t test was used.
(F) Distribution of hot and cold bins and TADs in chromatin compartment A or B.
SHM susceptibility and resistance of TADs, as measured by the ratio between GFP− and Total sequence read numbers, did not reveal distinct groupings of TADs but rather was a continuous property (Figure 2C). To facilitate the identification of features that distinguish SHM-susceptible and SHM-resistant portions of the genome, we focused subsequent analyses on 70 hot and 137 cold high-confidence non-Ig TADs (Figure 2C; Tables S2C and S2D). The high-confidence hot TADs contain 120 (69%) of the 175 hot bins. SHM susceptibility drops at the boundaries of hot TADs while the opposite takes place at the boundaries of cold TADs (Figure 2D). However, sequence read numbers in the Total cell population drop substantially at the boundaries of both hot and cold TADs (Figure 2D). Hence, the regions flanking hot and cold TADs tend to be poorly covered, limiting our ability to assess SHM susceptibility in those regions. We note also that read numbers in the Total population are higher on average in hot TADs than in cold TADs (Figure S3F).
Comparison of the DIVAC-trap HTISA and Hi-C data reveal numerous examples of the correspondence between regions of SHM-susceptibility/resistance and TAD or sub-TAD boundaries (Figures 3 and S4; Data S1). Both hot and cold TADs contain substantial transcriptional activity and active enhancers, as evidenced by substantial global run-on sequencing (GRO-seq) and H3K4me1 chromatin immunoprecipitation (ChIP)-seq signals, respectively. In some instances, regions spanning only a few megabytes contained both a hot TAD and a cold TAD, illustrating the separation of SHM susceptibility and resistance into distinct domains (Data S1). Notably, hot TADs encompass a substantial fraction of loci previously identified as targets of SHM in diffuse large B cell lymphoma (Khodabakhshi et al., 2012; Figure S5A). Hot TADs are also enriched in loci previously identified in Ramos as targets of AID-mediated mutation (Qian et al., 2014; Figure S5A), with the weaker overlap likely due to the fact that the analysis by Qian et al. (2014) was performed in cells deficient in base-excision repair and mismatch repair, which substantially expands the regions of the genome susceptible to mutation accumulation (Liu et al., 2008; data not shown). These results argue that SHM susceptibility and resistance are properties of at least some TADs and raise the possibility that TAD boundaries restrict the spread of SHM susceptibility.
Figure 3. Examples of SHM-Susceptible and SHM-Resistant TADs.

(A–C) Hi-C matrices are shown above DIVAC-trap HTISA data for cold (blue; A) and hot (red; B and C) TADs; HC TADs are indicated by asterisks. CTCF motif orientations (sense, blue; antisense, orange) overlapping with CTCF ChIP-seq peaks for the GM12878 human lymphoblastic cell line are indicated, with other data tracks as in Figure 1. Juxtaposed hot TADs (B) and ~1.2 Mb TAD encompassing and upstream of BCL6 composed of six HC hot sub-TADs (C). Positions of candidate DIVAC-enhancer elements analyzed in Figure 5, red arrows.
Susceptibility to the action of AID has previously been linked with convergent transcription (Meng et al., 2014). If convergent transcription also contributed to SHM susceptibility of our reporter vector, then vector insertions in strongly transcribed genes should be biased toward the antisense versus sense orientation in GFP− cells. No such bias could be detected (Figure 2E), arguing that the SHM susceptibility of our vector is driven by processes other than convergent transcription.
In addition to contact domains, the genome can also be divided into A and B compartments that preferentially self-associate in Hi-C analyses and are enriched in active and inactive chromatin, respectively (Lieberman-Aiden et al., 2009; Nuebler et al., 2018; Wang et al., 2016). As expected, given the preference of HIV-based vectors to insert into transcriptionally active regions, the vast majority of covered bins and TADs, as well as hot and cold bins and TADs, reside in compartment A (Figure 2F). Hence, the differences identified in our analyses between SHM-susceptible and -resistant regions of the genome are not driven by differences in genomic compartment.
TAD SHM Susceptibility Is Associated with a Specific Epigenetic Environment
Analysis of transcriptional activity by GRO-seq did not find a significant difference between hot and cold TADs (Figure 4A), indicating that integration of the reporter vector into a transcriptionally highly active region is not sufficient to yield SHM susceptibility. However, this does not rule out the possibility that reporter vectors inserted into hot regions of the genome are transcribed at higher levels than when inserted into cold regions. We analyzed 836 clones of cells infected with the no-DIVAC-GFP7 HIV vector for GFP mean fluorescence intensity (MFI; a measure of vector transcriptional activity) and GFP fluorescence loss. No correlation was observed between these two parameters (Figure 4B), arguing strongly that the increased mutation of no-DIVAC-GFP7 in hot versus cold TADs is not driven by elevated levels of vector transcription.
Figure 4. Epigenetic Environment Associated with SHM Susceptibility.

(A) Transcriptional activity (GRO-seq) in hot and cold TADs (reads per kilobase per million mapped reads [RPKM]). Two-tailed t test was used.
(B) GFP mean fluorescence intensity (a measure of vector transcriptional activity) and GFP fluorescence loss in 836 cell clones containing the no-DIVAC GFP7 vector (inset, same data plotted on different axes).
(C) Enrichment analysis in hot versus cold TADs. The signal for each parameter or factor (log(RPKM)) was compared between hot and cold TADs (two-tailed t test, with p values corrected for multiple hypothesis testing, Bonferroni correction). Corrected p values are plotted; NS, not significant; *p < 1.7 × 10e–3; **p < 1.7 × 10e–6; ***p < 1.7 × 10e–8. Data derived from Ramos: ChIP-seq for the indicated transcription or chromatin factors or modified histones, GRO-seq (GRO), and Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq). NPC, nuclear pore components; Pol II, total RNA polymerase II; S5P, serine-5-phosphorylated Pol II.
(D) Analysis of enhancers and super-enhancers in hot and cold TADs. Fraction of TADs that contain one or more super-enhancer is shown (left); fraction of the length of each TAD that is occupied by regular enhancers (middle), or super-enhancers (right). Data for all hot and cold TADs are shown. Fisher exact test and two-tailed t test were used.
(E) Composite graph of NIPBL binding intensity to regulatory elements in hot, cold, and all TADs.
(F) Distribution of NIPBL binding and SHM susceptibility in hot TADs. Hot TADs were clustered based on similarities in their distribution of normalized NIPBL intensities into six groups (rows). NIPBL distribution (top heatmap) and distribution of SHM susceptibility (bottom heatmap) is shown for the six groups of TADs. Scale bars at right.
(G) Plot of correlation coefficients obtained by comparing the distribution of NIPBL binding intensity and SHM susceptibility for each pair of rows in the data from (F).
To identify features that may distinguish SHM-susceptible and -resistant regions of the genome, we generated genome-wide datasets in Ramos to assess various chromatin properties, histone modifications, binding of transcription factors, total and serine-5-phosphorylated (S5P) Pol II, and factors involved in chromatin architecture and dynamics. We compared the abundance of each factor in high-confidence hot and cold TADs, with data displayed as statistical significance of enrichment (Figure 4C) or as fold enrichment (Figure S5B) in hot versus cold TADs. Levels of H3K4me3 were not significantly enriched in hot versus cold TADs (Figure 4C, bar 4), in keeping with the lack of difference in transcriptional activity. In contrast, H3K27Ac and especially H3K4me1 were significantly enriched in hot versus cold TADs (Figure 4C, bars 19 and 28), in agreement with a prior study showing enrichment of these marks in AID-dependent translocation hotspots (Wang et al., 2014). This finding is consistent with a role for enhancers or enhancer-like elements in SHM susceptibility. Notably, while hot and cold TADs contain equivalent densities of enhancers, hot TADs contain a markedly higher density of super-enhancers (Figure 4D), raising the possibility that the aggregation of enhancers into super-enhancers predisposes a region to SHM susceptibility. This idea is consistent with previous studies demonstrating that AID-mediated double-strand breaks and translocations occur predominantly within super-enhancers (Meng et al., 2014; Qian et al., 2014).
The most significant difference between hot and cold TADs was found in the occupancy of NIPBL (Figure 4C, bar 30), the major subunit of the cohesin loading complex (Gao et al., 2019; Visnes et al., 2014). Enrichment of the RAD21 cohesin subunit fell just short of statistical significance after correction for multiple hypothesis testing (bar 15). NIPBL-mediated cohesin loading and chromatin loop extrusion are thought to facilitate interactions between transcriptional regulatory elements (Matthews and Waxman, 2018; Merkenschlager and Nora, 2016; Vian et al., 2018). However, NIPBL also possesses cohesin-independent functions in transcription regulation, including interactions that influence Pol II pause release (Enervald et al., 2013; van den Berg et al., 2017; Zuin et al., 2014). Hence, enrichment of NIPBL in hot TADs is consistent with several possible mechanisms for SHM susceptibility, including roles for regulatory element interactions and transcriptional stalling.
A role for transcriptional regulatory elements was supported by a significant enrichment of binding of numerous transcription factors in hot as compared to cold TADs. Enriched factors included E2A, IRF4, PU.1, MEF2B, and nuclear factor κB (NF-κB), whose binding sites contribute to the DIVAC function of Ig enhancers (Buerstedde et al., 2014), as well as BCL6, Ikaros, and Aiolos, which play important roles in B cell development and function (Basso and Dalla-Favera, 2012; Cortés and Georgopoulos, 2004; Merkenschlager, 2010; Figure 4C). Transcription factor enrichment in hot TADs is accompanied by increased occupancy by total Pol II (Figure 4C, bar 17). However, as noted above, GRO-seq data indicate comparable levels of elongation-competent Pol II in hot and cold TADs. This apparent discrepancy can be resolved by the finding that hot TADs are markedly enriched in S5P-Pol II and Spt5, both of which are implicated in Pol II pausing/stalling (Figure 4C, bars 24 and 25). In addition, Spt5 is thought to play an important role in AID recruitment to DNA in SHM and CSR (Álvarez-Prado et al., 2018; Maul et al., 2014; Pavri et al., 2010). Our findings suggest that a larger proportion of Pol II is paused or stalled in hot versus cold TADs, which is consistent with current models in which AID acts on DNA in the context of a stalled Pol II complex (Methot and Di Noia, 2017; Sun et al., 2013).
The Location and Strength of NIPBL Binding Correlate with SHM-Targeting Activity
Analysis of individual distal regulatory elements, defined by the colocalization of NIPBL and H3K4me1, revealed that NIPBL forms sharp peaks over such elements in both hot and cold TADs, but with substantially more NIPBL bound in hot TADs (Figure 4E). We clustered hot TADs based on similarities in their distributions of NIPBL binding intensities, resulting in six groups (the rows of Figure 4F). A correlation is evident between the distribution of NIPBL binding and SHM susceptibility in these groups (Figure 4G; mean correlation coefficient of 0.7). Hence, while SHM susceptibility is high throughout hot TADs, it tends to peak in the vicinity of the strongest NIPBL binding.
Our findings led us to hypothesize that the susceptibility of hot TADs to SHM is driven, at least in part, by the presence of enhancer element(s) possessing DIVAC activity. We selected seven enhancer elements from hot TADs and two from the CD19 locus in a cold TAD based on features typical of active enhancers and clustered binding of transcription factors associated with B cell development and/or the germinal center reaction. These candidate enhancer elements were inserted into GFP7 and introduced into Ramos. Four of the candidate elements (ELF1e, MSH6e, ZCCHC7e, BCL6-2e) yielded a clear increase in GFP fluorescence loss above that of the no-DIVAC vector (Figure 5A; DIVAC-trap HTISA and Hi-C profiles for the TADs containing these enhancers are shown in Figures 3C, 6A, S4A, and S4B). The ELF1e element was the most active, displaying GFP fluorescence loss comparable to that of the strong IGH intronic enhancer (~80-fold above the no-DIVAC background). The four active elements tended to have stronger NIPBL binding than the inactive elements (Figure 5B). We also assessed NIPBL binding at three locations in the composite superDIVAC element, which had been inserted into a cold TAD by targeted integration (see below). In all three locations, the binding of NIPBL was comparable to that at the ELF1e element (Figure S5C). Increased GFP fluorescence loss correlated with small (up to 1.6-fold) increases in mean GFP fluorescence intensity, indicating increased transcription of the GFP7 cassette driven by the more active DIVAC elements (Figure S5D). However, little correlation exists between GFP fluorescence loss and mean fluorescence intensity for independent Ramos clones infected with the ELF1e-GFP7 vector (Figure S5E). Together with the data of Figure 4B, these data argue that the levels of transcription per se are not a dominant determinant of SHM susceptibility.
Figure 5. Identification and Characterization of Non-Ig DIVAC Elements.

(A) GFP fluorescence loss (3 weeks of culture) in Ramos clones infected with GFP7 containing no-DIVAC, IgHi, or candidate enhancer elements from loci, as indicated below the graph. Data presented as in Figure 1B. Data points outside the y axis range are in parentheses. Elements with detectable DIVAC activity, red arrows; schematic of GFP7, above.
(B) NIPBL ChIP-seq signal at the strongest NIPBL binding site in candidate enhancer elements in their endogenous context. IgHi and non-Ig elements exhibiting or lacking DIVAC activity, green, red, and blue, respectively.
(C) GFP fluorescence loss and mean GFP fluorescence intensity of clones infected with GFP7 containing ELF1e or ZCCHC7e. Vector integration sites in hot or cold TADs are indicated by red and blue bars, respectively. GFP fluorescence loss values are shown above the bars.
(D) ChIP-qPCR analysis of NIPBL binding in two independent Ramos clones harboring GFP7-ELF1e integrated in different cold TADs. Binding at endogenous ELF1e (endoELF1e) and vector ELF1e (vector ELF1e) was assessed within the major NIPBL peak; vector ELF1e contained two 5-bp substitutions to allow the design of primers specific for ectopic ELF1e. Each data point represents an independent measurement (average of duplicate technical replicas); bar indicates mean. C1, a NIPBL-non-binding region (based on NIPBL ChIP-seq data) in the TAD where the vector integrated in clone 1; H3, histone H3; IgG, control ChIP with non-specific antibody.
Figure 6. Deletion Analysis of ELF1e DIVAC Element.

(A) Hi-C and DIVAC-trap HTISA data for the ELF1 locus, presented as in Figure 3. Red arrow, location of ELF1e element.
(B) GFP fluorescence loss (3 weeks of culture) in Ramos clones infected with GFP7 containing no-DIVAC, ELF1e, or truncation mutants of ELF1e. Data are presented as in Figure 5A. Data points outside of the y axis range are in parentheses.
(C) Diagram of entire ELF1e element with tracks for NIPBL, H3K27Ac, H3K4me1, H3K4me3, and GRO-seq. Locations of binding motifs for transcription factors (Z, ZEB1; P, PU.1; M, MEF2; E, E2A; B, BCL6; Y, YY1) are marked. The region of ELF1e retained in the mutants in (B) or deleted from the mutants in (D) is indicated with color-coded bars.
(D) Analysis as in (B), but using GFP7 vectors in which small regions were deleted from full-length ELF1e. Deleted regions 1–3 are indicated in (C). Data points outside the y axis range are in parentheses.
This analysis of non-Ig enhancer elements suggests the possibility that non-Ig SHM activity is driven by enhancers that bind NIPBL strongly and have transcription factor binding profiles resembling those of Ig DIVAC enhancer elements. Comparing the four active non-Ig enhancers to the five with minimal DIVAC function revealed that the active enhancers were enriched for many of the same transcription factors and chromatin features as were hot TADs (Figure S5F, compare to Figure 4C), although because of the small number of enhancer elements analyzed, none of these differences reached statistical significance.
To address the possibility that the non-Ig DIVAC elements identified above activate SHM by altering vector integration preference and favoring insertion near a genomic DIVAC element or in hot TADs, we identified integration sites of vectors containing ELF1e or ZCCHC7e in infected single-cell clones. This demonstrated that both enhancers are able to drive substantial GFP loss in both hot and cold TADs (Figure 5C). Levels of GFP loss driven by these non-Ig DIVACs were comparable in range to those observed for the no-DIVAC GFP7 vector inserted into hot TADs (Figure S5G). These data argue that the newly identified non-Ig DIVAC elements are able to target SHM to the reporter vector regardless of integration site, as is the case for Ig DIVAC.
The strong 3.9-kb ELF1e DIVAC element contains an intense, sharp peak of NIPBL binding (Figure 6A). To determine whether this element is able to recruit NIPBL in the context of the reporter vector, we measured NIPBL binding to ELF1e in two single-cell clones containing the ELF1e-GFP7 vector inserted into different cold TADs. NIPBL bound at least as well to the ectopic ELF1e elements as to the endogenous ELF1e enhancer (Figure 5D). Hence, ELF1e contains sequences sufficient to mediate strong NIPBL binding at various sites in the genome.
The NIPBL-Binding Region Is Required but Not Sufficient for SHM-Targeting Activity
We performed a deletion analysis ELF1e to localize DNA sequences important for its SHM targeting function. We first tested regions of varying sizes (800, 390, and 250 bp) encompassing the NIPBL peak region in the GFP7 vector and observed a progressive decline in GFP fluorescence loss as the element was shortened, with the smallest fragment, containing only the core of the NIPBL peak, exhibiting almost no activity (Figure 6B). In the context of GFP7, this small 250-bp fragment was able to bind substantial amounts of NIPBL (Figure S6A), which is consistent with the idea that NIPBL binding is not sufficient for SHM targeting (cold TADs exhibit substantial NIPBL binding; Figures 1D, 3A, 4E, and S4A; Data S1) and with our previous findings that multiple sequences, often spread out over considerable distances, contribute to the DIVAC activity of any given element Buerstedde et al., 2014; Kohler et al., 2012; McDonald et al., 2013). Hence, the core NIPBL-binding region is not sufficient for efficient targeting of SHM. The full-length element contains numerous TFBSs identified in our analysis as correlating with SHM susceptibility (Figure 6C), many of which are shared with Ig DIVAC elements. The trimming of ELF1e resulted in the loss of most TFBSs and the loss of SHM-targeting activity, similar to the progressive loss of SHM targeting activity that accompanied sequential deletion of TFBSs in Ig DIVAC elements (Buerstedde et al., 2014). We then did a reciprocal experiment to determine whether the NIPBL-binding region was required for DIVAC activity of large enhancer fragments, deleting from the full-length enhancer fragment the same three regions that were retained in the trimming experiment. Even the smallest deletion (250 bp; del3) almost completely eliminated SHM-targeting activity (Figure 6D). These data indicate that the NIPBL-binding region is a critical component of the ELF1e DIVAC enhancer, and neither it nor the flanking regions containing numerous TFBSs are sufficient for substantial SHM-targeting activity.
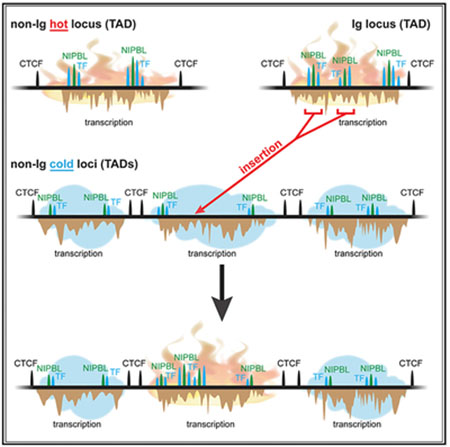
DIVAC Insertion Transforms a Cold TAD into an SHM-Susceptible Genomic Region
The data presented above lead to a number of important predictions regarding the mechanisms that regulate susceptibility and resistance to SHM, including (1) SHM-resistant and -susceptible regions are delineated by TAD boundaries; (2) SHM resistance or susceptibility is an intrinsic property of a TAD that is established in cis by properties of the TAD itself; and (3) the presence of an element(s) with DIVAC activity is an important, and perhaps vital, cis-acting property of a TAD for SHM susceptibility, with the further implication that DIVAC is able to act over long genomic distances, circumscribed by the insulating properties of TAD boundaries. To test these predictions, we used CRISPR-mediated homology-directed targeting to insert superDIVAC into a cold TAD located on chromosome 22 in WT Ramos cells. This TAD, which is 295 kb in size and contains 2 large genes, was selected because both it and its flanking TADs were well covered and resistant to SHM in our DIVAC-trap HTISA analysis, thereby allowing us to assess the effect of DIVAC insertion on the targeted TAD and potential spreading of effects to neighboring TADs. The strong superDIVAC element was chosen to provide a stringent test of the hypothesis that TAD boundaries limit the spread of SHM susceptibility. The resulting targeted cell line was infected with the no-DIVAC reporter vector and subjected to DIVAC-trap HTISA.
The results revealed that DIVAC insertion dramatically increased SHM in the modified TAD, converting it from cold to very hot (Figure 7A). SHM susceptibility is a property of much or all of the modified TAD, with reads in the GFP− cell population encompassing most of the TAD and closely mirroring the pattern of reads in the Total population. SHM susceptibility in the adjacent TADs increased much less than in the targeted TAD (Figure 7B). The fact that some increase is observed in the flanking TADs suggests that TAD boundaries can be “leaky,” which is consistent with a recent study that found substantial variation in TAD boundaries at the single-cell level (Bintu et al., 2018).
Figure 7. DIVAC Insertion Transforms a Cold TAD into a SHM-Susceptible Genomic Region.

(A) DIVAC-trap HTISA data before and after superDIVAC insertion into chromosome 22. Coverage (read numbers in the total cell population) was higher in the chromosome 22 superDIVAC insertion experiment than in the experiment with unmodified Ramos. Data presented as in Figure 3. Red arrow, location of superDIVAC insertion.
(B) SHM susceptibility (ratio between GFP− and Total sequence read numbers) data for modified TADs and their flanking TADs in unmodified Ramos (no-DIVAC) and Ramos with superDIVAC insertion in chromosome 22 (SD in chromosome 22 TAD) and chromosome 11 (SD in chromosome 11 TAD).
(C) Model for SHM susceptibility. We propose that SHM susceptibility arises in TADs that bind NIPBL strongly (green peaks) and contain one or more enhancers with DIVAC activity (purple oval) that bind transcription factors (yellow shapes) resembling those bound by Ig enhancers and interact with promoters (arrow) efficiently due to NIPBL-mediated recruitment of cohesin (red rings) and loop extrusion and/or through other mechanisms, such as phase separation, that could facilitate intra-TAD interactions. It is not known how DIVAC elements increase SHM, although one possibility raised by our findings is an increase in paused/arrested RNA Pol II, creating a favorable single-stranded DNA substrate for AID. Potential loop extrusion-independent roles for cohesin and cohesin-independent roles for NIPBL are not depicted. Arrowheads, convergently oriented CTCF binding sites.
To confirm and extend these results, we selected another cold TAD with somewhat different properties for targeted insertion of superDIVAC followed by DIVAC-trap HTISA. This TAD, located on chromosome 11, spans 110 kb, contains 12 genes and 1 super-enhancer, and is flanked by 2 nearby cold TADs. The results resembled those of the chromosome 22-modified line, with a large increase in SHM susceptibility of the targeted TAD and no detectable increase in the flanking TADs (Figures 7B and S6B). These results are consistent with our predictions and indicate that DIVAC insertion is able to convert a cold TAD into an SHM-susceptible TAD, with this susceptibility confined largely to the TAD containing DIVAC.
A Model for SHM Susceptibility
High overall levels of transcription in a TAD are not sufficient for SHM susceptibility; rather, our findings suggest that SHM susceptibility depends on two features: (1) strong binding of NIPBL and (2) the presence of enhancer elements with DIVAC activity. We propose that these two features work together to create SHM susceptibility. Strong binding of NIPBL is thought to promote high levels of chromatin loop extrusion, a process implicated in TAD formation and efficient interaction of enhancers and super-enhancers with transcription units (Vian et al., 2018). Such interactions are likely to be important for DIVAC function and may cooperate with the cohesin-independent functions of NIPBL, particularly those related to transcription pause release (van den Berg et al., 2017). Super-enhancers, with their high NIPBL occupancy (Dowen et al., 2013; Hnisz et al., 2013) and numerous component enhancers and TFBSs, are pre-disposed to contribute to SHM susceptibility, which is consistent with our findings and those of others (Meng et al., 2014; Qian et al., 2014; Wang et al., 2014). However, the presence of a super-enhancer is neither necessary nor sufficient for SHM susceptibility (Figure 4D; Meng et al., 2014; Qian et al., 2014; Wang et al., 2014).
Our identification of SHM-resistant domains of the genome represents a substantial advance over previous studies, which could not distinguish between two different explanations for a failure to detect AID/SHM activity in particular genomic regions: (1) those regions lacked suitable highly active endogenous transcription units or were within active genes but were too far from the transcription start site to be acted on by AID, or (2) those regions were intrinsically resistant to SHM. Our findings lead to the conclusion that the vast majority of the genome targeted by our vector, and probably the vast majority overall, is intrinsically resistant to SHM. Resistant regions are unable to mutate a highly expressed reporter, even though that same reporter is expressed at similar levels but mutated efficiently in susceptible regions. Our approach also provides a broader and more complete view of intrinsic SHM susceptibility than prior studies. In contrast to the focal sites of susceptibility identified with previous genome-wide approaches (Meng et al., 2014; Qian et al., 2014), our analysis reveals large SHM-susceptible domains sometimes spanning ≥1 Mb, as in the region upstream of BCL6 (Figure 3C). This contrast is illustrated in the region surrounding ZCCHC7, where we identified a >200-kb SHM-susceptible region spanning the entirety of the ZCCHC7 gene, while previous analyses of stimulated primary mouse B cells found small windows of vulnerability in the corresponding region of the mouse genome (Figure S7A). A similar picture emerges from a comparison of findings in the vicinity of REL (Figure S7B).
We propose a working model (Figure 7C) in which SHM susceptibility arises in TADs with high levels of NIPBL binding that also contain DIVAC-like enhancer(s) that bind an ensemble of transcription factors resembling those bound by Ig enhancers. In this model, transcription factors bound to DIVAC-like enhancers interact efficiently with transcription units in the TAD as a result of loop extrusion-mediated “scanning” of chromatin in the TAD (Vian et al., 2018) and/or diffusion-mediated collisions that occur efficiently in TADs and in domains, such as those attributed to super-enhancers, that are restricted to discrete nuclear volumes due to a high density of interacting factors (Hnisz et al., 2017). Cohesin interacts with AID and is required for efficient CSR (Thomas-Claudepierre et al., 2013), and hence may also contribute to SHM targeting by mechanisms distinct from loop extrusion. Similarly, it is plausible that the cohesin-independent functions of NIPBL, particularly that related to Pol II pausing (van den Berg et al., 2017), contribute to SHM susceptibility.
This model provides an appealing framework to explain the targeting of SHM to Ig loci as well as to susceptible non-Ig loci. For example, the TADs containing the Ig heavy-chain (IGH) locus and the highly SHM-susceptible region upstream of Bcl6 each contain powerful locus control region super-enhancers and elements with DIVAC function and exhibit particularly intense loop extrusion activity (Bunting et al., 2016; Rouaud et al., 2013; Vian et al., 2018). Ig loci appear to contain multiple, partially redundant, strong DIVAC elements (Buerstedde et al., 2014; Odegard and Schatz, 2006), which likely contributes to their highly efficient SHM of endogenous V gene regions and the integrated GFP7 vector. Given the abundance of super-enhancers in SHM-susceptible non-Ig TADs, it is possible that they also contain multiple elements with DIVAC activity. Other mechanisms, such as convergent transcription (Álvarez-Prado et al., 2018; Meng et al., 2014), likely also contribute to SHM susceptibility of particular endogenous sequences. Our findings indicate, however, that convergent transcription does not explain SHM of the GFP7 reporter, nor can it readily account for TADs as an organizational unit of SHM susceptibility and resistance.
A major unresolved issue remains the mechanism by which DIVAC elements stimulate SHM. Our model argues that answering this question will shed light on both “on-target” SHM of Ig V-regions and “off-target” SHM in hot TADs scattered across the genome.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Schatz (david.schatz@yale.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Ramos cell line - Human Caucasian Burkitt’s lymphoma (male). Derived from a Burkitt’s lymphoma which does not possess the EBV genome. The cells have B lymphocyte characteristics, with surface associated mu and kappa chains. Ramos cells were cultured in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS) (GIBCO), 1% Penicillin-Streptomycin (Sigma) in 5% CO2 atmosphere at 37° C.
293T cell line – human embryonic kidney epithelial (female), cell contains the SV40 T-antigen. 293T cells were grown in DMEM media (Sigma) supplemented with 10% FBS (GIBCO)), 1% Penicillin-Streptomycin (Sigma) in 5% CO2 atmosphere at 37°C.
DT40 cell line - avian leukosis virus (RAV-1) induced bursal lymphoma cell line derived from a Hyline SC chicken (female). Cell suspensions prepared from tumors that developed within the bursa of Fabricus were transferred intravenously into young syngeneic recipient chickens. After one transfer in vivo, the DT40 cell line was established. DT40 cells were cultured in RPMI-1640 media supplemented with 10% FBS (GIBCO), 1% chicken serum (GIBCO), 1% Penicillin-Streptomycin (Sigma) in 5% CO2 atmosphere at 40°C.
METHOD DETAILS
Plasmid construction
The GFP-IRES-Bsr cassette from pIgLGFP2 (Blagodatski et al., 2009) was amplified and inserted between NotI and SalI sites of the pCDH-CMV-MCS-EF1-Puro (System Biosciences) to generate the pLCGIB vector. The pLCGSIB vector was constructed by amplification of the 1252 bp XhoI-BamHI fragment from pRCASBP and insertion into SnaBI site in pLCGIB vector. SHM is known to be restricted to region up to 2 kb from the transcription start site (TSS) (Storb, 1996) hence we expect accumulation of mutations mostly in GFP and only minimum mutations in the IRES-Bsr region placed 2.6-3.6 kb from the TSS (Figure 1A). The design of pLCGSIB enables us to select analyzed cells with blasticidin thus removing cells containing a transcriptionally silenced vector and to quantify GFP-negative cells that lost GFP fluorescence due to coding sequence mutation. To increase the sensitivity of mutation detection, we designed a GFP-based fluorescence marker consisting of a spacer, hypermutation target sequence (HTS7) and brightness-optimized eGFP (GFPnovo2) (Arakawa et al., 2008) coding sequence. The HTS7 sequence was designed to include an array of AID hotspot motifs that when mutated by AID would cause in-frame stop codons and thus a loss of GFP translation. The HTS7 sequence was custom synthesized (Blue Heron Biotechnology). The fluorescence intensity of HTS7-GFP was not sufficient to reliably and efficiently sort GFP-positive cells without contamination with GFP-negative cells. Therefore we inserted T2A peptide between HTS7 and GFP to release the HTS7-encoded polypeptide from GFP during translation, thereby restoring GFP fluorescence intensity. The HTS7-T2A-GFP reading frame was preceded with a leader DNA sequence derived from mouse Rag1 intronic sequence, thereby positioning the 366 bp HTS7 region more than 250 bp downstream of the transcription start site of the CMV promoter, an optimal location for mutation. The final vector containing HTS7-T2A-GFP was named GFP7. We generated an ASLV-derived version of GFP7 by insertion of SpeI-SalI fragment from GFP7 into the ClaI site of pRV3 (Senigl et al., 2012) with U3 region deleted between bases 9 and 217. GFP7 was used for insertion of various enhancer elements into its unique HpaI site (HIV-derived) or SalI site (ASLV-derived) vector. SuperDIVAC and IgHi (Buerstedde et al., 2014; Williams et al., 2016) were amplified from plasmid DNA while candidate DIVAC elements were amplified from genomic DNA of wild-type Ramos cells. Deletion mutants of ELF1e were generated by amplification of the respective regions from the ELF1e-GFP7 plasmid and cloned into the HpaI site of GFP7 with the In-Fusion system (Clontech). All modifications of the GFP7 vector were verified by sequencing.
Flow cytometry
GFP expression was assessed by analysis with an LSRII cytometer (Becton Dickinson). All cultures were split one day before the analysis in order to analyze cells in exponential phase. Mean fluorescence intensity was compared only between cultures grown in parallel and analyzed at the same time to avoid bias caused by the cytometer alignment and settings.
Cell culture and virus propagation
Ramos cells were propagated in RPMI 1640 medium (Sigma) supplemented with 10% fetal calf serum (GIBCO) and antibiotic mixture (Sigma) in a 5% CO2 atmosphere at 37°C. DT40 cells were propagated in RPMI 1640 medium (Sigma) supplemented with 10% fetal calf serum and 1% chicken serum (GIBCO) and antibiotic mixture (Sigma) in a 5% CO2 atmosphere at 40°C. The AviPack packaging system was utilized for the ASLV-derived virus propagation and pseudotyping with vesicular stomatitis virus protein G (VSV-G) as described in Plachy et al. (2010). HIV-derived vector was produced by 293T cell line co-transfection (X-Treme HP, Roche) with 1 μg of GFP7 vector, 1 μg of psPAX2 (Addgene plasmid # 12260) and 1 μg of pVSV-G (Clontech) in a 6 cm Petri dish. Viral supernatants were collected, filtered through a 0.45 μm SFCA filter and stored at −80°C.
Infection and cloning of Ramos cells
4 × 10e6 Ramos cells were collected and infected with the retroviral vectors at MOI < 0.01 to obtain less than 1% GFP-positive cells. 200 μl of the suspension was applied and allowed to adsorb for 40 min at room temperature. After adsorption, 10 mL of fresh medium was added and cells were cultured at 37°C and 5% CO2. Two days post infection, the percentage of GFP-positive cells was analyzed by flow cytometry and blasticidin (final concentration 5 μg/ml) was added for two days. Seven days post infection, GFP-positive cells were sorted in a single-cell sort mode with an Influx cell sorter (Becton Dickinson) into 96-well tissue culture plates to obtain single-cell clones. Expanded clones were cultured for 17 days when blasticidin (final concentration 15 μg/ml) was added to the culture. Twenty-one days after cloning, the percentage of GFP-positive cells was assessed with an LSRII cytometer.
Candidate clones of Ramos cells infected with no-DIVAC GFP7 vector identified in the clonal DIVAC-trap assay were further subcloned to verify the extent of GFP-fluorescence loss. For each candidate clone, 12 subclones were isolated and analyzed.
Cloning and sequencing of provirus integration sites
Provirus-host genome DNA junction sequences were amplified using the splinkerette-PCR method (Senigl et al., 2012; Uren et al., 2009). Genomic DNA was isolated by phenol-chloroform extraction from individual clones and cleaved with either DpnII (ASLV-derived vector integrations) or NlaIII (HIV-derived vector integrations) restriction enzymes. The restriction fragments were ligated overnight at 15°C with a 10-fold molar excess of adaptors formed by annealing of HMspAa and HMspBb-Sau3AI or HMspBb-NlaIII oligonucleotides complementary to the particular cleavage site of the enzyme used for genomic DNA digestion. The ligation products were subsequently cleaved with Bsu36I (ASLV-derived vector integrations) or PvuII (HIV-derived vector integrations) to destroy undesirable products of adaptor ligation to the 3’LTRs. The resulting mixture of fragments was then purified with a High Pure PCR Cleanup Kit (Roche) and used as a template for nested PCR with primers specific for the retrovirus LTR and the splinkerette adaptor. Primary PCR was performed with primers Splink1 and spSIN-ASLV_R or spSIN-HIV_R as follows: 94°C for 3 min, 2 cycles of 94°C 15 s, 68°C 30 s, 72°C 2 min and 31 cycles of 94°C 15 s, 62°C 30 s, 72°C 2 min and final polymerization 72°C for 5 min. The secondary PCR used primers Splink2 and spinSIN-ASLV_R or spinSIN-HIV_R with the program setting: 94°C 3 min, 30 cycles of 94°C 15 s, 60°C 30 s, 72°C 2 min and final 72°C 5 min. The specific PCR products were sequenced and the resulting sequences adjacent to the 5′ LTR were aligned to the Human Genome assembly version hg19. All junction sequences containing the end of 5′ LTR and the unique cellular DNA sequence obtained from the splinkerette PCR were mapped to February 2009 human genome assembly (hg19) using BLAT from the UCSC Genome Browser website (http://genome.ucsc.edu/). Genomic coordinates of the LTR-proximal nucleotide of the obtained genomic sequences with a unique score were considered as the position of the integration sites.
DIVAC-trap assay
10e7 Ramos cells were infected (4 independent infections) with no-DIVAC GFP7 vector at low multiplicity resulting in 0.5%-1% GFP-positive cells 2 days after infection. Blasticidin (final concentration 5 μg/ml) was added 2 days post infection. Three days post infection the blasticidin concentration was increased to 6.5 μg/ml and kept until the 5th day. Seven days post infection, GFP positive cells were sorted and cultured for two days. Nine days post infection, GFP-positive cells were sorted again for GFP-positive cells to remove all traces of GFP-negative cells and produce a starting population containing ca. 100,000 vector integration sites. Each culture was split in half to create duplicate “A” and “B” cultures (allowing us to assess reproducibility and clonal drift during culture). During subsequent culture, SHM in the vector and GFP fluorescence loss occur more frequently in cells with the vector integrated in SHM-susceptible regions and rarely in most cells. After 20 days of propagation, the culture was selected with blasticidin (12 μg/ml) to remove cells with silenced vector. After 4 days of blasticidin selection, GFP-negative cells (containing mostly vectors integrated into SHM-susceptible sites) were then sorted from each of the 8 cultures (“A” and “B” duplicates of 4 infections) and sorted again 3 days later to remove the remaining GFP-positive cells. Total and GFP-negative cultures were harvested for genomic DNA isolation on the same day so that culture times after infection with GFP7 were the same. Genomic DNA was isolated using a salt-extraction method (Aljanabi and Martinez, 1997).
High-throughput insertion site analysis (HTISA)
HTISA libraries were prepared and sequenced from GFP-negative and Total populations (“B” culture genomic DNAs were combined to yield a GFP-negative “Pool” library and a Total “Pool” library, so that finally, we obtained one pooled and 4 separate GFP-negative samples and one pooled and 4 separate Total samples). We used a linear amplification–PCR protocol based on a previously published high-throughput, genome-wide, translocation sequencing (HTGTS) method (Frock et al., 2015). The adaptation of the protocol was as follows: Retroviral integration sites were linearly amplified from the vector 5′ LTR using biotinylated Bio_L7a (HIV-derived vector integrations) or Bio_A7a (ASLV-derived vector integrations) primers. Nested PCR was performed with barcoded inner primer I5_bar75_L7a (HIV-derived vector integrations) or I5_bar75_A7a (ASLV-derived vector integrations). The blocking digestion of the nested PCR product was omitted. The resulting PCR product were gel purified and sequenced using a NextSeq500 (Illumina) sequencer.
ChIP and ChIP-seq (H3, H3K27Ac, Pol2, Ser5P Pol2, Spt5)
The H3 ChIP was done using SimpleChIP Enzymatic Chromatin IP Kit according to manufacturer’s instructions (Cell Signaling Technology). H3K27Ac ChIP was performed essentially as described (Lee et al., 2006) with minor modifications. Cells were fixed in 1% formaldehyde for 15 min at room temperature and 1.7 × 10e7 cell equivalents were sonicated in a water bath sonicator (Diagenode Bioruptor) for 35 cycles (30 s on/30 s off). After clearing the sonicated material 1.5 × 10e7 cell equivalents were subjected to immunoprecipitation with 5 μg of antibody overnight. Magnetic beads were washed 6 times in 1 mL of RIPA buffer (50 mM HEPES, pH 7.5, 500 mM LiCl, 1 mM EDTA, 1% Igepal CA-630, 0,7% sodium deoxycholate) and once with TE buffer with 50 mM EDTA. The DNA was eluted in 100 μL of elution buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 1% SDS) by heating to 65°C for 15 min. After reversal of cross-links the DNA was purified by phenol-chloroform-isoamyl alcohol extraction.
PolII, Ser5P PolII and Spt5 ChIPs were done as described above with the following modifications. 1.7 × 10e7 fixed cells were lysed in SDS lysis buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 1% SDS) supplemented with Complete protease inhibitor cocktail (Roche) and diluted 4-fold with dilution buffer (16.4 mM Tris-HCl, pH 8.0, 167 mM NaCl, 1.2 mM EDTA, 0.01% SDS, 1.1% Triton X-100, supplemented with inhibitors). The lysate was sonicated using a water bath sonicator (Diagenode Bioruptor Pico) for 11 cycles (30 s on/30 s off). Sonicated material was further diluted 1.5-fold in dilution buffer and 1.7 × 10e7 cell equivalents were subjected to immunoprecipitation with 5 μg (α-PolII and α-Ser5 PolII) or 7 μg (α-Spt5) of antibodies. Magnetic beads were washed twice with low salt wash buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), twice with RIPA buffer and twice with TE buffer with 50 mM NaCl.
Two ChIP-seq libraries were prepared for each ChIP from three independent ChIP experiments using TruSeq ChIP Sample Preparation kit (Illumina) following manufacturer’s instructions. The libraries were sequenced using a NextSeq500.
The following antibodies were used for ChIP: H3 (D2B12) XP antibody was purchased from Cell Signaling Technology, H3K27Ac (ab4729) and Ser5P PolII (ab5131) antibodies was purchased from Abcam. PolII (N20x) and Spt5 (H-300x) were purchased from Santa Cruz.
ChIP-seq (transcription factors, Rad21)
Chromatin Immunoprecipitation was performed with the SimpleChIP Enzymatic Chromatin IP Kit (#9003) according to the manufacturer’s protocol with minor modifications. In brief, 40 × 10e6 Ramos cells were spun down at 90xg for 10 min before the pellet was reconstituted in 9 mL of RPMI with 2% FBS in a 15 mL conical tube. Then, 0.6 mL of 16% Formaldehyde (Pierce/ThermoFisher #28906) was added and the cells were placed on a rocker for 10 min at RT. One ml of 10x glycine solution was added to quench the reaction, and the cells were then returned to the rocker for 5 min at RT before being spun down at 300xg and washed 2x with PBS.
All following steps were performed on ice or at 4°C unless otherwise noted. Cells were reconstituted in 10 mL of Buffer A with protease inhibitors and allowed to rest for 10 min and the lysed cells were spun down at 2000xg for 5 min to pellet nuclei. Pelleted nuclei were washed with 10 mL of Buffer B, pelleted at 2000xg for 5 min, and then resuspended in 1 mL of Buffer B. 1.7 μl of micrococcal nuclease (CST #10011) was added to the nuclei and the tube incubated in a 37°C water bath for 20 min. The reaction was stopped with 100 μl of 0.5 mL of EDTA, and the nuclei pelleted at 16,000xg for 1 min. The pellet was then resuspended in 1 mL of 1x ChIP Buffer with protease inhibitors and sonicated in 200 μl aliquots in a Qsonica sonicator for 2 cycles of 15 son, 45 s off at 20% power. The lysate was then clarified at 10,000xg for 10 min before the supernatant was removed and diluted five-fold in 1x ChIP Buffer with protease inhibitors.
For each ChIP, 500 μl of chromatin was incubated with 1-2 μg of antibody in a 1.5 mL Eppendorf tube at 4°C overnight on a rotator. The next day, 30 μl of Protein G Magnetic beads (CST #70024) was added to each tube and the mixture incubated at 4°C for 2 h on a rotator. The beads were pelleted using a magnetic separation rack and the supernatant discarded. The beads were then washed 3x with a low-salt wash and 1x with a high-salt wash using 5 min incubations on a rotator. Chromatin was eluted from beads with the addition of 150 μl of 1x Elution Buffer and incubation at 1200 rpm on a thermal mixer for 2 h at 65°C. The beads were pelleted and the supernatant was moved to a new 1.5 mL Eppendorf tube and 2 μl of Proteinase K (CST #10012) added. The mixture was incubated for 2 hr at 65°C and then DNA was purified using SimpleChIP DNA Purification Buffers and Columns (CST #14209) and eluted in a volume of 50 μl.
ChIP-Seq libraries were prepared from 50 ng of eluted DNA using the Ultra II DNA Library Prep Kit for Illumina (NEB #E7645) following the manufacturer’s protocol.
The following antibodies were used for ChIP: Ikaros D10E5 Rabbit mAb (CST #9034), Aiolos D1C1E Rabbit mAb (CST #15103), E2A (CST #12258), ZEB1 (Proteintech #21544-1-AP), YY1 (SCBT #7341X), PU.1 (CST #2258), Helios D8W4X (CST #42427), IRF4 (CST 4964), MEF2B (Abcam #ab33540), BCL6 (CST #5650), IRF8 (CST #5628), NFKB1 (CST #12540), ELF1 (Bethyl laboratories A301-443A), ELF2 (Invitrogen #PA5-52247), p65 (CST #8242), c-Rel (CST #12659), Nuclear Pore Proteins (Abcam #ab24609), Rad21 (CST #4321), Pax5 (Novus #NBP2-29905), YY2 (Sigma #HPA030335.
ChIP-seq data for NIPBL, H3K4me1 and H3K4me3 were obtained from Qian et al. (2014). ChIP-seq data for MYC were obtained from Seitz et al. (2011).
NIPBL ChIP qPCR
NIPBL and H3 ChIP were performed using the SimpleChIP enzymatic Chromatin IP kit (Cell Signaling Technology, catalog number 9003) according to supplier’s protocol. Briefly, Ramos ELF1e or ELF1e-3 clones or Ramos cell with superDIVAC insertion in chr22 TAD were seeded at 3 × 10e5 cells/ml. Cells containing ELF1e or ELF1e-3 vectors were treated with 15 μg/ml blasticidin to remove cells containing inactivated lentiviral vector and cultured using standard cell culture techniques for 2 days. 4 × 10e6 cells per IP were collected and fixed with 1% formaldehyde for 10 min. After neutralization with glycine and washes with cold PBS, cell pellets were stored at −80°C. Frozen cell pellets were thawed on ice and nuclei were isolated. Following digestion with Micrococcal nuclease (0.85 μl micrococcal nuclease/20 × 10e6 cells; total volume 500 μl; 20 min at 37°C), nuclear pellets were sonicated using QSonica q800R sonicator (2 cycles; 20% amplitude; 15 s on/15 s off). Nuclear lysates were cleared by centrifugation and the prepared chromatin was subjected to ChIP using H3 (Cell Signaling Technology, #4620), NIPBL (Bethyl Laboratories; #A301-779A) and normal rabbit IgG (EMD Millipore, 12-370) antibodies. Protein G magnetic beads were used to pellet the immunoprecipitates and after 3 low salt washes and one high salt wash, bound chromatin was eluted and crosslinks reversed. Spin columns were used to purify DNA which was used as templates for qPCR reactions using iTaq universal SYBR green supermix (Bio-Rad; #1725121).
Omni-ATAC-Seq
Omini-ATAC-Seq was performed on 50,000 Ramos cells following a previously published protocol (Corces et al., 2017) with the following modifications: Following amplification, libraries were purified with Ampure XP beads using two-sided size selection to remove primer dimers and fragments >1000 bp.
CRISPR-Cas9 based Knock-Out in Ramos Cell Lines
Px458 was a gift from Feng Zhang (addgene: 48138). Px458 espCas9(1.1) GltRNA was derived from px458 by replacing SpCas9 with eSpCas9(1.1) (Slaymaker et al., 2016) and the U6 promoter with a glutamine tRNA promoter (Mefferd et al., 2015). Donor vectors for the knock-in constructs targeting superDIVAC to “cold” regions of chromosome 11 and 22 were made by insertion of a floxed superDIVAC-PGK-Hygro-SV40polyA cassette in place of the eukaryotic expression cassette in pExpress (Forman and Samuels, 1991). This vector was further subcloned by placing homology arms targeting regions of chromosome 11 or 22 on either side of the superDIVAC cassette.
Guide RNAs targeting human AICDA were designed using CRISPR Design (http://zlab.bio/guide-design-resources) and were cloned into px458. The gRNA plasmids were transiently transfected into 4×10e6 Ramos cells with Gene Pulser Electroporation Buffer (BioRad #1652676) using the Gene Pulser XCell Electroporation System (BioRad #1652660) before being returned to 10 mL of conditioned 20% FBS media to recover for 36-48 h. Bulk transfected cells were then single cell sorted for the GFP high population into 96 well plates containing 200 μl of 20% FBS conditioned media in each well.
After 14-21 days, colonies in 96 well plates were expanded in 24 well plates and a small aliquot of cells (~1,000-10,000 cells) digested in 20 μl of 1x Phusion PCR Buffer (NEB) with 1 mg/ml Proteinase K at 55°C for 1 h. One μl of the crude genomic DNA preparation was then subjected to PCR to amplify the targeted genomic region. Three μl of this PCR product was then combined with 3 μl of PCR product amplified from WT DNA. This mixture was incubated in a thermal cycler to form heteroduplex DNA using the following conditions: 95°C for 5 min followed by stepwise reduction in temperature 2.5°C/min to 25°C. A T7 Endonuclease assay was carried out on the heteroduplex reaction mixture by addition of 0.1 μl T7 endonuclease I (NEB #M0302), 1.5 μl Buffer 2 (NEB #B7002), and 7.4 μl of ddH2O and incubating for 37°C for 1 h. The T7 Assay products were run on a 2% Agarose Gel. Mismatches between the WT DNA and potential genome-edited clones were visualized as bands smaller than the primary amplicon.
PCR reactions from clones showing evidence of genome editing were TA-Cloned using the Topo TA Cloning Kit (Invitrogen #K4574) and Sanger sequenced. Clones showing evidence of large deletions or nonsense mutations were further expanded before 2-5 × 106 cells were collected, washed with PBS, and lysed in RIPA Buffer. Knockout of the gene of interest was confirmed by Western Blotting when commercially available antibodies were available.
CRISPR-Cas9 based Knock-In in Ramos Cell Lines
Guide RNAs were designed using the Broad Institute’s sgRNA designer (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) and were cloned into pX458 espCas9(1.1) GltRNA. These vectors were co-transfected with donor vectors targeting cold regions in chromosomes 11 or 22, generated as described above. Following four days of recovery, the transfected cells were diluted 10-fold in 20% FBS media with hygromycin (final concentration: 0.5 mg/ml) before 200 μl of the diluted cell mixture was added to each well of 5 96-well plates. Following 14-17 days, hygromycin resistant clones were expanded and genomic DNA was prepared using a salt-extraction method (Aljanabi and Martinez, 1997). Targeted knock-in was confirmed by PCR using primers inside the cassette and outside the homology arms.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Pre-processing Alignment and Filtering
For DIVAC-trap data, fastq raw reads files were split into the individual Total and GFP-negative libraries by their barcodes, using fastqx toolkit fastx_barcode_splitter.pl program with–bol–exact parameters (search exact match of barcode at the beginning of the read).
For each library, the viral LTR sequence spanning the first 41 base-pairs of the read was trimmed and processed reads were mapped to the human grch37/hg19 genome build using Bowtie (version 1.1.2) software with seed length 25 and 3 mismatches allowed. To reduce background, integration sites with only a single read were discarded. ChIP-seq, ATAC-seq and GRO-seq data were mapped by Bowtie (version 1.1.2) with seed length 50 and 2, 3, and 3 mismatches allowed, respectively.
Defining SHM-susceptibility of genomic regions
The genome was divided into 25 kb bins and the number of reads for each bin was counted. Covered bins were defined as those with at least 3 unique integration sites and total 50 or more reads. Bins that did not match these criteria were discarded from the analysis. For each covered bin, enrichment of reads in the GFP-negative population was determined as follows. First, the reads that were mapped to Ig loci were discarded. Next, the reads in the Total population were normalized to have the same total number of reads as the GFP-negative population. The resulting normalized number of reads per bin in the total population was used as the Poisson l parameter against which the number of reads in the GFP-negative population at the same bin were compared in the Poisson test. This generated an array of p value to which we applied Benjamini-Hochberg criteria using FDR of 0.05 to determine the significant GFP-negative enriched bins for this pair of Total/GFP-negative libraries. We repeated this procedure for four biological replicates, and a fifth replicate (“B” samples) consists of pooling all the libraries prior to sequencing. Finally, a significant “hot” bin was defined as a bin in which at least two libraries and the pool library show significant enrichment of reads in GFP-negative versus Total. Similarly, a cold bin was defined as a bin in which none of the libraries showed enrichment. A similar analysis was performed on Topologically Associated Domains (TADs) following loop calling from the Hi-C data (see details below).
In places where the GFP-negative and Total reads were compared (Figures 2C, 2D, and 7B), the reads from all 4 libraries + pool library were used.
Peak calling
Peaks were called using MACS 1.4.3 (Zhang et al., 2008). For ChIP-seq peaks, default parameters were used (p value cutoff for peak detection = 1x10e-5;–keep-dup = auto), with corresponding DNA input as a control. For GRO-seq and ATAC-seq peaks, no control was used, and the parameters were tuned to fit broader peaks (–nolambda,–nomodel) (Feng et al., 2011). NIPBL summits annotations were defined by MACS output.
Defining Hotness of genomic factors
Hotness of genomic factors (Figure 4C) was defined as the enrichment of a factor at hot TADs compared to cold TADs. To determine factor hotness, we first performed a peak calling to each factor, using the matched input DNA library as a control, when applicable (see Peak Calling section for details). Reads residing outside peaks were filtered out. After filtering, reads were counted within TADs and normalized to reads-per-million-per-kb (RPKM). A two-tailed t test was performed on log(RPKM) between hot and cold TAD. Hotness was defined as hotness = −log(p value)*is_hot, where is_hot = 1 if the averaged log(RPKM) of hot TADs is higher than the one of cold TADs, and −1 otherwise.
Bins clusters
Clustered covered/hot/cold bins (Figure 2A) were defined as bins with at least one neighboring bin from the same category. To determine the expected probability of covered/hot/cold bins to be clustered, the positions of these bins were randomized 100 times, and the averaged fraction of clustered bins was taken.
Data visualization
Aligned-reads bed files were first converted to bedgraph files using bedtools genomecov (Quinlan and Hall, 2010) following by bedGraphToBigWig to make a bigwig file (Kent et al., 2010). Visualization of genomic profiles was done by the UCSC browser (Kent et al., 2002). Heatmaps (Figure 4F) were produced using the R package pheatmap. For aggregate plot around NIPBL summit (Figure 4E), signal was smoothed using smooth.spline function in R.
Genomic Annotations
Genes were defined using RefSeq genes annotations taken from UCSC database (Karolchik et al., 2004). Annotations of Ramos enhancers and super-enhancers were taken from Qian et al. (2014).
Hi-C data – pre-processing and TAD calling, and downstream analysis
We mapped physical contacts between loci, in Ramos cells, using in situ Hi-C procedure, which combines DNA-DNA proximity ligation with high throughput sequencing, in intact nuclei (Rao et al., 2014). The maps allow reliable detection of compartment structures and loops, genome-wide, at 5 kb resolution. Juicer software was used to filter reads and subsequently normalize the ligation frequency matrices as previously published (Rao et al., 2014). All of the normalized data correspond to matrices balanced using the Knight-Ruiz algorithm as described (Rao et al., 2014). We next used the juicebox dump function to extract the normalized matrices from the inter_30.hic file (Durand et al., 2016). For this analysis, we used 5kb resolution matrices. We then used juicer software to call loops with the default parameters. To cluster TADs with respect to their NIPBL distribution (Figure 4F), TADs were first divided into 100 bins, to obtain a 100 length vector consisting of the mean NIPBL ChIP-seq value for each TAD, and then were divided into 6 clusters using k-means algorithm, implemented by pheatmap R function.
Statistical Analysis
Statistical analysis was performed using R version 3.3.1 (http://www.r-project.org). The statistical tests used are reported in the figure legends and main text.
DATA AND CODE AVAILABILITY
Data, code and materials used in this study can be made available upon request to the corresponding authors. All datasets generated during this study are available at GEO: GSE139810.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-H3 antibody | Cell Signaling Technology | Cat#4620; RRID: AB_1904005 |
| anti-H3K27Ac | Abcam | Cat#ab4729; RRID: AB_2118291 |
| anti-Ser5P PolII | Abcam | Cat#ab5131; RRID: AB_449369 |
| anti-NIPBL | Bethyl Laboratories | Cat#A301-779A; RRID: AB_1211232 |
| normal rabbit IgG | EMD Millipore | Cat#12-370; RRID: AB_145841 |
| PolII | Santa Cruz Biotechnology | N20x; Cat#sc-899; RRID: AB_632359 |
| Spt5 | Santa Cruz Biotechnology | H-300x; Cat#sc-28678; RRID: AB_668824 |
| Ikaros D10E5 Rabbit mAb | Cell Signaling Technology | Cat#9034; RRID: AB_2797691 |
| Aiolos D1C1E Rabbit mAb | Cell Signaling Technology | Cat#15103; RRID: AB_2744524 |
| E2A | Cell Signaling Technology | Cat#12258; RRID: AB_2797860 |
| ZEB1 | Proteintech | Cat#21544-1-AP; RRID: AB_10734325 |
| YY1 | Santa Cruz Biotechnology | Cat#sc-7341; RRID: AB_2257497 |
| PU.1 | Cell Signaling Technology | Cat#2258; RRID: AB_2186909 |
| Helios D8W4X | Cell Signaling Technology | Cat#42427; RRID: AB_2799221 |
| IRF4 | Cell Signaling Technology | Cat#4964; RRID: AB_10698467 |
| MEF2B | Abcam | Cat#ab33540; RRID: AB_2142738 |
| BCL6 | Cell Signaling Technology | Cat#5650; RRID: AB_10949970 |
| IRF8 | Cell Signaling Technology | Cat#5628; RRID: AB_10828231 |
| NFKB1 | Cell Signaling Technology | Cat#12540; RRID: AB_2687614 |
| ELF1 | Bethyl laboratories | Cat#A301-443A; RRID: AB_960983 |
| ELF2 | Invitrogen | Cat#PA5-52247; RRID: AB_2640985 |
| p65 | Cell Signaling Technology | Cat#8242; RRID: AB_10859369 |
| c-Rel | Cell Signaling Technology | Cat#12659; RRID: AB_2797983 |
| Nuclear Pore Proteins | Abcam | Cat#ab24609; RRID: AB_448181 |
| Rad21 | Cell Signaling Technology | Cat#4321; RRID: AB_1904106 |
| Pax5 | Novus | Cat#NBP2-29905 |
| YY2 | Sigma | Cat#HPA030335; RRID: AB_2673434 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ampure XP beads | Beckman Coulter | Cat#A63880 |
| Gene Pulser Electroporation Buffer | BioRad | Cat#1652676 |
| Proteinase K | Cell Signaling Technology | Cat#10012 |
| T4 DNA ligase | Promega | Cat.#M1804 |
| T7 endonuclease I | New England Biolabs | Cat#M0302 |
| Blasticidin | InvivoGen | Cat# ant-bl-5 |
| Dynabeads MyONE C1 streptavidin beads | Life Technologies | Cat#65002 |
| Hexammine cobalt (III) chloride | Sigma Life Sciences | Cat#H7891 |
| PEG8000 | Sigma Life Sciences | Cat#P2139 |
| micrococcal nuclease | Cell Signaling Technology | Cat#10011 |
| Protein G Magnetic beads | Cell Signaling Technology | Cat#70024 |
| SimpleChIP DNA Purification Buffers and Columns | Cell Signaling Technology | Cat#14209 |
| Hygromycin | EMD Millipore | Cat#400050 |
| Dynabeads Protein G beads (ChIP-seq of H3, H3K27Ac, Ser5P PolII, PolII and Spt5) | Invitrogen | Cat#10004D |
| Protease inhibitor cocktail complete, EDTA free | Roche Merck | Cat#5056489001 |
| Critical Commercial Assays | ||
| SimpleChIP enzymatic Chromatin IP kit | Cell Signaling Technology | Cat#9003 |
| iTaq universal SYBR green supermix | Bio-Rad | Cat#1725121 |
| Phusion Hot Start Flex DNA polymerase | New England Biolabs | Cat#M0535L |
| In-Fusion | Takara | Cat#638911 |
| TruSeq ChIP Sample Preparation kit | Illumina | Cat#IP-202-1012 |
| Ultra II DNA Library Prep Kit | New England Biolabs | Cat#E7645 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GSE139810 |
| ChIP-seq data for MYC | Seitz et al., 2011 | GSE30726 |
| ChIP-seq data for NIPBL, H3K4me1 and H3K4me3 | Qian et al., 2014 | GSE62063 |
| Experimental Models: Cell Lines | ||
| human: Ramos cell line | Laboratory of Michael Neuberger | N/A |
| human: 293T cell line | ATCC | CRL-3216 |
| chicken: DT40 cell line | Laboratory of Jean-Marie Buerstedde | N/A |
| Oligonucleotides | ||
| Primers for NIPBL ChIP, see Table S3 | This paper | N/A |
| Primers for splinkerette, see Table S3 | This paper | N/A |
| Primers for HTISA, see Table S3 | This paper | N/A |
| chr11 gRNA-1: AAACAATGTCCGCCTACCCT | This paper | N/A |
| chr11 gRNA-2: AGCTTCGGTGCCACACAACG | This paper | N/A |
| chr22 gRNA-1: CCTAATTCAGCATGCGTTGG | This paper | N/A |
| chr22 gRNA-2: AAGCCTAATTCAGCATGCGT | This paper | N/A |
| Recombinant DNA | ||
| Px458 | Ran et al. (2013) | Addgene plasmid #48138 |
| pExpress | Forman and Samuels, 1991 | N/A |
| pIgLGFP2 | Blagodatski et al., 2009 | N/A |
| pGFP7 | This paper | N/A |
| GFPnovo2 | Arakawa et al., 2008 | N/A |
| pRV3 | Senigl et al., 2012 | N/A |
| SuperDIVAC | Williams et al., 2016 | N/A |
| psPAX2 | unpublished | Addgene plasmid # 12260 |
| pVSV-G | Clontech | Cat#631530 |
| Software and Algorithms | ||
| CRISPR Design | http://zlab.bio/guide-design-resources | |
| Broad Institute’s sgRNA designer | Doench et al. (2016) | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| Bowtie 1.1.2 | Langmead et al. (2009) | https://sourceforge.net/projects/bowtie-bio/files/bowtie/1.1.2/ |
| MACS 1.4.3 | Zhang et al., 2008 | https://pypi.org/pypi/MACS/1.4.3 |
| UCSC database | Karolchik et al., 2004 | https://genome.ucsc.edu |
| UCSC Genome Browser | Kent et al., 2002 | https://genome.ucsc.edu |
| Bedtools | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| R | R Development Core Team (2008) | https://www.r-proiect.org/ |
Highlights.
A lentiviral-based assay was developed to map SHM-susceptible regions of the genome
SHM susceptibility and SHM resistance are confined within TADs
Robust transcriptional activity does not explain SHM susceptibility
SHM targeting elements present in the genome likely help explain SHM susceptibility
ACKNOWLEDGMENTS
The authors thank Zdenek Cimburek for help with cell sorting and Richard Frock for instruction in the use of HTGTS. This work was supported in part by grant R01 AI127642 (D.G.S.), grant 15-24776S from the Czech Science Foundation (F.S.), Praemium Academiae of the Czech Academy of Science (J.H.), the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and the National Institutes of Health (NIH) (R.C.), a Gruber Science Fellowship and National Science Foundation Graduate Research Fellowship (R.D.), and grants from the Sigrid Juselius Foundation, the Jane and Aatos Erkko Foundation, the Jenny and Antti Wihuri Foundation, the Ella and Georg Ehrnrooth Foundation, the Cancer Society of South-West Finland, and the Emil Aaltonen Foundation (J.A.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.039.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aljanabi SM, and Martinez I (1997). Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 25, 4692–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez-Prado AF, Pérez-Durán P, Pérez-García A, Benguria A, Torroja C, de Yébenes VG, and Ramiro AR (2018).A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. J. Exp. Med 215, 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa H, Kudo H, Batrak V, Caldwell RB, Rieger MA, Ellwart JW, and Buerstedde JM (2008). Protein evolution by hypermutation and selection in the B cell line DT40. Nucleic Acids Res. 36, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, and Dalla-Favera R (2012). Roles of BCL6 in normal and transformed germinal center B cells. Immunol. Rev 247, 172–183. [DOI] [PubMed] [Google Scholar]
- Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, and Zhuang X (2018). Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagodatski A, Batrak V, Schmidl S, Schoetz U, Caldwell RB, Arakawa H, and Buerstedde JM (2009). A cis-acting diversification activator both necessary and sufficient for AID-mediated hypermutation. PLoS Genet. 5, e1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerstedde JM, Alinikula J, Arakawa H, McDonald JJ, and Schatz DG (2014). Targeting of somatic hypermutation by immunoglobulin enhancer and enhancer-like sequences. PLoS Biol. 12, e1001831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting KL, Soong TD, Singh R, Jiang Y, Béguelin W, Poloway DW, Swed BL, Hatzi K, Reisacher W, Teater M, et al. (2016). Multi-tiered Reorganization of the Genome during B Cell Affinity Maturation Anchored by a Germinal Center-Specific Locus Control Region. Immunity 45, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, and Di Noia JM (2016). Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat. Rev. Immunol 16, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. (2011). Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortés M, and Georgopoulos K (2004). Aiolos is required for the generation of high affinity bone marrow plasma cells responsible for long-term immunity. J. Exp. Med 199, 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, and Mirny L (2016). The 3D Genome as Moderator of Chromosomal Communication. Cell 164, 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia JM, and Neuberger MS (2007). Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem 76, 1–22. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen JM, Bilodeau S, Orlando DA, Hübner MR, Abraham BJ, Spector DL, and Young RA (2013). Multiple structural maintenance of chromosome complexes at transcriptional regulatory elements. Stem Cell Reports 1, 371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enervald E, Du L, Visnes T, Björkman A, Lindgren E, Wincent J, Borck G Colleaux L, Cormier-Daire V, van Gent DC, et al. (2013). A regulatory role for the cohesin loader NIPBL in nonhomologous end joining during immunoglobulin class switch recombination. J. Exp. Med 210, 2503–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, and Zhang Y (2011). Using MACS to identify peaks from ChIP-Seq data. Curr. Protoc. Bioinformatics Chapter 2, Unit 2.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman BM, and Samuels HH (1991). pEXPRESS: a family of expression vectors containing a single transcription unit active in prokaryotes, eukaryotes and in vitro. Gene 105, 9–15. [DOI] [PubMed] [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, and Alt FW (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol 33, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Zhu B, Cao X, Zhang M, and Wang X (2019). Roles of NIPBL in maintenance of genome stability. Semin. Cell Dev. Biol 90, 181–186. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017).A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz S (2006). Myc translocations in B cell and plasma cell neoplasms. DNA Repair (Amst.) 5, 1213–1224. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, and Kent WJ (2004). The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keim C, Kazadi D, Rothschild G, and Basu U (2013). Regulation of AID, the B-cell genome mutator. Genes Dev. 27, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodabakhshi AH, Morin RD, Fejes AP, Mungall AJ, Mungall KL, Bolger-Munro M, Johnson NA, Connors JM, Gascoyne RD, Marra MA, et al. (2012). Recurrent targets of aberrant somatic hypermutation in lymphoma. Oncotarget 3, 1308–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler KM, McDonald JJ, Duke JL, Arakawa H, Tan S, Kleinstein SH, Buerstedde JM, and Schatz DG (2012). Identification of core DNA elements that target somatic hypermutation. J. Immunol 189, 5314–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothapalli N, Norton DD, and Fugmann SD (2008). Cutting edge: a cis-acting DNA element targets AID-mediated sequence diversification to the chicken Ig light chain gene locus. J. Immunol 180, 2019–2023. [DOI] [PubMed] [Google Scholar]
- Kothapalli NR, Collura KM, Norton DD, and Fugmann SD (2011). Separation of mutational and transcriptional enhancers in Ig genes. J. Immunol 187, 3247–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger PH, and de Laat W (2016). Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol 17, 771–782. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Johnstone SE, and Young RA (2006). Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc 1, 729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T , Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, and Schatz DG (2009). Balancing AID and DNA repair during somatic hypermutation. Trends Immunol. 30, 173–181. [DOI] [PubMed] [Google Scholar]
- Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, and Schatz DG (2008). Two levels of protection for the B cell genome during somatic hypermutation. Nature 451, 841–845. [DOI] [PubMed] [Google Scholar]
- Matthews BJ, and Waxman DJ (2018). Computational prediction of CTCF/cohesin-based intra-TAD loops that insulate chromatin contacts and gene expression in mouse liver. eLife 7, e34077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul RW, Cao Z, Venkataraman L, Giorgetti CA, Press JL, Denizot Y, Du H, Sen R, and Gearhart PJ (2014). Spt5 accumulation at variable genes distinguishes somatic hypermutation in germinal center B cells from ex vivo-activated cells. J. Exp. Med 211, 2297–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JJ, Alinikula J, Buerstedde JM, and Schatz DG (2013). A critical context-dependent role for E boxes in the targeting of somatic hypermutation. J. Immunol 191, 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefferd AL, Kornepati AV, Bogerd HP, Kennedy EM, and Cullen BR (2015). Expression of CRISPR/Cas single guide RNAs using small tRNA promoters. RNA 21, 1683–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, Meyers RM, Amor C, Wasserman CR, Neuberg D, et al. (2014). Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell 159, 1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager M (2010). Ikaros in immune receptor signaling, lymphocyte differentiation, and function. FEBS Lett. 584, 4910–4914. [DOI] [PubMed] [Google Scholar]
- Merkenschlager M, and Nora EP (2016). CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu. Rev. Genomics Hum. Genet 17, 17–43. [DOI] [PubMed] [Google Scholar]
- Methot SP, and Di Noia JM (2017). Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Adv. Immunol 133, 37–87. [DOI] [PubMed] [Google Scholar]
- Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, Ecker JR, and Bushman FD (2004). Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2, E234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müschen M, Re D, Jungnickel B, Diehl V, Rajewsky K, and Küppers R (2000). Somatic mutation of the CD95 gene in human B cells as a side-effect of the germinal center reaction. J. Exp. Med 192, 1833–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narezkina A, Taganov KD, Litwin S, Stoyanova R, Hayashi J, Seeger C, Skalka AM, and Katz RA (2004). Genome-wide analyses of avian sarcoma virus integration sites. J. Virol 78, 11656–11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuebler J, Fudenberg G, Imakaev M, Abdennur N, and Mirny LA (2018). Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 115, E6697–E6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig A, and Nussenzweig MC (2010). Origin of chromosomal translocations in lymphoid cancer. Cell 141, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegard VH, and Schatz DG (2006). Targeting of somatic hypermutation. Nat. Rev. Immunol 6, 573–583. [DOI] [PubMed] [Google Scholar]
- Orthwein A, and Di Noia JM (2012). Activation induced deaminase: how much and where? Semin. Immunol 24, 246–254. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Migliazza A, Fracchiolla N, William C, Neri A, Baldini L, Chaganti RS, Klein U, Küppers R, Rajewsky K, and Dalla-Favera R (1998). BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc. Natl. Acad. Sci. USA 95, 11816–11821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Küppers R, and Dalla-Favera R (2001). Hypermutation of multiple proto-on-cogenes in B-cell diffuse large-cell lymphomas. Nature 412, 341–346. [DOI] [PubMed] [Google Scholar]
- Pavri R, and Nussenzweig MC (2011). AID targeting in antibody diversity. Adv. Immunol 110, 1–26. [DOI] [PubMed] [Google Scholar]
- Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, et al. (2010). Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 143, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, Economides AN, and Basu U (2014). Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature 514, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plachy J, Kotáb J, Divina P, Reinisová M, Senigl F, and Hejnar J (2010). Proviruses selected for high and stable expression of transduced genes accumulate in broadly transcribed genome areas. J. Virol 84, 4204–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, Liang G Tang Z, Mathé E, Benner C, et al. (2014). B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 159, 1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2008). R: A language and environment for statistical computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, Bolland DJ, Chen HT, Corcoran AE, Nussenzweig A, and Nussenzweig MC (2008). AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell 135, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouaud P, Vincent-Fabert C, Saintamand A, Fiancette R, Marquet M, Robert I, Reina-San-Martin B, Pinaud E, Cogné M, and Denizot Y (2013). The IgH 3’ regulatory region controls somatic hypermutation in germinal center B cells. J. Exp. Med 210, 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2016). The three-dimensional genome: principles and roles of long-distance interactions. Curr. Opin. Cell Biol 40, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz V, Butzhammer P, Hirsch B, Hecht J, Gütgemann I, Ehlers A, Lenze D, Oker E, Sommerfeld A, von der Wall E, et al. (2011). Deep sequencing of MYC DNA-binding sites in Burkitt lymphoma. PLoS One 6, e26837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senigl F, Auxt M, and Hejnar J (2012). Transcriptional provirus silencing as a crosstalk of de novo DNA methylation and epigenomic features at the integration site. Nucleic Acids Res. 40, 5298–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, and Cavalli G (2015). The role of chromosome domains in shaping the functional genome. Cell 760, 1049–1059. [DOI] [PubMed] [Google Scholar]
- Shen HM, Peters A, Baron B, Zhu X, and Storb U (1998). Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science 280, 1750–1752. [DOI] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, and Zhang F (2016). Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storb U (1996). The molecular basis of somatic hypermutation of immunoglobulin genes. Curr. Opin. Immunol 8, 206–214. [DOI] [PubMed] [Google Scholar]
- Sun J, Rothschild G, Pefanis E, and Basu U (2013). Transcriptional stalling in B-lymphocytes: a mechanism for antibody diversification and maintenance of genomic integrity. Transcription 4, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Claudepierre AS, Schiavo E, Heyer V, Fournier M, Page A, Robert I, and Reina-San-Martin B (2013). The cohesin complex regulates immunoglobulin class switch recombination. J. Exp. Med 210, 2495–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren AG, Mikkers H, Kool J, van der Weyden L, Lund AH, Wilson CH, Rance R, Jonkers J, van Lohuizen M, Berns A, and Adams DJ (2009). A high-throughput splinkerette-PCR method for the isolation and sequencing of retroviral insertion sites. Nat. Protoc 4, 789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg DLC, Azzarelli R, Oishi K, Martynoga B, Urbán N, Dekkers DHW, Demmers JA, and Guillemot F (2017). Nipbl Interacts with Zfp609 and the Integrator Complex to Regulate Cortical Neuron Migration. Neuron 93, 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, Huang SC, El Khattabi L, Dose M, Pruett N, et al. (2018). The Energetics and Physiological Impact of Cohesin Extrusion. Cell 173, 1165–1178.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visnes T, Giordano F, Kuznetsova A, Suja JA, Lander AD, Calof AL, and Ström L (2014). Localisation of the SMC loading complex Nipbl/Mau2 during mammalian meiotic prophase I. Chromosoma 123, 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Oliveira T, Jankovic M, Silva IT, Hakim O, Yao K, Gazumyan A, Mayer CT, Pavri R, Casellas R, et al. (2014). Epigenetic targeting of activation-induced cytidine deaminase. Proc. Natl. Acad. Sci. USA 111, 18667–18672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Su JH, Beliveau BJ, Bintu B, Moffitt JR, Wu CT, and Zhuang X (2016). Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AM, Maman Y, Alinikula J, and Schatz DG (2016). Bcl6 Is Required for Somatic Hypermutation and Gene Conversion in Chicken DT40 Cells. PLoS One 11, e0149146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, and Ren B (2017). The Three-Dimensional Organization of Mammalian Genomes. Annu. Rev. Cell Dev. Biol 33, 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin J, Franke V, van Ijcken WF, van der Sloot A, Krantz ID, van der Reijden MI, Nakato R, Lenhard B, and Wendt KS (2014). A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet. 10, e1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data, code and materials used in this study can be made available upon request to the corresponding authors. All datasets generated during this study are available at GEO: GSE139810.